

Abstract
Neuropathic pain states and tolerance to opioids can result from system changes in the CNS, such as up-regulation of the NK1 receptor and substance P, which have anti-opioid effects in ascending or descending pain-signaling pathways. Bifunctional compounds, possessing both the NK1 antagonist pharmacophore and the opioid agonist pharmacophore with delta-selectivity, could counteract these system changes to have significant analgesic efficacy without undesirable side effects. As a result of the introduction of cyclic and topological constraints with penicillamines, 2 (Tyr-cyclo[D-Pen-Gly-Phe-Pen]-Pro-Leu-Trp-NH-[3′,5′-(CF3)2-Bzl]) was found as the best bifunctional compound with effective NK1 antagonist and potent opioid agonist activities, and 1400-fold delta-selectivity over the mu-receptor. The NMR structural analysis of 2 revealed that the relative positioning of the two connected pharmacophores as well as its cyclic and topological constraints might be responsible for its excellent bifunctional activities as well as its significant delta-opioid selectivity. Together with the observed high metabolic stability, 2 could be considered as a valuable research tool and possibly a promising candidate for a novel analgesic drug.
Keywords: bifunctional compound, opioid receptor agonists, neutokinin-1 receptor antagonists, NMR structure, membrane-compound interaction
Introduction
The clinical treatment of pain, especially prolonged and neuropathic pain, is still a major challenge, and the efficacy of current analgesic drugs, such as opioids, is often reduced by undesired dose-limiting side effects including development of tolerance and physical dependence. The mechanisms for these effects are still largely unclear, but it is clear that prolonged pain states lead to neuroplastic changes in both ascending and descending pathways in the spinal column in which there is increased release of neurotransmitters (e.g. substance P) that enhance pain and increased expression of the corresponding receptors for those newly released pain-causing ligands.1–3 Current treatment of prolonged pain generally can only modulate pain, and cannot counteract these induced neuroplastic changes. Thus, it is not surprising that current analgesic drugs do not work well in these pathological conditions.
To effectively address this problem, we have taken a new approach to design bifunctional ligands that act as agonists at opioid receptors and as antagonists at NK1 receptors to block the signals mediated by substance P, which is one of these newly expressed pain-causing neurotransmitters.4–7 Our developing ligands have synergistic biological activities and overlapping pharmacophores (Figure 1), and would not be expected to possess the undesirable side effects of current opioids and other pain-killing drugs, such as the development of tolerance. Indeed, previous reports have indicated that substance P plays a role as a mediator of signals induced by opioid stimulation.1–3 Combination of the agonist effects at opioid receptors together with the blocking of signals through NK1 receptors has led to enhanced antinociceptive effects in acute pain animal models and has prevented the opioid-induced tolerance in chronic trials.8–12 NK1 knockout mice did not show the rewarding properties of morphine.13 We also emphasize that designing these bifunctional compounds with multiple biological activities in a single ligand has additional advantages over a cocktail of individual drugs including easy administration, single biodistribution, no drug-drug interactions, simple pharmacokinetic profiles, and higher local concentration in the synaptic cleft, which may lead to significant synergies in potency and efficacy.14 However, obtaining a bifunctional ligand with desired bioactivity is a difficult challenge, since connected pharmacophores interfere with each other to change or even to reduce their affinities at the corresponding receptors.
Figure 1.
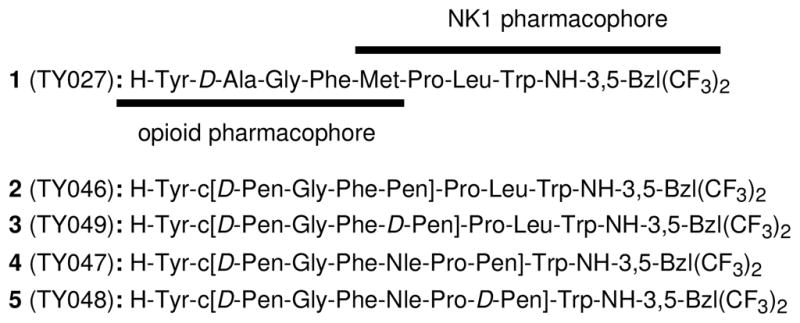
Sequences of opioid and NK1 receptor peptides.
In this report, we have focused on the δ/μ activity of targeted opioid receptors for the bifunctional compounds. The μ-opiates have been well-established to show strong analgesic potency with several disadvantages such as constipation, respiratory depression, dysphoria, opioid-induced tolerance development and addiction.15, 16 Selective δ-opioid agonists have several potential clinical advantages over their μ-counterparts, including reduced respiratory depression17 and constipation18 as well as a decreased potential for the development of physical dependence.19–21 However, selective δ-opioid agonists do not attenuate the same level of pain as μ-agonists, and no δ-opioid selective agonist ligand has been found to have clinical efficacy sufficient to be a useful agent for the treatment of severe pain.22 Therefore, bifunctional ligands possessing selective δ-opioid agonist activity but also some μ-opioid agonist activity together with the NK1 antagonist activity could be a promising strategy to find next-generation analgesics which have powerful pain controlling effects without development of tolerance, physical dependence and other toxicities.4, 5 In fact, our lead bifunctional compounds, TY005 (Tyr1-D-Ala2-Gly3-Phe4-Met5-Pro6-Leu7-Trp8-O-[3′,5′-(CF3)2-Bzl]) and TY027 (1: Tyr1-D-Ala2-Gly3-Phe4-Met5-Pro6-Leu7-Trp8-NH-[3′,5′-(CF3)2-Bzl]) possessing moderately δ-selective (up to 25-fold) δ/μ opioid agonist and potent NK1 antagonist activities (Table 1), have shown the powerful attenuation of neuropathic pain without producing the opioid-induced tolerance, to effectively prove our opioid agonist/NK1 antagonist bifunctional concept in the animal models of neuropathic pain.23, 24
Table 1.
Binding affinity, GTP binding assay and Emax % for bifunctional peptide derivative ligands at opioid and neurokinin receptors
| No. | Radioligand Bindinga,b |
[35S]GTPγS Bindinga,c |
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| δ | μ | hNK1 | rNK1 | δ | μ | |||||||||
| IC50 | Ki (nM) | IC50 | Ki (nM) | IC50 | Ki (nM) | IC50 | Ki (nM) | EC50 | EC50 (nM) | Emax (%) | EC50 | EC50 (nM) | Emax (%) | |
| 1d | −8.84 ± 0.07 | 0.66 | −7.44 ± 0.05 | 16 | −10.9 ± 0.10 | 0.0065 | −7.61 ± 0.03 | 7.3 | −8.07 ± 0.11 | 8.6 | 60 ± 2 | −8.16 ± 0.17 | 7.0 | 51 ± 3 |
| 2 | −8.44 ± 0.04 | 1.7 | −5.36 ± 0.27 | 2300 | −10.91 ± 0.03 | 0.0053 | −7.38 ± 0.04 | 10 | −8.50 ± 0.39 | 3.2 | 57 ± 5 | −7.54 ± 0.41 | 29 | 14 ± 2 |
| 3 | −7.14 ± 0.07 | 38 | −5.38 ± 0.08 | 2100 | −9.38 ± 0.03 | 0.18 | −7.74 ± 0.03 | 4.5 | −6.94 ± 0.32 | 120 | 49 ± 3 | −6.87 ± 0.22 | 130 | 28 ± 2 |
| 4 | −6.50 ± 0.14 | 150 | −5.43 ± 0.14 | 2000 | −6.87 ± 0.06 | 59 | −6.18 ± 0.05 | 160 | −6.27 ± 0.11 | 530 | 63 ± 2 | −5.67 ± 0.46 | 2900 | 15 ± 3 |
| 5 | −5.86 ± 0.04 | 720 | −5.69 ± 0.06 | 1000 | −8.37 ± 0.02 | 1.9 | −6.98 ± 0.05 | 26 | −6.91 ± 0.29 | 120 | 42 ± 3 | −6.87 ± 0.36 | 140 | 26 ± 4 |
| Tyr-DAla-Gly-Phe-Met-NH2 | −8.86 ± 0.05 | 0.66 | −8.90 ± 0.16 | 0.50 | ||||||||||
| DPDPEe | 0.5±0.1 | |||||||||||||
| DPDPEf | 1.6 | 610 | ||||||||||||
| DPDPEg | 10 | 3700 | ||||||||||||
| DPLPEg | 16 | 2800 | ||||||||||||
| Biphalin | 2.6h | 1.4h | −8.95 ± 0.17 | 1.1 | 83 ± 4 | |||||||||
| DAMGOi | 0.85±0.2 | −7.44 ± 0.19 | 37.0 | 150 | ||||||||||
| L-732, 138 | −8.83 ± 0.02 | 0.73 | −6.40 ± 0.03 | 134 | ||||||||||
The experiments were run in at least two independent experiments performed in duplicate.
IC50 values are expressed as logarithmic mean ± standard errors. Ki values were calculated from the anti-logarithmic mean IC50 values using the Cheng and Prusoff equation.
EC50 values are expressed as both logarithmic mean ± standard errors and anti-logarithmic value of the mean. Emax values were defined as [net [35S]GTPγS bound/basal [35S]GTPγS bound] × 100%, expressed as mean ± standard error.
From reference.5
Dissociation constant (KD, nM) of [3H]DPDPE at human δ opioid receptor expressed as mean ± S.E.M.
From reference.27
From reference.28
From reference.56
KD value (nM) of [3H]DAMGO at rat μ opioid receptor expressed as mean ± S.E.M.
In order to perform further biological optimization of the bifunctional ligands, we describe here the design, synthesis, structure-activity relationships (SAR) and further biological characterizations of bifunctional compounds which have significantly improved selectivity both for affinity and agonist activity for the δ-opioid over μ-opioid receptors, together with effective NK1 antagonist activity. In fact, we previously reported the finding of a selective δ-opioid activator, possessing two cysteine residues, in the conventional MVD and GPI assays, but its δ-opioid selectivity in terms of the binding affinity was relatively lower.25 While our earlier efforts led to the discovery that introduction of two topologically constrained L- or D-penicillamine (Pen or D-Pen) residues into several cyclic pentapeptide enkephalin analogues produced compounds with enhanced δ-selectivity through the reduction of opioid affinity at the μ-opioid receptor: Tyr-cyclo[D-Pen-Gly-Phe-D-Pen] (DPDPE) and Tyr-cyclo[D-Pen-Gly-Phe-Pen] (DPLPE) are the most typical δ-selective opioid agonists (Table 1 and 2).5, 26–28 Previous conformation-activity relationship studies suggested that the 14-membered ring structure with their geminal-dimethyl group at the second position has special importance to interfere with the binding at the μ-opioid receptor so as to greatly favor binding to the δ-opioid receptor.29, 30 We have now applied this modification to our lead bifunctional octapeptide analogue 1, in which Tyr1, Gly3, Phe4 and Trp8 were considered as the major “message” residues of each pharmacophore,5 and Pro6 had a positive effect on affinity at the NK1 receptors.31 Thus, the residues 2, 5 and 7 in 1 were selected as the sites to be cyclized to yield four bifunctional peptide derivatives possessing a 14 or 20-membered ring structure with two Pen or D-Pen residues (2–5, Figure 1). Subsequent conformational analysis was performed using NMR experiments in the presence of membrane-mimicking perdeuterated dodecylphosphocholine (DPC) micelles, since understanding membrane-bound structures of ligands and ligand-membrane interactions is indispensable for further evaluation of their diverse biological behaviors,5 and the elucidation of the conformational motif for δ-opioid selective binding affinities could provide important information in the discovery of novel drug candidates for next-generation neuropathic pain treatment.
Table 2.
Functional assay results for bifunctional peptide ligands at opioid and Substance P receptors
| No | Opioid agonist | SP antagonist | |
|---|---|---|---|
| MVD (δ) | GPI (μ) | GPI | |
| IC50 (nM)a | IC50 (nM)a | Ke (nM)b | |
| 1c | 15 ± 2 | 490 ± 29 | 10 ± 2 |
| 2 | 9.6 ± 3.5 | 26 % inh. at 1 uMd | 6.9 ± 1.8 |
| 3 | 130 ± 25 | 0.6 % inh. at 1 uMd | 8.2 ± 4.2 |
| 4 | 250 ± 30 | 6 % inh. at 1 uMd | 72 ± 7.2 |
| 5 | 1100 ± 250 | 7 % inh. at 1 uMd | 66 ± 35 |
| DPDPEe | 2.5 ± 0.03 | 2720 ± 50 | |
| DPLPEe | 2.2 ± 0.30 | 6930 ± 120 | |
| L-732,138 | 250 ± 87 | ||
Concentration at 50% inhibition of muscle concentration at electrically stimulated isolated tissues (n = 4).
Inhibitory activity against the Substance P induced muscle contraction in the presence of 1 μM naloxone (n = 4), Ke: concentration of antagonist needed to inhibit Substance P to half its activity.
reference.5
No antagonist activity was observed at the tested concentration.
reference.28
Results and Discussion
Peptide Synthesis
The peptide derivatives 2–5 were synthesized using a convergent peptide synthesis strategy,32 since the coupling of Tyr1 was difficult using the normal sequential method. First, the N-terminal fragment, Boc-Tyr(tBu)-D-Pen(Mob)-Gly-OH, was synthesized with Nα-Boc chemistry using a solution peptide synthesis strategy. The C-terminal fragments, H-Phe-Pen(Trt)-Pro-Leu-Trp(Boc), H-Phe-D-Pen(Trt)-Pro-Leu-Trp(Boc), H-Phe-Nle-Pro-Pen(Trt)-Trp(Boc) and H-Phe-Nle-Pro-D-Pen(Trt)-Trp(Boc) were prepared on 2-Chlorotrityl resin using Nα-Fmoc chemistry with HCTU as a coupling reagent. These two fragments were coupled on the resin in the presence of an excess amount of EDC and HOAt, followed by cleavage using 1 % TFA in DCM, to afford the complete sequence of the C-terminal-free linear peptide with its protecting groups. The introduction of 3′,5′-bistrifluoromethyl benzyl amide at the C-terminus was conducted by standard EDC/Cl-HOBt coupling chemistry using two equivalents of the amine with the protected C-terminal-free peptide. All of the acid-labile protecting groups were removed with the cleavage cocktail treatment (6 : 1 mixture of TFA and anisole to quench the highly stabilized carbocations released from the permanent protecting groups) at 80 °C for 30 min to give the final linear crude peptides, which were directly cyclized using the oxidizing agent K3Fe(CN)6.25 Oxidation was performed by slowly adding a solution of the peptide into a reaction vessel containing an excess of the oxidizing agent in aqueous solution. The concentration of the reduced peptide was regulated by controlling the speed of addition of the peptide with the help of an automated syringe pump.25 The crude oxidized peptides were concentrated using a solid-phase extraction technique with a C-18 reversed-phase silica gel column, and then purified with RP-HPLC followed by lyophilizion (≥ 95 %). The final purified peptides were characterized by analytical HPLC, 1H-NMR, HRMS and TLC.
Biological activities
The evaluation of the synthesized bifunctional ligands 2–5 was performed as previously described.4–7 Binding affinities were tested on cell membranes from cells that stably express the human δ-opioid, the rat μ-opioid, the human NK1 or the rat NK1 receptors (Table 1). The [35S]GTPγS binding assays were performed to characterize the functional activities for the δ and μ opioid agonists of 2–5 (Table 1). Tissue-based functional assays also were performed to evaluate δ/μ opioid agonist as well as the substance P antagonist activities in the guinea pig isolated ileum (GPI) and mouse vas deferens (MVD) (Table 2).
At the δ-opioid receptor, the cyclic peptide possessing a 20-memberd ring with D-Pen2 and Pen7 (4) had a Ki of 150 nM, which was 230-fold less binding affinity than 1 (Table 1). The δ-opioid affinity of its D-Pen7 derivative (5) also showed a decrease in affinity of 720 nM, indicating that introduction of D-Pen or Pen at the seventh residue in the sequence of 1 decreases affinity at the δ-opioid receptor. The Ki of the D-Pen2, D-Pen5 cyclic analogue 3 was 38 nM for the δ receptor, demonstrating that the cyclic peptide with a smaller 14-membered ring had enhanced affinity compared to the 20-membered-ring analogues. Interestingly, the cyclic D-Pen2, L-Pen5 analogue 2 had the best affinity among 2–5 for the δ-opioid receptor with Ki value of 1.7 nM. This is the closest to the KD value of DPDPE (0.5 ± 0.1 nM), which was used as a radioactive ligand of 1 nM in the competition assay on the cells membrane expressing the δ-opioid receptors (Table 1). Thus, the affinities of 2–5 for the δ-opioid receptor were different depending on the ring-size and the chirality of bridgehead penicillamine residues, and the combination of D-Pen2 and Pen5 clearly showed the best results. Interestingly, the affinity difference at the δ-opioid receptor between pentapeptides DPDPE and DPLPE was only 1.6-fold and DPDPE showed the better affinity,28 but in the case of the bifunctional compounds with the longer peptide sequences, the compound 2 (DPLPE-derivative) showed better affinity than 3 (DPDPE-derivative), and their affinity difference was 22-fold. This result apparently indicates that the extended sequence at the C-terminus (Pro-Leu-Trp-NH-3,5-Bzl(CF3)2) plays a critical role for the δ-opioid selectivity, and 2 has a preferable three-dimensional conformational motif for binding at the δ-opioid receptor compared to 3. For the linear pentapeptide Tyr-D-Ala-Gly-Phe-Met-NH2, the C-terminal elongation to the octapeptide derivative 1 resulted in a decrease in μ-opioid binding affinity with no shift in the affinity at the δ-receptor (Table 1). Consistent with the trend in the affinities at the δ-opioid receptor, the EC50 values of 3–5 were more than 100 nM in the GTPγS binding assay (120, 530 and 120 nM for 3, 4 and 5, respectively), while only 2 showed significantly better efficacy with a nanomolar level EC50 value (EC50 = 3.2 nM, Table 1). In accord with these results, 3–5 had relatively lower inhibitory activities in the MVD assays (IC50 = 130, 250 and 1100 nM for 3, 4, and 5, respectively), whereas 2 showed good activity (IC50 = 9.6 nM) which is improved from that of 1 (Table 2).
For the μ-opioid receptor, the cyclic peptides with a 14-membered ring (2 and 3) showed drastically reduced affinities (Ki = 2300 and 2100 nM for 2 and 3, respectively; Table 1), as expected from the results of DPDPE and DPLPE. The compounds with the larger 20-membered ring (4 and 5) also showed reduced affinity at the μ-opioid receptor (Ki = 2000 and 1000 nM for 4 and 5, respectively), confirming that the introduction of a disulfide ring possessing D-Pen2 is detrimental to the affinities at the μ-opioid receptor, regardless of the ring size of the octapeptide derivatives. In the GTPγS binding assays, the cyclic peptides 2–5 stimulate the μ-opioid receptor poorly with the Emax values less than 28 % (Table 1). In agreement with their low Ki and Emax values, 2–5 showed very weak μ-opioid-related agonist activities in the GPI assays (26, 0.6, 6 and 7 % inhibition at 1 μM for 2, 3, 4 and 5, respectively; Table 2). Therefore, only 2 showed significant δ-opioid selectivity over the μ-receptor (1400-fold) which is 58 times more δ-selective than 1 (24-fold), and 3.7 and 7.8 times more selective compared to DPDPE (380-fold) and DPLPE (180-fold), respectively (Table 1). The δ-selectivity of 3 was 55-fold, which is better than that of 1.
These results suggest that three important structural factors have an influence on the opioid affinity and bioactivity for these novel bifunctional compounds: 1) the 14-membered cyclic structure with D-Pen2; 2) the chirality of the fifth residue; and 3) the existence of the C-terminal NK1 pharmacophore. Apparently, introduction of a 14-membered ring with a D-Pen2 resulted in a limited shift in their δ-opioid binding affinities, but a more significant reduction in affinities at the μ-receptor, leading to the enhanced δ-receptor selectivity compared to DPDPE and DPLPE. Addition of the C-terminal NK1 pharmacophore to the cyclic opioid sequence in 2 resulted in improved affinity at the δ-receptor, while the combination with the N-terminus in 3 possessing a D-Pen5 resulted in a reduction in the δ-opioid binding affinity. These results clearly indicate that the NK1 pharmacophore works as an address region for the opioid agonist activities, and an appropriate combination of two connected pharmacophores can yield improved affinities and selectivities of the ligand. This is an important synergistic effect of our bifunctional concept.
Biological evaluations were also conducted at the NK1 receptors. Analogue 4 possessing a D-Pen2 and Pen7, showed reduced affinities at both the hNK1 and rNK1 receptors compared to those of 1 (Ki = 59 and 160 nM, respectively; Table 1). The Ki value of peptide 5 with a D-Pen7 was 1.9 nM at the hNK1 receptor and 26 nM at the rNK1 receptor, indicating that the D-Pen7 provided better affinities than L-Pen7 introduction. These affinities of compounds with 20-membered rings were drastically decreased from those of 1, and their antagonist activities on the GPI tissue were consistent with the observed affinities (Ke = 72 and 66 nM for 4 and 5, respectively; Table 2). However, the introduction of a ring structure between the second and seventh residues resulted in a reduction of biological activities at the NK1 receptors, suggesting an unfavorable topographical structure for the NK1R. Analogue 3, possessing a smaller 14-membered ring with the DPDPE sequence, has 28 times lower affinity for the hNK1 receptor than that of 1, but its Ki value at the rNK1 receptor was slightly improved from that of 1 (Ki = 0.18 and 4.5 nM for hNK1 and rNK1 receptors, respectively). The antagonist activity against substance P stimulation of 3 in the GPI assay was nearly comparable to that of 1 (Ke = 8.2 nM). The DPLPE derivative 2 showed similar antagonist activity against substance P stimulation at the GPI, with a Ke value of 6.9 nM (Table 2). 2 also showed the best affinity at the hNK1 receptor among the synthesized cyclic peptide derivatives 2–5 with a picomolar Ki value (Ki = 0.0053 nM).
For further biological characterization of 2, its metabolic stability was tested by incubation in rat plasma at 37 °C.6 Analogue 2 possesses significantly improved stability compared to the linear peptide derivative 1. The calculated half lives of the peptide derivatives (T1/2) were 4.8 h for 1 and 5.8 h for 2 (Figure 2). Consequently, c[D-Pen2, Pen5]TY027 (2) is a promising ligand with significant δ-selective opioid agonist activity, improved metabolic stability and nearly equivalent antagonist activities for NK1 receptors compared to those of the lead compound 1. The important conformational motif of the two pharmacophores for the bioactivities of 2 was next examined using NMR-based three-dimensional structural analysis.
Figure 2.
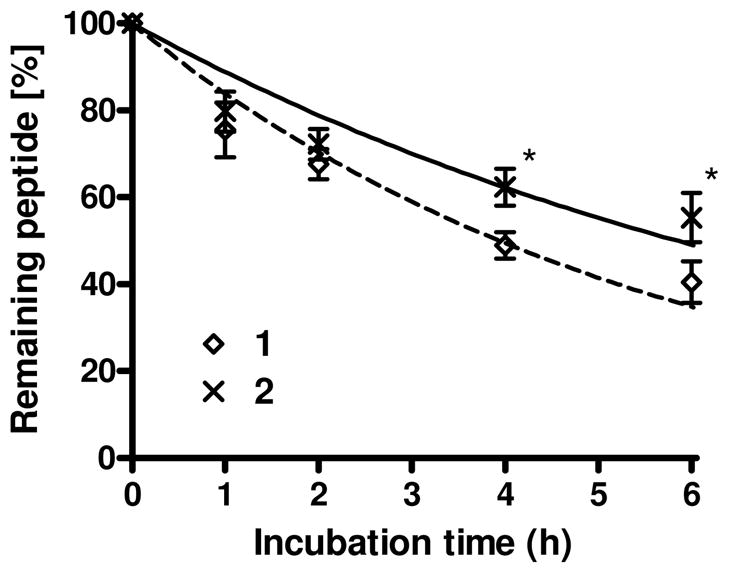
Comparison of the in vitro stability of peptide derivatives for 1 and 2 incubated in rat plasma at 37°C (*, p < 0.05).
Conformational analysis of cyclic peptide 2 based on assigned 1H NMR resonances
The ligand binding sites for opioid and NK1 receptors have been found in their transmembrane domain,33, 34 which is surrounded by the cell membrane, suggesting that the ligand binding event at these receptors occur near the membrane. The potential importance of membrane-bound conformations of ligands has increased recently.5, 6 The transfer from an aqueous environment to a cell membrane environment is an important step in receptor-ligand binding, and may be accompanied with a conformational change to a more biologically relevant form.17, 38 Thus, NMR structural analysis of 2 was performed to clarify the relationship between its biological activity and its conformation in a membrane-mimicking environment.
The NMR experiments and analysis of 2 were conducted using previously reported procedures.5, 6, 35, 36 The two-dimensional 1H-NMR studies included TOCSY, DQF-COSY and NOESY, and were performed in pH 4.5 buffer (45 mM CD3CO2Na/HCl, 1 mM NaN3, 90% H2O/10% D2O) solutions in the presence of a 40-fold excess of DPC micelles, which is a commonly-used membrane-like environment to determine the solution NMR structures of proteins and peptides.35–40 All 1H chemical shift assignments in aqueous media and other NMR data are found in the Supporting Information.
Conformational Calculations
The total number of NOE restraints for structural calculation of 2 was 184 including 63 intraresidual NOEs, 67 sequential NOEs, 49 medium-range NOEs (2–4 residues)and 5 long-range NOEs (> 4 residues). For the 3JHN-Hα values, only those greater than 8 Hz or less than 6 Hz were used as ϕ dihedral angle constraints. The 1H-NMR spectra in 2 showed a negligible amount of the minor cis/trans rotamer at the Pro6 residue. For conformational calculations, the Pen5-Pro6 bond of the major rotamer was fixed in the trans configuration based on the observation of Pen5 Hα to Pro6 Hδ sequential NOEs, together with the absence of sequential Pen5 Hα-Pro6 Hα NOEs.
Analysis and statistics of the calculated conformation for 2 were performed on the 10 structures with the lowest total energies after restrained molecular dynamics (rMD) refinement. The number of total NOE restraint violations was 25 with a maximum NOE violation of 0.15 Å, and no ϕ dihedral angle violations were found. The restraints energy derived from the amber force field was only 3.9 kcal mol−1 in the most stable 10 structures.
As shown in Figure 3A, the best 10 structures of 2 showed well-aligned superimposed conformations whose rmsd values were 0.15 ± 0.06 Ǻ; in an alignment with the backbone atoms (Table 3). Since these rmsd values were significantly improved from those of 1 (1.03 ± 0.57 Å for the best 10 structures; Supporting Information),5 the extensively structured backbone of 2 could be attributed to the cyclic and topological constraints induced by the disulfide bridge with the D-Pen2 and Pen5 residues. It should be noted that not only was the conformation of the N-terminal half, where the two penicillamines were introduced, but also the C-terminus, whose amino acid sequence is identical to 1, showed reduced rmsd values as well. The rmsd values of 2 for the alignment with all non-hydrogen atoms were also significantly reduced (0.52 ± 0.12 Ǻ, Table 3) indicating well-defined three-dimensional positions of the side-chains in 2 under the applied timescale in the NMR experimental (Figure 3B). Different from the observed well-defined side-chain conformation of 2, rather poorly-defined side-chains were previously observed for the linear compound 1. Thus, the constraints in ligand 2 due to the incorporation of two penicillamines provided improved conformation stability for the backbone atoms as well as for the side-chain atoms, including the two aromatic amino acid residues Tyr1 and Phe4 and the protonated N-terminal nitrogen, all of which have been reported as important pharmacophores in the binding of enkephalins to opioid receptors.41 The observed well-defined conformation of 2 suggests it has a structured conformation inside or close to the cell membrane, in close relationship with the improvement in activity and selectivity at the tested membrane-bound receptors.
Figure 3.
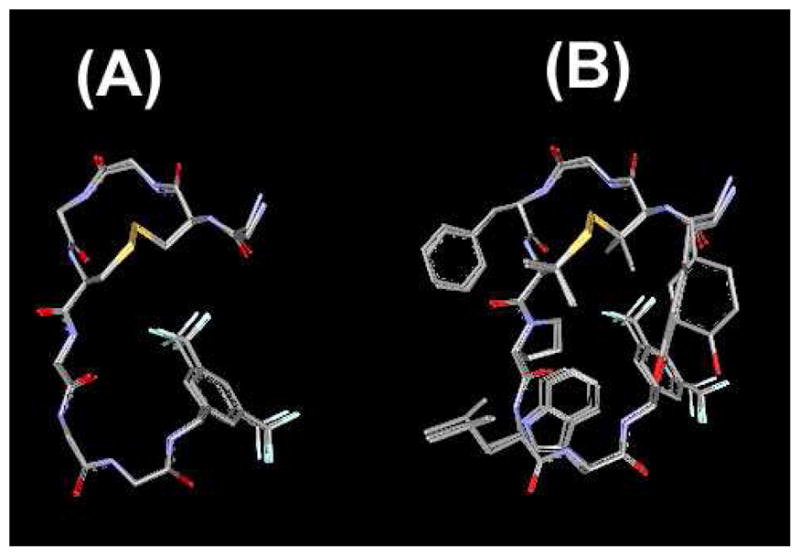
Ensembles of the best 10 calculated structures of 2 in 40-fold DPC micelle/pH 4.5 buffer with the lowest restraint energy, aligned on backbone atoms of residues 1–8. (A) Only backbone atoms in the aligned structures are illustrated with C-terminal benzyl moiety and disulfide bond. (B) All non-hydrogen atoms were displayed.
Table 3.
Atomic rmsd values (Å) for the final 9 conformers compared to the most stable conformer of bifunctional peptide derivatives 2.
| Aligned residues | Whole molecule | 1–4 resdues | 5–8 residues and C-terminus |
|---|---|---|---|
| backbone atoms (N, Cα, C′) | 0.15 ± 0.06 | 0.02 ± 0.04 | 0.14 ± 0.05 |
| all non- hydrogen atoms | 0.52 ± 0.12 | 0.35 ± 0.21 | 0.58 ± 0.11 |
As for the observed secondary structural elements, the NMR structure of 1 has two type IV β-turns at residues 2 to 5 and 6 to the C-terminus.5 Although the peptide sequence was different, the obtained NMR structure of ligand 2 had type IV β-turns at the same positions in all 10 of the best structures (Table 4), indicating that the introduction of constraints by D-Pen2 and Pen5 made the two β-turns more structured without changing the secondary structural elements of ligand. DPDPE also has a β-turn at residues 2–5 in its X-ray crystal structure,42 in the solution NMR structure43 and in the docking study at the δ opioid receptor,44 and thus, this turn has been hypothesized as a key structure to enhance the δ-selectivity of enkephalin analogues, through the lock-in of flexible pharmacophores at the appropriate positions (Figure 4A).
Table 4.
Number of β-turn structural elements and the distance between alpha carbons of i th and (i + 3)rd residues.a
| Residuesb | number of β-turns | distance (Å) |
|---|---|---|
| C2α-C5α | 10 | 5.36 ± 0.00 |
| C6α-Bzl | 10 | 6.43 ± 0.04 |
Out of the best 10 calculated structures. The distance is the mean distance between two alpha carbons ± standard deviation (SD). The sequences with less than 7 Å distance between alpha carbons of i th and (i + 3) rd residues without helical structure were considered as a β-turn.57 Bzl stands for the benzyl moiety at the C-terminus.
Only residues possessing β-turn structural element were displayed.
Figure 4.
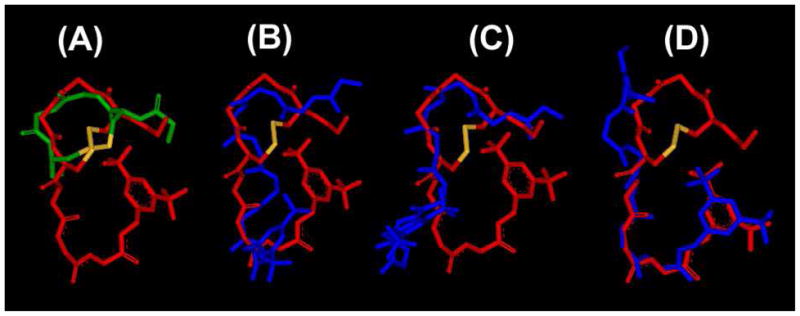
(A) Superimposed image of 2 (red) with the X-ray crystal structure of highly selective δ opioid agonist DPDPE (green).42 Superimposition was performed on all the backbone atoms of residues 1–5 (rmsd = 1.81 Å). Only backbone atoms are illustrated with C-terminal benzyl moiety and disulfide bond (yellow). Superimposed image of obtained NMR structure of 2 with the NMR structure of 15 (blue) at the lowest restraint energies. Superimposition was performed on all the backbone atoms of: (B) residues 1–8, rmsd = 2.34 Å; (C) residues 1–5, rmsd = 1.40 Å; (D) residues 5–8, rmsd = 0.65 Å.
Although 1 and 2 had the same secondary structural elements, their NMR structures did not overlap completely on alignment with the backbone atoms of the whole molecule (rmsd = 2.34 Å, Figure 4B). However, the rmsd value was decreased to 1.41 Å when the alignment was made only for the N-terminal opioid pharmacophore where the common β-turn structural elements for 1 and 2 were observed (Figure 4C). It should be mentioned that when the alignment was made on the N-terminus of 1 and 2, their corresponding C-terminal halves were directed differently and showed almost no overlap. As expected, when the alignment was made with the C-terminus (rmsd = 0.65 Å), the N-terminal halves of 1 and 2 showed almost no conformational overlap (Figure 4D). These results clearly indicated that the opioid and NK1 pharmacophores of ligands 1 and 2 independently possess nearly the same conformations in terms of their backbone atoms, but the three-dimensional positionings of the NK1 pharmacophore relative to the opioid pharmacophore are different. This could have an important connection with the different address effect of the C-terminus of 1 and 2 for the observed binding affinities at δ and μ opioid receptors.
Further structural examination was made on the torsional angles of the fifth residue, where the opioid and NK1 pharmacophores were overlapped (Figure 1), to examine the relative positioning of two pharmacophores. The NMR conformer of 1 had the Met5 residue with positive ϕ and ψ angles in all 10 of the best structures.5 Since the positive ϕ angle in an L-amino acid residue is energetically unfavorable, the backbone conformation of 1 tends to bend or to twist near this “distorted” site at the connecting position of two interacting pharmacophores. While Pen5 in all 10 structures of 2 had negative ϕ angles (Table 5). This difference in torsional angles for the fifth residue leads to conformational differences near the overlapping site, and modifies the positional relationship of the two connecting pharmacophores which may possibly account for the shift in biological activities at the tested receptors.
Table 5.
ϕ and ψ angle values of the bifunctional peptide derivative 2 in the observed NMR structure.a
| angle | Tyr1 | D-Pen2 | Gly3 | Phe4 |
|---|---|---|---|---|
| ϕ | −154.9 ± 2.3 | −85.0 ± 1.4 | −105.9 ± 0.6 | |
| ψ | −70.9 ± 0.5 | −173.6 ± 0.2 | −73.6 ± 0.9 | 128.3 ± 0.3 |
| angle | Pen5 | Pro6 | Leu7 | Trp8 |
| ϕ | −91.6 ± 0.6 | −86.6 ± 0.8 | −120.9 ± 4.7 | −146.1 ± 1.3 |
| ψ | 118.5 ± 0.6 | −102.7 ± 138.5 | 57.0 ± 1.1 | −41.7 ± 0.9 |
Average ± SD values out of the best 10 calculated structures were listed. The C-terminus was considered as residue 9.
Paramagnetic Broadening Studies on 1H NMR
In order to evaluate the interacting mode of 2 in a membrane-like environment, Mn2+ was used as a paramagnetic ion to eliminate the resonance intensities of solvent-exposed protons of 2 in the DPC micelles. The effects of the reagent were observed as an ensemble of cross-peaks belonging to the same residue spin system in TOCSY spectra measured with a 62.2 ms mixing time (Figure 6).5 Interestingly, most of the cross-peaks derived from the backbone NH protons of 2 were eliminated with Mn2+ addition. The only exception was the NH proton of Pen5 whose cross-peak was retained in the presence of Mn2+ ions, suggesting that the backbone of 2 was mostly exposed to the surface of micelles, and only the backbone atoms of Pen5 were buried inside of micelles (Figure 6). The side-chains of 2, most of which were lipophilic groups, were unchanged after the Mn2+ addition, implying that these side-chain groups interact with DPC micelles very well and are buried inside of them. Therefore, these results revealed that 2 locates in DPC micelles with the amino acid side-chain groups and backbone atoms of Pen5 buried inside of the micelles, indicating its strong interaction with the membrane. This observed interacting mode of 2 with membrane-like micelles was similar to that of 1,5 suggesting that the incorporation of a 14-membered ring structure with D-Pen2 and Pen5 did not affect the strong interaction of these peptides with membranes, but provides a more structured conformation which could modulate the affinity and selectivity of the ligand for the receptor in biological membranes.
Figure 6.

Typical effect of Mn2+ ions on TOCSY Spectra. 2 with DPC micelles (top row) and with 200 μM Mn2+ (bottom), for HN-Hα region (left column), aliphatic side-chain region (middle) and aromatic region (right). Preserved resonances (labeled) are shown by the addition of Mn2+. Spectra were compared from the same noise level.
Conclusions
Cyclic and topographical constraints with two penicillamine residues were introduced and optimized based on our developing bifunctional drug-design concept. As a result, the 14-membered ring structure with a Pen5 and a C-terminal NK1 pharmacophore in 2 was found as the best combination to show potent antagonist actions at NK1 receptors and potent opioid agonist actions with significantly improved δ-opioid affinity selectivity over the μ-opioid (1400-fold), than those of DPDPE (380-fold), DPLPE (180-fold) and the previously reported linear peptide derivative 1 (24-fold). NMR structural analysis showed that significant δ-opioid selectivity in 2 could be attributed to its well-defined conformation with cyclic and topographical constraints and appropriate positioning of opioid and NK1 pharmacophores. The torsional angle of the fifth residue plays a key role to modulate the address effect of the C-terminal NK1 pharmacophore for the δ/μ opioid selectivity of the ligand, by adjusting the three-dimensional positioning of the two connected pharmacophores. Together with its observed good metabolic stability, 2 could be considered as a valuable research tool and possibly a promising candidate for a novel analgesic drug.
Experimental Section
Materials
All amino acid derivatives and coupling reagents were purchased from EMD Biosciences (Madison, WI), Bachem (Torrance, CA), SynPep (Dublin, CA) and Chem Impex International (Wood Dale, IL). 2-Chlorotrityl resin was acquired from Iris Biotech GmbH (Marktredwitz, Germany). Perdeuterated DPC was purchased from C/D/N Isotopes (Quebec, Canada). ACS grade organic solvents were purchased from VWR Scientific (West Chester, PA), and other reagents were obtained from Sigma-Aldrich (St. Louis, MO) and used as obtained. The polypropylene reaction vessels (syringes with frits) were purchased from Torviq (Niles, MI). Myo-[2-3H(N)]-inositol; [Tyrosyl-3,5-3H(N)] D-Ala2-Mephe4-glyol5-enkephalin (DAMGO); [Tyrosyl-2,6-3H(N)]-(2-D-Penicillamine, 5-D-Penicillamine)enkephalin (DPDPE); [3H]-Substance P; and [35S]-guanosine 5′-(γ-thio) triphosphate (GTPγS) were purchased from Perkin Elmer (Wellesley, MA). Bovine serum albumin (BSA), protease inhibitors, Tris and other buffer reagents were obtained from Sigma (St. Louis, MO). Culture medium, Penicillin/Streptomycin and fetal calf serum (FCS) were purchased from Invitrogen (Carlsbad, CA).
Nα-Boc-Tyr(tBu)-D-Pen(Mob)-Gly-OH
Boc-D-Pen(Mob)-OH (1.50 g, 4.06 mmol) and H-Gly-OEt·HCl (677 mg, 4.87 mmol) were dissolved in DMF (5 mL). HOBt (657 mg, 4.87 mmol), HBTU (1.85 g, 4.87 mmol) and NMM (904 mg, 8.95 mmol) were added to the solution at 0° C. After stirring overnight at room temperature, saturated aqueous sodium bicarbonate was added to the solution and most of the organic solvent was removed under reduced pressure. The residue was extracted with ethyl acetate three times followed by washing with 5 % aqueous citrate and saturated aqueous sodium chloride. The solution was dried over sodium sulfate. The solvent was evaporated and the crude peptide was precipitated in cold petroleum ether, centrifuged and dried under reduced pressure. The obtained solid was dissolved in TFA (6 mL) at 0° C. After stirring for 3 h, the solution was concentrated. Ether (50 mL) and 4 M HCl in 1,4-dioxane (2 mL) were added to the solution to obtain 1.33 g (3.42 mmol, 84.4 %) of crude H-D-Pen(Mob)-Gly-OEt·HCl. The peptide was used for the next reaction without further purification. Boc-Tyr(tBu)-OH (1.38 g, 4.10 mmol) was next coupled in the presence of Cl-HOBt (694 mg, 4.10 mmol), HCTU (1.69 g, 4.10 mmol) and NMM (760 mg, 8.20 mmol) in DMF (5 mL). The reaction mixture was stirred overnight at room temperature. Saturated aqueous sodium bicarbonate was added to the residue and extracted with ethyl acetate three times followed by washing the combined solution with saturated aqueous sodium chloride. The solution was dried over sodium sulfate and concentrated under reduced pressure. The obtained residue was purified using silica gel chromatography (hexane : EtOAc = 10 : 1 to 2 : 1). The residue was dissolved in methanol (8 mL) and THF (8 mL), then 1 M NaOH (9 mL) was added at 0° C. After stirring for 1 h at room temperature, 1 M HCl (10 mL) was added at 0° C, then evaporated. The obtained residue was extracted with ethyl acetate three times followed by washing with 5% aqueous citrate and saturated aqueous sodium chloride. The solution was dried over sodium sulfate. The solvent was evaporated and the crude peptide was precipitated in cold ether. The precipitate was centrifuged, dried under reduced pressure to obtain the title compound (1.53 g, 84.1%).
1H-NMR (DMSO-d6) δ: 1.10–1.32(21H, m), 1.45(3H, s), 1.99(2H, s), 2.28–3.10 (2H, m), 3.68–3.92(5H, m), 4.03(1H, q, J=7.0Hz), 4.80(1H, d, J=9.5Hz), 6.84(4H, d, J=5.5Hz), 7.00(1H, d, J=8.5Hz), 7.18–7.26(4H, m), 8.22(1H, d, J=9.5Hz), 8.65–8.72(1H, m). MS (ESI) 646 (MH)+
Linear Peptide Synthesis
The peptide was synthesized manually by the Nα-Fmoc solid-phase methodology using HCTU as the coupling reagents as previously reported.5, 7 2-Chlorotrityl resin (500 mg, 0.78 mmol/g) was placed into a 50 mL polypropylene syringe with the frit on the bottom and swollen in DMF (10 mL) for 1 h. The resin was washed with DMF (3 × 15 mL) and then with DCM (3 × 15 mL). Fmoc-Trp(Boc)-OH (1.2 equiv.) was dissolved in 30 mL of DCM, and then DIEA (5 equiv.) was added. The reaction mixture was transferred into the syringe with the resin, and then shaken for 2 h. The resin was washed three times with DMF (15 mL) and three times with DCM (15 mL), and then with DMF (3 × 15 mL). The Nα-Fmoc protecting group was removed by 20% piperidine in DMF (10 mL, 1 × 2 min and 1 × 20 min). The deprotected resin was washed with DMF (3 × 15 mL), DCM (3 × 15 mL) and then with DMF (3 × 15 mL). The protected amino acid (3 equiv.) and HCTU (2.9 equiv.) were dissolved in 30 mL of DMF, then DIEA (6 equiv.) was added. Fmoc-D-Pen(Trt)-OH, Fmoc-Pen(Trt)-OH, Fmoc-Pro-OH, Fmoc-Nle-OH and Fmoc-Phe-OH were used for respective coupling as protected amino acids. The following coupling with BocTyr(tBu)-D-Pen(Mob)-Gly-OH (1.5 equiv.) were performed using EDC (2.25 equiv.), HOAt (2.25 equiv.) and NMM (3 equiv.) in DMF (10 mL). The coupling mixture was transferred into the syringe with the resin, and then shaken for 2 h. All the other amino acids were consecutively coupled using the procedure described above, using the TNBS test or chloranil test to check the extent of coupling. In case of a positive test result, the coupling was repeated until a negative test result was obtained. The resulting batch of the resin-bound protected peptide was carefully washed with DMF (3 × 15 mL), DCM (3 × 15 mL), DMF (3 × 15 mL), and DCM (3 × 15 mL), and dried under reduced pressure. The peptide was cleaved off the solid support with 1% v/v TFA in DCM (10 mL) for 30 min, and most of the organic solvent was removed under reduced pressure. The obtained protected peptides with free C-terminal were precipitated out by the addition of chilled petroleum ether (30 mL) to give a white precipitate. The suspensions were centrifuged for 20 min at 7000 rpm, and then the liquid was decanted off. The crude peptides were washed with petroleum ether (2 × 50 mL), and after another centrifugation, the peptides were dried under vacuum (2 h) to obtain the title compound. The purity of the protected peptides with a free C-termini (> 90%) were checked by analytical RP-HPLC using a Hewlett Packard 1100 system (230 nm) on a reverse phase column (Waters NOVA-Pak C-18 column, 3.9 × 150 mm, 5 μm, 60Å). The peptide was eluted with a linear gradient of aqueous CH3CN/0.1% CF3CO2H (10–90% in 40 minutes) at a flow rate of 1.0 mL/min. The crude peptide was used for the subsequent reactions without further purification.
The protected peptide with a free C-terminal (300 mg, 0.173 mmol) and Cl-HOBt (33.4 mg, 0.208 mmol) were dissolved in DMF (3 mL). 3,5-Bistrifluoromethylbenzyl amine (84.1 mg, 0.346 mmol) and EDC (39.7 mg, 0.208 mmol) were added to the solution at RT and stirred until the starting material (the protected peptide with free C-terminal) wasn’t detected by TLC; then saturated aqueous sodium bicarbonate (100 mL) was added. The reaction mixture was extracted with ethyl acetate (100 mL) three times. The combined organic phases were washed with 5% aqueous citrate and saturated aqueous sodium chloride (100 mL each), then dried over sodium sulfate. The solvent was evaporated off and the crude protected peptide was precipitated in cold petroleum ether (45 mL). The product was twice dispersed in cold petroleum ether, centrifuged and decanted, then dried under reduced pressure. The obtained protected peptide was treated with a 6 : 1 mixture of TFA and anisole at 80°C (3 mL, 30 min). The crude peptide was precipitated out by the addition of chilled petroleum ether (45 mL) to give a dark oil. The resulting peptide suspensions were centrifuged for 20 min at 7000 rpm, and the liquid was decanted. The crude peptides were washed with diethyl ether (2 × 45 mL) to give a precipitate, and after a final centrifugation, the peptides were dried under vacuum (2 h). The resulting peptides were directly used for cyclization.
Oxidative Cyclization to Disulfides.25, 45
A solution of K3Fe(CN)6 was prepared as follows: 1 mmol (330 mg) of K3Fe(CN)6 was dissolved in a mixture of water (100 mL) and CH3CN (100 mL). A saturated solution of ammonium acetate (20 mL) was added to it, and the pH was adjusted to 6.0 with glacial acetic acid. A solution of the linear peptide (0.173 mmol) in the mixture of CH3CN (40 mL), DMSO (5 mL) and H2O (5 mL) was added to the above solution dropwise overnight with the help of a syringe pump.25 After the overnight reaction, glacial acetic acid was added to the reaction mixture to obtain pH 5.0, followed by 20 mL of Amberlite IRA-68 anion-exchange resin (pre-equilibrated with 1 M HCl and extensively washed with distilled water), and the suspension stirred for 30 min until the solution turned colorless and the resin turned yellow. The resin was suction-filtered and the filtrate rotoevaporated to remove most of the organic solvent. The remaining solution was concentrated on a Sep-Pak C18 cartridge (10g, Waters, Milford, MA), then eluted with CH3CN. The obtained yellow solution was concentrated under reduced pressure for the final purification by preparative RP-HPLC, and then lyophilized. Preparative RP-HPLC was performed on a Waters Delta Prep 4000 with a Waters XTerra C-18 column (19 × 250 mm, 10 μm, a linear gradient of 33–53% or 40–60% acetonitrile/0.1% TFA at a flow rate of 15.0 mL/min).
The purified final peptides (≥95 %) were characterized by HRMS, TLC, analytical HPLC and 1H-NMR (all the data, charts and experimental details are available in the Supporting Information).
NMR Spectroscopy in DPC Amphipathic Media and Conformational Structure Determination
All of the conformational determinations were performed as follows,5, 6, 35, 36 based on the NMR spectra using a Bruker DRX600 600 MHz spectrometer.
The samples were prepared by dissolving the peptide (4.2 mM) in 0.5 mL of 45 mM sodium acetate-d3 buffer (pH 4.5) containing 40 equivalents of dodecylphosphocholine-d38 and 1 mM sodium azide (90% H2O/10% D2O) followed by sonication for 5 min. Two-dimensional double quantum filtered correlation (DQF-COSY), nuclear Overhauser enhancement spectroscopy46 (NOESY, mixing time = 450 ms), Rotating frame Overhauser Effect Spectroscopy (ROESY, mixing time = 150 ms) and total correlation spectra47 (TOCSY, MLEV-17 mixing time = 62.2 ms, spin-lock field = 8.33 kHz) were acquired using standard pulse sequences at 310 K. Coupling constants (3JNH-Hα) were measured from 2D DQF-COSY spectra by analysis of the fingerprint region with a curve-fitting using 5-parameter Levenberg-Marquardt nonlinear least-squares protocol to a general antiphase doublet.
For the conformational structure determination, the volumes of the assigned cross-peaks in the 2D NOESY spectrum were converted into upper distance bounds of 3.0, 3.8, 4.8, or 5.8 Å. For overlapping cross-peaks, the distance categories were increased by one or two levels, depending on the qualitative estimate of the extent of overlap. Pseudoatoms were created for nonstereospecifically assigned methylene protons with a correction of 1.0 Å applied to their upper bound distances.48 In addition to the distance constraints, ϕ dihedral angle constraints derived from 3JHN-Hα coupling constants were set to between −90 and 40° for 3JHN-Hα< 6 Hz and to between −150 and −90° for 3JHN-Hα> 8 Hz. Dihedral angle constraints of 180 ± 5° for peptide bonds (ω) were also used to maintain the planarity of these bonds.
Structures were calculated using a hybrid distance geometry-dynamical simulated annealing protocol, followed by energy refinement using restrained molecular dynamics (rMD), using the NOE-derived distance constraints and dihedral angle (ϕ) constraints. Distance geometry calculations were performed using the DGII49 program within the INSIGHT II package (Accelrys Inc., San Diego, CA). Solvent was not explicitly included in the calculations. The bounds for the inter-atomic distances were embedded in four dimensional spaces, followed by the optimization using a simulated annealing protocol with sigmoidal cooling schedule from a maximum temperature of 200 K in 10,000 steps at simulation steps of 2.5 ps. The fail level was set to 2.00. All the embedded structures successfully passed the simulated annealing step and were minimized using the consistent valency force field (CVFF) (Accelrys Inc.). The entire process was repeated for 100 times to generate 100 independently optimized structures.
Of those, the 50 structures with the lowest penalty function were further refined by two rounds of rMD using the all-atom AMBER force field with additional parameters for fluorine atom,44, 50–52 using the standalone DISCOVER ver. 2.98 program (Accelrys Inc.).35, 53–55 A 12.0 Å cutoff for nonbonded interactions and a distance-dependent dielectric constant (4r) were used. All amide bonds were constrained to trans conformation by a 100 kcal mol−1 rad−2 energy penalty. The distance constraints and dihedral angles (ϕ) constraints were applied with a force constant of 25 kcal mol−1 Å−2 and 100 kcal mol−1 rad−2 were applied, respectively. After 100 steps of steepest descents minimization and 1000 steps of conjugate gradient minimization on the initial structures, an rMD equilibration at 500 K was performed for 1.5 ps, during which a scale factor of 0.1 was applied to the experimental restraint force constants. During the next 2 ps, full values of the experimental restraint force constants were applied. A further 1 ps rMD simulation was run at 500 K, and the system was then cooled to 0 K over 3 ps. After another 1 ps at 0 K, 100 cycles of steepest descents and 2000 steps of conjugate gradient minimization were performed. The final 10 structures with the lowest energies were used for the analysis. All calculations were performed on a Silicon Graphics Octane computer.
Radioligand Labeled Binding Assay, [35S]GTP-γ-S Binding Assay, GPI and MVD in Vitro Bioassay
The methods were carried out according to that previously described.4–7 Briefly, the evaluation of the binding affinities of the synthesized bifunctional peptide derivatives at the human δ-opioid receptors and rat μ-opioid receptors were performed on the cell (HN9.10) membranes that stably express these corresponding receptors using [3H]-c[D-Pen2, D-Pen5]-enkephalin ([3H]DPDPE, 44 Ci/mmol) and [3H]-[D-Ala2, NMePhe4, Gly5-ol]-enkephalin ([3H]DAMGO, 47.2 Ci/mmol) as radioligands, respectively. The affinity of [3H]DAMGO at the μ-opioid receptor and that of [3H]DPDPE for the δ-opioid receptor was defined by the dissociation constant (KD) determined by saturation analysis as previously described.4–7. For competition analysis, ten concentrations of a test compound were each incubated with 50 μg of membranes from δ- or μ-opioid receptors expressing cells labeled with [3H]DPDPE (1.0 nM) or [3H]DAMGO (1.0 nM) respectively. Naloxone at 10 μM was used to define the non-specific binding of the radioligands in all assays. For the affinity at the human or rat NK1 receptors, binding assays utilized membranes from transfected CHO cells that stably express corresponding receptors, using [3H]-substance P (135 Ci/mmol, 0.3–0.4 nM) as the standard radioligand. The [3H] substance P concentration was selected based on the saturation binding experiments which showed a high affinity binding with KD values of 0.40 ± 0.17 for hNK1 and 0.16 ± 0.03 nM for rNK1, respectively. Substance P at 10 μM was used to define the non-specific binding. All data were determined from a minimum of 2 independent analyses. The data were analyzed by non-linear least squares regression analysis using GraphPad Prism.
The [35S]GTPγS binding assays were used to estimate the functional activities of the test compounds at the δ and μ opioid receptors expressed in the transfected cells. Data were analyzed by non-linear least squares analysis using GraphPad Prism to determine the EC50 and Emax values for each test compound. The isolated tissue-based functional assays also were used to evaluate opioid agonist activities in the GPI (μ) and MVD (δ). Dose effects of the test compounds against substance P stimulation were determined in isolated GPI in the presence of 1 μM naloxone.
In Vitro Stability of Peptide Derivatives in Rat Plasma6
Stock solution of compounds (50 mg/mL in DMSO) were diluted 1000-fold into rat plasma (Lot 24927, Pel-Freez Biologicals, Rogers, AK) to give an incubation concentration of 50 μg/mL. All samples were incubated at 37 °C and 200μL of aliquots were withdrawn at 1 h, 2 h, 4 h and 6 h. Then 300 μL of acetonitrile was added and the proteins were removed by centrifugation. The supernatant was analyzed for the amount of remaining parent compound by HPLC (Hewlett Packard 1090m with Vydac 218TP104 C-18 column; 4.6 × 250 mm, 10 μm, 300 Å). The samples were tested in three independent experiments (n = 3) and the mean values ± SD were used for the analysis. The statistical significances were evaluated with the Student t-test.
Supplementary Material
Figure 5.

The Ramachandran ϕ, ψ plots for 2 for residues 2–8 of 10 final structures.
Acknowledgments
The work was supported by grants from the USDHS, National Institute on Drug Abuse, DA-13449 and DA-06284. We thank Dr. Guangxin Lin for kind assistance and advice with the NMR measurements, Ms. Magdalena Kaczmarska for culturing cells, and the University of Arizona Mass Spectrometry Facility for the mass spectral measurements. We express appreciation to Ms. Margie Colie for assistance with the manuscript.
A List of Abbreviations
The following additional abbreviations are used:
- Boc
tert-butyloxycarbonyl
- Cl-HOBt
1-hydroxy-6-chlorobenzotriazole
- CHO
Chinese hamster ovary
- DCM
dichloromethane
- DIEA
diisopropylethylamine
- DMF
N, N-dimethylformamide
- DMSO
dimethylsulfoxide
- DPC
Dodecylphosphocholine
- DPDPE
Tyr-cyclo[D-Pen-Gly-Phe-D-Pen]-OH
- DPLPE
Tyr-cyclo[D-Pen-Gly-Phe-Pen]-OH
- DQF-COSY
double quantum filtered correlation spectroscoopy
- DAMGO
[D-Ala2, NMePhe4, Gly5-ol]-enkephalin
- Fmoc
fluorenylmethoxycarbonyl
- GPI
guinea pig isolated ileum
- HBTU
1-[Bis(dimethylamino)methylene]-1H-benzotriazolium 3-oxide hexafluorophosphate
- HCTU
1H-Benzotriazolium-1-[bis(dimethylamino)methylene]-5-chloro-hexafluorophosphate-(1-),3- oxide
- HRMS
high-resolution mass spectroscopy
- LMMP
longitudinal muscle with myenteric plexus
- Mob
p-methoxy benzyl
- MVD
mouse vas deferens
- NK1
Neurokinin-1
- NOESY
nuclear Overhauser enhancement
- rmsd
root mean square deviation
- NMM
N-methyl morpholine
- RP-HPLC
reverse phase high performance liquid chromatography
- TFA
trifluoroacetic acid
- TLC
thin-layer chromatography
- TOCSY
total correlation spectroscopy
- Trp-NH-[3′
5′-(CF3)2-Bzl], the 3′,5′-(bistrifluoromethyl)-benzyl amide of tryptophan
Footnotes
Supporting Information Available: 1H-NMR, HPLC and MS data of the cyclic peptide derivatives 2–5 and their intermediates. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Mantyh PW, Allen CJ, Ghilardi JR, Rogers SD, Mantyh CR, Liu H, Basbaum AI, Vigna SR, Maggio JE. Rapid endocytosis of a G protein-coupled receptor: substance P evoked internalization of its receptor in the rat striatum in vivo. Proc Natl Acad Sci U S A. 1995;92:2622–2626. doi: 10.1073/pnas.92.7.2622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kalso E. Improving opioid effectiveness: from ideas to evidence. Eur J Pain. 2005;9:131–135. doi: 10.1016/j.ejpain.2004.05.007. [DOI] [PubMed] [Google Scholar]
- 3.King T, Ossipov MH, Vanderah TW, Porreca F, Lai J. Is paradoxical pain induced by sustained opioid exposure an underlying mechanism of opioid antinociceptive tolerance? Neurosignals. 2005;14:194–205. doi: 10.1159/000087658. [DOI] [PubMed] [Google Scholar]
- 4.Yamamoto T, Nair P, Davis P, Ma SW, Navratilova E, Moye M, Tumati S, Vanderah TW, Lai J, Porreca F, Yamamura HI, Hruby VJ. Design, Synthesis and Biological Evaluation of Novel Bifunctional C-terminal Modified Peptides for δ/μ Opioid Receptor Agonists and Neurokinin-1 Receptor Antagonists. J Med Chem. 2007;50:2779–2786. doi: 10.1021/jm061369n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yamamoto T, Nair P, Jacobsen NE, Davis P, Ma SW, Navratilova E, Lai J, Yamamura HI, Vanderah TW, Porreca F, Hruby VJ. The Importance of Micelle-Bound States for the Bioactivities of Bifunctional Peptide Derivatives for δ/μ Opioid Receptor Agonists and Neurokinin 1 Receptor Antagonists. J Med Chem. 2008;51:6334–6347. doi: 10.1021/jm800389v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yamamoto T, Nair P, Jacobsen NE, Vagner J, Kulkarni V, Davis P, Ma SW, Navratilova E, Yamamura HI, Vanderah TW, Porreca F, Lai J, Hruby VJ. Improving metabolic stability by glycosylation: bifunctional peptide derivatives that are opioid receptor agonists and neurokinin 1 receptor antagonists. J Med Chem. 2009;52:5164–5175. doi: 10.1021/jm900473p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yamamoto T, Nair P, Vagner J, Davis P, Ma SW, Navratilova E, Moye M, Tumati S, Vanderah TW, Lai J, Porreca F, Yamamura HI, Hruby VJ. A Structure Activity Relationship Study and Combinatorial Synthetic Approach of C-Terminal Modified Bifunctional Peptides That Are δ/μ Opioid Receptor Agonists and Neurokinin 1 Receptor Antagonists. J Med Chem. 2008;51:1369–1376. doi: 10.1021/jm070332f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.King T, Gardell LR, Wang R, Vardanyan A, Ossipov MH, Malan TP, Jr, Vanderah TW, Hunt SP, Hruby VJ, Lai J, Porreca F. Role of NK-1 neurotransmission in opioid-induced hyperalgesia. Pain. 2005;116:276–288. doi: 10.1016/j.pain.2005.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ma W, Zheng WH, Kar S, Quirion R. Morphine treatment induced calcitonin gene-related peptide and substance P increases in cultured dorsal root ganglion neurons. Neuroscience. 2000;99:529–539. doi: 10.1016/s0306-4522(00)00226-8. [DOI] [PubMed] [Google Scholar]
- 10.Powell KJ, Quirion R, Jhamandas K. Inhibition of neurokinin-1-substance P receptor and prostanoid activity prevents and reverses the development of morphine tolerance in vivo and the morphine-induced increase in CGRP expression in cultured dorsal root ganglion neurons. Eur J Neurosci. 2003;18:1572–1583. doi: 10.1046/j.1460-9568.2003.02887.x. [DOI] [PubMed] [Google Scholar]
- 11.Misterek K, Maszczynska I, Dorociak A, Gumulka SW, Carr DB, Szyfelbein SK, Lipkowski AW. Spinal co-administration of peptide substance P antagonist increases antinociceptive effect of the opioid peptide biphalin. Life Sci. 1994;54:939–944. doi: 10.1016/0024-3205(94)00494-3. [DOI] [PubMed] [Google Scholar]
- 12.Gu G, Kondo I, Hua XY, Yaksh TL. Resting and evoked spinal substance P release during chronic intrathecal morphine infusion: parallels with tolerance and dependence. J Pharmacol Exp Ther. 2005;314:1362–1369. doi: 10.1124/jpet.105.087718. [DOI] [PubMed] [Google Scholar]
- 13.Ripley TL, Gadd CA, De Felipe C, Hunt SP, Stephens DN. Lack of self-administration and behavioural sensitisation to morphine, but not cocaine, in mice lacking NK1 receptors. Neuropharmacology. 2002;43:1258–1268. doi: 10.1016/s0028-3908(02)00295-2. [DOI] [PubMed] [Google Scholar]
- 14.Morphy R, Rankovic Z. Designed multiple ligands. An emerging drug discovery paradigm. J Med Chem. 2005;48:6523–6543. doi: 10.1021/jm058225d. [DOI] [PubMed] [Google Scholar]
- 15.Ling GS, Spiegel K, Lockhart SH, Pasternak GW. Separation of opioid analgesia from respiratory depression: evidence for different receptor mechanisms. J Pharmacol Exp Ther. 1985;232:149–155. [PubMed] [Google Scholar]
- 16.Paakkari P, Paakkari I, Vonhof S, Feuerstein G, Siren AL. Dermorphin analog Tyr-D-Arg2-Phe-sarcosine-induced opioid analgesia and respiratory stimulation: the role of mu 1-receptors? J Pharmacol Exp Ther. 1993;266:544–550. [PubMed] [Google Scholar]
- 17.Chang KJ, Rigdon GC, Howard JL, McNutt RW. A novel potent and selective nonpeptidic delta opioid receptor agonist BW373U86. J Pharmacol Exp Ther. 1993;267:852–857. [PubMed] [Google Scholar]
- 18.Sheldon RJ, Riviere PJ, Malarchik ME, Mosberg HI, Burks TF, Porreca F. Opioid regulation of mucosal ion transport in the mouse isolated jejunum. J Pharmacol Exp Ther. 1990;253:144–151. [PubMed] [Google Scholar]
- 19.Cowan A, Zhu XZ, Mosberg HI, Omnaas JR, Porreca F. Direct dependence studies in rats with agents selective for different types of opioid receptor. J Pharmacol Exp Ther. 1988;246:950–955. [PubMed] [Google Scholar]
- 20.Quock RM, Burkey TH, Varga E, Hosohata Y, Hosohata K, Cowell SM, Slate CA, Ehlert FJ, Roeske WR, Yamamura HI. The delta-opioid receptor: molecular pharmacology, signal transduction, and the determination of drug efficacy. Pharmacol Rev. 1999;51:503–532. [PubMed] [Google Scholar]
- 21.Rew Y, Malkmus S, Svensson C, Yaksh TL, Chung NN, Schiller PW, Cassel JA, DeHaven RN, Taulane JP, Goodman M. Synthesis and biological activities of cyclic lanthionine enkephalin analogues: delta-opioid receptor selective ligands. J Med Chem. 2002;45:3746–3754. doi: 10.1021/jm020108k. [DOI] [PubMed] [Google Scholar]
- 22.Coop A, MacKerell ADJ. The future of opioid analgesics. Am J Pharm Educ. 2002;66:153–156. [Google Scholar]
- 23.Largent-Milnes TM, Yamamoto T, Nair P, Navratrilova E, Davis P, Ma S-W, Hruby VJ, Yamamura HI, Lai J, Porreca F, Vanderah TW. Dual acting opioid agonist/NK1 antagonist peptide reverses neuropathic pain in an animal model without demonstrating common opioid unwanted side effects. International Association for the Study of Pain/12th World Congress on Pain; Glasgow, Scotland. 2008. [Google Scholar]
- 24.Largent-Milnes TM, Yamamoto T, Nair P, Hruby VJ, Yamamura HI, Lai J, Porreca F, Vanderah TW. Spinal or systemic TY005, a peptidic opioid agonist/neurokinin 1 antagonist, attenuates pain with reduced tolerance. Br J Pharmacol. doi: 10.1111/j.1476-5381.2010.00824.x. accepted with revision 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yamamoto T, Nair P, Davis P, Ma SW, Yamamura HI, Vanderah TW, Porreca F, Lai J, Hruby VJ. The biological activity and metabolic stability of peptidic bifunctional compounds that are opioid receptor agonists and neurokinin 1 receptor antagonists with a cystine moiety. Bioorg Med Chem. 2009;17:7337–7343. doi: 10.1016/j.bmc.2009.08.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Haaseth RC, Horan PJ, Bilsky EJ, Davis P, Zalewska T, Slaninova J, Yamamura HI, Weber SJ, Davis TP, Porreca F, Hruby VJ. [L-Ala3]DPDPE: a new enkephalin analog with a unique opioid receptor activity profile. Further evidence of delta-opioid receptor multiplicity. J Med Chem. 1994;37:1572–1577. doi: 10.1021/jm00037a007. [DOI] [PubMed] [Google Scholar]
- 27.Toth G, Russell KC, Landis G, Kramer TH, Fang L, Knapp R, Davis P, Burks TF, Yamamura HI, Hruby VJ. Ring substituted and other conformationally constrained tyrosine analogues of [D-Pen2, D-Pen5]enkephalin with delta opioid receptor selectivity. J Med Chem. 1992;35:2384–2391. doi: 10.1021/jm00091a006. [DOI] [PubMed] [Google Scholar]
- 28.Mosberg HI, Hurst R, Hruby VJ, Gee K, Yamamura HI, Galligan JJ, Burks TF. Bis-penicillamine enkephalins possess highly improved specificity toward delta opioid receptors. Proc Natl Acad Sci U S A. 1983;80:5871–5874. doi: 10.1073/pnas.80.19.5871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Flippen-Anderson JL, Hruby VJ, Collins N, George C, Cudney B. X-ray Structure of [D-Pen2,D-Pen5]enkephain, a Highly Potent, delta Opioid Receptor-Selective Compound: Comparisons with Proposed Solution Conformations. J Am Chem Soc. 1994;116:7523–7531. [Google Scholar]
- 30.Mosberg HI, Omnaas JR, Goldstein A. Structural requirements for delta opioid receptor binding. Mol Pharmacol. 1987;31:599–602. [PubMed] [Google Scholar]
- 31.Millet R, Goossens L, Bertrand-Caumont K, Chavatte P, Houssin R, Henichart JP. Synthesis and biological evaluation of tripeptide derivatives of Cbz-Gly-Leu-Trp-OBzl(CF3)2 as NK1/NK2 ligands. Lett Pep Sci. 1999;6:255–262. [Google Scholar]
- 32.Barlos K, Gatos D. Convergent Peptide Synthesis. In: Chan WC, White PD, editors. Fmoc Solid Phase Peptide Synthesis: A Practical Approach. Oxford University Press; USA: 2000. pp. 214–228. [Google Scholar]
- 33.D’Alagni M, Delfini M, Di Nola A, Eisenberg M, Paci M, Roda LG, Veglia G. Conformational study of [Met5]enkephalin-Arg-Phe in the presence of phosphatidylserine vesicles. Eur J Biochem. 1996;240:540–549. doi: 10.1111/j.1432-1033.1996.0540h.x. [DOI] [PubMed] [Google Scholar]
- 34.Deber CM, Behnam BA. Role of membrane lipids in peptide hormone function: binding of enkephalins to micelles. Proc Natl Acad Sci U S A. 1984;81:61–65. doi: 10.1073/pnas.81.1.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jacobsen NE, Abadi N, Sliwkowski MX, Reilly D, Skelton NJ, Fairbrother WJ. High-resolution solution structure of the EGF-like domain of heregulin-alpha. Biochemistry. 1996;35:3402–3417. doi: 10.1021/bi952626l. [DOI] [PubMed] [Google Scholar]
- 36.Ying J, Ahn JM, Jacobsen NE, Brown MF, Hruby VJ. NMR solution structure of the glucagon antagonist [desHis1, desPhe6, Glu9]glucagon amide in the presence of perdeuterated dodecylphosphocholine micelles. Biochemistry. 2003;42:2825–2835. doi: 10.1021/bi026629r. [DOI] [PubMed] [Google Scholar]
- 37.Braun W, Wider G, Lee KH, Wuthrich K. Conformation of glucagon in a lipid-water interphase by 1H nuclear magnetic resonance. J Mol Biol. 1983;169:921–948. doi: 10.1016/s0022-2836(83)80143-0. [DOI] [PubMed] [Google Scholar]
- 38.Thornton K, Gorenstein DG. Structure of glucagon-like peptide (7–36) amide in a dodecylphosphocholine micelle as determined by 2D NMR. Biochemistry. 1994;33:3532–3539. doi: 10.1021/bi00178a009. [DOI] [PubMed] [Google Scholar]
- 39.Karslake C, Piotto ME, Pak YK, Weiner H, Gorenstein DG. 2D NMR and structural model for a mitochondrial signal peptide bound to a micelle. Biochemistry. 1990;29:9872–9878. doi: 10.1021/bi00494a017. [DOI] [PubMed] [Google Scholar]
- 40.Arora A, Abildgaard F, Bushweller JH, Tamm LK. Structure of outer membrane protein A transmembrane domain by NMR spectroscopy. Nat Struct Biol. 2001;8:334–338. doi: 10.1038/86214. [DOI] [PubMed] [Google Scholar]
- 41.Hruby VJ, Gehrig CA. Recent developments in the design of receptor specific opioid peptides. Med Res Rev. 1989;9:343–401. doi: 10.1002/med.2610090306. [DOI] [PubMed] [Google Scholar]
- 42.Flippen-Anderson JL, Hruby VJ, Collins N, George C, Cudney B. X-ray Structure of [D-Pen2, D-Pen5]enkephain, a Highly Potent, delta Opioid Receptor-Selective Compound: Comparisons with Proposed Solution Conformations. J Am Chem Soc. 1994;116:7523–31. [Google Scholar]
- 43.Shenderovich MD, Kover KE, Nikiforovich GV, Jiao D, Hruby VJ. Conformational analysis of beta-methyl-para-nitrophenylalanine stereoisomers of cyclo[D-Pen2, D-Pen5]enkephalin by NMR spectroscopy and conformational energy calculations. Biopolymers. 1996;38:141–156. doi: 10.1002/(sici)1097-0282(199602)38:2<141::aid-bip2>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- 44.2007 February; http://amber.scripps.edu/Questions/fluorine.html.
- 45.Balse-Srinivasan P, Grieco P, Cai M, Trivedi D, Hruby VJ. Structure-activity relationships of novel cyclic alpha-MSH/beta-MSH hybrid analogues that lead to potent and selective ligands for the human MC3R and human MC5R. J Med Chem. 2003;46:3728–3733. doi: 10.1021/jm030111j. [DOI] [PubMed] [Google Scholar]
- 46.Kumar A, Ernst RR, Wüthrich K. A two-dimensional nuclear Overhauser enhancement (2D NOE) experiment for the elucidation of complete proton-proton cross-relaxation networks in biological macromolecules. Biochem Biophys Res Commun. 1980;95:1–6. doi: 10.1016/0006-291x(80)90695-6. [DOI] [PubMed] [Google Scholar]
- 47.Davis DG, Bax A. Assignment of complex proton NMR spectra via two-dimensional homonuclear Hartmann-Hahn spectroscopy. J Am Chem Soc. 1985;107:2820–2821. [Google Scholar]
- 48.Wüthrich KBM, Braun W. Pseudo-structures for the 20 common amino acids for use in studies of protein conformations by measurements of intramolecular proton-proton distance constraints with nuclear magnetic resonance. J Mol Biol. 1983;169:949–961. doi: 10.1016/s0022-2836(83)80144-2. [DOI] [PubMed] [Google Scholar]
- 49.Havel TF. An evaluation of computational strategies for use in the determination of protein structure from distance constraints obtained by nuclear magnetic resonance. Prog Biophys Mol Biol. 1991;56:43–78. doi: 10.1016/0079-6107(91)90007-f. [DOI] [PubMed] [Google Scholar]
- 50.Weiner SJ, Kollman PA, Case DA, Singh UC, Ghio C, Alagona GS, Profeta J, Weiner P. A New Force Field for Molecular Mechanical Simulation of Nucleic Acids and Proteins. J Am Chem Soc. 1984;106:765–784. [Google Scholar]
- 51.Weiner SJ, Kollman PA, Case DA. An all atom force field for simulations of proteins and nucleic acids. J Comput Chem. 1986;7:230–252. doi: 10.1002/jcc.540070216. [DOI] [PubMed] [Google Scholar]
- 52.Gough CA, DeBolt SE, Kollman PA. Derivation of fluorine and hydrogen atom parameters using liquid simulations. J Comp Chem. 1992;13:963–970. [Google Scholar]
- 53.Milbradt AG, Kerek F, Moroder L, Renner C. Structural characterization of hellethionins from Helleborus purpurascens. Biochemistry. 2003;42:2404–2411. doi: 10.1021/bi020628h. [DOI] [PubMed] [Google Scholar]
- 54.Renner C, Behrendt R, Sporlein S, Wachtveitl J, Moroder L. Photomodulation of conformational states. I. Mono- and bicyclic peptides with (4-amino)phenylazobenzoic acid as backbone constituent. Biopolymers. 2000;54:489–500. doi: 10.1002/1097-0282(200012)54:7<489::AID-BIP20>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 55.Fuhrmans M, Milbradt AG, Renner C. Comparison of Protocols for Calculation of Peptide Structures from Experimental NMR Data. J Chem Theory Comput. 2006;2:201–208. doi: 10.1021/ct050203j. [DOI] [PubMed] [Google Scholar]
- 56.Lipkowski AW, Misicka A, Davis P, Stropova D, Janders J, Lachwa M, Porreca F, Yamamura HI, Hruby VJ. Biological activity of fragments and analogues of the potent dimeric opioid peptide, biphalin. Bioorg Med Chem Lett. 1999;9:2763–2766. doi: 10.1016/s0960-894x(99)00464-3. [DOI] [PubMed] [Google Scholar]
- 57.Wagner G, Neuhaus D, Worgotter E, Vasak M, Kagi JH, Wuthrich K. Nuclear magnetic resonance identification of “half-turn” and 3(10)-helix secondary structure in rabbit liver metallothionein-2. J Mol Biol. 1986;187:131–135. doi: 10.1016/0022-2836(86)90413-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.