

SUMMARY
The ability to make specific perturbations to biological molecules in a cell or organism is a central experimental strategy in modern research biology. We have developed a general technique in which the stability of a specific protein is regulated by a cell-permeable small molecule. Mutants of the E. coli dihydrofolate reductase (ecDHFR) were engineered to be degraded, and when this destabilizing domain is fused to a protein of interest, its instability is conferred to the fused protein resulting in rapid degradation of the entire fusion protein. A small-molecule ligand trimethoprim (TMP) stabilizes the destabilizing domain in a rapid, reversible, and dose-dependent manner, and protein levels in the absence of TMP are barely detectable. The ability of TMP to cross the blood-brain barrier enables the tunable regulation of proteins expressed in the mammalian central nervous system.
INTRODUCTION
Numerous perturbation techniques have been developed to study gene function in biological systems. The Cre-Lox technology is a useful approach to inactivate genes in mice. Tissue- or cell-specific promoters can be used to drive Cre expression thus providing a degree of temporal and spatial control to the disruption of targeted genes (Gaveriaux-Ruff and Kieffer, 2007). However, this approach is not reversible, and identifying a suitable promoter can be difficult. Strategies to conditionally regulate the transcription of genes (e.g., the tetracycline-regulated transactivator) are widely used to control gene expression (Furth et al., 1994). Protein levels can also be regulated by targeting the mRNA transcripts using RNA interference (Xia et al., 2002). All of the techniques that target the precursor DNA or mRNA molecules encoding a protein of interest suffer from inevitable delays following the perturbation. Existing protein molecules must be degraded, so the experimental delay is dictated by the biological half-life of the protein under study.
Targeting proteins directly, rather than the DNA or mRNA molecules that encode them, is a more direct and rapid experimental strategy. In the past decade, a number of conditional protein regulation systems have been developed that utilize the cellular degradation machinery. In Varshavsky’s early system a temperature-sensitive mutant of mammalian DHFR (DHFRts) bearing N-terminal arginine residue, a destabilizing residue by the N-end rule, was found to be stable at a permissive temperature of 23 °C but unstable at 37 °C (Dohmen et al., 1994; Levy et al., 1999). Addition of methotrexate, a high-affinity ligand for mammalian DHFR, to cells expressing Arg-DHFRts partially inhibited degradation of the protein (Levy et al., 1999). This was an important demonstration that a small-molecule ligand can stabilize a protein otherwise targeted for degradation in cells.
In 2003, Crabtree and coworkers demonstrated the rapid and reversible regulation of the GSK-3β kinase fused to an unstable triple-mutant of the FRB domain (FRB*) using the rapamycin-derivative C20-MaRap (Stankunas et al., 2003). MaRap induces a heterodimerization event between the FRB*-GSK3β fusion and ubiquitously expressed FKBP12 to stabilize the FRB* fusion and restore the function of the fused kinase. The FRB*-GSK3β was inducibly stabilized in cells as well as in embryos in vivo (Liu et al., 2007). This system demonstrated that ligand-dependent stability represented an attractive strategy to regulate the function of a specific protein in a complex biological environment.
We previously developed a strategy in which a cell-permeable ligand is used in conjugation with a single genetically-encoded domain to regulate any protein of interest. A small domain can be fused to any gene, and a fusion protein is produced as a result of this specific genetic perturbation. Mutants of the human FKBP12 protein were engineered to be metabolically unstable in the absence of its high-affinity ligand, Shield-1 (Banaszynski et al., 2006). We call these mutants destabilizing domains (DDs), and observe that the instability of a DD conferred to any fused partner protein results in degradation of the entire fusion protein by the proteasome. Shield-1 binds to and stabilizes the DD in a dose-dependent manner. The genetic fusion of the DD to the gene of interest ensures specificity, and small-molecule control confers reversibility and dose-dependence to protein stability and function.
Building on our experiences with the FKBP-derived DDs, we chose E. coli dihydrofolate reductase (ecDHFR) as a candidate protein to engineer a second destabilizing domain with improved properties. The 159-residue enzyme catalyzes the reduction of dihydrofolate to tetrahydrofolate, a cofactor that is essential for several steps in prokaryotic primary metabolism (Schnell et al., 2004). Numerous inhibitors of DHFR have been developed as drugs (Schweitzer et al., 1990), and one such inhibitor, trimethoprim (TMP), inhibits ecDHFR much more potently than mammalian DHFR (Matthews et al., 1985). This large therapeutic window renders TMP “biologically silent” in mammalian cells. The specificity of the ecDHFR-TMP interaction, coupled with the commercial availability and attractive pharmacological properties of TMP, makes this protein-ligand pair ideal for development as a DD system as presaged in another context by Varshavsky (Dohmen et al., 1994; Johnston et al., 1995; Lévy et al., 1999).
RESULTS
Screening strategies to engineer DDs
To engineer mutants that display ligand-dependent stability, we designed a cell-based screen using yellow fluorescent protein (YFP) as a reporter for ecDHFR stability. The strategy was designed to identify mutants possessing the desired characteristics of a destabilizing domain: low protein levels in the absence of TMP (i.e., low basal stability), large dynamic range, robust and predictable dose-response behavior, and rapid kinetics of degradation. We used error-prone PCR (Atwood-Moore et al., 2005; Zaccolo et al., 1996) to generate libraries of ecDHFR mutants based on the parental sequence (Figures S1A-D) as well as two point mutants (Y100I and G121V) that are known to compromise enzymatic activity (Figures S1E and S1F) (Adams et al., 1991; Cameron et al., 1997). Two libraries were prepared with the ecDHFR mutants cloned in-frame at either the 5′- or 3′-ends of the YFP gene, and a retroviral expression system was used to stably transduce the libraries into NIH3T3 fibroblasts. Fluorescence-activated cell sorting (FACS) was used to screen the libraries of candidate DDs. Cells were first sorted in the absence of TMP, where we selected cells that exhibited low levels of YFP expression. This population was then treated with 10 μM TMP for 24 hr, at which point YFP-expressing cells were isolated by FACS. Cells were evenly split into two populations and dosed with either 1 μM or 0.1 μM TMP, at which point a third sort was performed. This step allowed us to isolate cells expressing mutants that were responsive to lower concentrations of TMP. The fourth and final step was designed to enrich for ecDHFR mutants exhibiting fast kinetics of degradation, as cells exhibiting low fluorescence levels were collected 4 hr following removal of TMP from the media. After four rounds of sorting the cells were allowed to recover, their genomic DNA was extracted, and candidate DDs were amplified by PCR and isolated.
Characterization of ecDHFR Mutants that Display TMP-Dependent Stability
Genes encoding individual ecDHFR-YFP fusions were transduced into NIH3T3 cells, and YFP fluorescence levels were measured in the absence and presence of 10 μM TMP. Mutants displaying low basal fluorescence levels and also high dynamic range were then sequenced (Tables S1A and S1B). From the screen using ecDHFR fused to the N-terminus of YFP we chose three mutants (R12Y/Y100I, R12L/Y100I and R98H/F103S) for further analysis, and two mutants (F103L and N18T/A19V) were chosen from the C-terminal DD screen. Treating cells with various concentrations of TMP caused YFP levels to increase in a dose-dependent manner as measured by analytical flow cytometry (Figures 1A and 1B). DDs fused to the N-terminus of YFP are fully stabilized by 1 μM TMP and display improved dynamic range. The R98H/F103S mutant is stabilized 100-fold upon treatment with TMP compared to a 31-fold increase in YFP levels for the Y100I mutant (Figure 1A). The enhanced dynamic range is largely the result of more effective destabilization under nonpermissive conditions. The C-terminal DD screen produced mutants with wide dynamic ranges of YFP expression (between 63- and 93-fold) that are stabilized by lower concentrations of TMP (Figure 1B). All of the DDs caused drastic destabilization of the YFP protein in the absence of TMP. YFP fluorescence, measured by analytical flow cytometry, was barely above the autofluorescence of untransduced NIH3T3 cells (Figure S1G).
Figure 1. Characterization of ecDHFR Mutants that Display TMP-Dependent Stability.

(A) NIH3T3 cells stably transduced with the indicated ecDHFR-YFP fusions were treated with three-fold dilutions of TMP (30 μM to 1 nM) or mock-treated with DMSO and monitored by analytical flow cytometry. Data are represented as the average median fluorescence intensity (± s.d.) and normalized against the average MFI of untransduced cells. (B) NIH3T3 cells stably transduced with the indicated YFP-ecDHFR fusions were treated as in panel A. (C) The R12Y/Y100I destabilizing domain was spliced into the YFP gene between the codons encoding residues 157 and 158. Linkers of varying length were used to flank the ecDHFR gene. These constructs were stably expressed in NIH3T3 cells, and cells were either mock-treated or dosed with 10 μM TMP for 24 h prior to analysis by flow cytometry.
We next investigated the kinetics of destabilized domain rescue and degradation. YFP fluorescence levels of each of the DDs increased at a similar rate upon treatment with TMP (Figures S1H and S1I). To measure the rates of protein degradation upon switching to nonpermissive conditions, mutants were stabilized with TMP for 24 hr at which time TMP was removed from the culture media and cells were assayed for YFP fluorescence. The majority of mutants reach basal fluorescence levels within four hours, as anticipated from the final step of the screening strategy (Figures S1J and S1K). To investigate the source of proteolytic activity, cells stably transduced with either the R12Y/Y100I-YFP or YFP-N18T/A19V fusion proteins were cultured in the presence or absence of 10 μM TMP. Cells were also treated with inhibitors of either proteasome- or lysosome-mediated degradation (Reusch et al., 1999). Only cells treated with the proteasome inhibitor MG132 exhibited reduced degradation of DD-YFP fusion protein upon switching to nonpermissive conditions, suggesting that degradation is mediated by proteasome (Figure S1L).
We have thus far demonstrated that ecDHFR-derived DDs destabilize YFP when fused to the terminus for which they were screened and identified. In situations where proteins may not functionally tolerate N- or C-terminal fusions, it would be preferable to have destabilizing domains that can regulate protein levels when spliced into a tolerant domain within the protein of interest. To investigate whether the ecDHFR-derived DDs could confer instability in this context, we spliced the sequences for either the R12Y/Y100I or the N18T/A19V DD between the codons encoding residues 157 and 158 of the YFP gene, which is a site within YFP known to tolerate insertions (Magliery et al., 2005). To accommodate the folding requirements of these globular proteins we tethered the N- and C-termini of the ecDHFR domain to the YFP splice site using linkers varying from 2 to 19 residues on either side of the ecDHFR domain. The internal R12Y/Y100I domain dramatically decreased YFP fluorescence levels in the absence of TMP, and YFP levels were stabilized upon addition of TMP (Figure 1C). Although the N18T/A19V DD is strongly destabilizing when fused to the C-terminus of YFP, this mutant was not as effective as the R12Y/Y100I mutant when spliced internally (Figure S1M).
FKBP- and ecDHFR-derived DDs are tunably and predictably regulated by their respective ligands
One of the goals of this study was to develop a DD system that, when used in combination with the FKBP-derived DD system (Banaszynski et al., 2006), would allow simultaneous regulation of two proteins or groups of proteins. To demonstrate the orthogonality of the FKBP-derived and ecDHFR-derived DD systems, we stably transduced both the FKBP-derived L106P DD fused to mCherry and the ecDHFR-derived R12Y/Y100I DD fused to YFP into NIH3T3 cells. Doubly transduced cells were treated with various concentrations of Shield-1 and TMP and analyzed over the course of one week (Figure 2A). Epifluorescence microscopy images of the doubly transduced cells dosed with indicated combinations of TMP and Shield-1 are also shown (Figure 2B). The close correspondence of the observed fluorescence levels to those predicted from dose-response experiments highlights the ability of this system to regulate two proteins of interest independently of one another. As expected, Shield-1 and TMP do not have any effects on their orthogonal DDs (Figures S2A and S2B).
Figure 2. FKBP- and ecDHFR-derived DDs are tunably and predictably regulated using their respective ligands.

(A) NIH3T3 cells doubly transduced with ecDHFR-derived R12Y/Y100I-YFP and the FKBP-derived L106P-mCherry were dosed with varying concentrations of TMP (black) and Shield-1 (red), and fluorescence was monitored by flow cytometry at the time points indicated. Predicted MFI values (thick lines) were generated using a previous dose-response study of the same cell line. Data are presented as the average MFI ± s.d. and normalized against the average MFI of cells dosed with 10 μM TMP and 3 μM Shield-1, which were set at 100%. (B) NIH3T3 cells described in panel A were plated onto 2-well chamber slides and treated for 24 h with concentrations of TMP and Shield-1 corresponding to the conditions used in panel B. Live cells were visualized by epifluorescence microscopy.
Regulation of Transmembrane Proteins
A previous attempt to regulate CD8α, a transmembrane glycoprotein found on the surface of T cells, resulted in modest levels of regulation at the cell surface (Banaszynski et al., 2006). Because N18T/A19V and R12Y/Y100I ecDHFR are both more potent DDs when fused to the C-terminus of YFP, we suspected that the ecDHFR-derived DDs might offer improved regulation of CD8α expression. To test this theory, we fused either the N18T/A19V or R12Y/Y100I DD to the C-terminus of CD8α and transduced these fusions into NIH3T3 cells. Flow cytometry was used to quantitate levels of CD8α at the cell surface, where we observed a significant decrease in CD8α expression in the absence of TMP. Upon treatment of cells with 10 μM TMP for 24 hr, the N18T/A19V fusion showed higher expression of CD8α, and therefore greater dynamic range, than the R12Y/Y100I fusion (Figure 3A). TMP-mediated stabilization for 24 hrs followed by withdrawal of the ligand for 24 hrs (+/− in Figure 3A) produced protein levels nearly as low as cells maintained under nonpermissive conditions. Encouraged by these results we tested the ability of these DDs to destabilize a more structurally complex transmembrane protein, the β2-adrenergic receptor (β2AR) (Cherezov et al., 2007), which is a member of the GPCR family of integral membrane proteins. Cells were stably transduced with either the N18T/A19V or R12Y/Y100I DDs fused to the 3′-end of the β2AR gene, and we observed low levels of β2AR expression in the absence of TMP. Addition of TMP caused protein levels to increase, and TMP-mediated stabilization for 24 hrs followed by 24 hrs under TMP-free nonpermissive conditions returned β2AR levels to the low background level (Figure 3B).
Figure 3. Regulation of Transmembrane Proteins.

(A) The ecDHFR mutants N18T/A19V and R12Y/ Y100I were fused to the 3′-end of the CD8α gene and were stably transduced into NIH3T3 cells. Cells were divided into three pools, and the first population (–) was mock-treated, and the second population (+) was treated with 10 μM TMP for 24 h. The third population (+/−) was treated with 10 μM TMP for 24 h, then washed with media and cultured for 24 h in the absence of TMP. Live cells were probed with a FITC-conjugated anti-CD8α antibody and assayed by flow cytometry. Data are presented as the average MFI ± s.d. and normalized against the average MFI of untransduced NIH3T3 cells that were also stained with antibody. (B) The N18T/A19V and R12Y/Y100I DDs were fused to the 3′-end of the β2AR gene and these fusions were stably expressed in NIH3T3 cells. As a positive control, unfused β2AR was stably expressed and analyzed alongside both fusions. The cells were divided into three pools as described in panel A and analyzed by flow cytometry. All β2AR genes encoded an N-terminal FLAG epitope. Live cells were probed with Alexa Fluor 488-labeled M1 anti-FLAG antibody, which only recognizes the epitope expressed at the cell surface.
Regulating DDs in rat brain
The commercial availability, oral bioavailability, and low cost of TMP make this ecDHFR-derived DD system particularly attractive for studies in living animals, particularly rodents (Banaszynski et al, 2008). To test the suitability of this new DD system for use within the central nervous system of living rats, we injected recombinant lentivirus encoding either R12Y/Y100I-YFP or YFP-N18T/A19V into the striatum of adult rats (n=6). Half of the animals received TMP in their drinking water (0.2 mg/mL final concentration), and the water was exchanged daily over three weeks. The animals were sacrificed three weeks after injection of the lentivirus, and the brain tissue was fixed and stained for YFP expression. Immunohistochemical analysis of samples from animals that received TMP showed strong induction of YFP expression (Figures 4B and 4D), whereas YFP was barely detectable in animals that received no TMP (Figures 4A and 4C). This experiment demonstrates the first use of the destabilizing domain technology to conditionally regulate the stability of transgenes expressed in the mammalian central nervous system.
Figure 4. Regulating DD-YFP and YFP-DD in rat brain.
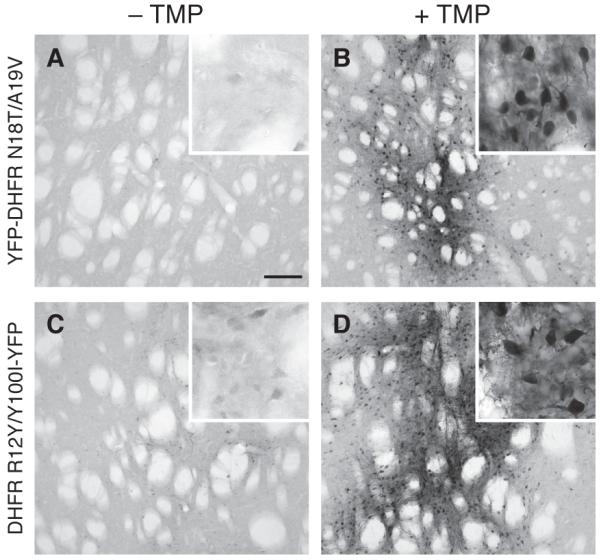
Adult rats received an injection of recombinant lentivirus (rLV) encoding R12Y/Y100I-YFP in the left striatum and YFP-N18T/A19V in the right striatum. Immunohistochemistry of rat brain tissue 3 weeks post rLV injection in animals that received (A) no TMP and (B) 0.2 mg/mL TMP in drinking water. rLV-ecDHFR R12Y/Y100I-YFP with (C) no TMP (D) 0.2 mg/mL TMP. The 40-μm coronal slices were probed with GFP antibody, followed by incubation in biotinylated secondary antibody. The tissues were visualized with avidin-biotin-peroxidase complex using 3,3-diaminobenzidine as chromogen. Scale bar in panel A represents 100 μm, and insets represent 20 μm.
DISCUSSION
The first DD technology was developed based on the human FKBP12 protein bearing a cavity-forming F36V mutation, as a family of FKBP ligands containing corresponding synthetic “bumps” has already been developed and found to be non-toxic in cultured cells and animals (Iuliucci et al., 2001). These high-affinity ligands bind to FKBP(F36V) more tightly than wild-type FKBP12 by three orders of magnitude (Clackson et al., 1998). Based on this protein-ligand pair we used error-prone PCR and FACS-based screening to identify dozens of FKBP(F36V) mutants whose stability can be modulated by a small-molecule ligand Shield-1. The resulting FKBP-derived DD system confers instability to a wide range of proteins and has been shown to confer ligand-dependent stability to kinases, cell cycle regulatory proteins and small GTPases expressed in cultured cells (Banaszynski, et al. 2006). The FKBP-derived DD was further demonstrated to work well in vivo, regulating the stability of secreted proteins in a dose-dependent manner in mice (Banaszynski et al, 2008). The FKBP system has also been used with success in other organisms, such as Toxoplasma (Herm-Götz et al., 2007) and Plasmodium (Russo et al., 2009; Dvorin et al., 2010).
Like the FKBP-derived DDs, the ecDHFR-derived DDs reported in this manuscript confer instability to a fused protein, resulting in rapid degradation of the protein upon withdrawal of the ligand TMP. Characterization of the ecDHFR-derived DDs revealed that the DDs behave very similarly to the FKBP-derived DDs, and the two DD systems work orthogonally as demonstrated in mammalian cells. However, there are notable differences between the two systems. The C-terminal ecDHFR-derived DDs are much more effective at destabilizing the fusion protein than the FKBP-derived L106P DD. Whereas the destabilizing effects of the L106P domain are reduced when fused at the C-terminus of YFP, the ecDHFR-derived DDs F103L and N18T/A19V are very successful at reducing the protein levels when fused to the C-terminus of YFP, with negligible protein levels observed in the absence of TMP.
Fusion of the FKBP-derived L106P DD to the C-terminus of a single-transmembrane protein CD8α is only modestly destabilizing, as a significant amount of protein is trafficked to the membrane in the absence of Shield-1 (Banaszynski et al., 2006). When we fuse DHFR N18T/A19V domain to CD8α, we observe far greater destabilization of CD8α relative to the L106P fusion. Encouraged by this result, we applied the DD technology to the more structurally complex and therapeutically relevant integral transmembrane protein, human β2AR, a G protein-coupled receptor (GPCR). TMP-dependent stability of β2AR was achieved with ~10-fold increase in β2AR-DD expression upon addition of the ligand. GPCRs represent an important class of drug targets for the pharmaceutical industry and currently 30% of marketed drugs act on this family of proteins (Hopkins and Groom, 2002). GPCRs play key roles in biological processes such as cellular metabolism, cell growth, inflammation, and neuronal signaling. Extensive research has taken place in the past decades to understand the function and pathways involved with these membrane proteins. These new ecDHFR-derived DDs may prove to be important tools for probing the functional roles of this large and therapeutically important family of cell surface receptors.
Another important advantage of using the ecDHFR-derived DD system is that the ligand is commercially available, inexpensive and possesses good pharmacological characteristics. The drug can be administered orally, a less invasive and more convenient method of drug administration than IP injection or IV administration. TMP-dosed rats presented no adverse effects such as loss of body weight or diarrhea during this study, even when dosed at 0.4 mg/mL (twice the dose used to regulate transgenes) for six weeks (data not shown). TMP crosses the blood-brain barrier (Barling and Selkon, 1978), allowing the conditional regulation of specific proteins within the central nervous system of living rodents. TMP also passes the placental barrier (Schulz 1972), with teratogenicity observed only when rats are given very high concentrations of TMP. Many genes misregulated in diseases such as cancer exhibit embryonic lethal phenotypes in knockout mouse models. For example, the disruption of genes involved in angiogenesis such as vascular endothelial growth factor or vascular endothelial receptor tyrosine phosphatase results in vascular defects, leading to embryonic lethality (Carmeliet et al., 1996; Dominguez et al., 2007) and precluding study of the roles of these proteins in tumor angiogenesis. The DHFR-derived DD system may be a suitable alternative for in vivo regulation of proteins that are required during embryonic development. Pregnant mice carrying fetuses encoding genes targeted with the ecDHFR-derived DD can be administered TMP during gestation to maintain protein levels necessary for proper development of the embryos.
The rapid switch-like behavior of the DD system makes it a powerful conditional perturbation method, and the ability to tunably regulate protein levels with a high degree of predictability makes the destabilizing domains valuable in broader contexts. For instance, a knowledge of the proteins comprising a signaling network is necessary but not sufficient for a full understanding of the behavior of the system. Researchers can utilize the DD systems to study how molecules comprising a signaling network are “wired” together through intermolecular interactions or diffusible second messengers by perturbing one member of a circuit and observing the effects of this perturbation on the other members of the circuit. This form of sensitivity analysis, performed on the proteins of an intact signaling circuit in a living cell, will enable researchers to deconvolute the interactions between signaling proteins and will ultimately lead to quantitative models of the signaling circuit that can both explain as well as predict the consequences of perturbations.
Thus far, all of the systems described above operate in a way where the protein level is maintained in the presence of ligand. Recently a conditional protein stability technology was developed by which proteins are rapidly depleted in the presence of the plant hormone, auxin (Nishimura et al 2009). In this system, the targeted protein is fused to the AUX/IAA transcriptional repressor (Aid). Degradation of Aid is mediated by the F-box transport inhibitor response 1 protein (TIR1), a plant F-box protein that is part of SCF E3 ubiquitin ligase complex. Addition of auxin promotes the interaction between SCF-TIR1 E3 ubiquitin ligase complex and Aid, resulting in degradation of Aid and any fused partner protein. This system represents a promising approach for using a ligand to destabilize (rather than stabilize) targeted proteins, and obviates the need to continuously administer the ligand until the desired experimental window. Depending on the system used the cost of maintenance dosing could be prohibitive, not to mention the effect of long-term administration of ligand to the animals. While the auxin-based system has the advantage of lacking off-target effects in mammalian cells, this system requires two genetic elements rather than one: researchers must introduce a transgene encoding the F-box protein, TIR1, as well as a second gene encoding the Aid-tagged gene of interest. A system requiring only a single genetic manipulation would be more desirable. Ideally, fusion of a single DD to a targeted protein would confer ligand-dependent stability, but in the opposite sense of the current DDs.
SIGNIFICANCE
We have developed a general method to regulate stability of a specific protein in a rapid, reversible, and tunable matter using a commercially available small molecule. Mutants of E. coli dihydrofolate reductase (ecDHFR) protein were engineered to be degraded when expressed in mammalian cells. When this destabilizing domain (DD) is fused to a protein of interest, its instability is conferred to the fused protein, resulting in rapid degradation of the entire fusion protein by the proteasome. Trimethoprim (TMP) is a high-affinity ligand for ecDHFR that stabilizes fusion proteins in a dose-dependent manner, and protein levels in the absence of TMP are negligible. This ecDHFR-TMP DD system works orthogonally to the existing FKBP/Shld1-based DD system, allowing simultaneous regulation of two proteins independently. The ecDHFR-DD system successfully regulates transmembrane proteins that were difficult to target with the FKBP-DD, such as a GPCR protein, beta2-adrenergic receptor. We have shown a successful regulation of transgenes expressed in rat brain by oral administration of TMP. The ability of TMP to cross the blood-brain barrier (BBB) enables the tunable regulation of proteins expressed in cells found within the mammalian central nervous system. Finally, TMP passes the placental barrier, possibly allowing tunable regulation of genes essential in embryonic development. The DHFR-derived DD system may prove useful in dissecting the role of genes involved in cancer and other diseases in vivo that previously were considered difficult due to essentiality of the genes in embryonic development. Controlling protein function directly using a small-molecule ligand is a more attractive approach than targeting precursor DNA or mRNA, as direct targeting of protein is rapid and is not limited by the intrinsic half-life of the targeted proteins.
EXPERIMENTAL PROCEDURES
ecDHFR Library Generation
Diversity in the ecDHFR sequence was generated using a combination of error-prone PCR (Atwood-Moore et al., 2005) and nucleotide analog mutagenesis (Zaccolo et al., 1996). Three ecDHFR genes were used as templates: parental, Y100I and G121V (see Figure S1 for a description of the parental sequence). For each of these templates, three mutagenesis conditions were used to generate diversity. Condition set A utilized the nonnatural nucleotides 8-oxo-dGTP and dPTP to encourage nucleotide misincorporation. Condition set B utilized 4 ng template, 0.5 μM of each oligonucleotide primer, 3.75 units of Taq polymerase, 5 mM MgCl2, 0.2 mM MnCl2, 0.4 mM dNTPs, and an excess 0.2 mM of dATP and dCTP. Condition set C was identical to B, but dGTP and dTTP were used in excess instead of dATP and dCTP.
Library Screening and Validation
Calculated library diversity was 3 × 104 members for the ecDHFR-YFP library and 1.5 × 105 members for the YFP-ecDHFR library. Screening was performed as described in the text. Upon completion of the fourth sort the cells were allowed to recover in media, and genomic DNA was obtained using Qiagen DNeasy kit. To isolate individual ecDHFR mutants from the pool of genomic DNA, the ecDHFR gene was PCR-amplified using primers that annealed to regions just outside the 5′- and 3′-end of the ecDHFR gene. PCR products were then subcloned into the pBMN YFP plasmids and transformed into E. coli Top10 cells. For the N-terminal ecDHFR DD library, 48 randomly chosen clones were further characterized in mammalian cells. The plasmid DNA was isolated from bacteria and stably transduced into NIH3T3 cells using the retroviral expression system described below. The cells were dosed with solvent or 10 μM TMP (1:1000 dilution of 10 mM stock in DMSO), and only the cells that displayed low YFP fluorescence in the absence of TMP and high levels of YFP expression in the presence of TMP were sequenced. Thirty-six clones were similarly isolated and validated from the C-terminal DD library. Sequence information is provided in Tables S1 and S2.
Cell Culture, Transfections, and Transductions
The NIH3T3 cell line was cultured in DMEM supplemented with 10% donor bovine serum (Invitrogen), 2 mM glutamine, 100 U/mL penicillin, and 100 μg/mL streptomycin. All stable cell lines were created using the Phoenix ecotropic packaging cell line that was transfected using a standard Lipofectamine 2000 protocol. Viral supernatants were harvested 48 hr posttransfection, filtered, and supplemented with 4 μg/mL polybrene. NIH3T3 cells were incubated with viral supernatant for 4 hr at 37 °C. Cells were cultured in growth media for 48 hr to allow for viral integration, then assayed as described. For fluorescence decay kinetics experiments, TMP was removed from the cells by first harvesting the cells with trypsin, then washing TMP from the cells by repeated centrifugation, aspiration, and resuspension in TMP-free media. Alternatively, washing adherent cells once with TMP-free media followed by incubating cells in media “conditioned” with bacterially expressed wild-type ecDHFR protein at 5 μM concentration is even more effective for switching to nonpermissive conditions.
Flow Cytometry
Twenty-four hours prior to analysis, transduced NIH3T3 cells were plated and treated with TMP as described. For cell lines expressing fluorescent proteins, cells were removed from the plate using trypsin and resuspended in growth media to avoid aggregation of the cells. Transmembrane protein expressing cell lines were removed from the plate using 2 mM EDTA in 1 × PBS, then treated with fluorescently-labeled antibody prior to analysis. Cells were analyzed at the Stanford Shared FACS Facility with 10,000 events represented.
Imaging
Tunability experiment was conducted on Zeiss Axioskop 2 epifluorescence microscope equipped with a QICAM FAST 1394 digital CCD camera.
Rat models
Lentiviral vectors were constructed using Gateway technology (Invitrogen). The human CMV promoter was placed upstream of the appropriate fusion gene in the lentiviral backbone 2k7neo (Suter et al., 2006). The vectors were produced and then titered using quantitative PCR as described previously (Nielsen et al., 2009). Adult female Sprague-Dawley rats were injected with lentivirus (6 × 104 TU in 2 μL vehicle) was injected into each striatum of adult (225 g) rats (n=6). rLV encoding the R12Y/Y100I-YFP fusion was injected into the left striatum and rLV encoding YFP-N18T/A19V was injected into the right striatum. The injections were performed in two 1-μL deposits along a single injection tract at the coordinates: anteroposterior = +0.5 mm; mediolateral = ± 3.0 mm relative to bregma and dorsoventral = −5.0 mm and −4.0 mm, calculated from the dural surface. The injection flow rate was 0.4 μL/min and the glass capillary was kept in place for 1 min between deposits and 3 min after the second deposit before retraction. Rats were housed at two or three per cage under a 12 hr light:dark cycle with ad libitum access to food and water. Half of the animals received TMP in the drinking water (TMP emulsion 10 mg/mL, with a final concentration of 0.2 mg/mL in water) for 3 weeks, upon which animals were sacrificed by sodium pentobarbital euthanasia and transcardially perfused with paraformaldehyde.
Immunohistochemistry
The fixed brain tissue was cut by freezing microtome into 40-μm coronal slices for free floating immunohistochemical staining for YFP expression. The sections were quenched in 3% H2O2 and 10% methanol, preincubated in 5% goat serum, and incubated overnight at room temperature with polyclonal GFP antibody that crossreacts with YFP.
HIGHLIGHTS.
Rapidly, reversibly, and tunably regulates the stability of a protein of interest
Regulates transmembrane proteins
Regulates transgenes expressed in the rodent brain by oral administration of trimethoprim
Works orthogonally to the existing FKBP/Shield-1-based DD system
Supplementary Material
Acknowledgments
We are grateful to Laura Banaszynski for suggestions and discussions that helped shape this project, L.-C. Chen for technical assistance, and the Kobilka lab for the β2AR gene and related reagents. We also thank M. Gilbert and T. Knaak of the Stanford FACS facility. This research was supported by the National Institutes of Health (GM068589).
Footnotes
This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Adams JA, Fierke CA, Benkovic SJ. The function of amino acid residues contacting the nicotinamide ring of NADPH in dihydrofolate reductase from Escherichia coli. Biochemistry. 1991;30:11046–11054. doi: 10.1021/bi00110a006. [DOI] [PubMed] [Google Scholar]
- Atwood-Moore A, Ejebe K, Levin HL. Specific recognition and cleavage of the plus-strand primer by reverse transcriptase. J. Virol. 2005;79:14863–14875. doi: 10.1128/JVI.79.23.14863-14875.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banaszynski LA, Chen LC, Maynard-Smith LA, Ooi AG, Wandless TJ. A rapid, reversible, and tunable method to regulate protein function in living cells using synthetic small molecules. Cell. 2006;126:995–1004. doi: 10.1016/j.cell.2006.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banaszynski LA, Sellmyer MA, Contag CH, Wandless TJ, Thorne SH. Chemical control of protein stability and function in living mice. Nat. Med. 2008;14:1123–1127. doi: 10.1038/nm.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barling RW, Selkon JB. The penetration of antibiotics into cerebrospinal fluid and brain tissue. J Antimicrob Chemother. 1978;4:203–227. doi: 10.1093/jac/4.3.203. [DOI] [PubMed] [Google Scholar]
- Carmeliet P, Ferreira V, Breier G, Pollefeyt S, Kieckens L, Gertsenstein M, Fahrig M, Vandenhoeck A, Harpal K, Eberhardt C, et al. Abnormal blood vessel development and lethality in embryos lacking a single VEGF allele. Nature. 1996;380:435–439. doi: 10.1038/380435a0. [DOI] [PubMed] [Google Scholar]
- Cameron CE, Benkovic SJ. Evidence for a functional role of the dynamics of glycine-121 of Escherichia coli dihydrofolate reductase obtained from kinetic analysis of a site-directed mutant. Biochemistry. 1997;36:15792–15800. doi: 10.1021/bi9716231. [DOI] [PubMed] [Google Scholar]
- Cherezov V, Rosenbaum DM, Hanson MA, Rasmussen SG, Thian FS, Kobilka TS, Choi HJ, Kuhn P, Weis WI, Kobilka BK, et al. High-resolution crystal structure of an engineered human beta2-adrenergic G protein-coupled receptor. Science. 2007;318:1258–1265. doi: 10.1126/science.1150577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clackson T, Yang W, Rozamus LW, Hatada M, Amara JF, Rollins CT, Stevenson LF, Magari SR, Wood SA, Courage NL, et al. Redesigning an FKBP-ligand interface to generate chemical dimerizers with novel specificity. Proc Natl Acad Sci U S A. 1998;95:10437–10442. doi: 10.1073/pnas.95.18.10437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dohmen RJ, Wu P, Varshavsky A. Heat-inducible degron: a method for constructing temperature-sensitive mutants. Science. 1994;263:1273–1276. doi: 10.1126/science.8122109. [DOI] [PubMed] [Google Scholar]
- Dominguez MG, Hughes VC, Pan L, Simmons M, Daly C, Anderson K, Noguera-Troise I, Murphy AJ, Valenzuela DM, Davis S, et al. Vascular endothelial tyrosine phosphatase (VE-PTP)-null mice undergo vasculogenesis but die embryonically because of defects in angiogenesis. Proc Natl Acad Sci U S A. 2007;104:3243–3248. doi: 10.1073/pnas.0611510104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dvorin JD, Martyn DC, Patel SD, Grimley JS, Collins CR, Hopp CS, Bright AT, Westenberger S, Winzeler E, Blackman MJ, et al. A plant-like kinase in Plasmodium falciparum regulates parasite egress from erythrocytes. Science. 2010;328:910–912. doi: 10.1126/science.1188191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furth PA, St. Onge L, Boger H, Gruss P, Gossen M, Kistner A, Bujard H, Hennighausen L. Temporal control of gene expression in transgenic mice by a tetracycline-responsive promoter. Proc. Natl. Acad. Sci. USA. 1994;91:9302–9306. doi: 10.1073/pnas.91.20.9302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaveriaux-Ruff C, Kieffer BL. Conditional gene targeting in the mouse nervous system: Insights into brain function and diseases. Pharmacol. Ther. 2007;113:619–634. doi: 10.1016/j.pharmthera.2006.12.003. [DOI] [PubMed] [Google Scholar]
- Herm-Gotz A, Agop-Nersesian C, Munter S, Grimley JS, Wandless TJ, Frischknecht F, Meissner M. Rapid control of protein level in the apicomplexan Toxoplasma gondii. Nat Methods. 2007;4:1003–1005. doi: 10.1038/nmeth1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopkins AL, Groom CR. The druggable genome. Nat Rev Drug Discov. 2002;1:727–730. doi: 10.1038/nrd892. [DOI] [PubMed] [Google Scholar]
- Iuliucci JD, Oliver SD, Morley S, Ward C, Ward J, Dalgarno D, Clackson T, Berger HJ. Intravenous safety and pharmacokinetics of a novel dimerizer drug, AP1903, in healthy volunteers. J Clin Pharmacol. 2001;41:870–879. doi: 10.1177/00912700122010771. [DOI] [PubMed] [Google Scholar]
- Johnston JA, Johnson ES, Waller PR, Varshavsky A. Methotrexate inhibits proteolysis of dihydrofolate reductase by the N-end rule pathway. J. Biol. Chem. 1995;270:8172–8178. doi: 10.1074/jbc.270.14.8172. [DOI] [PubMed] [Google Scholar]
- Lévy F, Johnston JA, Varshavsky A. Analysis of a conditional degradation signal in yeast and mammalian cells. Eur. J. Biochem. 1999;259:244–252. doi: 10.1046/j.1432-1327.1999.00024.x. [DOI] [PubMed] [Google Scholar]
- Liu KJ, Arron JR, Stankunas K, Crabtree GR, Longaker MT. Chemical rescue of cleft palate and midline defects in conditional GSK-3beta mice. Nature. 2007;446:79–82. doi: 10.1038/nature05557. [DOI] [PubMed] [Google Scholar]
- Magliery TJ, Wilson CG, Pan W, Mishler D, Ghosh I, Hamilton AD, Regan L. Detecting protein-protein interactions with a green fluorescent protein fragment reassembly trap: scope and mechanism. J. Am. Chem. Soc. 2005;127:146–157. doi: 10.1021/ja046699g. [DOI] [PubMed] [Google Scholar]
- Matthews DA, Bolin JT, Blurridge JM, Filman DJ, Volz KW, Kraut J. Dihydrofolate reductase. The stereochemistry of inhibitor selectivity. J. Biol. Chem. 1985;260:392–399. [PubMed] [Google Scholar]
- Nielsen TT, Jakobsson J, Rosenqvist N, Lundberg C. Incorporating double copies of a chromatin insulator into lentiviral vectors results in less viral integrants. BMC Biotechnol. 2009;9:13. doi: 10.1186/1472-6750-9-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimura K, Fukagawa T, Takisawa H, Kakimoto T, Kanemaki M. An auxin-based degron system for the rapid depletion of proteins in nonplant cells. Nat Methods. 2009;6:917–922. doi: 10.1038/nmeth.1401. [DOI] [PubMed] [Google Scholar]
- Reusch U, Muranyi W, Lucin P, Burgert HG, Hengel H, Koszinowski UH. A cytomegalovirus glycoprotein re-routes MHC class I complexes to lysosomes for degradation. EMBO J. 1999;18:1081–1091. doi: 10.1093/emboj/18.4.1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russo I, Oksman A, Vaupel B, Goldberg DE. A calpain unique to alveolates is essential in Plasmodium falciparum and its knockdown reveals an involvement in pre-S-phase development. Proc Natl Acad Sci U S A. 2009;106:1554–1559. doi: 10.1073/pnas.0806926106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnell JR, Dyson HJ, Wright PE. Structure, dynamics, and catalytic function of dihydrofolate reductase. Annu. Rev. Biophys. Biomol. Struct. 2004;33:119–140. doi: 10.1146/annurev.biophys.33.110502.133613. [DOI] [PubMed] [Google Scholar]
- Schulz R. Distribution and elimination of trimethoprim in pregnant and newborn rats. Naunyn Schmiedebergs Arch Pharmacol. 1972;272:369–377. doi: 10.1007/BF00501243. [DOI] [PubMed] [Google Scholar]
- Schweitzer BI, Dicker AP, Bertino JR. Dihydrofolate reductase as a therapeutic target. FASEB J. 1990;4:2441–2452. doi: 10.1096/fasebj.4.8.2185970. [DOI] [PubMed] [Google Scholar]
- Stankunas K, Bayle JH, Gestwicki JE, Lin YM, Wandless TJ, Crabtree GR. Conditional protein alleles using knockin mice and a chemical inducer of dimerization. Mol Cell. 2003;12:1615–1624. doi: 10.1016/s1097-2765(03)00491-x. [DOI] [PubMed] [Google Scholar]
- Suter DM, Cartier L, Bettiol E, Tirefort D, Jaconi ME, Dubois-Dauphisn M, Krause KH. Rapid generation of stable transgenic embryonic stem cell lines using modular lentivectors. Stem Cells. 2006;24:615–623. doi: 10.1634/stemcells.2005-0226. [DOI] [PubMed] [Google Scholar]
- Xia H, Mao Q, Paulson HL, Davidson BL. siRNA-mediated gene silencing in vitro and in vivo. Nat. Biotechnol. 2002;20:1006–1010. doi: 10.1038/nbt739. [DOI] [PubMed] [Google Scholar]
- Zaccolo M, Williams DM, Brown DM, Gherardi E. An approach to random mutagenesis of DNA using mixtures of triphosphate derivatives of nucleoside analogues. J. Mol. Biol. 1996;255:589–603. doi: 10.1006/jmbi.1996.0049. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.