

Abstract
Closed head injury is the leading cause of death in children less than 4 years of age, and is thought to be caused in part by rotational inertial motion of the brain. Injury patterns associated with inertial rotations are not well understood in the pediatric population. To characterize the physiological and pathological responses of the immature brain to inertial forces and their relationship to neurological development, toddler-age (4-week-old) piglets were subjected to a single non-impact head rotation at either low (31.6 ± 4.7 rad/sec2, n = 4) or moderate (61.0 ± 7.5 rad/sec2, n = 6) angular acceleration in the axial direction. Graded outcomes were observed for both physiological and histopathological responses such that increasing angular acceleration and velocity produced more severe responses. Unlike low-acceleration rotations, moderate-acceleration rotations produced marked EEG amplitude suppression immediately post-injury, which remained suppressed for the 6-h survival period. In addition, significantly more severe subarachnoid hemorrhage, ischemia, and axonal injury by β-amyloid precursor protein (β-APP) were observed in moderate-acceleration animals than low-acceleration animals. When compared to infant-age (5-day-old) animals subjected to similar (54.1 ± 9.6 rad/sec2) acceleration rotations, 4-week-old moderate-acceleration animals sustained similar severities of subarachnoid hemorrhage and axonal injury at 6 h post-injury, despite the larger, softer brain in the older piglets. We conclude that the traditional mechanical engineering approach of scaling by brain mass and stiffness cannot explain the vulnerability of the infant brain to acceleration-deceleration movements, compared with the toddler.
Key words: axonal injury, pediatric head injury, pig, toddler
Introduction
Head trauma in toddler-age children is generally marked by a pattern of neural tissue injury and time course that are distinct from adults and even infants (Bruce, 1990; Duhaime et al., 2000). This is likely the result of changes in the central nervous system as it undergoes a period of accelerated growth and development. Even during the first 2 years of development, the brain itself undergoes several function and compositional changes. On the cellular level, increases in dendritic and axonal branching in the cerebrum are accompanied by increases in DNA polymerase (DNA-P) content. Overall cell number more than quadruples, with primary increases in the cerebellum seen during the first 18 months (Dobbing and Sands, 1973). Steep increases in lipid content coupled with decreases in overall water content are evident from birth to 18 months, and plateau at 3 or 4 years after birth. The addition of lipid is the result of rapid myelination of axonal segments (Dobbing, 1968; Dobbing and Smart, 1974), and also contributes to the nearly threefold weight increase of the entire brain seen from birth to 18 months (Snyder et al., 1977). This gradual thickening of laminated sheaths around the axons improves transmission of action potentials and likely acts as a protective layer for the axon (Dobbing, 1981). The increase in lipid content also contributes to a 50% decrease in tissue stiffness from the newborn to the toddler brain, making the infant brain more resistant to deformation during contact and non-contact loads (Prange and Margulies, 2002). These distinctions likely affect the response of the brain to an applied load, and underscore the need for experimental and computational models that differentiate between the infant and toddler brain to identify age-specific mechanisms of head injury in the pediatric population.
Clinical evidence suggests that even within the pediatric population, age significantly affects the response of the immature brain to trauma. Cognitive and motor function deficits are more severe in children younger than 4 years of age compared to older children (Agran et al., 2003; Anderson et al., 2001; Levin et al., 1992; Verger et al., 2000). Also, closed head injury in infants often results in diffuse brain atrophy, which is rarely observed in older children (Duhaime and Raghupathi, 1999). Overall, children less than 4 years of age exhibit worse outcomes compared to older children and adults with head injury (Koskiniemi et al., 1995; Luerssen et al., 1988). Even among children <4 years of age, we demonstrated differences in head injuries between infants and toddlers after the same event (Ibrahim et al., in press). We presented a cohort study of infants and toddlers with accidental head injuries and found that infants sustain significantly more skull fractures after accidents, whereas toddlers presented with significantly more neurological signs, suggesting that development plays a role in the head-injury response of the immature brain.
Evidence of axonal injury following trauma supports a general age-dependent trend compared to adults, with infant piglets exhibiting more injured axons per unit area than the adult pig (Raghupathi and Margulies, 2002). Although the response of the infant brain has been shown to be distinct from that of adolescents and adults, experimental studies that have focused on differences between the infant and toddler offer disparate evidence for the effect of maturation on injury response. Specifically, porcine focal lesion traumatic brain injury (TBI) models have demonstrated that geometrically-scaled cortical contusions produce smaller lesions at 1 week and 30 days post-injury in the infant than in the toddler (Duhaime et al., 2000, 2003; Missios et al., 2009), but that infant piglets may sustain larger decreases in cerebral blood flow (Armstead and Kurth, 1994) in the first 3 h following TBI than toddler piglets. Rodent models have shown that infant-age animals are more susceptible to apoptotic neurodegeneration in the initial 6–24 h immediately following mass-scaled weight drops (Bittigau et al., 1999), chronic cognitive deficits several weeks after injury (Huh and Raghupathi, 2007), but similar white matter damage following contusive brain injury (Raghupathi and Huh, 2007) than toddler-age animals. Thus, while the literature supports the theory of an age-dependent response to head trauma, the evidence is supportive of both an increased and decreased vulnerability to brain injury in the infant compared to the toddler.
The objective of this study was to characterize the injury response of the toddler to rapid, non-impact rotations using a 4-week-old piglet model that represents a toddler-age human. We investigated acute (<6 h post-injury) physiological measures (such as electroencephalographic [EEG] recordings and duration of unconsciousness), and histopathological measures (such as hematoma, ischemia, and axonal injury), in response to low (31.6 ± 4.7 rad/sec2) or moderate (61.0 ±7.5 rad/sec2) rotational acceleration. Finally we compared the physiological and pathological brain injury response, including axonal injury and hemorrhage severity, in the 4-week-old animal to that seen in the 5-day-old animal (infant), at accelerations of 54.1 ± 9.6 krad/sec2, to determine age-related differences in these acute responses. According to mass scaling and brain mechanical properties, we would expect that the same rotational magnitude delivered to the heads of 5-day-old and 4-week-old animals will result in larger tissue deformations in the 4-week-old brain because of its larger mass and lower shear modulus. Because we suspect that there is increased axonal injury in the younger brain for the same tissue deformation, we hypothesize that for the same rotational acceleration magnitude, the 4-week-old and 5-day-old animals will have similar severities of physiological and histopathological injury, despite differences in mass and brain material properties that favor smaller deformations in the infant brain than in the toddler. We also speculate that this similarity in injury severity may indicate that the younger animal has a lower critical tissue deformation associated with axonal and hemorrhagic injury than the older animal.
Methods
Animal preparation
Female 4-week-old piglets (n = 13), corresponding in brain development and myelination to a 2- to 4-year-old human, were anesthetized with ketamine (20 mg/kg) and xylazine (2 mg/kg), followed by 4% isoflurane, and intubated. Heart rate (HR) and arterial oxygen saturation (Sao2) were monitored by pulse oximetry (Dinamap Model 8300; Critikon, Tampa, FL). End-tidal CO2 (etco2) and respiratory rate (RR) were monitored by capnometry (Vet/Cap; SDI, San Diego, CA). Body temperature was recorded by rectal thermometer and maintained between 36 and 39°C using a heating pad and heat lamp. HR, Sao2, etco2, RR, and body temperature were recorded every 15 min. Surface EEG recordings were obtained in a 6-channel bipolar montage (NicoletOne; Cardinal Health, Dublin, OH) on a subset of animals (n = 5) to assess severity, distribution of background abnormalities, and electrographic seizure onset. For animals in which EEG recordings were obtained, a non-depolarizing paralytic agent (0.3 mg/kg pancuronium) was administered 1 h prior to injury. After shaving the animal's head, 12 EEG leads were arranged on the animal's head as shown in Figure 1, and measurements were taken continuously throughout the 6-h survival interval.
FIG. 1.

Configuration of 12 electrodes placed on each piglet's head for continuous electroencephalographic monitoring.
Injury protocol
After placement on a biteplate and being secured with snout straps, the isoflurane was withdrawn, and the animals were subjected to a single rapid non-impact rotation in the axial plane such that the center of rotation was in the cervical spine (Fig. 2). Head rotation was performed using an inertial injury device that delivers controlled, reproducible rotational loads without impact by means of a pneumatic actuator. The device converts a linear impulsive motion into an angular motion, allowing the bite plate to rotate in the desired direction. The angular velocity was measured by an angular rate sensor (ATA Engineering, Inc., Herndon, VA) attached to the linkage sidearm, and angular acceleration was calculated by differentiating the angular velocity trace. Data were acquired at a frequency of 10 kHz using a Labview program and PC-based data acquisition system (National Instruments, Austin, TX).
FIG. 2.

Rapid head rotation performed using the inertial injury device.
The animals were divided into two injury groups: the first group of animals was injured at peak rotational accelerations of 61.0 ± 7.5 rad/sec2 (peak rotational velocities of 194.0 ± 14.8 rad/sec; moderate injury; n = 6), and the second group of 4-week-old piglets was injured at peak rotational accelerations of 31.6 ± 4.7 krad/sec2 (peak rotational velocities of 128.5 ± 12.6 rad/sec; low injury; n = 4). These rotational accelerations/velocities were chosen because the moderate acceleration matched the previous loads used to induce brain injury in 3- to 5-day-old piglets (Raghupathi and Margulies, 2002), and the mild acceleration represented the mass-scaled value required to achieve the same injury in the 4-week-old animals, according to the mass scaling principle proposed by Ommaya and Hirsch (Ommaya and Hirsch, 1971). The mass-scaling principle (Eq. 1) suggests that angular accelerations that result in similar injury patterns are related to the inverse ratio of the brain masses to the two-thirds power of the two animals. The mass-scaling principle assumes that the two animals have the same geometric head shape, same brain material properties, and same deformation threshold required to achieve injury.
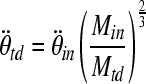 |
(1) |
where 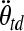 and Mtd are the rotational acceleration and brain mass, respectively, of the toddler, and where
and Mtd are the rotational acceleration and brain mass, respectively, of the toddler, and where 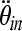 and Min are rotational acceleration and brain mass, respectively, of the infant. In the same way, the scaling principle suggests that the angular velocities which result in similar injury patterns are related to the inverse ratio of the brain masses to the one-third power of the two animals.
and Min are rotational acceleration and brain mass, respectively, of the infant. In the same way, the scaling principle suggests that the angular velocities which result in similar injury patterns are related to the inverse ratio of the brain masses to the one-third power of the two animals.
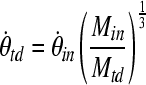 |
(2) |
Average brain masses for these two age groups are Mtd = 56.04 g (n = 6; 4-week-old piglets), and Min = 36.71 g (n = 9; 5-day-old piglets; Eucker, 2009).
Immediately prior to injury, anesthesia was withdrawn and the rapid head rotation was induced. Following injury, the piglet was removed from the bite plate and allowed to breathe spontaneously. Consciousness was assessed by reflexive withdrawal to pinch stimulus every 30–60 sec. If apnea occurred, the animal was immediately ventilated mechanically. Once the animal responded to the pinch reflex, isoflurane was resumed for the remainder of the 6-h survival, and vital signs were recorded every 15 min.
At 6 h post-injury the piglets were sacrificed via pentobarbital overdose. The brains were perfusion-fixed using 0.9% saline followed by 10% neutral buffered formalin. The brains were removed and maintained in 10% formalin for subsequent pathological analysis.
Histological and pathological assessment
All gross pathological examinations were performed by a neuropathologist blinded to experimental group. First, macroscopic examination of whole brains was performed to assess subdural/subarachnoid hemorrhage. Each brain was sectioned in 3-mm coronal slices (typically 16 slices through the cerebrum and cerebellum), and the sections were photographed and examined macroscopically for regional pathology. The tissue sections were subsequently embedded in paraffin. From each 3-mm coronal section, one 6-μm-thick slice was cut for microscopic examination. Thin section staining included hematoxylin and eosin (H&E) and the β-amyloid precursor protein (β-APP) immunohistochemical marker, with all sections lightly counter-stained with Meyer's hematoxylin. Rather than use stereological sampling, all fields were examined microscopically in each 6-μm slice. Microscopic assessment included the degree and distribution of cellular injury and axonal damage. H&E sections were examined to identify regions of infarct and ischemia. Axonal injury was assessed using β-APP Immunohistochemistry, and in a large subset of coronal slices, neurofilament staining (NF68). These two markers of axonal injury were chosen in part to study their relative sensitivity to axonal injury. For each animal, the distributions of axonal and neuronal injury were documented on the coronal section photographs.
Ischemia and brainstem injury were scored on a scale of 0–3, for which 0 corresponded to no injury and 3 corresponded to severe injury. Severity of ischemia in each brain was graded as either absent, mild (≤2 subcortical lesions), moderate (3–5 subcortical lesions, or lesions limited to arterial boundary zones singly or in combination, with subtotal infarction in the distribution of the cerebral arteries), or severe (diffuse, multifocal, and large lesions within arterial territories), in a manner similar to the method of Graham and colleagues (Graham et al., 1989). Brainstem injury was classified from absent to mild, moderate, or severe (multifocal prominent staining), based on the extent of both ischemic and white matter (β-APP) injury. Subarachnoid hemorrhage (SAH) was classified on a scale of 0–5 (0 = no blood, 1 = slight amount of blood on the cerebrum or in the gyral folds, 2 = blood on only one cerebral hemisphere, 3 = blood on both cerebral hemispheres, 4 = blood only on the brainstem/ventral surface of the brain, and 5 = blood on cerebral hemispheres and brainstem/ventral surface of the brain), based on macroscopic observation. All animals in all acceleration groups were assigned a score for ischemia, brainstem injury, and subarachnoid hemorrhage, using the same scoring paradigm described above.
Axonal injury, total tissue section area, area positive for β-APP, and area positive for NF68 were quantified for each region using photographic image analysis with ImageJ software (National Institutes of Health). These areas were used to calculate percentages of axonal injury.
Statistical analysis
Average heart rate, temperature, and mean arterial pressure (MAP) were calculated for three time segments: pre-injury, in the first hour post-injury, and during the remaining 5 h post-injury. MAP was calculated for every time point with (Eq. 3):
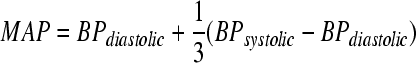 |
(3) |
The average percentage change from baseline (pre-injury) was calculated during the first hour and during the remaining 5 h post-injury, yielding an average value representing the time period immediately post-injury, and a second average value representing the time period several hours post-injury. A two-way analysis of variance (ANOVA) was used to determine the influence of time and rotational load on heart rate, temperature, MAP, and duration of unconsciousness.
Subarachnoid hemorrhage, ischemia, and brainstem injury scores were compared between the sham-injured, low-acceleration, and moderate-acceleration groups using the Mann-Whitney test for rank. Volume percentages of axonal injury, as calculated by regions stained with β-APP, were compared between the 4-week low- and moderate-acceleration groups to determine differences in axonal injury severity between groups using the Student's t-test. To assess the sensitivity of β-APP staining compared to NF68, an ANOVA was used to compare volume percentages of axonal injury calculated from these two methods separately for both the low- and moderate-acceleration groups. Furthermore, correlation comparisons were used to determine the relationship, if any, between angular acceleration and the percentage volume stained for axonal injury by β-APP and NF68. Finally, duration of unconsciousness and volume percentage of axonal injury were compared among the 4-week-old low-acceleration (mass-scaled), moderate-acceleration, and acceleration-matched 3- to 5-day-old animals using an ANOVA to determine the influence of age on injury response. For all quantitative statistics, significance was set at p < 0.05.
Results
Physiological measurements
We found no significant differences in the percentage changes of heart rate, temperature, or MAP between sham, low-, and moderate-acceleration toddler groups in the first hour post-injury, or during the remaining 5 h post-injury (Table 1). There were no significant differences in pre-injury measurements of heart rate, temperature, or MAP between any of the groups. After combining all three toddler groups, pre-injury baselines for heart rate, temperature, and MAP were 122.2 ± 17.9 bpm, 37.3 ± 0.6°C, and 98 ± 23 mm Hg, respectively. Because of the differences in heart rate, temperature, and MAP that occur naturally as a result of development, we present the percentage changes in these physiological variables from baseline to facilitate comparisons to the 5-day-old animals (see below) in a subsequent analysis. We noted a great deal of animal-to-animal variability in heart rate and MAP, such that any differences were difficult to distinguish. For example, in the moderate-acceleration group, heart rate increased on average immediately post-injury in three animals, while heart rate decreased on average in the other three moderate-acceleration animals. Large variability was also seen in MAP immediately post-injury in the same group, ranging from a 10% decrease to a 63% increase. A similar variability was noted in the low-acceleration group, with heart rate increasing in one animal and decreasing in three animals post-injury, and MAP increasing in three animals and decreasing in one animal post-injury. Thus, trends, if any, in heart rate and MAP resulting from rapid rotation were not distinguishable due to the variability seen among animals.
Table 1.
Percentage Change from Pre-injury Baseline of Heart Rate, Mean Arterial Pressure (MAP), and Body Temperature (Mean ± SEM)
| |
|
Heart rate (bpm) |
Δ Temperature (°C) |
Δ MAP (mm Hg) |
|||
|---|---|---|---|---|---|---|---|
| Group | n | 0–1 h post | 1–6 h post | 0–1 h post | 1–6 h post | 0–1 h post | 1–6 h post |
| Sham | 2–3 | −19.2 ± 6.7 | −11.1 ± 9.1 | −2.4 ± 1.3 | −0.7 ± 2.2 | 39.5 ± 1.7 | |
| 4-week-old low | 4 | −10.3 ± 5.0 | 16.1 ± 14.5 | −0.9 ± 0.5 | 2.9 ± 0.6a | 15.2 ± 12.9 | 14.4 ± 7.4 |
| 4-week-old moderate | 6 | 0.4 ± 4.3 | 23.2 ± 6.8a | −1.1 ± 0.4 | 0.9 ± 0.5a | 28.2 ± 12.2 | 5.4 ± 11.0 |
No significant differences were noted between any groups, but temperature was significantly increased at 1−6 h post-injury compared to 0-1 h post-injury in 4-week-old low- and moderate-acceleration groups. Heart rate was significantly increased at 1−6 h post-injury compared to 0−1 h post-injury in the 4-week-old moderate-acceleration groups.
Significantly different from 0−1 h post-injury; p < 0.05.
SEM, standard error of the mean; bpm, beats per minute.
All (100%) moderate-acceleration animals were apneic post-injury, compared to 50% of low-acceleration animals and none of the sham animals (p < 0.05; Table 2). Duration of unconsciousness was not significantly affected by rotational acceleration. Immediately post-injury, there was pronounced amplitude suppression in the EEG recordings from the moderate-acceleration group compared to the low-acceleration group (Fig. 3). Although EEGs were not measured in sham animals, the low-acceleration group had very minor changes in EEG amplitude and frequency between pre- and post-injury measurements. Representative recordings in two channels (F3-C3 and C3-P3) are shown in Figure 3.
Table 2.
Peak Rotational Velocity, Peak Rotational Acceleration, Duration of Unconsciousness, and Rate of Apnea for All Injury Groups (Mean ± SEM)
| Group | n | Peak angular velocity (rad/sec, ± SD) | Peak angular acceleration (krad/sec2, ±SD) | Unconsciousness time (min, ±SEM) | % Apneic |
|---|---|---|---|---|---|
| Sham | 2 | 0 | 0 | 24.4 ± 3.4 | 0 |
| 4-week-old low | 4 | 129 ± 13 | 31.6 ± 4.7 | 7.7 ± 2.6 | 50 |
| 4-week-old moderate | 6 | 194 ± 15 | 61.0 ± 7.5 | 16.0 ± 5.8 | 100a |
Significantly different from sham; p < 0.05.
SEM, standard error of the mean; SD, standard deviation.
FIG. 3.

Representative electroencephalogram (EEG) traces pre- and post-injury from 4-week-old animals in the low-acceleration (right) and moderate-acceleration (left) groups (ISO, isoflurane).
Histological and pathological differences
We found significant differences in histological and pathological outcomes between sham, low-acceleration, and moderate-acceleration animals (Table 3). Animals who received moderate-acceleration rotation had significantly more subarachnoid bleeding than shams (p < 0.05), while low-acceleration animals were statistically similar to shams. Moderate-acceleration animals also had more extensive regions of ischemia compared to both shams and low-acceleration animals (p < 0.05). Likewise, differences were noted in percentage volume of axonal injury detected by β-APP between groups. A larger percentage volume of β-APP staining for axonal injury was found in moderate-acceleration animals compared to shams and low-acceleration animals (p < 0.05). Figure 4 shows representative images of regions of β-APP staining for axonal injury in a moderate-acceleration animal.
Table 3.
Histological and Pathological Scoring for Subarachnoid Hemorrhage (SAH), Ischemia, Brainstem (BS), and Axonal Injury
| Group | SAH score | Ischemia score | BS score | % Volume axonal injury (β-APP) |
|---|---|---|---|---|
| Sham | 0 | 0 | 0 | 0 |
| 4-Week-old low | 2.25 ± 1.32 | 0.50 ± 0.29b | 0.25 ± 0.25 | 0.20 ± 0.03b |
| 4-Week-old moderate | 4.17 ± 0.65a | 2.17 ± 0.48a | 1.0 ± 0.37 | 0.94 ± 0.16a |
Significantly different from sham.
Significantly different from moderate-acceleration group; p < 0.05.
Ischemia and brainstem injury were scored on a scale of 0–3, with 0 corresponding to no injury and 3 corresponding to severe injury. SAH was classified on a scale of 0–5, with 0 corresponding to no blood, and 5 corresponding to blood in the cerebral hemispheres and brainstem/ventral surface of the brain, based on macroscopic observation. Mean ± SEM.
β-APP, β-amyloid precursor protein.
FIG. 4.

(A) Lateral ventricle with β-APP stained axons (10 × magnification). (B) Axonal spheroids stained for β-APP (60 × magnification; β-APP, β-amyloid precursor protein).
Although percentage volume injured varied between injury groups, consistent regional trends in the distribution of axonal injury in the rostral-caudal direction were noted. The average area of injury per slice was highest in the diencephalon and midbrain regions, and lowest in the extreme rostral and extreme caudal (pons) sections of the cerebrum (Fig. 5). No significant differences were found in brainstem injury across all groups.
FIG. 5.

Average axonal injury distribution by β-APP staining in the cerebrum over all injured animals in the rostral-caudal direction (mean ± standard error of the mean; β-APP, β-amyloid precursor protein).
When comparing methods of detection of axonal injury, β-APP- and NF68-stained sections were evaluated. Representative slices stained with both antibodies are shown in Figure 6. In the moderate-acceleration group, a paired t-test showed that β-APP immunostaining identified a significantly greater volume of white matter injury than NF68 (p < 0.005). In the low-acceleration group no NF68 staining was detected, and a paired t-test showed a significantly greater volume of white matter injury by β-APP than NF68 (p < 0.001; Fig. 7). Furthermore, β-APP staining demonstrated a significant difference in axonal injury volume between the low- and moderate-acceleration groups (0.22 ± 0.02 versus 0.92 ± 0.20; p < 0.006), while for NF68 the differences did not reach statistical significance (0 ± 0 versus 0.07 ± 0.03; p = 0.16). Finally, the correlation between rotational acceleration and percentage volume stained was calculated for both methods. Immunostaining by β-APP correlated better with rotational acceleration than NF68 (Corrβ-APP = 0.65, CorrNF68 = 0.29; Fig. 8). The correlation of β-APP with rotational acceleration reached significance (p = 0.022), whereas the correlation with NF68 did not (p = 0.37). Although not shown here, angular velocity correlated with brain injury similarly to acceleration, such that velocity significantly correlated with β-APP but not NF68 (Corrβ-APP = 0.64 and p = 0.024; CorrNF68 = 0.30 and p = 0.34). Using a Student's paired t-test, the percentage volume stained by β-APP was significantly greater than NF68. When NF68 detected any injury at all, β-APP detected 5 to 10 times larger volumes of injured axons than NF68 (Fig. 9). However, for 7 of 10 animals (n = 4 low-acceleration and n = 3 moderate-acceleration), NF68 did not detect any injured axons, yet β-APP detected regions of damaged white matter. In fact, NF68 was unable to detect volumes of injury less than 0.56%, whereas β-APP was able to distinguish volumes as small as 0.17% (Fig. 9).
FIG. 6.

Representative section from a moderate-acceleration animal stained with both NF68 (left) and β-APP (right; β-APP, β-amyloid precursor protein; NF68, neurofilament staining).
FIG. 7.

Axonal injury staining by β-APP and NF68 in the low- and moderate-acceleration groups. Brackets indicate groups that are significantly different (p < 0.05; mean ± standard deviation; β-APP, β-amyloid precursor protein; NF68, neurofilament staining).
FIG. 8.

Correlation between angular acceleration and percentage volume stained for axonal injury by β-APP and NF68 (β-APP, β-amyloid precursor protein; NF68, neurofilament staining).
FIG. 9.

Percentage volume of the whole brain stained by β-APP and NF68. β-APP detected 5 to 10 times larger volumes of injured axons than NF68 (β-APP, β-amyloid precursor protein; NF68, neurofilament staining).
Comparison with 5-day-old piglet injuries
Physiological and histopathological outcomes in the low- (31.6 ± 4.7 krad/sec2) and moderate-acceleration (61.0 ± 7.5 krad/sec2) 4-week-old animals were compared to previously observed outcomes in 5-day-old animals subjected to axial acceleration rotations (54.1 ± 9.6 krad/sec2; Table 4; Eucker, 2009). Evaluation of animals subjected to these specific loading conditions was performed to compare the injury response across acceleration-matched age groups, and across mass-scaled acceleration age groups. The acceleration-matched groups were considered as those receiving head rotations of 61.0 ± 7.5 krad/sec2 and 54.1 ± 9.6 krad/sec2 (not significantly different; p > 0.15), and the mass-scaled groups were those receiving head rotations of 31.6 ± 4.7 krad/sec2 and 54.1 ± 9.6 krad/sec2, scaled with average brain masses of 56.0 g (n = 4; low-acceleration group) and 36.7 g (n = 9) for the 4-week-old and 5-day-old animals, respectively.
Table 4.
Loading Conditions and Percentage Volume Axonal Injury by β -APP Immunostaining for 4-Week-Old Low- and Moderate-Acceleration, and 5-Day-Old Moderate-Acceleration Groups
| Group | Pig no. | Peak angular velocity (rad/sec) | Average peak angular acceleration (krad/sec2) | % β-APP staining |
|---|---|---|---|---|
| 4-Week-old low-acceleration group | 050420 | 112 | 38.6 | 0.24 |
| 050504 | 142.6 | 29.3 | 0.19 | |
| 050714 | 129.5 | 29.4 | 0.19 | |
| 050913 | 130 | 29.0 | 0.17 | |
| 4-Week-old moderate-acceleration group | 041021 | 210 | 71.0 | 0.38 |
| 050106 | 175 | 51.7 | 0.89 | |
| 050111 | 177 | 52.9 | 1.70 | |
| 050303 | 195 | 61.8 | 0.56 | |
| 050310 | 201 | 63.7 | 0.84 | |
| 050315 | 206 | 65.2 | 1.29 | |
| 5-Day-old moderate-acceleration group | 60419 | 207 | 61.2 | 1.05 |
| 60810 | 212 | 75.2 | 1.10 | |
| 70621 | 180 | 50.0 | 0.64 | |
| 70626 | 188 | 44.7 | 0.21 | |
| 70710 | 209 | 54.2 | 1.10 | |
| 70717 | 200 | 54.3 | 1.11 | |
| 70718 | 183 | 43.2 | 0.55 | |
| 70815 | 203 | 50.5 | 0.95 | |
| 71011 | 197 | 53.3 | 0.58 |
β-APP, β-amyloid precursor protein.
No significant differences in temperature change were noted between the 5-day-old group and either 4-week-old group post-injury, likely due to proper maintenance of body temperature between 37 and 38°C with heating pads and lamps. In both 4-week-old low and 5-day-old groups heart rate increased nearly 15% above pre-injury levels in the last 5 h of survival, while in 4-week-old moderate animals heart rate increased 23% above pre-injury levels, although these differences across age did not reach significance. No trends were noted in heart rate during the first hour post-injury among the three groups. We were unable to compare MAP across age because a therapeutic intervention was used in the 5-day-old animals to maintain MAP that was not used in any of the 4-week-old animals. Average duration of unconsciousness in the 5-day-old animals was 7.1 ± 1.8 min (mean ± SEM). This was not significantly different from the durations of unconsciousness for either the 4-week-old low or 4-week-old moderate groups (7.7 ± 2.6 min and 16.0 ± 5.8 min, respectively). Of the nine 5-day-old animals, 67% were apneic post-injury compared to 50% of 4-week-old low and 100% of 4-week-old moderate-acceleration animals. The rate of apnea post-injury was not significantly different across age, when comparing either scaled or unscaled acceleration group pairs. EEG recordings of 5-day-old animals showed similar amplitude suppression immediately post-injury, and for the remainder of the 6-h survival period (Fig. 10), similarly to the amplitude suppression seen in 4-week-old matched-acceleration animals (Fig. 3, left), but were larger suppressions than those seen in the 4-week-old scaled acceleration animals (Fig. 3, right).
FIG. 10.

Representative electroencephalogram recordings in two channels (F3-C3 and C3-P3) from a 5-day-old moderate-acceleration animal show that immediately post-injury there was pronounced amplitude suppression that was sustained for the 6-h survival period, similar to that observed in the 4-week-old moderate-acceleration animals (ISO, isoflurane).
Just as the EEG responses of the 5-day-old piglets were more similar to the acceleration-matched 4-week-old piglet group, histopathology was more similar across age when comparing acceleration-matched groups (Fig. 11). Specifically, average subarachnoid hemorrhage scores in the 5-day-old animals were not significantly different from the acceleration-matched 4-week-old piglet scores (5.0 ± 0 versus 4.17 ± 0.65), but significantly larger than 4-week-old scaled-acceleration animals (2.25 ± 1.32; p < 0.02). In addition, brain volume of axonal injury was not significantly different between acceleration-matched groups (4-week-old moderate = 0.94 ± 0.16%, and 5-day-old = 0.81 ± 0.11%; p = 0.53), but was significantly larger in 5-day-old animals compared to the mass-scaled 4-week-old low-acceleration group (4-week-old low = 0.20 ± 0.14%; p < 0.004; Fig. 11). No significant differences were found in the extent of ischemia or brainstem injury between either 4-week-old animal groups and the 5-day-old animals.
FIG. 11.

Representative coronal sections with regions of β-APP immunostaining for axonal injury in 4-week-old scaled, 4-week-old matched (moderate), and 5-day-old moderate-acceleration groups. Matched-acceleration groups in both ages had similar severity of white matter damage, while the mass-scaled acceleration group had significantly less axonal injury (β-APP, β-amyloid precursor protein).
Improved scaling law
Our piglet data refute the hypothesis that scaling rotational loads by brain mass as proposed by Ommaya and colleagues (Ommaya et al., 1967; Ommaya and Hirsch, 1971) results in similar brain injuries in 4-week-old and 5-day-old animals. In the unscaled rotations, we expected more severe subarachnoid and axonal injury in 4-week-old than in 5-day-old animals because of the larger, softer brain (Prange and Margulies, 2002) in the 4-week-old animals; instead we saw similar injuries. Furthermore, other studies comparing unscaled injury magnitudes also show that the infant-age animal often experiences similar or more severe injury in the acute period following the injury event, despite a smaller, stiffer brain (Gefen et al., 2003; Prange and Margulies, 2002). The previous scaling relationship assumed similar brain material properties and deformation required for injury. We hypothesize that this disparity in axonal injury volume between infants and toddlers is strongly influenced by age-related differences in brain tissue material properties and vulnerability to deformation, or critical strain. We propose that the scaling relationship between infants and toddlers must include differences in brain mass, material properties, and tissue vulnerability (defined as the strain threshold required to produce injury in the tissue) in the following manner:
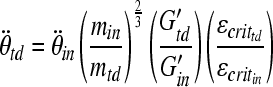 |
(4) |
where min and mtd are the brain masses, 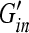 and
and 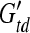 are brain elastic moduli,
are brain elastic moduli, 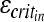 and
and 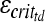 are the critical strain thresholds, and
are the critical strain thresholds, and 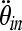 and
and 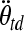 are the rotational accelerations in the infant and toddler, respectively, required to produce similar severity of axonal injury in these two age groups.
are the rotational accelerations in the infant and toddler, respectively, required to produce similar severity of axonal injury in these two age groups.
This new relationship builds on the previous proposed scaling relationships by Ommaya and associates (Ommaya et al., 1967), and later by Thibault and Margulies (Thibault and Margulies, 1998), by adding a critical strain factor that accounts for differences in tissue susceptibility to axonal injury between infants and toddlers. If we substitute into Eq. 4 the average rotational accelerations of the 5-day-old and 4-week-old moderate-acceleration groups that produced similar injury severity (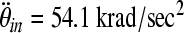 and
and 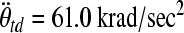 ), the respective brain elastic moduli (
), the respective brain elastic moduli (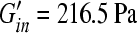 and
and 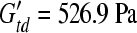 ; Prange and Margulies 2002), and the average masses of the 5-day-old (n = 9) and 4-week-old (n = 6) moderate-acceleration animals (min = 36.7 g and mtd = 55.0 g), an estimated ratio of the critical strains can be calculated as
; Prange and Margulies 2002), and the average masses of the 5-day-old (n = 9) and 4-week-old (n = 6) moderate-acceleration animals (min = 36.7 g and mtd = 55.0 g), an estimated ratio of the critical strains can be calculated as
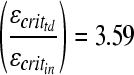 |
(5) |
Thus we determined that the toddler brain can withstand over three times greater strains than the infant before axonal injury results.
In summary, our data can be better described (lower sum of squares, SS) by this mass-modulus-critical-strain scaling relationship (Fig. 12) than by mass-scaling alone (SS: 1.0 × 109 versus 3.6 × 109). In Figure 12 the asterisks represent the average brain mass and rotational acceleration of 5 day and 4 week old animals with similar severity of axonal injury. Axonal injury (β-APP) volume percent (labeled next to each data point) from 4 week and 5 day animals moderate acceleration animals is shown. Furthermore, we validated this relationship with n = 7 additional 5-day-old low-acceleration animals (average axonal injury 0.18 ± 0.2% and acceleration 31.6–3.5 krad/sec2 (Table 5).
FIG. 12.

Mass-scaled injuries from 5-day-old moderate and 4-week-old moderate-acceleration groups plotted with the proposed theoretical relationship that predicts accelerations that result in similar injury severity based on brain mass, material properties, and critical tissue strain. Volume percentage of axonal injury is labeled next to each data point (asterisks represent the average mass and acceleration of the 5-day-old and 4-week-old groups).
Table 5.
Seven 5 day old low acceleration animals used for validation of the proposed mass-modulus critical strain scaling relationship
| Pig # | Brain Mass (g) | Rotational Acceleration (krad/s2) | % β-APP |
|---|---|---|---|
| 30619 | 34.7 | 30.6 | 0 |
| 90114 | 34.51 | 28.6 | 0.17 |
| 90127 | 36.09 | 25.8 | 0.05 |
| 90129 | 36.62 | 32.6 | 0 |
| 70726 | 37.94 | 32.9 | 0.59 |
| 70814 | 36.82 | 35.4 | 0.15 |
| 70925 | 35.72 | 35.2 | 0.29 |
| Mean ± S.D. | 36.06 ± 1.2 | 31.6 ± 3.5 | 0.18 ± 0.2 |
Discussion
In the current study, we demonstrated that increasing head rotational accelerations in the 4-week-old (toddler) piglet produce graded physiological and histopathological outcomes. Larger rotational acceleration and velocities resulted in decreased EEG amplitude, more severe subarachnoid hemorrhage, more areas of ischemia, and larger volumes of injured axons than lower magnitudes. Comparing mass-scaled rotational loads between the 4-week-old and 5-day-old piglets revealed more severe subarachnoid hemorrhage and more extensive β-APP-labeled white matter damage in the 5-day-old animals than the 4-week-old animals. Finally, we showed that immunostaining by β-APP identified larger regions of axonal injury than NF68, suggesting that β-APP is a more sensitive tool for assessing white matter damage.
The differences we observed in immunostaining for axonal injury may be related to the specific cytoskeletal proteins that bind to β-APP and NF68. The β-amyloid precursor protein (β-APP) is normally present in neurons and is thought to have diverse roles, including association with the endosomal/lysosomal system (Ferreira et al., 1993; Haass et al., 1992), and fast anterograde axonal transport (Koo et al., 1990). The 68-kDa neurofilament protein (NF68) is found along the length of the axon and is associated with regulation of the internal structure and axonal diameter (Alberts, 2002). β-APP has often been associated with axonal flow disruption, while NF68 is associated with structural damage to the axon itself (Posmantur et al., 1994, 1996; Sherriff et al., 1994). When an axon is injured, proteins and other substances normally carried by anterograde axonal transport begin to accumulate in the shaft, which causes an interruption of axonal flow and concentration of organelles. Axonal swelling occurs as a direct result, and finally disconnection from the distal part of the axon. Because of this step-wise process, the concentration of accumulating protein may therefore reach a detectable level by β-APP, even before signs of axonal structural damage occur that can be detected by NF68. Although both β-APP and NF68 have been shown to appear as early as 1–3 h post-injury in human and rodent models (Posmantur et al., 1994; Sherriff et al., 1994; Yam et al., 1998), NF68 appears in regions of dark shrunken neurons suggestive of impending cell death or irreversible damage (Posmantur et al., 1996), while β-APP is thought to label axons with functional transport deficits or possible reversible injury. Regardless of their mechanism, β-APP has been shown to be a more sensitive and specific marker of axonal injury in adult humans surviving 2.5 h to 2 weeks after head trauma (Sherriff et al., 1994). Our data in a porcine model support β-APP as a more sensitive marker for axonal dysfunction.
The trend we observed of increasing axonal and hemorrhagic injury with increasing rotational acceleration or velocity magnitude in 4-week-old piglets is consistent with reports of other types of brain injury and increasing magnitude of rotational acceleration. Previously published primate data in the rhesus (brain mass 70–100 g) and the squirrel (brain mass 20–27 g) monkey revealed that concussion was associated with larger velocities and accelerations, and not with smaller loads (Ommaya et al., 1966, 1967; Ommaya and Hirsch, 1971). Non-impact rotational loads in the adult mini-pig demonstrated that increasing peak angular velocity correlated positively with coma severity and the number of damaged axons (Smith et al., 2000). Although duration of unconsciousness did not correlate with rotational load magnitude in our study, we did find a positive relationship between several histopathological measures (subarachnoid hemorrhage, ischemia, and axonal injury) and rotational load (velocity and acceleration). Other animal models of traumatic brain injury such as fluid percussion (McIntosh et al., 1987, 1989; Vink et al., 1988; Zhang et al., 1999), cortical impact (Elliott et al., 2008; Igarashi et al., 2007; Lighthall, 1988; Saatman et al., 2006) and weight drop (Foda and Marmarou, 1994; Marmarou et al., 1994; Ucar et al., 2006) injuries also support our finding of increasing injury severity with increasing mechanical load. Within a single age group, the observed relationship between higher mechanical load and increasing severity of certain brain injuries may be directly related to functional or structural damage to the tissue (Bain and Meaney, 2000; Bain et al., 2001; Lusardi et al., 2003; Meaney et al., 1995; Smith et al., 1999; Wolf et al., 2001). Although the severity of several different types of brain injuries have been shown to correlate positively with magnitude of mechanical load, we have chosen to focus on axonal injury in our study of 5-day-old and 4-week-old piglets because it is an easily quantifiable injury that is a pathological hallmark of moderate-to-severe TBI in children.
In our study, we found an age-dependent injury response to scaled rapid rotational loads (expected to create similar injury severity), such that the 5-day-old infant animals sustained more severe injuries than 4-week-old toddler animals when evaluated 6 h after injury than would have been expected based on brain mass and stiffness. Several researchers have investigated the response of the immature brain in comparison to the adult in rodent (Adelson et al., 2001; Biagas et al., 1996; Fan et al., 2003; Prins et al., 1996) and porcine (Raghupathi and Margulies, 2002) models, highlighting both the vulnerability and plasticity of the immature brain. However, few investigators have explored age-related differences in brain injury response within the pediatric age range (i.e., between infants and toddlers).
Depending on the brain injury type, the animal model literature offers conflicting evidence for increased and decreased vulnerability in the toddler compared to the infant. Relative brain lesion volumes following scaled controlled cortical impact (matched percentage of brain deformed) were similar in 4-week-old and 5-day-old piglets at 24 h after injury, but were larger in the 4-week-old piglets than in the 5-day-old piglets at 7 days post-injury (Duhaime et al., 2000, 2003). Taken together with our data showing vulnerability in the infant at 6 h post injury, these results suggest a continuum, with more severe lesions to scaled loads in the infant, but a trend towards enhanced tissue repair and recovery in this youngest age group compared to the toddler. Studies of cerebral blood flow in the piglet following TBI offer conflicting trends with age. In a fluid percussion model, cerebral blood flow decreased in both ages within 10 min following injury, but returned to baseline only in the 4-week-old animals, and not in the 5-day-old animals within 3 h (Armstead and Kurth, 1994). However, in a cortical contusion model, cerebral blood flow increased in 5-day-old piglets and decreased in 4-week-old piglets 1–3 h following traumatic brain injury (Armstead and Kurth, 1994; Durham et al., 2000). In our unscaled rapid non-impact rotations, we saw a significant decrease in cerebral blood flow in both infant (Eucker, 2009) and toddler piglets (unpublished data), that was sustained for 6 h post-injury.
When we compare across age groups for the same (acceleration-matched) loads, we observed similar severity of axonal injury in 5-day-old (infant) and 4-week-old (toddler) animals, despite the larger softer brains of the toddlers (Prange and Margulies, 2002). Our results are supported by studies in rodents in which unscaled controlled cortical impact in PND11 (infant) and PND17 (toddler) rats resulted in similar severities of traumatic axonal injury (Raghupathi and Huh, 2007) in the white matter and thalamus below the impact site. However, younger animals exhibited reactive gliosis, atrophy in multiple brain regions, and larger fractions of tissue loss and learning deficits than older animals. Scaled and unscaled weight-drop injuries in PND7 (infant) and PND14 (toddler) rats also showed increased apoptotic cell death in younger compared to older animals up to 24 h post-injury, suggesting an increased vulnerability in the youngest animals (Bittigau et al., 1999). In our study of infant and toddler piglets, subarachnoid hemorrhage scores were more severe in infant piglets than in toddler piglets subjected to scaled accelerations, but similar to toddler piglets subjected to the same accelerations.
To determine if clinical data also suggest an initial vulnerability to brain injury in the infant immediately post-injury, perhaps followed by a period of enhanced recovery and repair compared to the toddler, we previously examined injury patterns in a cohort of 285 infants and toddlers <4 years of age who were admitted to the Children's Hospital of Philadelphia for a head injury from a fall over a 6-year period (Ibrahim et al, in press). Of the 285 patients, 17 infants and 44 toddlers were admitted for a fall within 24 h of the event with no external evidence of head impact (no scalp or facial soft tissue injury and no skull fracture). For these cases, we assumed that any head impact sustained during the fall was minimal and likely did not contribute to the resulting injury patterns. Instead, we assumed that any injuries probably resulted from inertial effects during the event. Injury Severity Scores (ISS) were significantly higher in infants compared to toddlers in this group (7.8 ± 1.1 versus 4.7 ± 0.7; p < 0.03), and although there was no difference in the incidence of altered mental status (lethargy, sluggishness, unexplained irritability) between infants and toddlers, more toddlers had a reported loss of consciousness following the event compared to infants (47.4% versus 12.5%; p = 0.01). The incidence of intracranial hemorrhage was not statistically significantly different across age groups (p = 0.1), but excluding cases with extradural hematoma, revealed that the incidence of all other types of intracranial hemorrhage combined is significantly lower among toddlers than in infants (7.1%; p < 0.04). In summary, these clinical data suggest that although toddlers may exhibit loss of consciousness more frequently in an event with minimal head impact, infants may be more susceptible to cerebral hemorrhage. Our piglet data also support an increased vulnerability to hemorrhage in the infant. Despite the smaller brain mass and likely smaller strains experienced in the infant head, similar hemorrhage severity for the same rotational acceleration magnitude suggests that the infant may be more vulnerable to hemorrhage than the toddler, but further studies are needed to investigate the role of age in subarachnoid hemorrhage.
Finally, our piglet data support the hypothesis that scaling rotational load by mass alone does not produce similar axonal injuries in infant and toddler piglets. We proposed an enhanced axonal injury scaling relationship between the infant and toddler that incorporates brain mass, brain material properties, and critical strain associated with axonal injury. Previous scaling laws utilized in the field of TBI have relied on geometry (length scaling), mass, and/or material property scaling in order to predict loads for two different subjects that would result in the same injury outcome (Irwin and Mertz 1997; Ommaya et al., 1967; Thibault and Margulies, 1998). When these scaling relationships have been applied to the pediatric population to predict comparable loads to those in the adult, younger subjects with smaller, stiffer brains have often been treated as miniature adults with differences only in head mass or size. However, this approach is reasonable only if brain shape, material properties, and tissue injury strain thresholds are similar for adults and children, and ample data exist refuting these assumptions (Danelson et al., 2008; Gefen et al., 2003; Prange and Margulies, 2002; Raghupathi and Margulies, 2002; Thibault and Margulies, 1998). Recent efforts in scaling and modeling of the pediatric head have tried to address these concerns by incorporating head shape changes that occur between childhood and adulthood (Danelson et al., 2008), but no studies have considered differences in tissue vulnerability. In the current study we show that tissue vulnerability or the critical strain required for injury may also play an important role in injury outcome across subjects, especially across toddler and infant subjects, with some evidence that the decrease in vulnerability with age may even extend beyond the toddler to the pre-adolescent.
In conclusion, graded physiological and histopathological injuries were produced in the 4-week-old piglet in a novel model of non-impact rotational injury. Animals subjected to larger rotational acceleration and velocity magnitudes had EEG amplitude reductions, severe subarachnoid hemorrhage, and regions of ischemia and extensive axonal injury more than animals subjected to lower magnitudes. β-APP immunostaining for axonal injury in the 4-week-old animals was more sensitive than NF68 immunostaining, likely due to detectable axonal transport functional differences rather than cytoskeletal structural rearrangements in the axon. Unexpectedly, scaling rotational accelerations from the 4-week-old toddler animals to the 5-day-old infant animals by brain mass alone resulted in more severe subarachnoid hemorrhage and white matter damage in the younger animals. We hypothesized that the younger, smaller, stiffer brain is more vulnerable to axonal injury than the older toddler brain, such that the strain required for the same severity of axonal injury is more than three times lower in the infant brain than in the older toddler brain. We developed a new scaling relationship incorporating brain mass, material properties, and this age-dependent vulnerability, and validated it with an independent group of animal studies.
Acknowledgments
This work was funded by the National Institute of Neurological Disorders and Stroke (NIH).
Author Disclosure Statement
No competing financial interests exist.
References
- Adelson P.D. Jenkins L.W. Hamilton R.L. Robichaud P. Tran M.P. Kochanek P.M. Histopathologic response of the immature rat to diffuse traumatic brain injury. J. Neurotrauma. 2001;18:967–976. doi: 10.1089/08977150152693674. [DOI] [PubMed] [Google Scholar]
- Agran P.F. Anderson C. Winn D. Trent R. Walton-Haynes L. Thayer S. Rates of pediatric injuries by 3-month intervals for children 0 to 3 years of age. Pediatrics. 2003;111:e683–e692. doi: 10.1542/peds.111.6.e683. [DOI] [PubMed] [Google Scholar]
- Alberts B. Molecular Biology of the Cell. Garland Science; New York: 2002. [Google Scholar]
- Anderson V.A. Catroppa C. Haritou F. Morse S. Pentland L. Rosenfeld J. Stargatt R. Predictors of acute child and family outcome following traumatic brain injury in children. Pediatr Neurosurg. 2001;34:138–148. doi: 10.1159/000056009. [DOI] [PubMed] [Google Scholar]
- Armstead W.M. Kurth C.D. Different cerebral hemodynamic responses following fluid percussion brain injury in the newborn and juvenile pig. J. Neurotrauma. 1994;11:487–497. doi: 10.1089/neu.1994.11.487. [DOI] [PubMed] [Google Scholar]
- Bain A.C. Meaney D.F. Tissue-level thresholds for axonal damage in an experimental model of central nervous system white matter injury. J. Biomech. Eng. 2000;122:615–622. doi: 10.1115/1.1324667. [DOI] [PubMed] [Google Scholar]
- Bain A.C. Raghupathi R. Meaney D.F. Dynamic stretch correlates to both morphological abnormalities and electrophysiological impairment in a model of traumatic axonal injury. J. Neurotrauma. 2001;18:499–511. doi: 10.1089/089771501300227305. [DOI] [PubMed] [Google Scholar]
- Biagas K.V. Grundl P.D. Kochanek P.M. Schiding J.K. Nemoto E.M. Posttraumatic hyperemia in immature, mature, and aged rats: autoradiographic determination of cerebral blood flow. J. Neurotrauma. 1996;13:189–200. doi: 10.1089/neu.1996.13.189. [DOI] [PubMed] [Google Scholar]
- Bittigau P. Sifringer M. Pohl D. Stadthaus D. Ishimaru M. Shimizu H. Ikeda M. Lang D. Speer A. Olney J.W. Ikonomidou C. Apoptotic neurodegeneration following trauma is markedly enhanced in the immature brain. Ann. Neurol. 1999;45:724–735. doi: 10.1002/1531-8249(199906)45:6<724::aid-ana6>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- Bruce D. Current problems in pediatrics. Year Book Medical Publishers; Chicago: 1990. Head injuries in the pediatric population. [DOI] [PubMed] [Google Scholar]
- Danelson K.A. Geer C.P. Stitzel J.D. Slice D.E. Takhounts E.G. Age and gender based biomechanical shape and size analysis of the pediatric brain. Stapp Car Crash J. 2008;52:59–81. doi: 10.4271/2008-22-0003. [DOI] [PubMed] [Google Scholar]
- Dobbing J. Sands J. Quantitative growth and development of human brain. Arch. Dis. Child. 1973;48:757–767. doi: 10.1136/adc.48.10.757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobbing J. Smart J.L. Vulnerability of developing brain and behaviour. Br. Med. Bull. 1974;30:164–168. doi: 10.1093/oxfordjournals.bmb.a071188. [DOI] [PubMed] [Google Scholar]
- Dobbing J. The development of the blood-brain barrier. Prog. Brain Res. 1968;29:417–427. doi: 10.1016/S0079-6123(08)64172-2. [DOI] [PubMed] [Google Scholar]
- Dobbing J. The later development of the brain and its vulnerability. In: Davis J., editor; Dobbings J., editor. Scientific Foundations of Paediatrics. Heinemann Medical; London: 1981. [Google Scholar]
- Duhaime A.C. Raghupathi R. Age-specific therapy for traumatic injury of the immature brain: experimental approaches. Exp. Toxicol. Pathol. 1999;51:172–177. doi: 10.1016/S0940-2993(99)80091-8. [DOI] [PubMed] [Google Scholar]
- Duhaime A.C. Hunter J.V. Grate L.L. Kim A. Golden J. Demidenko E. Harris C. Magnetic resonance imaging studies of age-dependent responses to scaled focal brain injury in the piglet. J. Neurosurg. 2003;99:542–548. doi: 10.3171/jns.2003.99.3.0542. [DOI] [PubMed] [Google Scholar]
- Duhaime A.C. Margulies S.S. Durham S.R. O'Rourke M.M. Golden J.A. Marwaha S. Raghupathi R. Maturation-dependent response of the piglet brain to scaled cortical impact. J. Neurosurg. 2000;93:455–462. doi: 10.3171/jns.2000.93.3.0455. [DOI] [PubMed] [Google Scholar]
- Durham S.R. Raghupathi R. Helfaer M.A. Marwaha S. Duhaime A.C. Age-related differences in acute physiologic response to focal traumatic brain injury in piglets. Pediatr. Neurosurg. 2000;33:76–82. doi: 10.1159/000028980. [DOI] [PubMed] [Google Scholar]
- Elliott M.B. Jallo J.J. Tuma R.F. An investigation of cerebral edema and injury volume assessments for controlled cortical impact injury. J. Neurosci. Methods. 2008;168:320–324. doi: 10.1016/j.jneumeth.2007.10.019. [DOI] [PubMed] [Google Scholar]
- Eucker S.A. Bioengineering. Ph.D. dissertation. University of Pennsylvania; Philadelphia: 2009. Effect of head rotation direction on closed head injury in neonatal piglets. [Google Scholar]
- Fan P. Yamauchi T. Noble L.J. Ferriero D.M. Age-dependent differences in glutathione peroxidase activity after traumatic brain injury. J. Neurotrauma. 2003;20:437–445. doi: 10.1089/089771503765355513. [DOI] [PubMed] [Google Scholar]
- Ferreira A. Caceres A. Kosik K.S. Intraneuronal compartments of the amyloid precursor protein. J. Neurosci. 1993;13:3112–3123. doi: 10.1523/JNEUROSCI.13-07-03112.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foda M.A. Marmarou A. A new model of diffuse brain injury in rats. Part II: Morphological characterization. J. Neurosurg. 1994;80:301–313. doi: 10.3171/jns.1994.80.2.0301. [DOI] [PubMed] [Google Scholar]
- Gefen A. Gefen N. Zhu Q. Raghupathi R. Margulies S.S. Age-dependent changes in material properties of the brain and braincase of the rat. J. Neurotrauma. 2003;20:1163–1177. doi: 10.1089/089771503770802853. [DOI] [PubMed] [Google Scholar]
- Graham D.I. Ford I. Adams J.H. Doyle D. Teasdale G.M. Lawrence A.E. McLellan D.R. Ischaemic brain damage is still common in fatal non-missile head injury. J. Neurol. Neurosurg. Psychiatry. 1989;52:346–350. doi: 10.1136/jnnp.52.3.346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haass C. Koo E.H. Mellon A. Hung A.Y. Selkoe D.J. Targeting of cell-surface beta-amyloid precursor protein to lysosomes: alternative processing into amyloid-bearing fragments. Nature. 1992;357:500–503. doi: 10.1038/357500a0. [DOI] [PubMed] [Google Scholar]
- Huh J.W. Raghupathi R. Chronic cognitive deficits and long-term histopathological alterations following contusive brain injury in the immature rat. J. Neurotrauma. 2007;24:1460–1474. doi: 10.1089/neu.2006.3787. [DOI] [PubMed] [Google Scholar]
- Ibrahim N.G. Bioengineering. Ph.D. dissertation. University of Pennsylvania; Philadelphia: 2009. Head injury biomechanics in toddlers: Integrated clinical, anthropomorphic dummy, animal, and finite element model studies–Implications for age-dependence. [Google Scholar]
- Igarashi T. Potts M.B. Noble-Haeusslein L.J. Injury severity determines Purkinje cell loss and microglial activation in the cerebellum after cortical contusion injury. Exp. Neurol. 2007;203:258–268. doi: 10.1016/j.expneurol.2006.08.030. [DOI] [PubMed] [Google Scholar]
- Irwin A. Mertz H. Biomechanical Bases for the CRABI and HYBRID III Child Dummies. Society of Automotive Engineers 1997 [Google Scholar]
- Koo E.H. Sisodia S.S. Archer D.R. Martin L.J. Weidemann A. Beyreuther K. Fischer P. Masters C.L. Price D.L. Precursor of amyloid protein in Alzheimer disease undergoes fast anterograde axonal transport. Proc. Natl. Acad. Sci. U.S.A. 1990;87:1561–1565. doi: 10.1073/pnas.87.4.1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koskiniemi M. Kyykka T. Nybo T. Jarho L. Long-term outcome after severe brain injury in preschoolers is worse than expected. Arch. Pediatr. Adolesc. Med. 1995;149:249–254. doi: 10.1001/archpedi.1995.02170150029004. [DOI] [PubMed] [Google Scholar]
- Levin H.S. Aldrich E.F. Saydjari C. Eisenberg H.M. Foulkes M.A. Bellefleur M. Luerssen T.G. Jane J.A. Marmarou A. Marshall L.F. Severe head injury in children: experience of the Traumatic Coma Data Bank. Neurosurgery. 1992;31:435–443. doi: 10.1227/00006123-199209000-00008. discussion 443–444. [DOI] [PubMed] [Google Scholar]
- Lighthall J.W. Controlled cortical impact: a new experimental brain injury model. J. Neurotrauma. 1988;5:1–15. doi: 10.1089/neu.1988.5.1. [DOI] [PubMed] [Google Scholar]
- Luerssen T.G. Klauber M.R. Marshall L.F. Outcome from head injury related to patient's age. A longitudinal prospective study of adult and pediatric head injury. J. Neurosurg. 1988;68:409–416. doi: 10.3171/jns.1988.68.3.0409. [DOI] [PubMed] [Google Scholar]
- Lusardi T.A. Smith D.H. Wolf J.A. Meaney D.F. The separate roles of calcium and mechanical forces in mediating cell death in mechanically injured neurons. Biorheology. 2003;40:401–409. [PubMed] [Google Scholar]
- Marmarou A. Foda M.A. van den Brink W. Campbell J. Kita H. Demetriadou K. A new model of diffuse brain injury in rats. Part I: Pathophysiology and biomechanics. J. Neurosurg. 1994;80:291–300. doi: 10.3171/jns.1994.80.2.0291. [DOI] [PubMed] [Google Scholar]
- McIntosh T.K. Noble L. Andrews B. Faden A.I. Traumatic brain injury in the rat: characterization of a midline fluid-percussion model. Cent. Nerv. Syst. Trauma. 1987;4:119–134. doi: 10.1089/cns.1987.4.119. [DOI] [PubMed] [Google Scholar]
- McIntosh T.K. Vink R. Noble L. Yamakami I. Fernyak S. Soares H. Faden A.L. Traumatic brain injury in the rat: characterization of a lateral fluid-percussion model. Neuroscience. 1989;28:233–244. doi: 10.1016/0306-4522(89)90247-9. [DOI] [PubMed] [Google Scholar]
- Meaney D.F. Smith D.H. Shreiber D.I. Bain A.C. Miller R.T. Ross D.T. Gennarelli T.A. Biomechanical analysis of experimental diffuse axonal injury. J. Neurotrauma. 1995;12:689–694. doi: 10.1089/neu.1995.12.689. [DOI] [PubMed] [Google Scholar]
- Missios S. Harris B.T. Simoni M.K. Dodge C.P. Costine B.A. Quebada P.B. Hillier S.C. Adams L.B. Duhaime A.C. Lee Y.L. Scaled cortical impact in immature swine: effect of age and gender on lesion volume. J. Neurotrauma. 2009;26:1943–1951. doi: 10.1089/neu.2009.0956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ommaya A.K. Hirsch A.E. Tolerances for cerebral concussion from head impact and whiplash in primates. J. Biomech. 1971;4:13–21. doi: 10.1016/0021-9290(71)90011-x. [DOI] [PubMed] [Google Scholar]
- Ommaya A.K. Hirsch A.E. Martinez J. The role of ‘whiplash’ in cerebral concussion; Proc. 10th Stapp Car Crash Conf.; 1966. pp. 191–203. [Google Scholar]
- Ommaya A. Yarnell P. Hirsch A. Harris E. Scaling of experimental data on cerebral concussion in sub-human primates to concussion threshold for man. Proc. 11th Stapp Car Crash Conf. SAE; Los Angeles. 1967. [Google Scholar]
- Posmantur R. Hayes R.L. Dixon C.E. Taft W.C. Neurofilament 68 and neurofilament 200 protein levels decrease after traumatic brain injury. J. Neurotrauma. 1994;11:533–545. doi: 10.1089/neu.1994.11.533. [DOI] [PubMed] [Google Scholar]
- Posmantur R.M. Kampfl A. Liu S.J. Heck K. Taft W.C. Clifton G.L. Hayes R.L. Cytoskeletal derangements of cortical neuronal processes three hours after traumatic brain injury in rats: an immunofluorescence study. J. Neuropathol. Exp. Neurol. 1996;55:68–80. doi: 10.1097/00005072-199601000-00007. [DOI] [PubMed] [Google Scholar]
- Prange M.T. Margulies S.S. Regional, directional, and age-dependent properties of the brain undergoing large deformation. J. Biomech. Eng. 2002;124:244–252. doi: 10.1115/1.1449907. [DOI] [PubMed] [Google Scholar]
- Prins M.L. Lee S.M. Cheng C.L. Becker D.P. Hovda D.A. Fluid percussion brain injury in the developing and adult rat: a comparative study of mortality, morphology, intracranial pressure and mean arterial blood pressure. Brain Res. Dev. Brain Res. 1996;95:272–282. doi: 10.1016/0165-3806(96)00098-3. [DOI] [PubMed] [Google Scholar]
- Raghupathi R. Huh J.W. Diffuse brain injury in the immature rat: evidence for an age-at-injury effect on cognitive function and histopathologic damage. J. Neurotrauma. 2007;24:1596–1608. doi: 10.1089/neu.2007.3790. [DOI] [PubMed] [Google Scholar]
- Raghupathi R. Margulies S.S. Traumatic axonal injury following closed head injury in the neonatal pig. J. Neurotrauma. 2002;19:843–853. doi: 10.1089/08977150260190438. [DOI] [PubMed] [Google Scholar]
- Saatman K.E. Feeko K.J. Pape R.L. Raghupathi R. Differential behavioral and histopathological responses to graded cortical impact injury in mice. J. Neurotrauma. 2006;23:1241–1253. doi: 10.1089/neu.2006.23.1241. [DOI] [PubMed] [Google Scholar]
- Sherriff F.E. Bridges L.R. Gentleman S.M. Sivaloganathan S. Wilson S. Markers of axonal injury in post mortem human brain. Acta Neuropathol. 1994;88:433–439. doi: 10.1007/BF00389495. [DOI] [PubMed] [Google Scholar]
- Smith D.H. Nonaka M. Miller R. Leoni M. Chen X.H. Alsop D. Meaney D.F. Immediate coma following inertial brain injury dependent on axonal damage in the brainstem. J. Neurosurg. 2000;93:315–322. doi: 10.3171/jns.2000.93.2.0315. [DOI] [PubMed] [Google Scholar]
- Smith D.H. Wolf J.A. Lusardi T.A. Lee V.M. Meaney D.F. High tolerance and delayed elastic response of cultured axons to dynamic stretch injury. J. Neurosci. 1999;19:4263–4269. doi: 10.1523/JNEUROSCI.19-11-04263.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder R.C. Schneider L.W. Owings C.L. Reynolds H.M. Golomb D.H. Schork M.A. Anthropometry of infants, children and youth to age 18 for product safety design. UM-HSRI-77 Final Report Contract CPSC-C-75-0068. Prepared for Consumer Product Safety Commission; Bethesda, MD.1977. [Google Scholar]
- Thibault K. Margulies S. Age-dependent material properties of the porcine cerebrum: effect on pediatric inertial head injury criteria. J. Biomechanics. 1998;31:1119–1126. doi: 10.1016/s0021-9290(98)00122-5. [DOI] [PubMed] [Google Scholar]
- Ucar T. Tanriover G. Gurer I. Onal M.Z. Kazan S. Modified experimental mild traumatic brain injury model. J. Trauma. 2006;60:558–565. doi: 10.1097/01.ta.0000209172.75637.db. [DOI] [PubMed] [Google Scholar]
- Verger K. Junque C. Jurado M.A. Tresserras P. Bartumeus F. Nogues P. Poch J.M. Age effects on long-term neuropsychological outcome in paediatric traumatic brain injury. Brain Inj. 2000;14:495–503. doi: 10.1080/026990500120411. [DOI] [PubMed] [Google Scholar]
- Vink R. McIntosh T.K. Yamakami I. Faden A.I. 31P NMR characterization of graded traumatic brain injury in rats. Magn. Reson. Med. 1988;6:37–48. doi: 10.1002/mrm.1910060105. [DOI] [PubMed] [Google Scholar]
- Wolf J.A. Stys P.K. Lusardi T. Meaney D. Smith D.H. Traumatic axonal injury induces calcium influx modulated by tetrodotoxin-sensitive sodium channels. J. Neurosci. 2001;21:1923–1930. doi: 10.1523/JNEUROSCI.21-06-01923.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yam P.S. Dewar D. McCulloch J. Axonal injury caused by focal cerebral ischemia in the rat. J. Neurotrauma. 1998;15:441–450. doi: 10.1089/neu.1998.15.441. [DOI] [PubMed] [Google Scholar]
- Zhang Y. Wang B. Liao Z. Xin J. Wu J. Wu M. [The establishment of a modified lateral fluid percussion model of brain injury in rat and the pertinent pathologic changes] Hua Xi Yi Ke Da Xue Xue Bao. 1999;30:363–367. [PubMed] [Google Scholar]