

Abstract
Genomic instability is a major feature of neoplastic development in colorectal carcinoma and other cancers. Specific genomic instability events, such as deletions in chromosomes and other alterations in gene copy number, have potential utility as biologically relevant prognostic biomarkers. For example, genomic deletions on chromosome arm 18q are an indicator of colorectal carcinoma behavior and potentially useful as a prognostic indicator. Adapting a novel genomic technology called molecular inversion probes which can determine gene copy alterations, such as genomic deletions, we designed a set of probes to interrogate several hundred individual exons of >200 cancer genes with an overall distribution covering all chromosome arms. In addition, >100 probes were designed in close proximity of microsatellite markers on chromosome arm 18q. We analyzed a set of colorectal carcinoma cell lines and primary colorectal tumor samples for gene copy alterations and deletion mutations in exons. Based on clustering analysis, we distinguished the different categories of genomic instability among the colorectal cancer cell lines. Our analysis of primary tumors uncovered several distinct categories of colorectal carcinoma, each with specific patterns of 18q deletions and deletion mutations in specific genes. This finding has potential clinical ramifications given the application of 18q loss of heterozygosity events as a potential indicator for adjuvant treatment in stage II colorectal carcinoma.
Introduction
Genomic instability is a phenotype associated with the development of cancer in which preneoplastic and tumor cells accumulate various genetic changes, including mutations, large-scale genomic deletions, and amplifications of genomic regions (1). For example, genomic deletions of individual exons or larger regions represent one of the critical events of a two-hit model resulting in tumor suppressor inactivation (2). Traditionally, hemizygous genomic deletions have been detected using genetic microsatellite markers to identify a loss of a single allele with the other remaining intact, otherwise known as a loss of heterozygosity (LOH). Likewise, genomic amplifications also contribute to the development of cancer (2). For example, increases in gene copy number can result in overexpression of oncogenes, such as MYC, and initiate malignant proliferation (3). Specific genomic instability events have potential utility as prognostic biomarkers in the clinical management of certain malignancies, such as colorectal carcinoma (4–7).
In colorectal neoplastic development, genomic instability is a hallmark of malignant development and is broadly defined into two categories (2). One category is called chromosomal instability (CIN), the accumulation of significant genetic aberrations, such as loss of entire chromosome arms, large genomic deletions, insertions, and rearrangements (8). The other is characterized by the presence of microsatellite instability (MSI), a phenomenon whereby genetic mutations occur at significantly elevated rates in specific repetitive sequence tracts (9). MSI is a result of loss of DNA mismatch repair. For example, colorectal carcinomas exhibiting MSI typically accumulate 1- to 2-bp insertion and deletion mutations in genes, such as transforming growth factor receptor type II (TGFBR2), which is involved in colonic growth inhibition (10). The categories of CIN and MSI in colorectal carcinoma are considered to be distinct. Colorectal carcinoma tumors with MSI show significantly fewer chromosomal changes as determined by cytogenetics than CIN tumors (11). However, there is increasing evidence that tumors with MSI also exhibit genomic deletions, suggesting some degree of overlap with CIN (12, 13).
In CIN, genomic deletions in the 18q21 region are frequent and can affect cancer genes in the region, including the DCC gene (deleted in colorectal carcinoma gene) and SMAD2 and SMAD4 (mothers against decapentaplegic homologue 2 and 4; ref. 14). However, genomic deletions of SMAD2 or SMAD4 are not sufficient to account for the prominence of other 18q21 LOH events (15). This suggests that other candidate colorectal cancer genes may exist in the 18q21 region (16).
For stage II and III colorectal cancer patients, several retrospective studies have shown strong correlations between LOH events on chromosome arm 18q and reduced survival for patients with colorectal carcinoma (5–7). In contrast, other studies have failed to identify this correlation between 18q LOH and prognosis (19, 20).
Herein, we describe the novel application of molecular inversion probe (MIP) genomic technology for detecting genomic instability-related gene copy alterations in colorectal carcinoma cell lines and primary tumor samples (21, 22). A MIP is a single oligonucleotide that recognizes and hybridizes to a genomic DNA target with two inverted recognition sequences at the flanks (21). The total length of the genomic sequence that is queried ranges from 41 to 61 bp. After the probe specifically hybridizes to the target DNA, a single base-pair gap exists in the middle of the two recognition sequences. This gap can be either a single nucleotide polymorphism or a nonpolymorphic nucleotide. With a series of specific enzymatic steps and the addition of the appropriate nucleotide, a circle is formed. Incorporated into the MIP oligonucleotide is a unique sequence barcode tag. If the probe anneals to its specific complementary genomic sequence, undergoes the appropriate intramolecular rearrangement, and forms a circle, the barcode sequence can be queried via a microarray (21). This enables multiplexing to >10,000 individual probes per a reaction. MIP technology has significant advantages in terms of designing probes. One can use nearly any unique sequence and choose specific exons or other interesting sequences without the design constraints of other gene copy analysis platforms, such as array comparative genomic hybridization (CGH). In comparison with microsatellite genetic markers, MIPs are not dependent on having informative alleles to discern LOH events caused by genomic deletions. Expanding or refining a gene copy analysis for different genomic regions is simply a matter of designing new oligonucleotides and does not require custom design of a new microarray. This technology is particularly well-suited to identifying genomic deletion mutations at a very high resolution.
We adapted MIP technology to detect genomic deletions and measure other gene copy number alterations in cancer. For this initial study, a probe set was designed using sequences from the exons of a large number of cancer genes with an overall distribution covering all chromosome arms. An analysis was carried out on colorectal carcinoma cell lines and primary tumor samples from patients with stage II and III disease.
Materials and Methods
Genomic DNA
We obtained genomic DNA samples from the Coriell Institute for Medical Research (Camden, NJ; Na06985, Na06991, Na10838, Na12003, Na12004, Na12753, Na12762, Na12763, Na02623, Na01416, and Na06061). Additional normal genomic DNA samples were extracted from peripheral leukocytes. The study was approved previously by the institutional review board at Stanford University School of Medicine. Patients signed informed consent regarding their blood samples. Colorectal cancer cell lines (CACO2, COLO320, DLD1, HCT116, HCT15, LOVO, LS174T, LS180, NCIH508, NCIH747, RKO, SKCO1, SW1116, SW403, SW48, SW480, SW620, and SW837) were obtained from the American Type Culture Collection (Manassas, VA) and grown under the recommended conditions according to the manufacturer’s instructions. Genomic DNA was isolated from peripheral leukocytes using the Gentra genomic DNA preparation kit (Minneapolis, MN). Genomic DNA was isolated from cell lines using the Qiagen genomic DNA preparation kit (Valencia, CA). Colon tumor genomic DNA was obtained from frozen tumor specimens (Oncomatrix, Inc., San Diego, CA). Each sample was composed of >70% tumor tissue.
Designing the MIPs
Exon sequences of 228 cancer-related genes and genomic sequences encompassing 108 microsatellite genetic markers, which did not occur in within any known gene, were used in designing the genomic homology regions of the probes. To approximate the location of an individual microsatellite marker, the MIP sequence was selected within 100 nucleotides of an individual microsatellite sequence. For this application, we chose a 61-nucleotide nonpolymorphic sequence for each individual MIP with a “G” nucleotide in the middle. This sequence was divided into two 30-nucleotide inverted flanks of the probe. Repetitive genomic sequences, which had the potential to hybridize to multiple genomic locations, were eliminated using MEGABLAST (23). The exact nucleotide location and uniqueness of all the MIP sequences were confirmed using the National Center for Biotechnology Information Human Genome Build 35.1. To identify probes with optimal annealing, we eliminated those sequences that had extreme melting temperature >58°C. We also filtered out any genomic homology region sequences that could lead to potential self-annealing termini hairpin or primer-dimer structures. This was done by eliminating those genomic homology sequences that had complementary sequence, which could anneal to each other. The other components of the probe sequence (tag barcode and PCR primer sites) were subsequently added and the probes were produced by Parallele Bioscience (South San Francisco, CA). The initial number of probes was 972.
MIP assay
Methods for conducting the MIP assay and primer sequences are as described previously (21). In brief, for the initial annealing reaction, 400 ng genomic DNA, 12 amol of each of the 667 probes, 0.0625 units Ampligase (Epicentre, Madison, WI), and 0.5 units Stoffel fragment DNA polymerase (Applied Biosystems, Foster City, CA) in 9 μL of 20 mmol/L Tris-HCl (pH 8.3), 25 mmol/L KCl, 10 mmol/L MgCl2, 0.5 mmol/L NAD, and 0.01% Triton X-100 were incubated for 4 minutes at 20°C, 5 minutes at 95°C, and overnight at 58°C. To complete the gap-fill reaction, dGTP was added and incubated for 10 minutes at 60°C and then 1 minute at 37°C. Subsequently, 10 units exonuclease I and 200 units exonuclease III (U.S. Biochemical, Cleveland, OH) were added and the mixture was incubated for 14 minutes at 37°C, 2 minutes at 95°C, and 1 minute at 37°C. Following exonucleolysis, the reaction was subjected to uracil depurination and cleavage with 2 units uracil-N-glycosylase (New England Biolabs, Ipswich, MA) in 25 μL of 1.6 mmol/L MgCl2, 10 mmol/L Tris-HCl (pH 8.3), and 50 mmol/L KCl and incubated for 9 minutes at 37°C and 20 minutes at 95°C. To amplify the barcode, 2 units AmpliTaq Gold (Applied Biosystems), 16 pmol primer 1 (5′-CCGAATAGGAACGTTGAGCCGT-3′), and 16 pmol primer 2 (5′-GCAAATGTTATCGAGGTCCGGC-3′) in 25 μL of 1.6 mmol/L MgCl2, 10 mmol/L Tris-HCl (pH 8.3), 50 mmol/L KCl, and 112 μmol/L deoxynucleotide triphosphate (dNTP) were added to the genotyping reactions.
The reactions were amplified in 28 cycles of 95°C for 20 seconds, 65°C for 45 seconds, and 72°C for 10 seconds. Additional barcode processing required adding 20 units exonuclease I and 10 units HaeIII (New England Biolabs) to 60 μL of each amplification product at 37°C for 1 hour and 80°C for 30 minutes.
Microarray hybridization
For hybridization to a barcode array (Truetage 5K, Affymetrix, Santa Clara, CA), 90 μL of the barcode product were added. Staining with streptavidin-phycoerythrin was carried out as per the manufacturer’s recommendations using the MegaAllele genotyping kit (Parallele Bioscience). Barcode arrays were washed as recommended by the manufacturer. Microarray images were analyzed using an Agilent (Palo Alto, CA) scanner at a wavelength of 570 nm.
PCR genotyping
For PCR confirmation of gene copy alterations, 10 ng genomic DNA from each sample (10 ng) was added to a PCR solution containing a final concentration of 2 μmol/L MgSO4, 200 μmol/L dNTP, 0.2 units AmpliTaq Gold, and a primer concentration of 20 nmol/L. Primers used include AFM074ZA9 (5′-TGGCTCTCTGAATATGACCTT-3′ and 5′-AGTTTAAAACCTGTAGGGGCT-3′), D18S500 (5′-CATGAGGTTATAGTGTACGAAGG-3′ and 5′-AGTTTCCTCTTTGCTGTTCATA-3′), and D18S8 (5′-TTGCACCATGCTGAAGATTGT-3′ and 5′-ACCCTCCCCCTGATGACTTA-3′). The cycles of the PCR were an initial incubation for 10 minutes followed by 35 cycles at 95°C, 55°C for 1 minute, and 72°C for 1 minute. The final step was at 72°C for 7 minutes. PCR products were analyzed on 4% Metaphor agarose gel (Cambrex, Rockland, ME) to resolve different alleles.
Data analysis
The Affymetrix Microarray Suite software generated the data files for signal intensities of each probe tag feature. Scaling normalization was conducted on the raw intensity data from the arrays. After normalization, the signal intensity values obtained from analysis of 21 normal genomic samples were averaged to obtain a mean and SD for each probe at diploid copy number. The gene copy ratio was calculated by dividing the observed signal by the mean signal from normal diploid samples. A log 2 transformation was used for all subsequent plotting and statistical analysis.
To characterize the performance of the probes, we analyzed 21 normal genomic DNA samples as a control for normal diploid gene copy number. For each probe, the mean signal intensity was determined from the data set generated from all of the normal samples. Data from the analysis of normal samples were used for rejection of outlier probes based on the following: (a) probe failure due to corresponding low signal intensity and (b) high variance. Probes that showed mean average signal intensity less than the mean of background plus 3 SDs were designated as failed (<600 signal intensity). For each probe, the coefficient of variation was calculated by dividing the SD with the average signal intensity. If an individual probe showed a coefficient of variation of >0.75, it was eliminated. The 667 probes used in this analysis passed all of these performance criteria. This represented 69% of the probes that were originally designed.
For additional genomic data and statistical analysis, we used the R-Bioconductor package. The Cluster along Chromosomes (CLAC) algorithm was modified for analyzing MIP data for significant gene copy changes from both the cell line and the primary colorectal cancer samples (24). CLAC creates a hierarchical cluster based on the fixed physical chromosomal order of each MIP. Significance of gains and losses are determined by applying a null distribution based on the data set generated from the normal genomic DNA samples. For MIP analysis, the false discovery rate (FDR) was set at 0.01. Given the relatively narrow region of genomic DNA that each MIP interrogates, no smoothing function was used in the analysis of the data. For our MIP-specific modification of the CLAC algorithm, this translated in using a window size of 1.
After CLAC analysis, hierarchical clustering was done on the separate MIP data sets from the cancer cell lines and colorectal cancer samples. Dissimilarities between cell lines primary tumor samples were calculated using Euclidean distances (root sum-of-squares of differences), and linkage was established by the average method (25). Clustering results were displayed as a heat map, graphically depicting the relationship among the cell lines and tumor samples based on the log 2 ratio data as well as displaying the data on a green (losses) versus red (gains) color scale. The statistical significance of cluster assignment versus pathologic or clinical characteristics was conducted using two-sided Fisher’s exact test and the Student’s t test when appropriate.
Results
Design of MIPs for cancer
A total of 667 MIPs were used for this analysis. The majority of probe sequences were derived from the exon sequences of 227 genes involved in cancer and other cellular processes. For this application, we chose nonpolymorphic sequences. Overall, 516 different exons are represented. Many of these genes directly contribute to colorectal cancer development, such as TP53 and MSH2. As a control, several genes were chosen in which gene copy number changes have been measured previously in cancer cell lines or other samples through quantitative PCR or array CGH. To investigate gene copy alterations, such as genomic deletions on chromosome arm 18q, we designed 106 probes to approximate the location of microsatellite genetic markers, otherwise called sequence tandem repeats. We selected a group of markers on chromosome 18 (d18s51, d18s535, d18s5474, d18s55, d18s57, d18s58, d18s61, d18s64, d18s65, d18s67, and d18s70) that were used in clinical studies of 18q LOH events (5, 7, 20, 26, 27). Additional chromosome 18 markers were included to increase the average density to one MIP per 711 kb. The remaining probes are located within 1 kb of a known gene.
Standard curve
As a quantitative measurement of the assay performance, we analyzed genomic DNA obtained from individuals with increasing numbers of X chromosomes. Using the probes specific for the X chromosome, we measured copy number from normal male genomic DNA (X), normal female genomic DNA (2X), and individuals showing chromosomal aberrations, including trisomy (3X), tetrasomy (4X), and pentasomy (5X). Relying on the data subset of normal control samples from females, the mean log 2 ratio was calculated for the four MIPs on the X chromosome (Fig. 1). The experimental log 2 ratio was plotted in comparison with the theoretical value. A regression correlation of 0.97 was shown.
Figure 1.
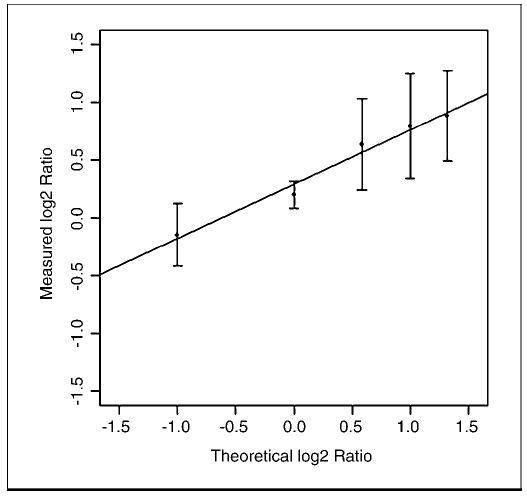
Standard curve for MIP quantitation using X chromosome variants. Genomic DNA samples were analyzed, which had different copy numbers of the X chromosome. Using the average intensity determined from normal female genomic DNA samples, the mean log 2 ratio was calculated for the four MIPs on the X chromosome. Y axis, measured log 2 ratio; X axis, theoretical log 2 ratio based on the known X chromosome variant.
Analysis of known gene copy number alterations
To assess quantitative detection of gene copy changes, we analyzed several DNA samples and cancer cell lines with known genomic deletions and amplifications that are listed in Table 1. The log 2 ratio values generally agreed with the predicted gene copy number change of known genomic deletion events and were elevated for amplifications. For example, homozygous genomic deletions had a negative log 2 ratio value exceeding 3 SDs for a given probe. The 17q12-q21 amplification in the breast cancer cell line BT474 has been characterized through fluorescence in situ hybridization (FISH), quantitative PCR, and array CGH methods (28–30). This amplification contains the gene ERBB2, also known as HER-2/neu. We designed nine probes, each representing separate exons of the gene ERBB2. We analyzed genomic DNA from BT474, averaged the value from all ERBB2 probes, and showed an increased gene copy number. The colorectal cancer cell line COLO320 contains a gene amplification of the gene MYC, also known as c-Myc or v-myc myelocytomatosis viral oncogene homologue (31). A variety of methods, including array CGH, have confirmed the MYC gene amplification (29, 32). We designed several probes specific to MYC. Analysis of COLO320 showed that the MYC gene amplification could be readily detected. The colorectal cancer cell line LOVO contains a homozygous gene deletion of MSH2, which involves exons 4 to 8, as determined by exon-specific PCR and other methods (33, 34). Gene copy alterations of 8 exons of MSH2 were measured. Probes representing exons 5 and 8 showed log 2 ratios below the theoretical copy number change for a homozygous deletion, whereas the flanking probes showed a diploid copy number. At the loci 17p13.1, the lung cancer cell line NCIH358 has a highly characterized 120-kb hemizygous deletion and, within this interval, a homozygous deletion of TP53 (35). MIPs were designed to represent the exons 1, 4, 6, 7, 9, and 11 from TP53. Three replicates of gene copy analysis were carried out on this sample. Analysis with the cancer probe set readily discriminated the homozygous deletion in exons 4, 6, 7, 9, and 11. We analyzed genomic DNA from an individual (Na16447) with a hemizygous 18q terminal deletion identified previously via karyotyping and FISH using λ phage clones that mapped to the 18q22.1 (36). This 18q deletion was used as a control for detecting hemizygous genomic deletions. This is analogous to LOH in tumors. Overall, our mapping of the deletion corresponded to the deletion identified previously with traditional microsatellite marker genotyping. The deletion was found to be distal of the probe approximating the location of the microsatellite marker D18S1269, which is located within the 18q22.1 cytogenetic region. All probes telomeric of this probe showed a hemizygous deletion.
Table 1.
Analysis of known genomic deletions and amplifications from cancer cell lines and other samples
| Amplifications | |||||
|---|---|---|---|---|---|
| Cell line | Gene | No. probes | Log 2 ratio average | SD | Estimated gene copy number |
| BT474 | ERBB2 | 9 | 1.28 | 0.42 | 4.85 |
| COLO320 | MYC | 3 | 2.42 | 0.61 | 10.69 |
| Deletions | |||||
|---|---|---|---|---|---|
| Cell line | Gene | Exon | Log 2 ratio | SD | Estimated gene copy number |
| LOVO | MSH2 | 1 | 0.05 | 0.36 | 2.06 |
| 5 | −3.41 | 0.85 | 0.19* | ||
| 8 | −4.17 | 0.73 | 0.11* | ||
| 9 | −0.13 | 0.16 | 1.83 | ||
| 11 | 0.05 | 0.23 | 2.07 | ||
| 12 | 0.06 | 0.43 | 2.08 | ||
| 15 | 0.16 | 0.61 | 2.24 | ||
| 16 | 0.54 | 0.80 | 2.91 | ||
| NCIH358 | TP53 | 1 | 0.46 | 0.34 | 2.76 |
| 4 | −4.01 | 0.00 | 0.12* | ||
| 6 | −3.80 | 0.79 | 0.14* | ||
| 7 | −5.06 | 0.59 | 0.06* | ||
| 9 | −5.62 | 1.00 | 0.04* | ||
| 11 | −5.82 | 0.47 | 0.04* | ||
| Sample | Ideogram interval | No. probes | Log 2 ratio | SD | Estimated gene copy number |
| Na16447 | 18q22.1– telomere of 18q | 12 | −0.91 | 0.74 | 1.06 |
Values less than 0.2 indicate a homozygous genomic deletion.
Analysis of colorectal cancer cell lines
We analyzed 18 colorectal cancer cell lines, 16 of which had been characterized previously for the presence of either MSI (LOVO, LS180, LS174T, RKO, DLD1, HCT15, HCT116, and SW48) or CIN (COLO320, SW837, SW403, CACO2, SW480, SW1116, SW620, and SKCO1; Fig. 2A; refs. 12, 37–40). Among these cell lines were LOVO and COLO320, which were employed as positive controls. Colorectal cancer cell lines were designated as CIN if there was evidence of aneuploidy via karyotyping, aneuploidy via FISH, or two or more LOH events without evidence of MSI. The cell lines NCIH508 and NCIH747 were characterized previously as microsatellite stable, but there were no published data relevant to their CIN status (41).
Figure 2.
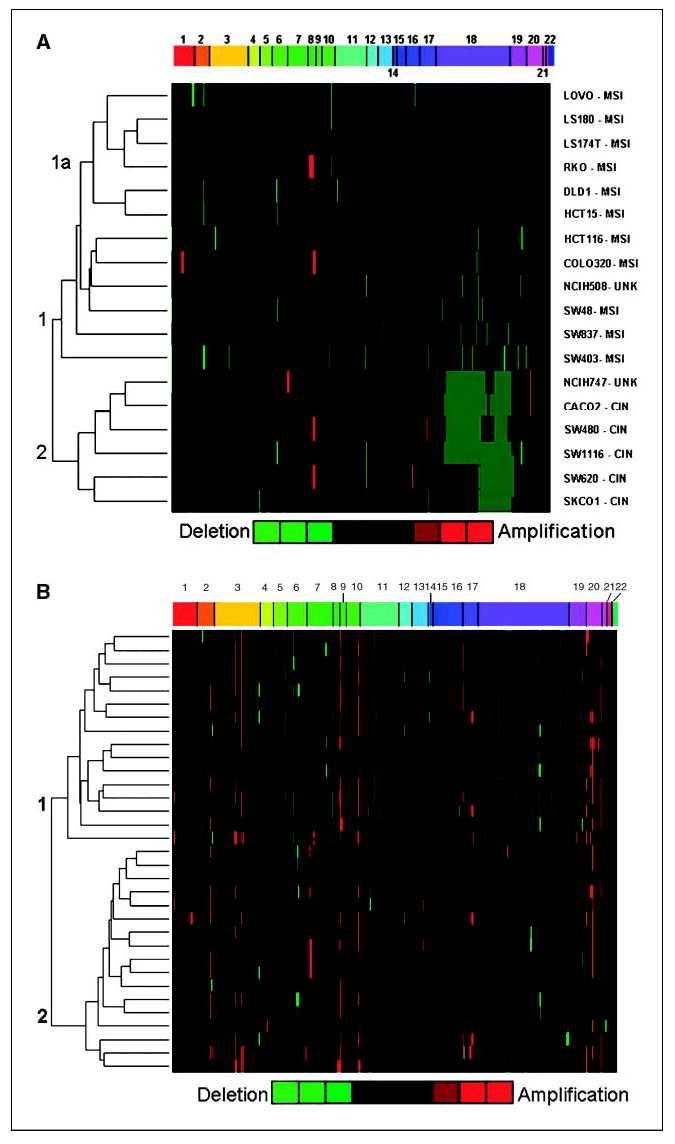
Heat map of cluster from significant gene copy alterations in colorectal cancer cell lines and carcinoma samples. Unsupervised hierarchical clustering was done on the data set of significant gene copy regions as determined by CLAC. The dendrogram from the clustering algorithm was used to generate a heat map, graphically depicting the relationship among the colorectal cancer cell lines. The heat map of the data uses a green (losses) versus red (gains) color scale. Each column represents an individual MIP arranged by chromosome and physical location in increasing order from left to right. A, each row represents a separate colorectal cancer cell line. The two topmost branches are designated as clusters 1 and 2. A subcluster branch representing MSI cell lines is labeled as 1a. B, each row represents a separate colorectal carcinoma sample. The two topmost clusters are labeled as clusters 1 and 2.
The log 2 ratio of each MIP was calculated as described previously. To determine the statistical significance of log 2 ratio data, a modified version of the CLAC algorithm was chosen for analyzing MIP data (24). CLAC creates a hierarchical cluster based on the fixed physical chromosomal order of each MIP. Significance of gains and losses are determined by applying a null distribution based on the data set generated from the normal genomic DNA samples. CLAC reports significant regions as the average of the log 2 ratio of the affected interval. CLAC was able to detect known gene copy alterations in our positive controls. Several methods to analyze array CGH data have been published, which are applicable to the MIP data set (42). However, we chose CLAC because it is one of the few approaches that employ a FDR for calling significant gene copy alterations (42). Hierarchical clustering analysis was done on the data set of significant gene copy regions as determined by CLAC. The clustering was displayed as a heat map, graphically depicting the relationship among the colorectal cancer cell lines (Fig. 2A). Individual rows represent separate samples. The columns represent probes, ordered based on chromosome number and nucleotide location, thus facilitating the identification of gene copy changes in contiguous probes.
Hierarchical clustering revealed two first-level branches, which we designated as cluster 1 and 2. Cluster 1 contained 12 cell lines and cluster 2 contained 6 cell lines. Among the cluster 1 cell lines, 8 were confirmed to have MSI, 3 had molecular features consistent with CIN, and 1 had no previous classification per our review of the literature (Fig. 2A). Within cluster 1, a second level branch, designated as subcluster 1a, contained 6 cell lines, all exclusively exhibit MSI. For cluster 2, 5 of the colon cancer cell lines were confirmed to have CIN using the previously mentioned molecular criteria and one cell line (NCIH747) had no previous classification but was known to be microsatellite stable. The genomic instability status (CIN versus MSI) of the cell lines was used to determine whether there was an association between the two major cluster groups. A Fisher’s exact test was used to determine if there was a statistically significant association between cluster designation (1 or 2) and genomic instability status (CIN and MSI). The clustering showed a statistically significant association with genomic instability status with a two-tailed P of 0.0256. Specifically, cluster 1 was associated with MSI and cluster 2 was associated with CIN.
For each cluster, we identified the highest frequency gene copy changes among the affected samples, which are listed in Table 2. Summing the number of affected probes, cluster 2 (CIN) was significantly higher with an average of 105.2 probes with altered gene copy changes per cell line as opposed to cluster 1 (MSI) with 16.5 per cell line. A larger genomic interval was assumed if contiguous probes with the same gene copy alteration fulfilled the following criteria: (a) the interval between probes was <10 kb, (b) the same gene was affected, or (c) there were three or more probes showing the same gene copy alteration. Minimal common deletion intervals were determined by identifying the deleted contiguous probes and represented among the highest number of samples.
Table 2.
Gene copy alterations from colorectal cancer cell lines and samples
| Cluster 1 frequency (%) | Cluster 2 frequency (%) | No. probes | Chromosome | Nucleotide | Ideogram | Genes or genetic markers | |
|---|---|---|---|---|---|---|---|
| Cell line deletions | |||||||
| 33.0 | 0.0 | 1 | 2 | 216062244 | — | 2q35 | FN1 |
| 33.0 | 0.0 | 1 | 2 | 216882889 | — | 2q35 | XRCC5 |
| 33.0 | 0.0 | 1 | 10 | 104582361 | — | 10q24.32 | CYP17A1 |
| 0.0 | 100.0 | 5 | 18 | 48943142 | 49190846 | 18q21.2 | DCC |
| 50.0 | 50.0 | 1 | 18 | 54383666 | — | 18q21.31 | D18S881 |
| 50.0 | 50.0 | 1 | 18 | 54850718 | — | 18q21.32 | D18S531 |
| 0.0 | 100.0 | 27 | 18 | 59194458 | 70195190 | 18q21.33-18q22.3 | VPS4B, SERPINB12, SERPINB13, SERPINB4, SERPINB3, SERPINB7, SERPINB2, SERPINB10, SERPINB8, CDH7, CDH19, TXNDC10, CD226, RTTN, SOCS6, GTSCR1, CBLN2, NETO1, FBXO15, CYB5 |
| Cell line amplifications | |||||||
| 16.7 | 16.7 | 1 | 8 | 102034675 | — | 8q22.3 | YWHAZ |
| 16.7 | 16.7 | 1 | 8 | 128820024 | — | 8q24.21 | MYC |
| 0.0 | 33.3 | 2 | 20 | 45684415 | 45686101 | 20q13.12 | NCOA3 |
| Primary tumor deletions | |||||||
| 94.0 | 0.0 | 1 | 5 | 133685387 | — | 5q31.1 | CDKL3 |
| 94.0 | 0.0 | 1 | 5 | 137921004 | — | 5q31.2 | HSPA9B |
| 94.0 | 12.0 | 1 | 6 | 57154822 | — | 6p12.1 | BAG2 |
| 0.0 | 35.3 | 2 | 6 | 144323338 | 144329507 | 6q24.2 | PLAG1 |
| 94.0 | 12.0 | 1 | 8 | 31058227 | — | 8p12 | WRN |
| 62.0 | 0.0 | 1 | 11 | 77456756 | — | 11q14.1 | THRSP |
| 62.0 | 0.0 | 1 | 11 | 101725901 | — | 11q22.2 | BIRC2 |
| 62.0 | 0.0 | 1 | 11 | 101744386 | — | 11q22.2 | BIRC2 |
| 94.0 | 6.0 | 1 | 18 | 27126956 | — | 18cen | D18S521 |
| 94.0 | 6.0 | 1 | 18 | 28078724 | — | 18q12.1 | D18S518 |
| 94.0 | 6.0 | 14 | 18 | 64574936 | 70195190 | 18q12.2-18q21.1 | NPM1P1, PIK3C3, RIT2SYT4, SETBP1, SLC14A2, SLC14A1, CD33L3, KIAA1632, PSTPIP2 |
| 94.0 | 24.0 | 1 | 18 | 47940111 | — | 18q21.1 | D18S89 |
| 94.0 | 24.0 | 1 | 18 | 48085326 | — | 18q21.1 | D18S88 |
| 62.5 | 35.3 | 1 | 18 | 54383666 | — | 18q21.31 | D18S881 |
| 62.5 | 35.3 | 1 | 18 | 54850718 | — | 18q21.32 | D18S531 |
| 100.0 | 6.0 | 4 | 18 | 66883827 | 68156526 | 18q22.2-18q22.3 | Q14754_HUMAN, ENSG00000196512 |
| Primary tumors amplifications | |||||||
| 25.0 | 53.0 | 1 | 2 | 113305137 | — | 2q13 | ILB1 |
| 25.0 | 53.0 | 1 | 2 | 191688469 | — | 2q32.2 | STAT1 |
| 56.0 | 35.0 | 1 | 3 | 41255650 | — | 3p21 | CTNNB1 |
| 56.0 | 35.0 | 1 | 3 | 47428610 | — | 3p12.31 | PTPN23 |
| 56.0 | 35.0 | 2 | 3 | 52410179 | 52414177 | 3p21.31-p21.2 | BAP1 |
| 87.5 | 35.3 | 1 | 8 | 102034675 | — | 8q22.3 | YWHAZ |
| 87.5 | 35.3 | 1 | 8 | 128820024 | — | 8q24.21 | MYC |
| 75.0 | 47.0 | 2 | 10 | 104154700 | 104155176 | 10q24 | PSD |
| 24.0 | 56.3 | 1 | 20 | 44075483 | — | 20q13.12 | MMP9 |
| 24.0 | 56.3 | 2 | 20 | 45684415 | 45686101 | 20q13.12 | NCOA3 |
| 58.0 | 6.0 | 2 | 20 | 48891730 | 48926584 | 20q13.13 | BCAS4 |
Unique to cluster 1 were three probes that showed hemizygous deletions. These deletions were not present in cluster 2. On chromosome 2, one probe was located in exon 45 of the gene FN1 and the other was located in exon 1 of XRCC5; on chromosome 10, a probe for exon 6 of CYP17A1 was deleted. Another cluster 1 deletion pattern found in 50% of the cell lines was located on chromosome 18, and involved two adjacent probes representing microsatellite markers. This deletion pattern was also found in cluster 2. Known genes between these microsatellite markers include ALPK2, an α-kinase, MALT1, involved in activation of nuclear factor-κB–inducing kinase, and ZNF532, a nucleic acid–binding protein. The remaining deletions in cluster 1 were generally unique to specific cell lines. Among the cluster 1 cell lines, several amplifications were observed, including MYC, in RKO and COLO320. In general, gene amplifications were specific to an individual cell line and no prominent patterns appeared frequently within either cluster.
Cluster 2 was associated with CIN. Unique to cluster 2, two minimal common deletion intervals occurred on 18q where adjacent probes showed hemizygous deletions. One of these intervals contains the gene DCC, which has been implicated in the development of colon cancer by numerous LOH studies. Multiple genes were contained in the chromosome 18 nucleotide interval of 59295188 to 69976639. Among the cluster 2 cell lines, several amplifications were observed, including MYC, which was observed in cell lines SW480 and SW620.
Analysis of colorectal cancer samples
We analyzed 33 primary colorectal adenocarcinoma samples from patients with stage II and III disease. Through previous molecular characterization, the tumors were all found to be microsatellite stable, thus lacking MSI. The clinical and pathologic characteristics are summarized in Table 3. The log 2 ratio was calculated and a CLAC analysis was conducted to determine significant gene copy changes. Hierarchical clustering revealed two distinct first level clusters, called 1 and 2, among the primary colorectal carcinoma samples (Fig. 2B). Sixteen of the colorectal samples were in cluster 1 and 17 were in cluster 2. We focused on the highest frequency gene copy changes that occurred in individual clusters, which are detailed in Table 2. The frequency of deletions and amplifications were plotted between clusters 1 and 2 (Fig. 3). Summing the number of affected probes, cluster 1 had a significantly higher number of gene copy alterations with an average of 102.4 per sample compared with cluster 2, which had 27 per sample. Hemizygous deletions in chromosome 18 for the colorectal samples were confirmed by PCR-based genotyping. We used primers specific for the microsatellite markers D18S8, D18S500, and AFM074ZA9 (data not shown). Genotyping-matched normal and tumor pairs with these microsatellite markers showed the presence of one allele, confirming the presence of hemizygous deletions on chromosome 18. Multiple gene amplifications were detected on chromosomes 2, 3, 8, 10, and 20 and occurred in both clusters. As with the cell lines, we determined the minimal common interval in each cluster by identifying those single or contiguous probes that showed a genomic deletion or other alteration among the highest number of samples within a cluster (Table 2).
Table 3.
Clinical features and analysis of colorectal carcinoma samples
| Clinical feature | Total (33) | Cluster 1 (16) | Cluster 2 (15) | P (statistic) |
|---|---|---|---|---|
| Stage | ||||
| II | 19 | 11 | 8 | 0.296 (Fisher’s two-tailed) |
| III | 14 | 5 | 9 | |
| Location | ||||
| Ascending colon | 6 | 3 | 3 | 1.000 (Fisher’s two-tailed) |
| Descending colon | 27 | 13 | 14 | |
| Sex | ||||
| Male | 21 | 10 | 11 | 1.000 (Fisher’s two-tailed) |
| Female | 12 | 6 | 6 | |
| Age | ||||
| Average (y) | 64.55 | 67.50 | 61.76 | 0.118 (unpaired t test) |
| SD | 10.01 | 10.46 | ||
| Size | ||||
| cm | 4.66 | 4.56 | 4.77 | 0.728 (unpaired t test) |
| SD | 1.60 | 1.64 | ||
| Differentiation | ||||
| Moderate | 26 | 10 | 16 | 0.039 (Fisher’s two-tailed) |
| Poor | 7 | 6 | 1 | |
| Level of invasion | ||||
| Serosal | 17 | 12 | 5 | 0.015 (Fisher’s two-tailed) |
| Pericolic | 16 | 4 | 12 | |
Figure 3.
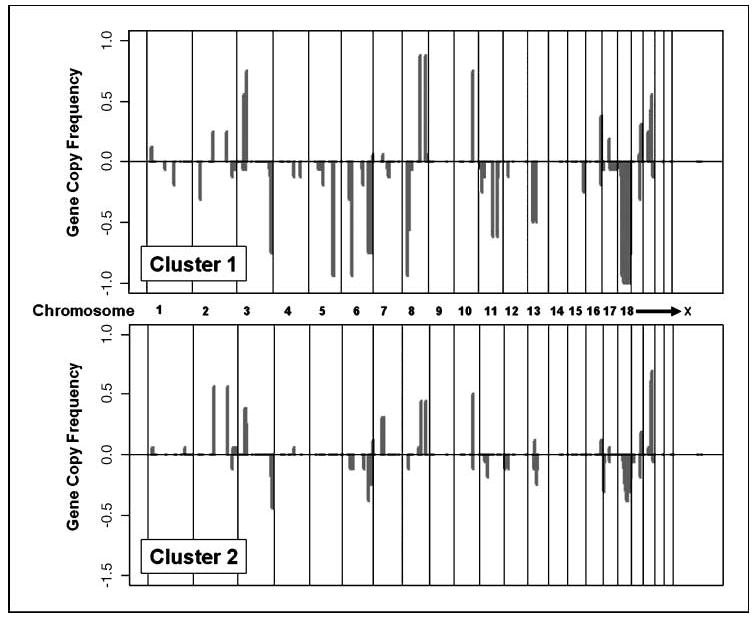
Overall frequency of deletions and amplifications of clusters 1 and 2 classification of primary colorectal carcinomas. The gene copy frequency between cluster 1 and 2 is separately plotted. Y axis, overall frequency of gene copy changes for individual MIP; X axis, location of probe as arranged by chromosome and their nucleotide location.
CRC samples: cluster 1
Cluster 1 was characterized by several minimal common deletions on chromosomes 5, 6, 11, and 18. These hemizygous genomic deletions were either not present or in low frequency in cluster 2. Cluster 1 gene deletions included exon 5 of CDKL3, exon 14 of HSPA9B, exon 2 of BAG2, and exon 9 of WRN. Particularly interesting were two minimal common deletion intervals on chromosome 18, which were found in >90% of the cluster 1 tumors but in <10% of the cluster 2 tumors. One of these intervals was located in 18q12.3 region and mapped to nucleotides spanning 34158149 to 41897329. Two probes representing 18q clinical microsatellite markers (d18s535 and d18s65) are within this deletion interval. A large number of genes are in this region, including PIK3C3, a phosphatidylinositol 3-kinase, and several genes involved in GTPase-mediated signal transduction. The other 18q hemizygous deletion interval involved the 18q 22.2 region. Two probes representing 18q clinical microsatellite markers (18s58 and d18s61) are within this second minimal deletion interval. The genes SMAD2 and SMAD4 have been implicated in colorectal cancer involvement. One probe representing exon 11 of SMAD2 was deleted in 25% of cluster 1 colorectal carcinomas but not in cluster 2 samples. A probe mapping to exon 3 of SMAD4 was found to be deleted in 50% of cluster 1 samples and 25% of samples in cluster 2.
Several probes showed gene amplifications that were found in both clusters. For cluster 1, the highest frequency amplification involved two adjacent probes located in exon 1 of YWHAZ and exon 2 of MYC on chromosome 8. Given the significant distance between these probes, they may represent separate amplicons. For cluster 2, the second most frequent amplification involved two adjacent probes located in exons 13 and 15 of PSD in chromosome region 10q24.
CRC samples: cluster 2
The most frequent hemizygous deletion unique to cluster 2 occurred in exons 3 and 4 of PLAGL1. The other frequent hemizygous deletion involved two probes on chromosome 18 representing microsatellite markers D18S881 and D18S531. These deletions overlapped with deletions in the colorectal cancer cell line. For cluster 2, the most frequent amplification involved three contiguous probes mapping to ideogram region 20q13. Multiple genes exist in this interval, including multiple transcription regulatory genes, such as ZNF334, NCOA5, and NCOA3 (43). The 20q region was notable for an additional gene amplification that occurred at lower frequency. The second most frequent amplification was noted in 53% of tumors involving two probes on chromosome 2, ideogram region 2q13-2q32.2, and spanning nucleotides 113305137 and 191688469. This interval contains the genes IL1B and STAT1.
Clinical correlation
We explored the relationship of tumor phenotype with the two major cluster designations (Table 3). Among the clinical and pathologic variables examined, an association was observed with the cluster designation and differentiation of the tumor (P < 0.05). Cluster 2 was associated with moderately differentiated adenocarcinoma, whereas cluster 1 was associated with poorly differentiated adenocarcinoma. Level of invasion also showed a correlation. Serosal level associated with cluster 1 and pericolic invasion associated with cluster 2 (P < 0.02).
Discussion
In this study, we examined genomic instability in colorectal carcinoma cell lines and primary samples applying a novel genomic technology using MIPs. For this study, sequences from 512 exons representing 227 cancer genes were examined for genomic deletions and other gene copy changes, such as gene amplifications. In addition, we designed a set of MIPs, which query sequences within 100 bp of microsatellite genetic markers used previously for LOH studies. We focused on the chromosome arm 18q given its clinical significance in colorectal carcinoma. In our review of the current literature of the prognostic value of 18q LOH events in colorectal carcinoma, only several microsatellite markers were used consistently among studies. The lack of a common set of microsatellite markers routinely employed from study to study prevents comparison of these results unless one assumes that a complete loss of the 18q arm has occurred. Furthermore, these individual markers often span an enormous gap of genomic sequence (in megabases). We used MIPs to refine the analysis of chromosome arm 18q deletions and incorporated the locations of multiple genetic microsatellite markers that have been used in various clinical studies of 18q LOH in colorectal cancer.
Our data show that MIP technology can quantitatively detect genomic deletion mutations and other gene copy alterations. The quantitative ability of these probes has been confirmed in a separate study (22). MIP gene copy quantitation was able to resolve hemizygous and homozygous deletions at exon resolution level as shown using samples with known genomic deletions. For example, in the cell line LOVO, we precisely identified a MSH2 homozygous deletion mutation of exons 4 to 8. MIP analysis was able to identify the ERBB2 gene amplification in the breast cancer cell line BT474 and the MYC gene amplification in the colon cancer cell line of COLO320. However, MIPs underestimated the overall gene copy number; thus, it is a semiquantitative method in this regard. This limitation is partly a result of the scaling normalization that was applied to the raw signal intensity data sets. In the future, we will improve the methods of normalization. MIPs have several advantages compared with array CGH. There are fewer constraints on the composition of the probe, whereas array CGH probes are specifically selected to perform optimally under one set of hybridization conditions. This constrains the type of sequences that can be placed on an array. In comparison, MIPs have significantly fewer constraints in regards to their composition. For example, multiple nonrepetitive sequences from any exon sequence could be represented without the limitations of the sequence annealing properties not performing well under a standard array CGH condition.
To investigate genomic instability in colorectal carcinoma, we analyzed a panel of 18 colorectal cancer cell lines to detect deletion mutations and measure other gene copy changes. In general, we were able to distinguish different categories of genomic instability based on our analysis of gene copy alterations and subsequent hierarchical clustering of the gene copy data. These results suggest that CIN category can be classified based on a quantitative scale using the frequency of deletion mutations and alterations in gene copy.
The two topmost clusters distinguished MSI (cluster 1) versus CIN (cluster 2) cell lines. There were several prominent gene copy changes that were specific for individual clusters and are thus associated with specific forms of genomic instability. Cluster 2 had two common minimal deletions regions located on chromosome arm 18q. One of the common deletion regions includes the gene DCC, which has been the subject of considerable research regarding its role in colorectal carcinoma development. Initially identified as a tumor suppressor, DCC was discovered to play a role as a receptor for the ligand Netrin-1, which mediates neuronal navigation during nervous system development (44). Given its role in neuronal embryonic development, the possibility of dual roles of DCC was difficult to reconcile. However, DCC belongs to a family of receptors that can induce apoptosis in the absence of its ligand. This function would support a role as a tumor suppressor. Within the second common minimal interval, a large of number of potential cancer genes exist, including several members of the cadherin gene family, which are involved in cell adhesion. Interestingly, deletions in an overlapping 4.9-Mb region of 18q have been implicated in the development of head and neck squamous cell carcinoma, although no candidate cancer genes have been identified (45). These lines of evidence support the existence of an important cancer gene in this region. For future studies, our efforts will focus on characterizing this region in greater detail and delineating a smaller interval to facilitate the discovery of candidate cancer genes.
We analyzed a cohort of MSI-negative colorectal carcinoma primary tumors from stage II and II patients. Using unsupervised clustering analysis, we discovered distinct categories of microsatellite stable colorectal carcinomas based on their profile of genomic deletions. Cluster 1 was remarkable for having a high number of deletions throughout the genome, consistent with a CIN pattern. To determine the biological and clinical significance of our analysis, we examined pathologic and clinical variables for association with the cluster designation. In particular, colorectal carcinoma extent of invasion showed an association with cluster designation. This association suggests that the multiple genomic instability events we observed influence the invasive behavior of colorectal carcinoma.
On chromosome arm 18q, two common deletion regions were identified in nearly all of the samples from cluster 1 but not frequently present in cluster 2 tumors. Four 18q microsatellite markers with clinical prognostic value were located within these two intervals. These patterns of deletion on 18q represent a potential molecular classifier in CIN colorectal carcinomas. This finding also suggests that a colorectal cancer gene exists within these 18q intervals. The prognostic significance of 18q LOH events is being determined in an ongoing prospective clinical trial through the Eastern Cooperative Group 5202 in which stage II patients with evidence of 18q LOH will be categorized as at high risk for cancer recurrence and subsequently receive adjuvant chemotherapy. Our analysis indicates that there are discrete patterns of 18q deletions that may influence tumor behavior and affect the results of this trial. We are currently evaluating the clinical significance of this finding.
We identified several gene exon deletions specific to cluster 1, including WRN, a DNA helicase and involved in DNA repair and maintenance of genomic stability. Another gene that is a frequently deleted in cluster 1 is BAG2, which is involved in apoptosis and protein folding. These genes have not been widely reported to be the target of mutations or LOH events in colorectal carcinoma development. Their high frequency of deletions in cluster 1 suggests that they play a role in a subset of CIN colorectal carcinomas. Our analysis of primary colorectal carcinomas identified amplifications events on chromosomes 2, 3, 8, 10, and 20 (43). As already noted, MIP measurements of gene amplifications are semiquantitative. Nevertheless, our data are consistent with other studies that have analyzed primary colorectal carcinomas for amplifications. For example, we identified multiple amplifications on 20q13. Amplifications on 20q13 have been identified in breast and ovarian cancer and may have prognostic significance (46). In the case of colorectal carcinoma, there are several reports of amplifications in the 20q11-13 region that includes the gene NCOA3 (47, 48). In these studies, 20q13 amplifications are correlated with worse outcomes. Likewise, our data showed multiple discrete regions of amplification in 20q11-13, which had been noted previously in array CGH analysis in colorectal carcinoma (48).
In summary, we have applied a novel genomic technology using MIPs to identify genomic deletions and other gene copy alterations in colorectal carcinoma. This technology has the ability to delineate hemizygous and homozygous deletions at the resolution of individual exons and microsatellite markers. To further improve the technology, we are expanding the set of probes to include all exons from a larger number of cancer genes. The benefits of this system for translational studies identifying gene copy alterations in cancer has been shown by our finding of identifying specific categories of colorectal carcinoma with CIN. It will be of significant value in identifying deletion mutants of genes in cancer. For projects, such as the recently announced Cancer Genome Initiative sponsored by the NIH, this technology will be of major benefit in providing information of deletion mutation in cancer in a cost-effective manner.
Acknowledgments
We thank Craig Giacomini and Dr. Jonathan Pollack (Stanford University) for providing genomic DNA from cancer cell lines, Dr. Sheng Zhong (University of Illinois), Dr. Tae Ji (University of Kentucky), and Dr. Mostafa Ronaghi (Stanford University) for participation in discussions, and Lisa Diamond for bioinformatics support.
Footnotes
Grant support: NIH grants CA96879 and 2P30DK056339 (H. Ji), CA109190 (J.M. Ford), GM-07365 (K. Salari), and 2P01HG000205 (R.W. Davis and J. Kumm).
References
- 1.Beckman RA, Loeb LA. Genetic instability in cancer: theory and experiment. Semin Cancer Biol. 2005;15:423–35. doi: 10.1016/j.semcancer.2005.06.007. [DOI] [PubMed] [Google Scholar]
- 2.Lengauer C, Kinzler KW, Vogelstein B. Genetic instabilities in human cancers. Nature. 1998;396:643–9. doi: 10.1038/25292. [DOI] [PubMed] [Google Scholar]
- 3.Cohn SL, Tweddle DA. MYCN amplification remains prognostically strong 20 years after its “clinical debut. Eur J Cancer. 2004;40:2639–42. doi: 10.1016/j.ejca.2004.07.025. [DOI] [PubMed] [Google Scholar]
- 4.Stoehlmacher J, Lenz HJ. Implications of genetic testing in the management of colorectal cancer. Am J Pharmacogenomics. 2003;3:73–88. doi: 10.2165/00129785-200303020-00001. [DOI] [PubMed] [Google Scholar]
- 5.Watanabe T, Wu TT, Catalano PJ, et al. Molecular predictors of survival after adjuvant chemotherapy for colon cancer. N Engl J Med. 2001;344:1196–206. doi: 10.1056/NEJM200104193441603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lanza G, Matteuzzi M, Gafa R, et al. Chromosome 18q allelic loss and prognosis in stage II and III colon cancer. Int J Cancer. 1998;79:390–5. doi: 10.1002/(sici)1097-0215(19980821)79:4<390::aid-ijc14>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 7.Ogunbiyi OA, Goodfellow PJ, Herfarth K, et al. Confirmation that chromosome 18q allelic loss in colon cancer is a prognostic indicator. J Clin Oncol. 1998;16:427–33. doi: 10.1200/JCO.1998.16.2.427. [DOI] [PubMed] [Google Scholar]
- 8.Rajagopalan H, Nowak MA, Vogelstein B, Lengauer C. The significance of unstable chromosomes in colorectal cancer. Nat Rev Cancer. 2003;3:695–701. doi: 10.1038/nrc1165. [DOI] [PubMed] [Google Scholar]
- 9.Boland CR. Molecular genetics of hereditary non-polyposis colorectal cancer. Ann N Y Acad Sci. 2000;910:50–9. 59–61. doi: 10.1111/j.1749-6632.2000.tb06700.x. [DOI] [PubMed] [Google Scholar]
- 10.Ji HP, King MC. A functional assay for mutations in tumor suppressor genes caused by mismatch repair deficiency. Hum Mol Genet. 2001;10:2737–43. doi: 10.1093/hmg/10.24.2737. [DOI] [PubMed] [Google Scholar]
- 11.Kleivi K, Teixeira MR, Eknaes M, et al. Genome signatures of colon carcinoma cell lines. Cancer Genet Cytogenet. 2004;155:119–31. doi: 10.1016/j.cancergencyto.2004.03.014. [DOI] [PubMed] [Google Scholar]
- 12.Tomlinson I, Ilyas M, Johnson V, et al. A comparison of the genetic pathways involved in the pathogenesis of three types of colorectal cancer. J Pathol. 1998;184:148–52. doi: 10.1002/(SICI)1096-9896(199802)184:2<148::AID-PATH986>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 13.Li LS, Kim NG, Kim SH, et al. Chromosomal imbalances in the colorectal carcinomas with microsatellite instability. Am J Pathol. 2003;163:1429–36. doi: 10.1016/S0002-9440(10)63500-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fearon ER. Molecular genetics of colorectal cancer. Ann N Y Acad Sci. 1995;768:101–10. doi: 10.1111/j.1749-6632.1995.tb12114.x. [DOI] [PubMed] [Google Scholar]
- 15.Kirley SD, D’Apuzzo M, Lauwers GY, et al. The cables gene on chromosome 18q regulates colon cancer progression in vivo. Cancer Biol Ther. 2005;4:861–4. doi: 10.4161/cbt.4.8.1894. [DOI] [PubMed] [Google Scholar]
- 16.Takagi Y, Koumura H, Futamura M, et al. Somatic alterations of the SMAD-2 gene in human colorectal cancers. Br J Cancer. 1998;78:1152–5. doi: 10.1038/bjc.1998.645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jernvall P, Makinen MJ, Karttunen TJ, Makela J, Vihko P. Loss of heterozygosity at 18q21 is indicative of recurrence and therefore poor prognosis in a subset of colorectal cancers. Br J Cancer. 1999;79:903–8. doi: 10.1038/sj.bjc.6690144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Font A, Abad A, Monzo M, et al. Prognostic value of K-ras mutations and allelic imbalance on chromosome 18q in patients with resected colorectal cancer. Dis Colon Rectum. 2001;44:549–57. doi: 10.1007/BF02234328. [DOI] [PubMed] [Google Scholar]
- 19.Carethers JM, Hawn MT, Greenson JK, Hitchcock CL, Boland CR. Prognostic significance of allelic lost at chromosome 18q21 for stage II colorectal cancer. Gastroenterology. 1998;114:1188–95. doi: 10.1016/s0016-5085(98)70424-x. [DOI] [PubMed] [Google Scholar]
- 20.Cohn KH, Ornstein DL, Wang F, et al. The significance of allelic deletions and aneuploidy in colorectal carcinoma. Results of a 5-year follow-up study. Cancer. 1997;79:233–44. [PubMed] [Google Scholar]
- 21.Hardenbol P, Baner J, Jain M, et al. Multiplexed genotyping with sequence-tagged molecular inversion probes. Nat Biotechnol. 2003;21:673–8. doi: 10.1038/nbt821. [DOI] [PubMed] [Google Scholar]
- 22.Wang Y, Moorhead M, Karlin-Neumann G, et al. Allele quantification using molecular inversion probes (MIP) Nucleic Acids Res. 2005;33:e183. doi: 10.1093/nar/gni177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang Z, Schwartz S, Wagner L, Miller W. A greedy algorithm for aligning DNA sequences. J Comput Biol. 2000;7:203–14. doi: 10.1089/10665270050081478. [DOI] [PubMed] [Google Scholar]
- 24.Wang P, Kim Y, Pollack J, Narasimhan B, Tibshirani R. A method for calling gains and losses in array CGH data. Biostatistics. 2005;6:45–58. doi: 10.1093/biostatistics/kxh017. [DOI] [PubMed] [Google Scholar]
- 25.Struyf A, Hubert M, Rousseeuw PJ. Integrating robust clustering techniques in S-PLUS. Comput Stat Data Anal. 1997;26:17–37. [Google Scholar]
- 26.Choi SW, Lee KJ, Bae YA, et al. Genetic classification of colorectal cancer based on chromosomal loss and microsatellite instability predicts survival. Clin Cancer Res. 2002;8:2311–22. [PubMed] [Google Scholar]
- 27.Diep CB, Thorstensen L, Meling GI, et al. Genetic tumor markers with prognostic impact in Dukes’ stages B and C colorectal cancer patients. J Clin Oncol. 2003;21:820–9. doi: 10.1200/JCO.2003.05.190. [DOI] [PubMed] [Google Scholar]
- 28.Kallioniemi A, Kallioniemi OP, Piper J, et al. Detection and mapping of amplified DNA sequences in breast cancer by comparative genomic hybridization. Proc Natl Acad Sci U S A. 1994;91:2156–60. doi: 10.1073/pnas.91.6.2156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pollack JR, Perou CM, Alizadeh AA, et al. Genome-wide analysis of DNA copy-number changes using cDNA microarrays. Nat Genet. 1999;23:41–6. doi: 10.1038/12640. [DOI] [PubMed] [Google Scholar]
- 30.Snijders AM, Nowak N, Segraves R, et al. Assembly of microarrays for genome-wide measurement of DNA copy number. Nat Genet. 2001;29:263–4. doi: 10.1038/ng754. [DOI] [PubMed] [Google Scholar]
- 31.Alitalo K, Schwab M, Lin CC, Varmus HE, Bishop JM. Homogeneously staining chromosomal regions contain amplified copies of an abundantly expressed cellular oncogene (c-myc) in malignant neuroendocrine cells from a human colon carcinoma. Proc Natl Acad Sci U S A. 1983;80:1707–11. doi: 10.1073/pnas.80.6.1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Heiskanen MA, Bittner ML, Chen Y, et al. Detection of gene amplification by genomic hybridization to cDNA microarrays. Cancer Res. 2000;60:799–802. [PubMed] [Google Scholar]
- 33.Umar A, Boyer JC, Thomas DC, et al. Defective mismatch repair in extracts of colorectal and endome-trial cancer cell lines exhibiting microsatellite instability. J Biol Chem. 1994;269:14367–70. [PubMed] [Google Scholar]
- 34.Liu B, Nicolaides NC, Markowitz S, et al. Mismatch repair gene defects in sporadic colorectal cancers with microsatellite instability. Nat Genet. 1995;9:48–55. doi: 10.1038/ng0195-48. [DOI] [PubMed] [Google Scholar]
- 35.Takahashi T, Nau MM, Chiba I, et al. p53: a frequent target for genetic abnormalities in lung cancer. Science. 1989;246:491–4. doi: 10.1126/science.2554494. [DOI] [PubMed] [Google Scholar]
- 36.Strathdee G, Zackai EH, Shapiro R, Kamholz J, Overhauser J. Analysis of clinical variation seen in patients with 18q terminal deletions. Am J Med Genet. 1995;59:476–83. doi: 10.1002/ajmg.1320590414. [DOI] [PubMed] [Google Scholar]
- 37.Rowan AJ, Lamlum H, Ilyas M, et al. APC mutations in sporadic colorectal tumors: a mutational “hotspot” and interdependence of the “two hits. Proc Natl Acad Sci U S A. 2000;97:3352–7. doi: 10.1073/pnas.97.7.3352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gayet J, Zhou XP, Duval A, et al. Extensive characterization of genetic alterations in a series of human colorectal cancer cell lines. Oncogene. 2001;20:5025–32. doi: 10.1038/sj.onc.1204611. [DOI] [PubMed] [Google Scholar]
- 39.Abdel-Rahman WM, Katsura K, Rens W, et al. Spectral karyotyping suggests additional subsets of colorectal cancers characterized by pattern of chromosome rearrangement. Proc Natl Acad Sci U S A. 2001;98:2538–43. doi: 10.1073/pnas.041603298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Douglas EJ, Fiegler H, Rowan A, et al. Array comparative genomic hybridization analysis of colorectal cancer cell lines and primary carcinomas. Cancer Res. 2004;64:4817–25. doi: 10.1158/0008-5472.CAN-04-0328. [DOI] [PubMed] [Google Scholar]
- 41.Shibata D, Peinado MA, Ionov Y, Malkhosyan S, Perucho M. Genomic instability in repeated sequences is an early somatic event in colorectal tumorigenesis that persists after transformation. Nat Genet. 1994;6:273–81. doi: 10.1038/ng0394-273. [DOI] [PubMed] [Google Scholar]
- 42.Lai WR, Johnson MD, Kucherlapati R, Park PJ. Comparative analysis of algorithms for identifying amplifications and deletions in array CGH data. Bioinformatics. 2005;21:3763–70. doi: 10.1093/bioinformatics/bti611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Anzick SL, Kononen J, Walker RL, et al. AIB1, a steroid receptor coactivator amplified in breast and ovarian cancer. Science. 1997;277:965–8. doi: 10.1126/science.277.5328.965. [DOI] [PubMed] [Google Scholar]
- 44.Mehlen P, Furne C. Netrin-1: when a neuronal guidance cue turns out to be a regulator of tumorigenesis. Cell Mol Life Sci. 2005;62:2599–616. doi: 10.1007/s00018-005-5191-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Blons H, Laccourreye O, Houllier AM, et al. Delineation and candidate gene mutation screening of the 18q22 minimal region of deletion in head and neck squamous cell carcinoma. Oncogene. 2002;21:5016–23. doi: 10.1038/sj.onc.1205626. [DOI] [PubMed] [Google Scholar]
- 46.Ried T, Knutzen R, Steinbeck R, et al. Comparative genomic hybridization reveals a specific pattern of chromosomal gains and losses during the genesis of colorectal tumors. Genes Chromosomes Cancer. 1996;15:234–45. doi: 10.1002/(SICI)1098-2264(199604)15:4<234::AID-GCC5>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 47.Aust DE, Muders M, Kohler A, et al. Prognostic relevance of 20q13 gains in sporadic colorectal cancers: a FISH analysis. Scand J Gastroenterol. 2004;39:766–72. doi: 10.1080/00365520410003191. [DOI] [PubMed] [Google Scholar]
- 48.Xie D, Sham JS, Zeng WF, et al. Correlation of AIB1 overexpression with advanced clinical stage of human colorectal carcinoma. Hum Pathol. 2005;36:777–83. doi: 10.1016/j.humpath.2005.05.007. [DOI] [PubMed] [Google Scholar]