

Summary
Characterizing structural variants in the human genome is of great importance, but a genome wide analysis to detect interspersed repeats has not been done. Thus, the degree to which mobile DNAs contribute to genetic diversity, heritable disease, and oncogenesis remains speculative. We perform transposon insertion profiling by microarray (TIP-chip) to map human L1(Ta) retrotransposons (LINE-1 s) genome-wide. This identified numerous novel human L1(Ta) insertional polymorphisms with highly variant allelic frequencies. We also explored TIP-chip's usefulness to identify candidate alleles associated with different phenotypes in clinical cohorts. Our data suggest that the occurrence of new insertions is twice as high as previously estimated, and that these repeats are under-recognized as sources of human genomic and phenotypic diversity. We have just begun to probe the universe of human L1(Ta) polymorphisms, and as TIP-chip is applied to other insertions such as Alu SINEs, it will expand the catalog of genomic variants even further.
Introduction
Following completion of the human genome reference sequence, comparative genomics across and within species is identifying functional elements and establishing relationships between genetic variation and phenotypic diversity. The HapMap project addresses interindividual human SNP variation (International HapMap Consortium, 2003). Recent studies have shown the human genome also contains extensive structural variants (SVs), encompassing in aggregate greater nucleotide content than SNPs, and with potential relationships to genetic diseases (Stankiewicz and Lupski, 2010). A more extensive and specialized toolkit of molecular methods is required to fully appreciate this dynamic dimension of our genomes.
Studies of SVs, mainly focused on copy number variations (CNVs), rely on fosmid library paired-end sequencing and comparative genomic hybridization (CGH) (Scherer et al., 2007). These techniques are somewhat biased against the discovery of those SVs that are less than several kb long and high in copy number; these account for the majority of human SV sequence. Therefore, important gaps remain in understanding the full spectrum of human genetic variation. SV of high copy interspersed repeats or insertion sequence variations (ISVs) (Gresham et al., 2008) are relatively uncharacterized. These sequences, most of which are derived from “copy-and-paste” retroelements, differ in structure, copy number, and location. They pose a significant challenge even to whole-genome sequencing, and are often underrepresented in genome assemblies.
Besides representing a major class of structural variant, ISVs can serve as sites for nonallelic homologous recombination to create CNVs. Large genome-wide studies have found statistical enrichment of mobile DNAs near CNVs and translocation or inversion breakpoints (Cooper et al., 2007; Korbel et al., 2007). Examples of L1 or Alu involved in disease related deletions and translocation junctions are also well-documented (Gu et al., 2008; Kolomietz et al., 2002; Morisada et al., 2010); however most were discovered by CGH and related methods which are blind to new insertions.
ISVs reflecting polymorphic mobile element insertions have significant functional impact. Short interspersed elements (SINEs, such as Alus) frequently serve as gene enhancers and promoters or affect transcript structure (i.e., being incorporated into exons or used as sites for alternative mRNA splicing; (Cordaux and Batzer, 2009). Similarly, evidence suggests that long interspersed elements 1 (LINE1/L1) can alter mRNA splicing of target transcripts (Belancio et al., 2008; Speek, 2001), result in transcript initiation or truncation/reinitiation (Wheelan et al., 2005), and may play roles in generating neuronal diversity (Muotri et al., 2005). Intronic L1 insertions can also damp or otherwise subtly alter gene expression (Chen et al., 2006; Han and Boeke, 2005; Han et al., 2004). Indeed, intronic insertions can lead to decreased transcript levels and loss of gene function in humans (Schwahn et al., 1998; Ustyugova et al., 2006) and other mammals (Credille et al., 2009; Yajima et al., 1999). Therefore, such intronic L1 insertions might well predispose to complex traits in humans. There are also examples of exonic mobile element insertions causing genetic diseases by germline or early embryonic integration (Kazazian et al., 1988; Van de Water et al., 1998) and transforming mutations in cancer by somatic insertion (Miki et al., 1992). It is unknown whether the relative rarity of such reports reflects mobile element quiescence in the context of effective host defense mechanisms or systematic biases against their discovery. Even in a healthy individual, we still do not know the number of mobile element copies of each type present or the frequencies of these alleles in the general population. For all these reasons, we developed an effective general tool, TIP-chip, for genome-wide discovery of human insertion polymorphisms.
We describe here primarily our findings using TIP-chip to identify polymorphic L1 insertions. Most ISV families (L1, Alu, and SVA) continue to accumulate in our genomes through the activity of L1, specifically, the youngest L1 family, L1PA1, also known as the “transcribed L1, subset a” or L1(Ta)s (Skowronski et al., 1988). In vitro assays unequivocally demonstrate the retrotransposition capacity of full-length (6 kb) intact L1(Ta)s (Brouha et al., 2003; Moran et al., 1996); nearly all known human L1 polymorphisms are related to L1(Ta) insertions.
Results
Transposon Insertion Profiling by Microarray Method for L1(Ta)
Our approach for identifying L1(Ta) ISV depends on a ligation-mediated PCR (Arnold and Hodgson, 1991; Wheelan et al., 2006). In this method, partially complementary oligonucleotides (vectorettes) are ligated to restriction enzyme (RE) digested genomic DNA. This requires first strand PCR priming from known (transposon end) sequence. The 3′ terminus of the first strand primer hybridizes to a three base pair sequence unique to the L1(Ta) subset (Skowronski et al., 1988). In subsequent cycles, the 3′ end of the first strand pairs with a second primer allowing exponential amplification. The resulting amplicons include the extreme 3′ end of the L1(Ta) and unique downstream DNA sequence. The amplicon mixture is fluorescently labeled and hybridized to tiling microarrays (Figure 1). TIP-chip data consist of small numbers of high intensity probes (Figure S1 available online) recognizable as peaks formed by contiguous probes when associated with corresponding genomic locations. Multiple PCR templates are generated for each genomic L1(Ta) by parallel RE digests prior to vectorette ligation. The combination of REs used maximizes genomic coverage; an insertion lies within 1–5 kb of at least one 3′ RE site in approximately 92% of the genome (Experimental Procedures). The interprobe distance on the tiling arrays used is such that for approximately 90.5% of the genome, there are three probes (average of 7) within 3 kb of an insertion. Since sequences closer to the 3′ end of an insertion are included in more RE fragments, and shorter templates amplify better, there is an inverse relationship between probe fluorescence intensity and distance from the L1(Ta). Thus, peak shapes reflect both insertion position and orientation.
Figure 1. Transposon Insertion Profiling Chip Method.
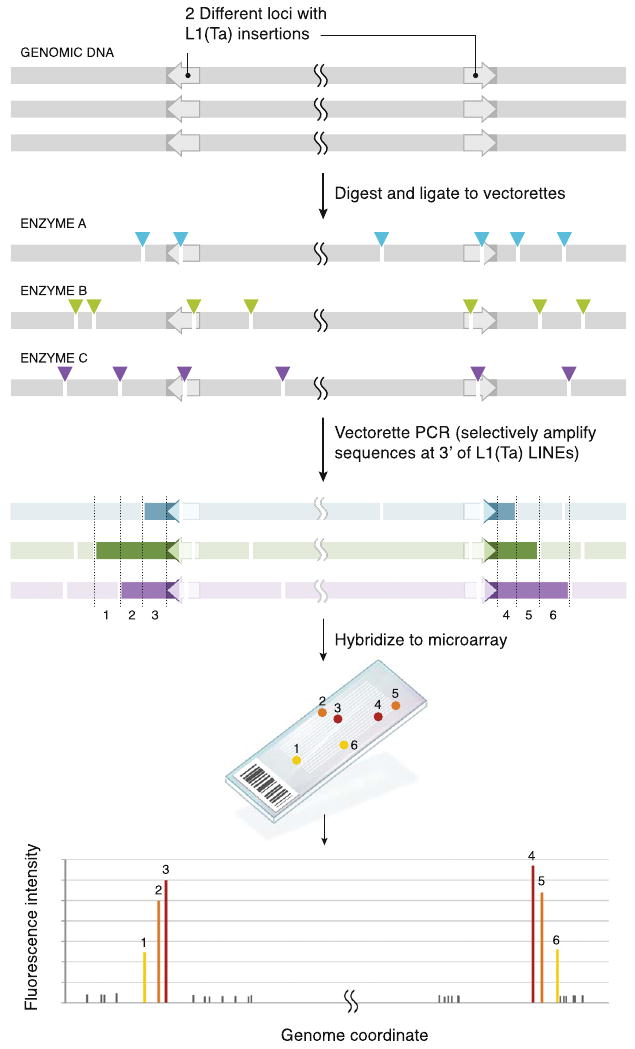
Human genomic DNA contains numerous L1(Ta) insertions (arrows 5′ → 3′); minus (left) and plus strand (right) insertion are illustrated here. Multiple copies of genomic DNA are digested in parallel with different REs (colored arrows, sites; each color is a different RE), and vectorette linkers (data not shown) are ligated to fragments. Vectorette PCR then specifically amplifies 3′ L1(Ta) sequence and unique genomic sequence 3′ of the L1(Ta) insertions (resulting amplicons are denoted by colored fragments). The cuts create a series of variable-length PCR templates for each L1(Ta) insertion. Genomic DNA fragments lacking L1(Ta)insertions are not amplified. Amplicons are labeled and hybridized to genomic tiling microarrays, generating peaks of signal intensity at probes (1–6) corresponding to genomic locations immediately adjacent to L1(Ta) insertions. For each peak, probes closest to L1(Ta) have highest fluorescence intensity with a gradient of diminishing signal seen downstream of the insertion because proximal probes are represented in more PCR products and shorter PCR products including them amplify more efficiently. Thus, slope of the signal gradient (±) opposes insertion orientation.
See also Figure S1.
TIP-Chip for Alu and HERV-K
To evaluate the general applicability of TIP-chip to map other human mobile elements, we designed primers specific to SINEs that have been recently expanding in humans through the activity of L1(Ta), AluYa5/8 families and AluYb8/9 families, and a family of autonomous endogenous retrovirus, Hs_a HERV-K (Experimental Procedures). In all three cases we were able to detect numerous insertions of those types included in the March 2006 human reference sequence (hs_ref [hg18] NCBI Build 36.1) (Figure S1). HERV-K TIP-chip showed relatively lower numbers of insertions and levels of polymorphism, though we were able to discover nonreference LTRs. Alu insertions in contrast are abundant and highly polymorphic. In addition to intergenic and intronic Alus, we discovered a polymorphic exonic AluYb in the complement component 7 (C7) locus in the first sample we analyzed (Xing et al., 2009). Thus, TIP-chip appears a robust method for identifying insertions of a wide variety of transposable element types. For the remainder of the paper, we focus on our experience with L1(Ta) detection by TIP-chip.
L1(Ta) Discovery and Inheritance Patterns on the X Chromosome
To test L1(Ta) TIP-chip utility and reproducibility, we used 385K feature X chromosome genomic tiling arrays. As expected, numerous peak positions corresponded to one of the 38 known L1(Ta)s exactly matching our forward primer sequence the hs_ref (Figure 2). In a family of 4, we identified 28 peaks reflecting reference L1(Ta)s, and correctly identified orientation in 84% of these. No non-hs_ref L1(Ta) insertions cataloged in the database of human retrotransposon insertion polymorphisms (dbRIP) (Wang et al., 2006) or included in alternate genome assemblies (Levy et al., 2007; Venter et al., 2001) were detected (Table S1). Importantly, however, this experiment showed 6 previously unknown L1(Ta)s which were verified by 3′ junction PCR analyses (Table S1). Of the 34 L1(Ta)s seen in this family, 13 were polymorphic. All showed sex-linked inheritance.
Figure 2. Inheritance Pattern of X Chromosome L1s.
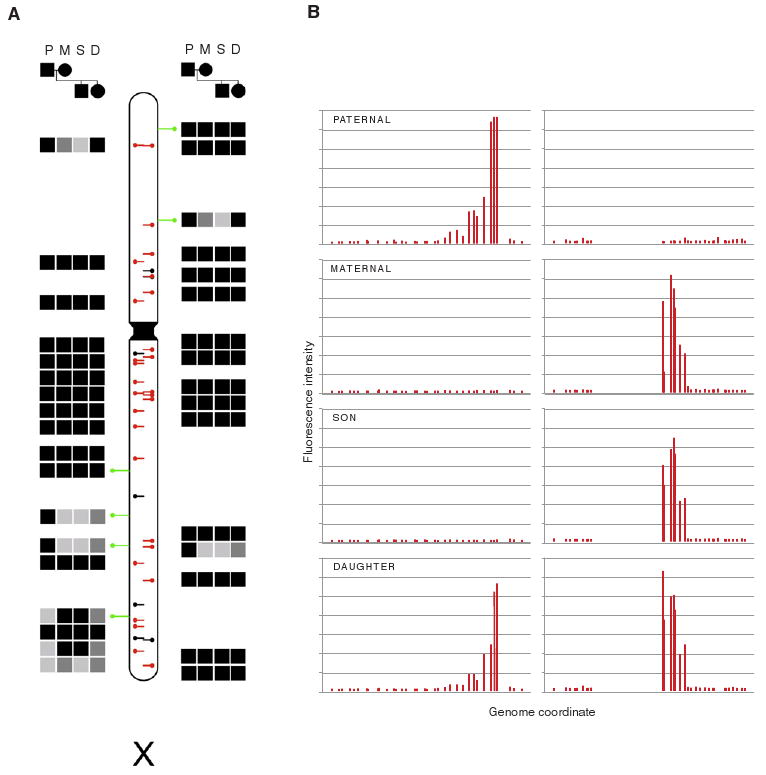
(A) L1(Ta) insertion profiles were generated for a family by TIP-chip using X chromosome microarrays. Presence (filled squares) or absence (empty squares) of peaks is indicated in paternal (P), maternal (M), son (S), and daughter (D) samples. Black or gray filled squares indicate an L1(Ta) detected at a specific site, as opposed to no fill; gray indicates inferred heterozygosity. Lollipops on the ideogram correspond to insertion coordinates. Black lines in center mark L1(Ta) incorporated in hs_ref NCBI Build 36.1. These are overlaid with red where observed. Green lines are PCR-verified novel insertions. Side represents insertion orientation (left = plus strand). In this family, 6 L1(Ta)s are paternal, nonmaternal; 4 are maternal, nonpaternal; and 4 additional maternal L1(Ta)s were not passed to her son, indicating maternal heterozygosity. Thus at least 33.33% of insertions found are polymorphic in this family.
(B) Raw intensity data of two representative reference L1(Ta) insertions (one in each orientation) across four family members. x axis indicates genomic coordinate. Probe fluorescence intensity is shown on y axis. Each bar represents one array probe.
Genome-wide L1(Ta) Identification
We tested whether TIP-chip could comprehensively map L1(Ta)s on a whole-genome tiling microarray (four 2.1M feature arrays). For data analysis, we developed a Hidden Markov Model (HMM) for recognizing characteristically asymmetric peaks and imposed a multivariate cutoff algorithm for retaining peaks (Experimental Procedures). Figure 3A illustrates distribution of L1(Ta) peaks in peripheral blood leukocytes (PBL) DNA from a healthy individual (sample 1); data from other representative samples are included in Figure S2. In these examples, we recognize a range of total peak numbers in excess of Ne, the expected number of different L1(Ta) alleles per diploid human genome (515; see Experimental Procedures) and impose a cutoff based on this value. In the sample illustrated in Figure 3A, we retained 514 peaks, 323 of which correspond to reference L1(Ta)s. Of the 191 candidate non-hs_ref L1(Ta)s identified in the sample, 49 were in dbRIP (Wang et al., 2006) or included in the alternate genome assemblies, 3 were confirmed by data in Beck et al. (2010) [this issue of Cell]. We attempted to verify 139 others by site-specific PCR crossing the 3′ junction of the L1 or spanning the insertion (Table S1) and recovered amplicons consistent with 91 insertions. Of a sequenced subset, 22 were sequence-verified, a recovery which allowed us to estimate that 56 reflect true L1 insertions. Thus, of novel peaks retained by the cutoff algorithm, 108 appear to represent true insertions verifiable by data mining or PCR validation, for an overall assay positive predictive value of 84%. Additionally, in this sample, we were able to sequence verify an additional seven insertions among peaks that did not meet the cutoff. Thus, cutoff criteria can be relaxed to maximize new L1(Ta) discovery, but is kept close to Ne here to conservatively reflect the expected number of true positives.
Figure 3. Genome-wide Mapping of L1(Ta) Insertions in an Individual.
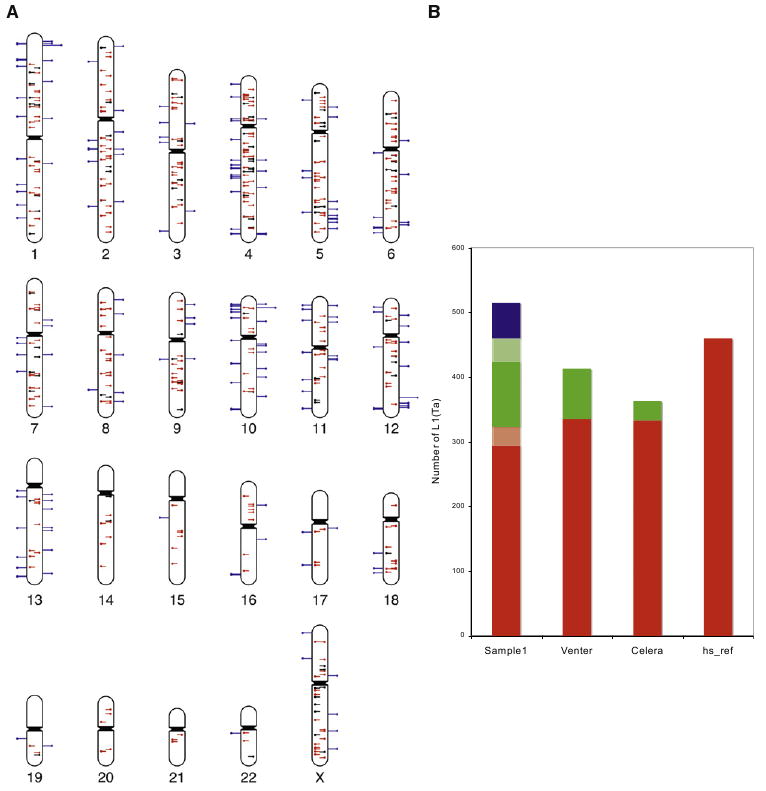
(A) Ideogram illustrates TIP-chip peaks in an individual; 514 peaks are included after imposing the cutoff (Experimental Procedures). Marks show predicted positions of L1(Ta) insertions on the plus (left side) and minus strands. Central lines similarly illustrate position and orientation of L1(Ta)s in the human reference sequence (hs_ref NCBI Build 36.1). These are color coded to indicate those identified by TIP-chip in this individual (red, n = 323) and those not seen in this sample (black). Blue lines on the outside of the chromosome correspond to nonreference insertions (n = 191). In addition to reference L1(Ta)s, 52 were considered true positives because they correspond to insertions included in dbRIP (n = 25) or were described by human sequencing projects (n = 24), as well as 3 by Beck and Moran (Beck et al., 2010). As described further in the text, additional TIP-chip peaks were verified by PCR and sequencing.
(B) TIP-chip and whole-genome sequencing in identifying L1(Ta) insertions. The y axis shows the L1(Ta) count in each sample. Sample1 was profiled by TIP-chip, whereas the other three samples are from different whole-genome sequencing approaches. Insertions present in hs_ref are displayed in red. Verified nonreference L1(Ta) insertions are shown in green. Lighter shades of red reflect reference insertions that were not retained after the imposed cutoff, while that of green reflects 3′ PCR verified insertions that might not become sequence verified. Candidate novel L1(Ta) insertions identified by TIP-chip after the cutoff and awaiting further verification, are marked in blue. The ability of TIP-chip to identify L1(Ta) insertions is comparable to whole-genome sequencing.
See also Figure S2 and Table S1.
Identification of L1(Ta)s included in hs_ref serves as a quality metric; most high quality TIP-chip data sets identify about 300 of 460 perfect matches to our L1(Ta) primer present in the reference genome. This value is comparable to numbers of reference L1(Ta)s included in the alternate genome assemblies (Figure 3B). Unidentified reference L1(Ta)s can be ascribed to polymorphic insertions absent from an individual (true negatives) and undetected L1(Ta)s [false negatives, e.g., due to < 3 probes in the 1 kilobase downstream of the L1(Ta) 3′ end]. In whole-genome TIP-chip studies of 15 unrelated individuals, there are 56 reference L1(Ta)s undetected in any individual; 47 fall in this ‘probe poor’ category. Forty of these lie in repeat-rich regions [>900bp of the 1 kilobase following the L1(Ta) 3′ end are annotated by RepeatMasker (Smit et al., 2004) (Figure S3)]. For the remaining 9 insertions, insertion allele frequencies are reported for five of them in dbRIP, with four sufficiently infrequent that their absence in this sample set is expected (insertion allele frequencies 0.019–0.051). Twelve ‘probe poor’ reference L1(Ta)s are found on the X chromosome. Of these, 9 were detected on the 385K chromosome X array platform, indicating that detection difficulty on the whole-genome array does not reflect failure to amplify these sequences and could be solved by improved probe content (Figure S3).
We compared L1(Ta) identification by TIP-chip directly with assembled whole-genome sequencing data for Hs_alt_huref (Venter) DNA. Xing et al. (2009) found 49 nonreference Hs_alt_ huref insertions by analyzing indel-containing contigs. We found 40 more in the Hs_alt_huref assembly deposited at NCBI and an additional 32 sequence verified insertions by TIP-chip. (Table S1).
To assess whole-genome TIP-chip reproducibility and address the hypothesis that L1(Ta) insertions commonly occur in early stages of human embryonic development so as to create significant somatic mosaicism (Kano et al., 2009; van den Hurk et al., 2007), we performed whole-genome TIP-chip analysis on PBLs (4 paired samples) or lymphoblastoid lines (1 paired sample) of 5 phenotypically discordant monozygotic twin sets. We find high agreement between L1(Ta) TIP-chip peaks in comparing these samples (Figure 4). No peak discrepancies (i.e., peak presence versus absence) were found in pairwise comparisons. We attempted PCRs at 89 peak positions showing differences in peak height between twins and discovered no insertions unique to one individual in a twin pair.
Figure 4. High Reproducibility of Whole-Genome TIP-chip.
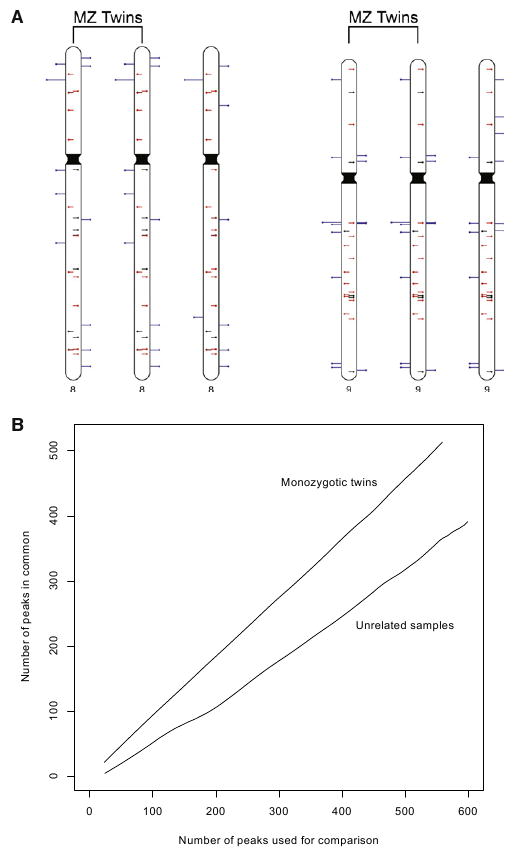
(A) Ideogram illustrating TIP-chip peaks on chromosomes 8 and 9 in a monozygotic twin pair and an unrelated individual. Marks on chromosomes show predicted positions of L1(Ta) insertions on the plus (left side) and minus strands. Central lines similarly illustrate position and orientation of L1(Ta)s in hs_ref. These are color-coded to indicate L1(Ta)s identified by TIP-chip in these individuals (red) and those not seen in this sample (black). Blue lines on the outside of the chromosome correspond to candidate nonreference L1(Ta)s. When our automated peak identification program is complemented by visual inspection of the raw data, twins have identical peak patterns while displaying many polymorphisms as compared to the unrelated individual (right most).
(B) Correspondence at the top (CAT) (Irizarry et al., 2005) plot illustrating consistency in the data obtained from a monozygotic twin pair as compared to that of two unrelated individuals at the whole-genome level. The x axis shows the number of the peaks used for comparison, taken in rank order. The y axis indicates the number of peaks in common between the two samples. Twins share far more high-ranking peaks than unrelated individuals.
See also Figure S3.
Insights into L1(Ta) Insertion Allele Frequencies from TIP-Chip
TIP-chip enables assessment of L1(Ta) genotypes in numerous samples and thus is useful for determining population-based allele frequencies. Given this, we can estimate average allele frequency of the L1(Ta) complement present in any individual; a parameter we call F̄i. We determined F̄i using two independent methods, the first based on TIP-chip as the sole means of genotyping 75 males for X chromosome insertions and the second was based on whole-genome analyses (below). On the X chromosome, 161 high scoring peaks served as the basis for F̄i calculation. Of these, 33 correspond to L1(Ta) in hs_ref, and extensive validation PCRs for 10 samples on this array platform indicate a positive predictive value of 80.5% for non-hs_ref insertions (Figure S4). Nonreference L1(Ta)s showed an extremely broad range of allele frequencies (0.013 and 0.987, average 0.58; Figure 5A, Table S2). The average L1(Ta) allele frequency F̄i, was determined to be 0.75 (Figure 5B). This parameter defines genome-wide variation of this class of ISV.
Figure 5. Polymorphism of X chromosome L1(Ta)s.
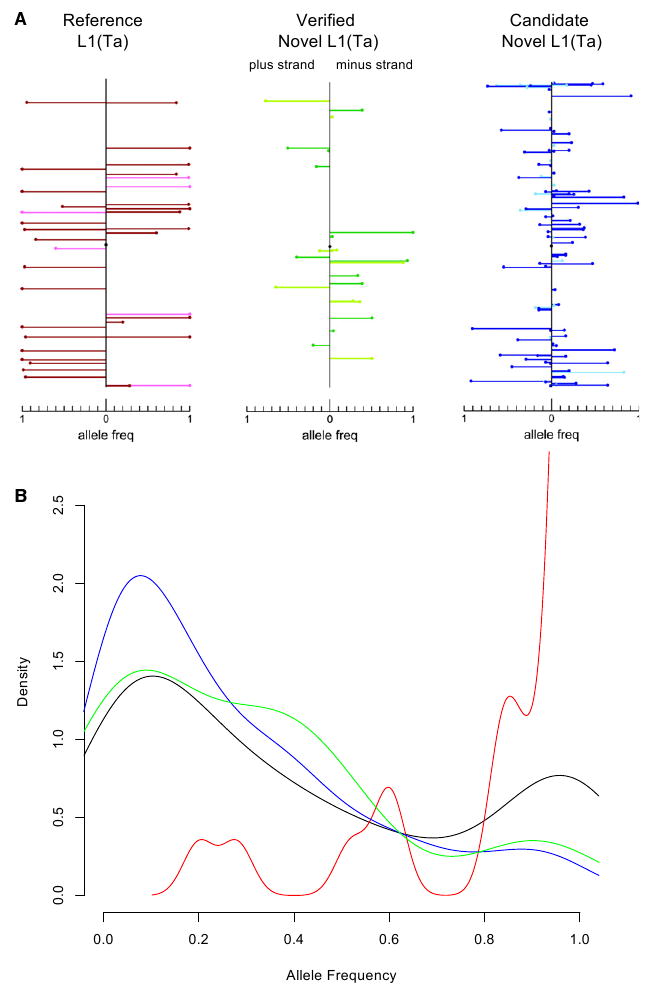
(A) Each mark represents a L1(Ta) insertion. y axis denotes position along the X chromosome and the x axis reflects allele frequencies for L1(Ta) insertions on the plus (left) and minus strands (i.e., % of males with respective insertion). In total, 75 unrelated clinical male samples collected in the United States were included in this analysis; samples were not selected based on ethnic background. As a generalization, L1(Ta)s included in hs_ref (reference L1(Ta)s, red; leftmost panel) had higher allele frequencies (0.896 ± 0.202) than novel L1(Ta)s identified (0.263 ± 0.266, green and blue for PCR verified and not yet verified, respectively, see Table S2). No significant difference in allele frequencies were observed comparing intergenic L1(Ta)s (darker hue) with intronic/intragenic insertions (lighter hue).
(B) Probability density function of allele frequencies of L1(Ta) insertions on the X chromosome. The area under each curve equals one. The x axis denotes the allele frequency ranging from 0 to 1 (present in all samples tested). Allele frequencies are calculated using X chromosome TIP-chip profiles of 75 unrelated males. The red curve shows the probability density function for insertions in hs_ref. The green curve depicts verified insertions. The blue curve displays TIP-chip peaks not yet verified. Black indicates the combined total of all three classes described above.
See also Figure S4 and Table S2.
We also evaluated new insertion discovery rates and L1(Ta) insertion characteristics on the X chromosome. Discovery rates for potential novel L1(Ta)s were highest in the first 10 samples, reflecting high frequency polymorphisms, and thereafter decreased with sample number, although low allele frequency insertions continued to be found at ∼0.8 insertions per sample throughout (samples 11–75; Figure S4). Insertion-spanning PCRs were designed to estimate L1(Ta) length for 16 novel insertions, of which 7 (44%) appear full-length (FL;6kb). In combination with reference L1(Ta)s detected (n = 49), allele frequency and L1(Ta) size were uncorrelated (p = 0.11), concordant with earlier work (Boissinot et al., 2004). Similarly, allele frequencies were similar in intergenic (0.76, sd 0.33) and intragenic (0.67, sd 0.38) L1(Ta)s(p=0.41). When considered with hs_ref L1(Ta) insertion lengths on the X chromosome and compared to autosomal reference insertions, we found more FL L1(Ta)s on chromosome X but the difference was statistically insignificant (37.9% FL on X; 26.2% FL on autosomes, sd = 18.2%).
Our second assessment of F̄i based on whole-genome data reflects a weighted average of reference and non-hs_ref L1(Ta) allele frequencies in proportions reflective of one individuals genome. Whole-genome TIP-chip data for 15 unrelated individuals provided an allele frequency for the reference L1(Ta)s (average = 0.94). The non-hs_ref L1(Ta) allele frequency was based on: (1) non-hs_ref L1(Ta)s with allele frequency data in dbRIP (Myers et al., 2002) (a.f. range 0.15–0.83; average 0.47) and (2) genotyping panels of individuals for 8 novel L1(Ta)s found in sample 1 (a.f. 0–0.82, average 0.38, n = 196; Figure 6). Genotype distributions for these insertions departed in varying degrees from Hardy-Weinberg equilibrium values as expected for studies of heterogeneous ethnic populations. Even low frequency insertions were found in multiple ethnic groups; one was absent from African individuals. These data give an estimate for F̄i of 0.83. Thus, the two values of F̄i are in relatively good agreement. For the following sections, we used the X chromosome-derived value of 0.75 as it used the highest quality array platform, was hemizygous-based, and surveyed the largest population.
Figure 6. Polymorphism of L1(Ta)s.
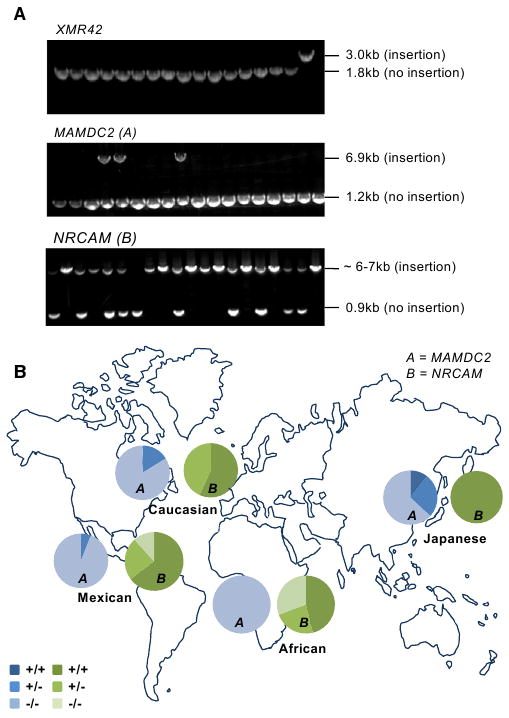
(A) Agarose gel images showing genotyping PCR products for three different L1(Ta) insertion sites in 17 individuals. In each case, primers were designed to flank the L1(Ta) insertion position identified by TIP-chip. Top panel shows a 1.2 kb insertion unique to the proband studied; about 600 other individuals were homozygous or hemizygous for the empty allele (i.e., lacking this X chromosome L1(Ta)). Middle gel shows an intronic L1(Ta) insertion in MAMDC2 on chromosome 7. Three genotypes were observed: (1) homozygous present (one band at 6.9 kb); (2) heterozygous (two bands, 6.9 kb and 1.2 kb); (3) homozygous absent (a single band at 0.9 kb). The third insertion site shown is within NRCAM on chromosome 9 (∼6–7 kb amplicon represents insertion allele, 0.9 kb represents empty allele).
(B) Pie charts indicate genotype distribution for two representative nonreference L1(Ta)s (not included in hs_ref) identified by TIP-chip studies of an individual (see Figure 3) across two human ethnic diversity panels. The total sample size of both diversity panels is 198 people. The Caucasian, Mexican and Japanese sample groups were represented most highly (n = 37, 17 and 18 respectively) and were used for Hardy-Weinberg calculations. For Locus A (MAMDC2) the allele frequencies for each population, as well as the chi square values for the biggest population groups are as follows: Caucasians (0.08; χ2 = 0.29); Mexican (0.03; χ2 = 0.02); Japanese (0.25; χ2 = 1.41); African (0.00, n = 13). For Locus B (NRCAM) the allele frequencies for each population, as well as the chi-square values for the biggest population groups are as follows: Caucasians (0.79; χ2 = 2.82); Mexican (0.77; χ2 = 2.04); Japanese (1.00); African (0.58, n = 13).
See also Figure S5 and Table S3.
Chromosome Distribution, GC Content, and Gene Loci Surrounding L1(Ta)s
The chromosomal distribution of TIP-chip peaks largely reflects chromosome size (Figures S5A and S5B). A 2-fold enrichment on the X chromosome for L1(Ta) elements is observed across the 3 published haploid sequence assemblies, consistent with the elevated overall density of older L1s on the X (Bailey et al., 2000). The tendency of L1(Ta)s to accumulate in AT-rich regions has been described previously (Gasior et al., 2006). GC content analysis of genomic intervals surrounding candidate and verified novel L1(Ta)s found by TIP-chip confirm this observation (Figure S5C).
Although genes are enriched in GC-rich genomic intervals, we identified many L1(Ta)s within genes. Intragenic sequences comprise 41% of the genome (1% exons and 40% introns; UCSC known genes definitions). In the genome-wide L1(Ta) TIP-chip survey in Figure 3A, 201 (39%) reflect intronic insertions, and 313 (61%) are intergenic. No exonic insertions of L1(Ta) were identified; however we did find an exonic Alu insertion by TIP-chip (above).
Because intronic insertions can affect gene function, we evaluated intragenic insertions further. Gene category studies were based on distribution of associated molecular functions, biological processes, and pathways as annotated in PANTHER (Thomas et al., 2003). Intragenic insertions were most frequently in genes categorized as “unclassified” molecular function and/or process, though neither group was overrepresented statistically. Also of note, no L1(Ta)s (or Alus) were identified in the four homeobox gene clusters, HOXA, HOXB, HOXC and HOXD, a megabase region relatively devoid of interspersed repeats (Lander et al., 2001). Reference and candidate L1(Ta)s inferred from peaks within or near (<5 kb) genes were enriched in antisense orientation relative to target gene (p < 0.0001, Figures S6A and S6B).
L1(Ta)s in Patients with X-Linked Disorders
To identify possible genetic etiologies for X-linked disease, insertions were profiled on the X chromosome in 10 males with unexplained muscular dystrophy or X-linked cardiomyopathy and 69 unrelated male probands with presumptively X-linked intellectual disability. No novel dystrophin insertions were seen in the first group. However, several novel L1(Ta) X chromosome insertions were discovered in the latter cohort; 6 were low frequency insertions based on genotyping (Table S4). Three insertions were “private” (unique to the proband) based on screening ∼600 individuals of ethnically diverse backgrounds.
Two L1(Ta) low-frequency alleles are shown in Figure 7 (see also Figure S6). Each is intronic and in antisense orientation relative to the gene; one is located in the Nance-Horan syndrome (NHS) gene, the other in DACH2. NHS is caused by protein-truncating mutations and characterized by congenital ocular anomalies and partially penetrant intellectual disability. The L1(Ta) insertion is a 206 bp sequence in the first intron. This allele was found in 5 of 361 control males (allele frequency 1.38%) without intellectual disability, so its clinical significance is unclear. The insertion in the DACH2 locus is private and consists of a 368bp L1 sequence, is located in the second intron of DACH2 and accompanied by a 4bp target deletion. The DACH2 Drosophila ortholog dachshund regulates neuronal differentiation (Martini et al., 2000). Mammalian Dach2 is highly expressed in fetal brain relative to other tissues (Kent et al., 2002), and mapping studies have implicated it as a potential locus for intellectual disability. Though functional effects of this intronic insertion are as yet unknown, it illustrates how L1(Ta) mapping can identify infrequent or private insertions meriting further study in the context of disease.
Figure 7. L1(Ta) Insertions Found in Clinical Genetics Patients.
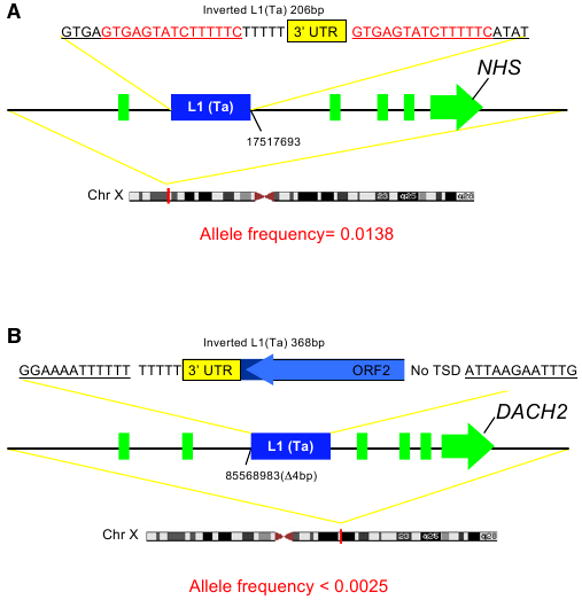
(A) NHS locus insertion. In this case, a 206bp L1(Ta) inserted into the first intron of the NHS gene.
(B) DACH2 locus insertion. A 368bp 5′ truncated L1(Ta) is inserted into the second intron of DACH2, deleting 4bp of the flanking sequence. This insertion was unique to the proband studied and not seen in 400 other samples.
See also Figure S6 and Table S4.
Discussion
L1s and other ISVs reflect an important source of human genetic diversity. They are understudied because conventional genomic approaches generally exclude high copy number, large repeats. In silico studies mining human genome sequencing data for novel L1(Ta) insertions and their characterization in demographically diverse samples have provided important insights into L1(Ta) activity (Bennett et al., 2004; Boissinot et al., 2000; Konkel et al., 2007; Myers et al., 2002; Witherspoon et al., 2006; Xing et al., 2009). Thus far, this approach has limited novel L1(Ta) discovery to relatively few individuals and/or loci. As an alternative, several one-sided PCR methods have been described to clone insertion sites of L1(Ta) elements, but these have not readily lent themselves to high-throughput L1(Ta) mapping. Thus reliable identification of infrequent or somatic insertions has been untenable, and even common insertions are poorly characterized. Similarly, direct measures of ongoing L1(Ta) activity have been difficult to accomplish experimentally.
In our assessment, TIP-chip represents the first method to comprehensively and quickly map retroelement insertions. Using TIP-chip, we discovered numerous novel L1(Ta) insertion alleles, including high frequency alleles, in many demographics. In a typical individual, TIP-chip identifies over 100 L1(Ta) insertions absent from hs_ref. In Hs_alt_huref DNA, the method was able to detect 32 novel insertions not incorporated into the assembly of whole-genome shotgun sequencing reads. These findings underscore the incompleteness of reference genome assemblies with respect to ISVs.
In papers submitted in parallel, Beck et al. (2010) and Iskow et al. (2010) used fosmid end-sequence mapping and deep sequencing approaches to generate genome-wide L1(Ta) insertion datasets. All the methods have advantages and disadvantages. Primary advantages of the fosmid method include its unbiased ability to identify large indels, its utility to detect insertions in repetitive DNA, and its low false positive rate. Its main disadvantages are that it is low throughput and cannot identify small insertions; many L1(Ta) insertions are < 1 kb. Short-read deep sequencing approaches can detect precise insertion positions – a major advantage. Challenges include optimizing amplicon sizes and sequencing coverage to allow multiplex runs and thereby reduce cost per sample. Like TIP-chip, insertions in repetitive regions are difficult or impossible to map, though we have shown this disadvantage of TIP-chip can be mitigated in part by increasing the length of the vectorette PCR amplicons with no additional cost and improved probe and array design. TIP-chip is the fastest, and we believe, the most cost effective method today. It is also especially valuable when describing polymorphisms in specific genomic regions is desirable (i.e., single chromosomes, candidate gene loci) as these can be easily tiled on small custom arrays and run at low cost on many samples. Moreover, once more complete maps of transposon insertions are available, small but genome-wide transposon genotyping arrays can be designed for association studies. Finally, TIP-chip effectively detects many types of ISVs, including SINEs, and the two-color platform allows distinguishing two element types on one array.
We have re-examined several properties of L1(Ta)s with the most comprehensive data set now available. While quality metrics have varied between and within these multiarray runs, we have no evidence that total L1(Ta) burden varies substantially between individuals. The chromosomal distribution of TIP-chip peaks largely reflects chromosome size, and shows a modest albeit not statistically significant enrichment of L1(Ta)s on chromosome 4, like the distribution of L1(Ta)s in hs_ref (Figures S5A and S5B). A 2-fold enrichment on the X chromosome for L1(Ta) elements is observed across the published haploid sequence assemblies, consistent with elevated overall density of older L1s on the X (Bailey et al., 2000).
We also observe a predilection for L1(Ta)s to accumulate in AT-rich regions, reflecting either mechanism of ORF2p mediated insertion and/or selection against insertions in proximity to genes (Gasior et al., 2007). Thus far, we have found verified novel insertions only in intergenic or intronic regions; no exonic L1(Ta) insertion (or otherwise obviously deleterious to gene function) were observed. These are consistent with prior in silico analyses of polymorphic L1(Ta) integrations, but contrast with studies of Alu insertions which are seen frequently in proximity to genes and occasionally in exons (Xing et al., 2009), perhaps providing selective advantage (Lander et al., 2001). In a single sample, genome-wide mapping of AluYa5/8 and AluYb8/9 insertions by TIP-chip, we observed an exonic insertion, and we expect features of exonic sequence (GC content and uniqueness) will make for especially effective probe coverage and high quality TIP-chip peaks in these areas. Of L1(Ta) elements inserted within or near (<5000bp) genes, we noticed a statistically significant enrichment for antisense orientation, both considering reference L1(Ta)s or all candidate L1(Ta) insertions identified by TIP-chip. These results and other analyses (Figures S6A and S6B) suggest that L1(Ta)s inserted in antisense orientation relative to host genes are less deleterious overall, consistent with the hypothesis that sense insertions can lead to polymerase elongation defects and/or premature polyadenylation (Han et al., 2004; Perepelitsa-Belancio and Deininger, 2003). Presumably, such a bias against sense insertions is more obvious in reference L1(Ta)s and L1(pre-Ta)s (older elements), due to increased selection time. Mechanisms for target gene dysregulation by L1(Ta)s in both orientations have been posited, however (Belancio et al., 2008; Han and Boeke, 2005; Han et al., 2004; Speek, 2001; Wheelan et al., 2005).
We have gained insights into the prevalence of polymorphic L1(Ta)s by performing X chromosome directed screens in large numbers of males and by genome-wide TIP-chip L1(Ta) discovery followed by genotyping human genetic diversity panels by site-specific PCR. Our X chromosome data suggest that across all L1(Ta) insertions in one human, the average insertion allele frequency is about 0.75. Many novel insertions we describe in this study show high allele frequencies across different populations. This suggests that, despite the status of various human genome projects, we are in the early phases of describing these important ISVs. Additionally, we found many uncommon alleles, some of which are likely private insertions unique to a limited kindred or individual.
The sheer quantity and low allele frequency of many novel insertions described suggest L1(Ta)s remain highly active in modern humans. Indeed, TIP-chip data provide an experimental basis for revisiting estimates of L1 activity (i.e., occurrences of de novo insertions in the general population). By comparing the Hs_alt_ huref L1(Ta) profile as discovered with TIP-chip and in silico analysis to the hs_ref profile, we revise the current estimate of L1(Ta) insertion rate from 1 insertion in every 225 births to approximately 1 in 108 (Experimental Procedures). This number is a conservative estimate, as we have not exhaustively PCR verified TIP-chip peaks in this sample and excluded many peaks from consideration. That we readily identified one low-frequency insertion absent from African individuals in one sample and three potential private insertions in a single chromosome study of a clinical cohort (see below) also suggests L1(Ta) activity—and the LINE and SINE ISVs it enables—may have been previously underestimated.
Finally, although TIP-chip can be employed for ISVs discovery throughout the entire genome, the method has the unique advantage that it can be used to efficiently characterize relatively rare insertions over narrower intervals in surveying large populations. This feature may make TIP-chip especially useful in clinical genetics. Here we examine X chromosome L1(Ta) sites in 69 males with clinically defined X-linked intellectual disability, and verified 6 novel, relatively uncommon L1(Ta) insertions and 3 private insertions within this group (insertion allele frequencies < 0.0018–0.0025). Three are in or near brain-expressed genes or genes with known roles in central nervous system development. Though the biological effect of these particular intronic L1(Ta) insertions remains uncertain, the study shows how knowledge of L1(Ta) positions can identify candidate risk alleles meriting further study.
In summary, we have developed a high-throughput method, TIP-chip, for mapping an active group of mobile DNAs in humans. We show the technique is readily generalized to other interspersed repeats. We illustrate initial insights it has provided into L1(Ta) genomic distribution and the dynamics of these repeats in our genomes. Genome-wide TIP-chip studies of several individuals show that L1(Ta)s are extremely polymorphic and an underappreciated type of SV underlying human genetic diversity. Future L1(Ta) and ISV mapping by TIP-chip and similar methods will continue to expand our understanding of the human genomic diversity and play an increasingly important role in identifying causes of genetic disease.
Experimental Procedures
L1(Ta) TIP-Chip
Aliquots of high Mr genomic DNA were digested in parallel with six REs (AseI, BspHI, BstYI, HindIII, NcoI, and PstI) chosen by a greedy algorithm to maximize genomic fragments 1–5 kb long. Sticky ends are ligated to vectorette adapters. Vectorette PCR was performed using a touchdown PCR program and ExTaq polymerase (Takara Bio; Shiga, Japan). Amplicons were purified and concentrated using Microcon columns (Millipore; Billerica, MA) and digested with REs to generate smaller fragments. These fragments are labeled with Cy3-dUTP or Cy5-dUTP (Enzo Biochem; New York, NY) using exo− Klenow polymerase-mediated (New England Biolabs; Ipswich, MA) extension from random 9-mers (Stratagene-Agilent Technologies; Santa Clara, CA). After additional clean up and concentration using Microcon columns, labeled amplicon fragments were hybridized to 2.1M feature HD2 whole-genome economy-type microarrays or 385K feature single chromosome arrays (Nimblegen/Roche Applied Science; Madison, WI) according to manufacturer's instructions. Arrays were hybridized in MAUI mixers (Biomicro Systems; Salt Lake City, UT), washed, and scanned using a Genepix (Molecular Devices; Sunnyvale, CA). A detailed description may be found in Supplemental Experimental Procedures.
Peak Recognition
Probe coordinates and fluorescence intensity values (.gff files) were generated using Nimblescan (Roche Nimblegen). Peaks corresponding to candidate transposon insertion site are identified by custom L1 Signal Analysis (LISA-map) software (Huang, et al. in preparation, available on request). Peak positions that overlap with the insertions found in the hs_ref genome (referred to as reference peaks) were used as a quality control measurement. The software detects peaks based on a HMM incorporating probe intensity and peak morphology. Peaks are ranked by the sum of the posterior probability of each probe being in a peak. The best cutoff of each sample was determined by systemically varying four different parameters after exclusion of peaks identified as vectorette PCR background. Peaks were removed (i) after the ith number of reference peaks in the ranked list, (ii) if the region showed ‘noisy’ background (variance = j), (iii) if the peak was made up of less than k number of consecutive probes (allowing 1 failed probe within the peak interval), and (iv) if local background intensity (defined by a 40 probe window flanking the peak) was above threshold m. Finally, peaks were reranked based on maximum probe intensity and peaks below the last reference peak are deleted. Cutoff values for each variable were imposed to target a total peak number closest to the expected number of L1(Ta) insertion positions per diploid human, Ne = 535 (see below), while removing the fewest reference L1(Ta) peaks. Reference L1(Ta)s that did not make the cutoff (on average < 12% per sample) are retained in the final list.
Calculation of Expected Number of Different L1(Ta) Alleles in a Diploid Individual
The expected number of different L1(Ta) alleles per diploid human genome (Ne; i.e., true, unique TIP-chip peaks) was estimated assuming that total L1(Ta) number does not vary significantly between individuals. In the three sequenced haploid genome assemblies (hs_ref, hs_alt_HuRef, hs_alt_celera from ftp://ftp.ncbi.nih.gov/genomes/), the L1(Ta) counts are 413, 363, and 460 respectively. We used the average of these values (412) as an estimate of L1(Ta) insertions per haploid genome. Determining the diploid L1(Ta) content then requires an estimate of zygosity, derived from the average allele frequency for L1(Ta)s found in any single individual (F̄i, see text). This value is assumed to be constant and invariant among chromosomes. Allele frequencies of 161 candidate novel L1(Ta) insertions found by chromosome X TIP-chip were defined based on 75 male samples profiled (allele frequency = number of TIP-chip peaks found at that genomic location divided by 75, Figure 5A, Table S2). Then, F̄i was determined for each of the individuals by averaging the allele frequencies for each insertion on their X chromosome; the mean of the 75 F̄i values was 0.75 (0.95 for hs_ref L1(Ta)s; 0.58 for nonreference L1s). Defining this average frequency value on a per individual genome basis is fundamental to both our derivation and application of this estimate. If one instead considers the universe of L1(Ta) insertion allele frequencies, the average allele frequency value would asymptotically approach zero as more people are profiled and rarer and rarer insertion alleles discovered. The product of the number of insertions in a haploid genome times the average allele frequency (412*0.75 = 309) provides the number of expected homozygous insertions. Therefore, the expected number of distinct L1(Ta) alleles per diploid human genome, Ne, is 412*2 – 309 = 515.
Estimate of L1(Ta) Activity in Modern Humans
We followed the method described by Xing et al. (2009). These authors used SNPs to estimate divergence of the haploid genomes Hs_alt_huref and the NCBI reference hs_ref build at 18,483 generations. The authors then cataloged nonreference L1s in indel-containing contigs without ‘N’ nucleotides from the diploid Venter genome; their resulting estimate for L1(Ta) new alleles is 1 in 225 individuals (1 in 212 considering all L1). This value is based on both L1(Ta) retrotransposition events and establishment of homozygosity. Our group analyzed the haploid assembly (Hs_alt_huref) of the Venter genome at NCBI and identified additional L1(Ta)-containing reads by searching for exact matches to the primer used in our vectorette PCR. In addition, TIP-chip identified 32 more that were subsequently verified by sequencing. This sums to 121 nonreference L1(Ta) insertions in the Hs_alt_huref genome, a value higher than previously recognized. Using the nonreference F̄i derived above, 0.58, weestimated the ratio of homozygous to heterozygous insertions at 41:59 [F̄iˆ2: 2*F̄i (1-F̄i)], giving a total number of non-hs_ref L1(Ta) insertions of 85 in the haploid genome. This provides a basis for revising the estimate of L1(Ta) insertions upwards to one insertion per 108 individuals.
Supplementary Material
Acknowledgments
Supported in part by NIH grant P01-CA16319, RC1 HG005359 and grants from the Brain Science Institute at Johns Hopkins University School of Medicine and the Goldhirsh Foundation (J.D.B.), and NIH grant K08-CA134746 and a Career Award for Medical Scientists from the Burroughs Wellcome Foundation (K.H.B.). We thank Jon Alder, Joe Costello, Lisa Scheifele, Daniel Yuan, Syntyche Walker, Ed Davis, Kate O'Donnell, Lixin Dai, Wengfeng An, and Christina Schrum for helpful discussions and Audrey Hendley, Naera El-Sharkawy, Lisa Scheifele, and Daniel Yuan for technical assistance. We thank Robert B. Weiss and Kevin M. Flanigan for X-linked dilated cardiomyopathy and Becker muscular dystrophy patient samples; Cindy Skinner, Cassandra Obie, and Abby Adamczyk for assistance in providing X-linked intellectual disability genomic DNA samples; and Pei-Lung Chen, Darci Ferrer, Sarah E. Ritter, and Gary Cutting for familial and twin genomic DNA. Finally, we thank Bang Wong at ClearScience and Cheng Lai Victor Huang for assistance with artwork.
Footnotes
Supplemental Information: Supplemental Information includes Extended Experimental Procedures, four tables, six figures, and Supplemental References and can be found with this article online at doi:10.1016/j.cell.2010.05.026.
References
- Arnold C, Hodgson IJ. Vectorette PCR: A novel approach to genomic walking. PCR Methods Appl. 1991;1:39–42. doi: 10.1101/gr.1.1.39. [DOI] [PubMed] [Google Scholar]
- Bailey JA, Carrel L, Chakravarti A, Eichler EE. Molecular evidence for a relationship between LINE-1 elements and X chromosome inactivation: the Lyon repeat hypothesis. Proc Natl Acad Sci USA. 2000;97:6634–6639. doi: 10.1073/pnas.97.12.6634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck CR, Collier P, Macfarlane C, Malig M, Kidd JM, Eichler EE, Badge RM, Moran JV. LINE-1 retrotransposition activity in human genomes. Cell. 2010;141:1159–1170. doi: 10.1016/j.cell.2010.05.021. this issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belancio VP, Roy-Engel AM, Deininger P. The impact of multiple splice sites in human L1 elements. Gene. 2008;411:38–45. doi: 10.1016/j.gene.2007.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett EA, Coleman LE, Tsui C, Pittard WS, Devine SE. Natural genetic variation caused by transposable elements in humans. Genetics. 2004;168:933–951. doi: 10.1534/genetics.104.031757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boissinot S, Chevret P, Furano AV. L1 (LINE-1) retrotransposon evolution and amplification in recent human history. Mol Biol Evol. 2000;17:915–928. doi: 10.1093/oxfordjournals.molbev.a026372. [DOI] [PubMed] [Google Scholar]
- Boissinot S, Entezam A, Young L, Munson PJ, Furano AV. The insertional history of an active family of L1 retrotransposons in humans. Genome Res. 2004;14:1221–1231. doi: 10.1101/gr.2326704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brouha B, Schustak J, Badge RM, Lutz-Prigge S, Farley AH, Moran JV, Kazazian HH., Jr Hot L1s account for the bulk of retrotransposition in the human population. Proc Natl Acad Sci USA. 2003;100:5280–5285. doi: 10.1073/pnas.0831042100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Rattner A, Nathans J. Effects of L1 retrotransposon insertion on transcript processing, localization and accumulation: lessons from the retinal degeneration 7 mouse and implications for the genomic ecology of L1 elements. Hum Mol Genet. 2006;15:2146–2156. doi: 10.1093/hmg/ddl138. [DOI] [PubMed] [Google Scholar]
- Cooper GM, Nickerson DA, Eichler EE. Mutational and selective effects on copy-number variants in the human genome. Nat Genet. 2007;39:S22–S29. doi: 10.1038/ng2054. [DOI] [PubMed] [Google Scholar]
- Cordaux R, Batzer MA. The impact of retrotransposons on human genome evolution. Nat Rev Genet. 2009;10:691–703. doi: 10.1038/nrg2640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Credille KM, Minor JS, Barnhart KF, Lee E, Cox ML, Tucker KA, Diegel KL, Venta PJ, Hohl D, Huber M, et al. Transglutaminase 1-deficient recessive lamellar ichthyosis associated with a LINE-1 insertion in Jack Russell terrier dogs. Br J Dermatol. 2009;161:265–272. doi: 10.1111/j.1365-2133.2009.09161.x. [DOI] [PubMed] [Google Scholar]
- Gasior SL, Preston G, Hedges DJ, Gilbert N, Moran JV, Deininger PL. Characterization of pre-insertion loci of de novo L1 insertions. Gene. 2007;390:190–198. doi: 10.1016/j.gene.2006.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasior SL, Wakeman TP, Xu B, Deininger PL. The human LINE-1 retrotransposon creates DNA double-strand breaks. J Mol Biol. 2006;357:1383–1393. doi: 10.1016/j.jmb.2006.01.089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gresham D, Dunham MJ, Botstein D. Comparing whole genomes using DNA microarrays. Nat Rev Genet. 2008;9:291–302. doi: 10.1038/nrg2335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu W, Zhang F, Lupski JR. Mechanisms for human genomic rearrangements. Pathogenetics. 2008;1:4. doi: 10.1186/1755-8417-1-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han JS, Boeke JD. LINE-1 retrotransposons: modulators of quantity and quality of mammalian gene expression? Bioessays. 2005;27:775–784. doi: 10.1002/bies.20257. [DOI] [PubMed] [Google Scholar]
- Han JS, Szak ST, Boeke JD. Transcriptional disruption by the L1 retrotransposon and implications for mammalian transcriptomes. Nature. 2004;429:268–274. doi: 10.1038/nature02536. [DOI] [PubMed] [Google Scholar]
- International HapMap Consortium. The International HapMap Project. Nature. 2003;426:789–796. doi: 10.1038/nature02168. [DOI] [PubMed] [Google Scholar]
- Irizarry RA, Warren D, Spencer F, Kim IF, Biswal S, Frank BC, Gabrielson E, Garcia JG, Geoghegan J, Germino G, et al. Multiple-laboratory comparison of microarray platforms. Nat Methods. 2005;2:345–350. doi: 10.1038/nmeth756. [DOI] [PubMed] [Google Scholar]
- Iskow RC, McCabe MT, Mills RE, Torene S, Pittard WS, Neuwald AF, Van Meir EG, Vertino PM, Devine SE. Natural mutagenesis of human genomes by endogenous retrotransposons. Cell. 2010;141:1253–1261. doi: 10.1016/j.cell.2010.05.020. this issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kano H, Godoy I, Courtney C, Vetter MR, Gerton GL, Ostertag EM, Kazazian HH., Jr L1 retrotransposition occurs mainly in embryogenesis and creates somatic mosaicism. Genes Dev. 2009;23:1303–1312. doi: 10.1101/gad.1803909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazazian HH, Jr, Wong C, Youssoufian H, Scott AF, Phillips DG, Antonarakis SE. Haemophilia A resulting from de novo insertion of L1 sequences represents a novel mechanism for mutation in man. Nature. 1988;332:164–166. doi: 10.1038/332164a0. [DOI] [PubMed] [Google Scholar]
- Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, Haussler D. The human genome browser at UCSC. Genome Res. 2002;12:996–1006. doi: 10.1101/gr.229102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolomietz E, Meyn MS, Pandita A, Squire JA. The role of Alu repeat clusters as mediators of recurrent chromosomal aberrations in tumors. Genes Chromosomes Cancer. 2002;35:97–112. doi: 10.1002/gcc.10111. [DOI] [PubMed] [Google Scholar]
- Konkel MK, Wang J, Liang P, Batzer MA. Identification and characterization of novel polymorphic LINE-1 insertions through comparison of two human genome sequence assemblies. Gene. 2007;390:28–38. doi: 10.1016/j.gene.2006.07.040. [DOI] [PubMed] [Google Scholar]
- Korbel JO, Urban AE, Affourtit JP, Godwin B, Grubert F, Simons JF, Kim PM, Palejev D, Carriero NJ, Du L, et al. Paired-end mapping reveals extensive structural variation in the human genome. Science. 2007;318:420–426. doi: 10.1126/science.1149504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, Devon K, Dewar K, Doyle M, FitzHugh W, et al. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- Levy S, Sutton G, Ng PC, Feuk L, Halpern AL, Walenz BP, Axelrod N, Huang J, Kirkness EF, Denisov G, et al. The diploid genome sequence of an individual human. PLoS Biol. 2007;5:e254. doi: 10.1371/journal.pbio.0050254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martini SR, Roman G, Meuser S, Mardon G, Davis RL. The retinal determination gene, dachshund, is required for mushroom body cell differentiation. Development. 2000;127:2663–2672. doi: 10.1242/dev.127.12.2663. [DOI] [PubMed] [Google Scholar]
- Miki Y, Nishisho I, Horii A, Miyoshi Y, Utsunomiya J, Kinzler KW, Vogelstein B, Nakamura Y. Disruption of the APC gene by a retrotransposal insertion of L1 sequence in a colon cancer. Cancer Res. 1992;52:643–645. [PubMed] [Google Scholar]
- Moran JV, Holmes SE, Naas TP, DeBerardinis RJ, Boeke JD, Kazazian HH., Jr High frequency retrotransposition in cultured mammalian cells. Cell. 1996;87:917–927. doi: 10.1016/s0092-8674(00)81998-4. [DOI] [PubMed] [Google Scholar]
- Morisada N, Rendtorff ND, Nozu K, Morishita T, Miyakawa T, Matsumoto T, Hisano S, Iijima K, Tranebjaerg L, Shirahata A, et al. Branchio-oto-renal syndrome caused by partial EYA1 deletion due to LINE-1 insertion. Pediatr Nephrol. 2010;25:1343–1348. doi: 10.1007/s00467-010-1445-x. [DOI] [PubMed] [Google Scholar]
- Muotri AR, Chu VT, Marchetto MC, Deng W, Moran JV, Gage FH. Somatic mosaicism in neuronal precursor cells mediated by L1 retrotransposition. Nature. 2005;435:903–910. doi: 10.1038/nature03663. [DOI] [PubMed] [Google Scholar]
- Myers JS, Vincent BJ, Udall H, Watkins WS, Morrish TA, Kilroy GE, Swergold GD, Henke J, Henke L, Moran JV, et al. A comprehensive analysis of recently integrated human Ta L1 elements. Am J Hum Genet. 2002;71:312–326. doi: 10.1086/341718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perepelitsa-Belancio V, Deininger P. RNA truncation by premature polyadenylation attenuates human mobile element activity. Nat Genet. 2003;35:363–366. doi: 10.1038/ng1269. [DOI] [PubMed] [Google Scholar]
- Scherer SW, Lee C, Birney E, Altshuler DM, Eichler EE, Carter NP, Hurles ME, Feuk L. Challenges and standards in integrating surveys of structural variation. Nat Genet. 2007;39:S7–S15. doi: 10.1038/ng2093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwahn U, Lenzner S, Dong J, Feil S, Hinzmann B, van Duijnhoven G, Kirschner R, Hemberger M, Bergen AA, Rosenberg T, et al. Positional cloning of the gene for X-linked retinitis pigmentosa 2. Nat Genet. 1998;19:327–332. doi: 10.1038/1214. [DOI] [PubMed] [Google Scholar]
- Skowronski J, Fanning TG, Singer MF. Unit-length line-1 transcripts in human teratocarcinoma cells. Mol Cell Biol. 1988;8:1385–1397. doi: 10.1128/mcb.8.4.1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smit AFA, Hubley R, Green P. RepeatMasker: Open-3.0. 1996–2004. 2004 http://www.repeatmasker.org.
- Speek M. Antisense promoter of human L1 retrotransposon drives transcription of adjacent cellular genes. Mol Cell Biol. 2001;21:1973–1985. doi: 10.1128/MCB.21.6.1973-1985.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stankiewicz P, Lupski JR. Structural variation in the human genome and its role in disease. Annu Rev Med. 2010;61:437–455. doi: 10.1146/annurev-med-100708-204735. [DOI] [PubMed] [Google Scholar]
- Thomas PD, Kejariwal A, Campbell MJ, Mi H, Diemer K, Guo N, Ladunga I, Ulitsky-Lazareva B, Muruganujan A, Rabkin S, et al. PANTHER: a browsable database of gene products organized by biological function, using curated protein family and subfamily classification. Nucleic Acids Res. 2003;31:334–341. doi: 10.1093/nar/gkg115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ustyugova SV, Lebedev YB, Sverdlov ED. Long L1 insertions in human gene introns specifically reduce the content of corresponding primary transcripts. Genetica. 2006;128:261–272. doi: 10.1007/s10709-005-5967-2. [DOI] [PubMed] [Google Scholar]
- Van de Water N, Williams R, Ockelford P, Browett P. A 20.7 kb deletion within the factor VIII gene associated with LINE-1 element insertion. Thromb Haemost. 1998;79:938–942. [PubMed] [Google Scholar]
- van den Hurk JA, Meij IC, Del Carmen Seleme M, Hoefsloot LH, Sistermans EA, de Wijs IJ, Plomp AS, de Jong PT, Kazazian HH, Cremers FP. L1 retrotransposition can occur early in human embryonic development. Hum Mol Genet. 2007;16:1587–1592. doi: 10.1093/hmg/ddm108. [DOI] [PubMed] [Google Scholar]
- Venter JC, Adams MD, Myers EW, Li PW, Mural RJ, Sutton GG, Smith HO, Yandell M, Evans CA, Holt RA, et al. The sequence of the human genome. Science. 2001;291:1304–1351. doi: 10.1126/science.1058040. [DOI] [PubMed] [Google Scholar]
- Wang J, Song L, Grover D, Azrak S, Batzer MA, Liang P. dbRIP: a highly integrated database of retrotransposon insertion polymorphisms in humans. Hum Mutat. 2006;27:323–329. doi: 10.1002/humu.20307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheelan SJ, Aizawa Y, Han JS, Boeke JD. Gene-breaking: a new paradigm for human retrotransposon-mediated gene evolution. Genome Res. 2005;15:1073–1078. doi: 10.1101/gr.3688905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheelan SJ, Scheifele LZ, Martinez-Murillo F, Irizarry RA, Boeke JD. Eukaryotic Transposable Elements and Genome Evolution Special Feature: Transposon insertion site profiling chip (TIP-chip) Proc Natl Acad Sci USA. 2006;103:17632–17637. doi: 10.1073/pnas.0605450103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witherspoon DJ, Marchani EE, Watkins WS, Ostler CT, Wooding SP, Anders BA, Fowlkes JD, Boissinot S, Furano AV, Ray DA, et al. Human population genetic structure and diversity inferred from polymorphic L1(LINE-1) and Alu insertions. Hum Hered. 2006;62:30–46. doi: 10.1159/000095851. [DOI] [PubMed] [Google Scholar]
- Xing J, Zhang Y, Han K, Salem AH, Sen SK, Huff CD, Zhou Q, Kirkness EF, Levy S, Batzer MA, et al. Mobile elements create structural variation: analysis of a complete human genome. Genome Res. 2009;19:1516–1526. doi: 10.1101/gr.091827.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yajima I, Sato S, Kimura T, Yasumoto K, Shibahara S, Goding CR, Yamamoto H. An L1 element intronic insertion in the black-eyed white (Mitf[mi-bw]) gene: the loss of a single Mitf isoform responsible for the pigmentary defect and inner ear deafness. Hum Mol Genet. 1999;8:1431–1441. doi: 10.1093/hmg/8.8.1431. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.