

Abstract
Crush to the mammalian spinal cord leads to primary mechanical damage followed by a series of secondary biomolecular events. The chronic outcomes of spinal cord injuries have been well detailed in multiple previous studies. However, the initial mechanism by which constant displacement injury induces conduction block is still unclear. We therefore investigated the anatomical factors that may directly contribute to electrophysiological deficiencies in crushed cord. Ventral white matter strips from adult guinea pig spinal cord were compressed 80%, either briefly or continuously for 30 min. Immunofluorescence imaging and coherent anti-Stokes Raman spectroscopy (CARS) were used to visualize key pathological changes to ion channels and myelin. Compression caused electrophysiological deficits, including compound action potential (CAP) decline that was injury-duration-dependent. Compression further induced myelin retraction at the nodes of Ranvier. This demyelination phenomenon exposed a subclass of voltage-gated potassium channels (Kv1.2). Application of a potassium channel blocker, 4-aminopyridine (4-AP), restored the CAP to near pre-injury levels. To further investigate the myelin detachment phenomenon, we constructed a three-dimensional finite element model (FEM) of the axon and surrounding myelin. We found that the von Mises stress was highly concentrated at the paranodal junction. Thus, the mechanism of myelin retraction may be associated with stress concentrations that cause debonding at the axoglial interface. In conclusion, our findings implicate myelin disruption and potassium channel pathophysiology as the culprits causing compression-mediated conduction block. This result highlights a potential therapeutic target for compressive spinal cord injuries.
Key words: acute spinal cord injury, demyelination, potassium ion channel, sustained compression
Introduction
Compression is a major mode of trauma in spinal cord injury (SCI; Banerjee et al., 2004; Ng et al., 1999), and the biomechanics leading to primary and secondary damage have been studied in multiple animal models (Casha et al., 2001; Ma et al., 2001; Scheff et al., 2003; Young, 2002). However, most studies have focused primarily on the role of damage from acute or instantaneous mechanical insult (Fehlings and Nashmi, 1997; Ghasemlou et al., 2005; Morino et al., 2003; Shi and Borgens, 1999; Tysseling-Mattiace et al., 2008). While indicative of certain injuries, these types of experiments do not capture potential temporal effects associated with longer-duration compression. Situations of sustained compression are also clinically relevant, as bone fragments may impinge upon the cord following trauma. In such cases, surgical decompression is necessary, although few studies have investigated its benefits (Carlson et al., 2003; Shields et al., 2005). Thus, early decompression is recommended, even though it does not always result in optimum recovery (Fehlings and Perrin, 2005; Fehlings et al., 2001; Fehlings and Tator, 1999; Lim and Tow, 2007).
In this work we were interested in the mechanisms responsible for conduction dysfunction in spinal cord subjected to continuous crush. There is key evidence that crush causes myelin disruption in compressed cords (Kamencic et al., 2001; Li et al., 1999; Takahashi et al., 1996), but it is unclear whether simple demyelination or other deleterious factors contribute to conduction block. For instance, we found that in stretch injury, demyelination at the juxtaparanodal interface exposes a subpopulation of potassium voltage-gated channels that are generally covered by myelin (unpublished data). Activation of these potassium channels can inhibit action potential propagation. We hypothesize that a similar effect might be occurring at the compressed lesion, whereby ion channel derangement may play a factor in conduction block. Additionally, we have shown that membrane damage is only a small contributor in quasi-static compressive trauma (Ouyang et al., 2009). These clues direct us towards other mechanisms of damage, possibly related to ion channel disruptions. Of specific interest was the spatial distribution of juxtaparanodal (i.e., Kv1.2) voltage-gated potassium channels pre- and post-crush. We used label-free coherent anti-Stokes Raman spectroscopy (CARS) and immunofluorescence imaging to examine tissue deformation and the integrity of the axoglial junction. Electrophysiology as a function of crush duration was monitored. We further investigated the role of potassium channels in conduction pathophysiology via application of 4-aminopyridine (4-AP), a potassium channel blocker. Finally, we employed finite element analysis to explore tissue level stresses, and to provide a potential biomechanical explanation of functional impairment.
Methods
Isolation of spinal cord from guinea pig
The experimental protocol was approved by the Purdue University Animal Care and Use Committee, and the procedures were similar to those used in previous studies (Ouyang et al., 2008; Shi et al., 2000; Shi and Blight, 1996). A total of 20 female adult guinea pigs weighing 260–360 g were used in this study. The animals were first anesthetized with 60 mg/kg ketamine and 10 mg/kg xylazine, and then perfused with cold oxygenated Krebs solution. The vertebral column was removed after perfusion and a complete laminectomy was performed. The spinal cord was then extracted from the spinal column carefully with bone cutters, and separated in half by cutting longitudinally along the medial sagittal plane (Fig. 1A). Finally, ventral white matter strips were separated by cutting each hemi-cord radially. To enhance tissue viability, the ventral white matter strips were immediately immersed in oxygenated Krebs solution for at least an hour before further testing.
FIG. 1.

Experimental setup. (A) Schematic describing the extraction of ventral white matter from adult guinea pigs. (B) Multimodal measuring system used for concurrent mechanical testing and electrophysiology. The cord specimens were compressed by a soft-tissue indenter/actuator that was computer controlled. The force generated from compression rod to the specimen was recorded by the load cell. The electrophysiological response was recorded by the double sucrose gap chamber. (C) Transverse views of ventral white matter before compression, while being compressed at 80%, and after rod release. (D) Top view of the sucrose gap chamber used for electrophysiological recording. A 320-mM sucrose stock was perfused through the gaps between isotonic KCl and oxygenated Krebs solution. The nerve preparation was stimulated (left) and recorded (right) with a pair of Ag/AgCl electrodes. (Color image is available online at www.liebertonline.com/neu.)
Sustained compression and decompression of ventral white matter and electrophysiological recording
Compressive deformation was performed with the multimodal soft tissue tester developed in previous studies (Ouyang et al., 2008, 2009). A standard double sucrose gap chamber was implemented in the testing system to record the electrophysiological response of ventral white matter during mechanical loading (Shi and Borgens, 1999; Shi and Whitebone, 2006; Fig. 1B). A 4-cm ventral white matter strip was positioned in the central compartment of the sucrose gap chamber with the two free ends placed in the side compartments (Fig. 1D). The central well was perfused with oxygenated Krebs solution kept at 37°C, and two side compartments were filled with isotonic (120 mM) KCl. Two gaps, each positioned between the central and end wells, were circulated with sucrose (320 mM) to eliminate ion exchange between the central and side compartments. To prevent solution from leaking into the sucrose gaps from both the central and side compartments, the ventral white matter strip was sealed with vacuum grease and thin pieces of plastic on both sides of each sucrose gap. The cord in one end of the chamber was stimulated and the corresponding compound action potentials (CAPs) were recorded from the other end with Ag/AgCl wire electrodes. Supramaximal stimuli in the form of 0.1-msec-duration constant current pulses were delivered. The CAP responses were recorded at 25 kHz acquisition frequency with a Neurocorder (Neurodata Instruments Corp., New York, NY) interfaced to LabVIEW (National Instruments Corp., Austin, TX). For mechanical compression, the white matter strip was positioned on the tissue holding stage located in the central compartment of the sucrose gap chamber (Fig. 1B). The movement and position of the compression rod was monitored with the aid of a computer and LabVIEW software. Spinal cord samples were first compressed at a rate of 0.05 mm/sec to 80% (or 20% of its original thickness; Fig. 1C). In one group of experiments, the rod was advanced into the white matter and quickly retracted (no sustained compression). In the other group, compression was maintained for 30 min. To examine the functional recovery after sustained crush, the CAP response was monitored for an additional 30 min after compression rod retraction.
Voltage sweeps
Voltage tests were performed on injured cords subjected to 80% sustained compression. A range of stimulus intensity ranging from 1.85 to 6.5 V was used to stimulate cords before compression (pre), 30 min after sustained compression (recovery), at the end of 4-AP application, and at the end of the wash period.
4-AP treatment
4-Aminopyridine (4-AP; Sigma-Aldrich, St. Louis, MO) was dissolved in Krebs solution to make 10 mM stock concentration. On the day of the experiment, 4-AP stock was dissolved in the Krebs buffer to make 100 μM working solution. At 30 min after the compression rod was removed, the cord was perfused with 100 μM 4-AP maintained at 37°C for 30 min. The CAP response of the injured cord was continuously monitored during 4-AP treatment.
Coherent anti-Stokes Raman scattering (CARS) imaging
Detailed descriptions of how CARS functions has been explained in prior studies (Fu et al., 2007, 2008). In brief, a pump field (ωp) and a Stokes field (ωs) are generated by lasers and the interaction of these two fields results in an anti-Stokes field (ωas). An anti-Stokes field amplifies the signal from CH2 groups found in lipids. Since myelin is approximately 70% lipid by weight (Morell and Quarles, 1999), CARS is a highly sensitive tool for myelin visualization (Fig. 2). Immediately following compression experiments, the white matter specimens were fixed with 4% paraformaldehyde for at least 24 h prior to CARS imaging. All the imaging experiments were performed at room temperature (23°C). To assess myelin damage, we introduced a quantity termed the node ratio, which is the ratio of nodal distance over nodal diameter (Fig. 2G). The nodal distance was defined as the exposed myelin distance at the node, while diameter was taken to be the axonal diameter at the node. A larger node ratio corresponds to an elongated node morphology.
FIG. 2.

Coherent anti-Stokes Raman spectroscopy (CARS) and immunohistochemistry images of normal and damaged myelin in ventral white matter strips. (A) CARS image of normal myelin. A node of Ranvier is highlighted in the dashed box. (B) Immunofluorescent image of fast potassium voltage channels Kv1.2 clustered in the juxtaparanodal area. (C) Merged image of CARS (A) and immunofluorescence (B) depicting Kv1.2 channels that are normally shielded by myelin. (D) CARS image of damaged myelin and a node of Ranvier. The dashed white box delineates the node of Ranvier. (E) Immunofluorescence of Kv1.2 channels. (F) Merged image of CARS (D) and immunostaining (E) showing partial Kv1.2 exposure following 80% sustained compression. (G) The node ratio was used to quantify elongation of the nodal region. The node ratio was defined as the nodal distance divided by the nodal diameter. Larger node ratios indicate more myelin disruption (scale bar = 5 μm). (H) Scatterplot of the node ratio for compressed (30 min sustained) and uninjured cord as a function of axon diameter. Node morphology analysis showed an increase in the node ratio following compression. (Color image is available online at www.liebertonline.com/neu.)
Immunofluorescence imaging
Following compression testing and electrophysiological recording, the ventral white matter strips were fixed in 4% paraformaldehyde overnight. The strips were then immersed in glycerol solution (20% glycerol and 2% paraformaldehyde) for cryoprotection for 48 h. Then 50-μm tissue sections were transversely cut from the injured site of the strips using a vibratome. The tissue sections were subsequently placed in 10% BSA solution with 0.05% Triton-X 100 for 1 h. After being washed (three times for 10 min each), the tissue sections were incubated with primary antibody anti-Kv1.2 (1:100 working dilution, host: rabbit; Alomone Labs, Jerusalem, Israel) in 1% BSA solution with 0.1% Triton-X 100 overnight. After another wash (three times for 10 min each), the tissue sections were incubated with secondary antibody Alexa Fluor 488 goat anti-rabbit (Invitrogen, Carlsbad, CA; 1:100 working dilution) in 1% BSA solution with 0.1% Triton-X 100 for 1 h. After the final wash (three times for 10 min each), the tissue sections were stored in darkness until imaging. All the solutions used for wash and storing were diluted in phosphate-buffered saline (PBS). The immunofluorescence images of tissue sections were obtained by a laser scanning confocal microscope (FV300/IX70; Olympus, Inc., Center Valley, PA) at an excitation wavelength of 488 nm.
Finite element analysis
A three-dimensional, large deformation finite element model (FEM) was developed to investigate the stress levels at the paranode. This model was constructed by inputting material properties from prior experiments, as well as considering the myelin and axon to be separate entities. Details of the finite element model parameters and geometry can be found in the Appendix.
Statistical analysis
One-way ANOVA with Tukey's post-hoc test and a Student's t-test were used for appropriate comparisons. In both cases, p values < 0.05 were considered statistically significant. All data are shown in the form of means ± standard error.
Results
Myelin damage revealed by immunofluorescence and label-free CARS imaging
Using coherent anti-Stokes Raman scattering imaging, we were able to achieve dye-free labeling of the myelin sheaths. The results clearly show the high contrast of the myelin due to the CH2-rich groups present in lipids (Fig. 2A). Nodes of Ranvier are seen as distinct breaks along the longitudinal direction. Immunofluorescence of voltage-gated potassium channels (Kv1.2; Fig. 2B), and the image merged with CARS (Fig. 2C) demonstrated Kv1.2 channels clustered in juxtaparanodal regions, and were covered by myelin under normal conditions. After sustained compression (80% for 30 min), the myelin sheaths showed fraying and delamination from the axon shaft. In addition, the exposed nodal regions (node ratio) were considerably lengthened following compression (Fig. 2H). This observation was consistent for the entire range of measured axon calibers. A merged image of the immunofluorescent (Fig. 2E) and CARS (Fig. 2D) channels showed Kv1.2 exposure after sustained compressive injury (Fig. 2F).
Sustained compression decreases compound action potential conduction and increases effective nodal distance
An example of the CAP recording history is shown in Figure 3A. In brief compression, the rod was first used to indent the cord and then retracted after crushing 80%. At this maximum crush magnitude, the CAP was found to be 54.2 ± 5.1% of the pre-crush value (n = 7; p < 0.01; Fig. 3B). The CAP amplitude of injured cords after 30 min of recovery was 84.3 ± 2.4% of the pre-injured condition (n = 7). Additionally, we found that node ratios following brief compression (2.94 ± 0.3; n = 67; p < 0.001) were significantly larger than the uninjured values (0.74 ± 0.04; n = 63; Fig. 3C and Table 1). At 30 min post-crush, the node ratio recovered to 1.14 ± 0.10 (n = 64), which was not significantly different from pre-crush. However, electrophysiological and myelin recovery was inhibited during delayed decompression (Fig. 4A and B). CAP amplitudes of injured cords subjected to sustained 30-min compression was only 8 ± 1.8 % of pre-injury values (n = 7). Moreover, the node ratio for this group of cords was 3.52 ± 0.25 (n = 66), which was significantly different than the uninjured case (p < 0.001; Fig. 4C). Even after rod retraction and an additional 30-min rest period, the node ratio (3.0 ± 0.2; n = 65) was still markedly different than the uninjured case (p < 0.001).
FIG. 3.

Electrophysiological responses and myelin damage of cords experiencing brief compression. (A) Example of compound action potential (CAP) profile in a cord with 80% compression. The CAP was continuously monitored before compression (pre) and 30 min after rod retraction (recovery). The CAP amplitudes were normalized to the CAP prior to compression. (B) CAP comparisons between brief injury and following the recovery phase. (C) Bar graph showing node ratios for the different experimental conditions (**p < 0.01; ***p < 0.001).
Table 1.
Node Ratio Parameters
| Condition | n | Nodal diameter (μm) | Nodal length (μm) | Nodal ratio |
|---|---|---|---|---|
| Control | 63 | 3.07 ± 0.13 | 2.31 ± 0.23 | 0.74 ± 0.04 |
| Injured (brief) | 67 | 2.14 ± 0.10 | 6.13 ± 0.65 | 2.94 ± 0.30 |
| Recovery from brief injury | 64 | 3.54 ± 0.13 | 4.01 ± 0.45 | 1.14 ± 0.10 |
| Injured (30 min SC) | 66 | 2.59 ± 0.13 | 9.55 ± 0.95 | 3.52 ± 0.25 |
| Recovery from 30 min SC | 65 | 3.97 ± 0.20 | 11.81 ± 0.99 | 3.00 ± 0.20 |
Mean ± Standard error.
SC, sustained compression.
FIG. 4.

Electrophysiological responses and myelin damage in cords with 80% sustained compression. (A) A sample compound action potential (CAP) history of the experimental protocol. (B) CAP comparison after 30 min of sustained compression versus the recovery phase. The CAP magnitudes were normalized with respect to the uninjured condition. (C) Comparisons of node ratio in cords with 80% nominal sustained compression before and after a 30-min recovery period (***p < 0.001).
Sustained compressive injury affects large and small axons
The CAP response of pre- and post-crushed cord was compared at various stimulus intensities (ranging from 1.85 to 6.5 V). Cords recovering from compression had significantly lower CAP amplitudes than uninjured samples for stimulus levels in the 3 to 6.5 V range (Figs. 5A and 6B; n = 7; p < 0.05). However, the minimum activation voltages for both uninjured and injured cords were comparable (approximately 3 V), and the slope at each stimulus value was similar. A plot of the injured CAP versus the uninjured CAP revealed a linear trend (Fig. 5C). The congruence of slopes and the similarity in activation thresholds suggest both large and small axons were equally affected by compression (McBride et al., 2007). Any bias damage towards an axon size group would cause a change in the slope or induce a horizontal shift in the activation thresholds (for example, a rightward shift if larger axons were damaged preferentially). Thus, we conclude that compression damages large and small axons in equal proportion. In all cases, the CAP amplitudes were normalized to the respective peak CAP at maximum stimulus (6.5 V).
FIG. 5.

The relation between stimulus intensity and corresponding compound action potential (CAP) amplitude before and after 80% sustained compression. (A) Examples of individual CAP responses to a range of stimulus intensities before and after compression. (B) Stimulus intensities ranged from 1.85 to 6.5 V. Thirty minutes of continuous compression significantly decreased CAP amplitude at all stimulus intensities (*p < 0.05; n = 7). (C) Normalized CAP amplitudes before and after compression are plotted on the same graph. CAP amplitudes were normalized with respect to the peak CAP and maximum stimulus (6.5 V). The linear relationship implies that sustained compression had no difference in changing conduction properties of large- or small-caliber axons.
FIG. 6.

Diagram showing the sequence of mechanical compression, release, and application of 4-aminopyridine (4-AP). (A) Each ventral white matter strip was compressed to 80% (20% of its original thickness) and the compression was maintained for 30 min. Then decompression was performed by removing the compression rod from the tissue. Thirty minutes after decompression, the cord was treated with 100 μM 4-AP for an additional 30 min. The compound action potential (CAP) amplitude was continuously monitored from pre-compression to 30 min after 4-AP was washed away with oxygenated Krebs solution. In one category of the experiment, the ventral white matter strip was compressed briefly (i.e., decompression was performed immediately after the strip was compressed to 80%, without sustained compression; # denotes when voltage tests were performed throughout the series of experiments). (B) Examples of individual CAP amplitude recorded in pre, sustained compression, recovery, 4-AP, and wash periods.
4-Aminopyridine enhances axonal conduction after sustained compression
To examine the effect of 4-AP on axonal conductivity, we compared the CAP amplitude in injured cords before and after 4-AP treatment (Fig. 6). Stimulus intensities ranged from 1.85 to 6.5 V. Application of 100 μM 4-AP significantly increased CAP amplitude in stimulus intensities ranging from 3 to 6.5 V (Fig. 7B; 7 samples; p < 0.05). We also plotted CAP amplitudes of injured cords before and after 4-AP treatment (Fig. 7C). CAP amplitude was normalized as a percentage of CAP response at a stimulus intensity of 6.5 V (max.). Again, a linear relationship demonstrated that large- and small-diameter axons equally benefitted from 100 mM 4-AP application in enhancing the CAP amplitude.
FIG. 7.

The relationship between stimulus intensity and corresponding compound action potential (CAP) amplitude with 100 μM 4-aminopyridine (4-AP) treatment. (A) Examples of individual CAP responses before and after 4-AP treatment. (B) Stimulus intensities ranged from 1.85 to 6.5 V. Application of 100 μM 4-AP significantly increased CAP amplitude at all stimulus intensities (*p < 0.05; n = 7). (C) Normalized CAP amplitude before 4-AP treatment (after recovery from compression) was plotted against the CAP amplitude after 4-AP treatment. All CAP amplitudes were normalized with respect to the maximum CAP value at peak stimulus (6.5 V). The linear relationship suggests 4-AP treatment had no bias in altering the conduction properties of large- or small-caliber axons.
Finite element analysis of myelinated axons in compression
As expected, the maximum von Mises stress was found near the node of Ranvier for both 60% and 80% nominal compression of the white matter (Fig. 8). At 60% nominal compression, the peak stress was approximately 6500 Pa, 50% greater than the maximum stress far from the node of Ranvier. Similarly, at 80% nominal compression, the peak von Mises stress was nearly 50,000 Pa, more than double the peak stress far from the node of Ranvier.
FIG. 8.

Finite element simulation of compressive injury at the axonal level. (A) Two-dimensional schematic of the myelinated axon and node of Ranvier (* denotes the spatial starting point for tabulation of the von Mises stress values). (B) The simulated peak von Mises stress as a function of distance (along the axon) from the node center. (C) Transverse plane of the finite element model depicting the axon shaft (circles, denoted by a), and myelin (region outside the circles, denoted by m). The gray area denotes the region of simulation. (D) Transverse distribution of von Mises stress at 60% compression at the location highlighted by * in part A. (E) Distribution of von Mises stress in the transverse plane at location * in part A at 80% compression. (Color image is available online at www.liebertonline.com/neu.)
Discussion
Paranodal demyelination is a consequence of compression
In a prior study, we established that sustained compression resulted in a series of biophysical events that included membrane damage, conduction dysfunction, and permanent tissue deformation (Ouyang et al., 2009). Those findings corroborate other acute injury models that showed reduced axonal excitability and CAP decreases (Nashmi and Fehlings, 2001). Using 80% crush as the starting point for severe compression, we have conducted a more detailed analysis of the temporal anatomical and electrophysiological responses. Specifically, we were interested in the mechanism by which compression leads to conduction pathophysiology. Multiple in vivo models point to demyelination as an outcome of SCI. Chronic demyelination has been shown to persist up to 450 days post-injury in rats (Totoiu and Keirstead, 2005), and this loss of conductive pathways contributes to debilitating sensorimotor deficits (Nashmi and Fehlings, 2001). However, we postulate there may be a similar, but earlier form of myelin disorganization. Normally, myelin sheaths attach to axons through putative cell-cell interactions at paranodal axo-glial junctions where glial neurofascin 155 from myelin binds with axonal contactin-associated protein (Caspr) and contactin (Bhat et al., 2001; Sherman and Brophy, 2005). When compression is applied to white matter, tissue deformation occurs in the loading direction as well as in the lateral directions (Poisson effect). We hypothesize that tension in the orthogonal directions or shear effects at the myelin-axolemma interface may disrupt the anchoring bonds, and induce myelin sheath retraction. In healthy tissue, intact myelin covers subpopulations of physiologically-silent potassium ion channels (Kv1.1, Kv1.2, and Kvβ2.1; Poliak and Peles, 2003; Poliak et al., 2003). This unique structural arrangement was observed ex vivo via concurrent CARS and immunofluorescence staining. However, paranodal stripping would cause morphological changes at the node. We subsequently used the node ratio as a metric for assessing the severity of demyelination. It was evident that following compression, the axo-glial interface was compromised. The myelin sheaths detached from the nodes and the effective node length increased. Since saltatory conduction occurs through current loops moderated primarily by sodium channels, exposure of the potassium channels would cause current leakage and fix the membrane close to its resting potential (Ritchie and Chiu, 1981; Waxman, 1982). Consequently, spike generation may be impeded. In crushed cord samples, we observed CAP reductions to be consistent with the simultaneous failure of paranodal myelin.
Sustained compression induces additional irreversible dysfunction
The combined electrophysiological and imaging assessment clearly shows that continued compression exacerbates the primary injury in spinal cord white matter. In cases of sustained crush, the cord samples regained only 50% of the uninjured CAP, whereas in brief compression, the CAP was 84.3 ± 2.4% of the pre-injury value. Others have also reported a similar CAP block during continuous crush (Carlson et al., 2003). Moreover, the demyelination event and node ratio elongation were less severe for short-duration compression. This is clearly shown in the CARS signal of myelin, where the node ratio of briefly compressed cord was 3.0 versus 3.5 for prolonged crush. Node ratio recovery following crush was more prominent after brief compression. Thus, sustained compression not only increases immediate damage, but also delays or obstructs the recovery mechanism. Interestingly, we previously demonstrated that axonal membrane damage also occurred during the onset of compression. Continued compression, however, impaired the CAP despite membrane resealing (Ouyang et al., 2009). Results from the current study corroborate the hypothesis that myelin damage, rather than axonal membrane disruption, might be the primary factor that leads to functional deficits in the early phase of sustained crush.
Potassium channel blockers enhance functional recovery
4-AP is a well-studied potassium channel blocker, and has been shown to alleviate conduction block in multiple spinal cord injury paradigms (Blight, 1989; Blight et al., 1991; Hayes et al., 1994; Jensen and Shi, 2003). In this study, we used 4-AP to provide further support that abnormally-exposed potassium channels are the culprit leading to action potential block. We found that 4-AP immediately restored axonal excitability and CAP amplitudes when administered after injury. Activation curves (voltage) also followed the same shape pre- and post-crush, suggesting that no subpopulation of axons were either biased in initial damage or in recovery. Washing away the 4-AP eliminated its beneficial effects, resulting in CAP responses comparable to pre-treatment levels. This implicates potassium channels as a key player in compression-related functional impairment. We should note that 4-AP is a broad-spectrum potassium channel blocker, and that other channels not targeted in this study may also have been inhibited.
Von Mises stress is highly localized at the paranode
A three-dimensional finite element model of the axon and surrounding myelin was constructed to evaluate the white matter stress distribution. Two severity indices (60% and 80% compression) were used to track the trends in the von Mises stress. The von Mises stress is an aggregate stress metric commonly employed in engineering failure analysis. We previously reported that this parameter was highly correlated with anatomical damage in bulk spinal cord tissue (Ouyang et al., 2008). Thus we postulated that such relationship may also exist at the axonal level. The computational data showed a considerable increase in the von Mises stress near the axo-glial junction. In the 60% compression simulation, the stresses near the node (within 3 μm) were approximately double compared to values far from the node (>3 μm). At 80% compression, von Mises stress increased at a much faster rate near the node center. We should note that 80% deformation represents an extreme case, and the stress estimates need to be validated with further experimentation. Nonetheless, the FE model shows stress concentrations co-localized with myelin detachment at putative axolemma anchoring regions. We believe these findings provide a potential biomechanical explanation of the demyelination event.
Clinical implications
We show for the first time that demyelination at the juxtaparanodal region is a key consequence of acute crush. Moreover, the severity of myelin retraction and the corresponding node ratio are exacerbated with longer injury durations. These data provide insight into time-dependent effects that may warrant early decompression following crush. We caution that the current model does not take into account the role of secondary biochemical cascades, but we ultimately demonstrated that the biomechanics of compression leads to potassium channel exposure at the paranodal junction. This phenomenon may explain many of the acute functional deficiencies observed in crushed spinal cord.
Finally, we note that Kv1.2 exposure is not singular to compressive injury. Non-mechanical disturbances such as anoxia or disease states (e.g., multiple sclerosis) may cause a similar denuding of the axon (Waxman et al., 1992). These cumulative data highlight the vulnerability of the axo-glial interface and its role in regulating saltatory conduction. In this regard, agents aimed at blocking physiologically-silent potassium channels may ameliorate conduction block. As shown in this study, the application of 4-AP following compression leads to restoration of the compound action potential. Additional research with this class of drugs may prove useful in the therapeutic treatment of demyelinating pathologies.
Appendix
Finite element analysis
In order to determine the stress distribution in the axons and myelin, a large-deformation, three-dimensional finite element model (FEM) was developed. For the purposes of this model, it was necessary to consider the axons and myelin separately. With measured values of axonal stiffness (Bernal et al., 2007), and whole white matter segments (Ouyang et al., 2008), it was possible to calibrate the FEM and use it to estimate the stress levels that lead to demyelination. The model consisted of a central axon surrounded by myelin, except for a 2.1-μm region in the middle, which represented the node of Ranvier. We further assumed that the surrounding axons were arranged in an “X” configuration. Previous work clearly demonstrated that transverse compression of guinea pig white matter generated no appreciable load until the samples reached 40% nominal compression (Ouyang et al., 2008). With the pia mater removed, compression of the white matter drives tissue fluid out until the axon-myelin bundles become compact. At this point, the sample begins to take load. We chose this point as our reference configuration and evaluated the stress and strain distribution at 20% and 40% model compression (corresponding to 60% and 80% nominal compression of the white matter, respectively).
The average axon diameter was assumed to be 3 μm, with a G-ratio, 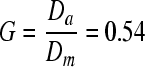 , where Da is the axon diameter and Dm is the outside diameter of the myelin bundle. For these values of Da and G, the corresponding value of Dm = 5.56 μm, and the equivalent square area has a length, Lm = 4.93 μm (Fig. 3). The model length was varied from 20 to 200 μm in order to determine the minimum length required to prevent the stress concentrations around the node of Ranvier from propagating to the ends of the model. The final length chosen for these simulations was 25 μm. These parameters produced a three-dimensional model of a central axon with an unmyelinated node of Ranvier in the middle, surrounded by completely myelinated axons in a repeating unit cell configuration (Fig. 9). The symmetry of the problem was exploited so that only ⅛ of the original geometry had to be meshed and solved (Comsol v3.5). A convergence study was performed and the final converged mesh consisted of 315,969 tetrahedral elements, 24,149 triangular boundary elements, and 1,330,527 degrees of freedom. The minimum element quality was 0.177, and a stationary direct solver (SPOOLES) with a relative tolerance of 1 × 10−6 was used to perform the analysis.
, where Da is the axon diameter and Dm is the outside diameter of the myelin bundle. For these values of Da and G, the corresponding value of Dm = 5.56 μm, and the equivalent square area has a length, Lm = 4.93 μm (Fig. 3). The model length was varied from 20 to 200 μm in order to determine the minimum length required to prevent the stress concentrations around the node of Ranvier from propagating to the ends of the model. The final length chosen for these simulations was 25 μm. These parameters produced a three-dimensional model of a central axon with an unmyelinated node of Ranvier in the middle, surrounded by completely myelinated axons in a repeating unit cell configuration (Fig. 9). The symmetry of the problem was exploited so that only ⅛ of the original geometry had to be meshed and solved (Comsol v3.5). A convergence study was performed and the final converged mesh consisted of 315,969 tetrahedral elements, 24,149 triangular boundary elements, and 1,330,527 degrees of freedom. The minimum element quality was 0.177, and a stationary direct solver (SPOOLES) with a relative tolerance of 1 × 10−6 was used to perform the analysis.
FIG. 9.

Geometric model used in the large deformation, three-dimensional finite element model of a myelinated axon. This repeating unit cell configuration contained a central axon with an unmyelinated node of Ranvier. Away from the node, the axon was surrounded by myelin. The bare axon diameter was 3 μm, and the length of the transverse square of the model was 4.93 μm. The node of Ranvier was 2.1 μm long, and the simulation depth of the model was 25 μm. (Color image is available online at www.liebertonline.com/neu.)
Each axon and the surrounding myelin was modeled as a nonlinear, neo-Hookean material with a strain energy function, W,
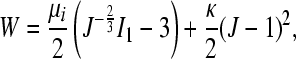 |
where μi denotes the stiffness parameter for the axons or myelin and the bulk modulus, and κ was assumed to be 1 × 106 Pa for both. For the axons, the stiffness parameter, μaxon, was fixed at 12,000 Pa (Bernal et al., 2007). The stiffness parameter for the myelin, μmyelin, was varied until the average stress in the tissue at 80% nominal compression reached 10,000 Pa, as demonstrated by previous experiments (Ouyang et al., 2008). The final calibrated value for μmyelin was 4100 Pa. The unmyelinated region around the node of Ranvier was assumed to be a weak extracellular matrix with a stiffness parameter an order of magnitude less than that of the myelin.
Acknowledgments
We thank Gary Leung for performing guinea pig isolation surgery, Michel Schweinsberg for figure illustrations, and Sean Connell for proofing the manuscript. We also wish to acknowledge support from National Institutes of Health (NIH) and the State of Indiana.
Author Disclosure Statement
No competing financial interests exist.
References
- Banerjee R. Palumbo M.A. Fadale P.D. Catastrophic cervical spine injuries in the collision sport athlete, part 2: principles of emergency care. Am. J. Sports Med. 2004;32:1760–1764. doi: 10.1177/0363546504268931. [DOI] [PubMed] [Google Scholar]
- Bernal R. Pullarkat P.A. Melo F. Mechanical properties of axons. Phys. Rev. Lett. 2007;99:018301. doi: 10.1103/PhysRevLett.99.018301. [DOI] [PubMed] [Google Scholar]
- Bhat M.A. Rios J.C. Lu Y. Garcia-Fresco G.P. Ching W., St. Martin M. Li J. Einheber S. Chesler M. Rosenbluth J. Salzer J.L. Bellen H.J. Axon-glia interactions and the domain organization of myelinated axons requires neurexin IV/Caspr/Paranodin. Neuron. 2001;30:369–383. doi: 10.1016/s0896-6273(01)00294-x. [DOI] [PubMed] [Google Scholar]
- Blight A.R. Effect of 4-aminopyridine on axonal conduction-block in chronic spinal cord injury. Brain Res. Bull. 1989;22:47–52. doi: 10.1016/0361-9230(89)90126-3. [DOI] [PubMed] [Google Scholar]
- Blight A.R. Toombs J.P. Bauer M.S. Widmer W.R. The effects of 4-aminopyridine on neurological deficits in chronic cases of traumatic spinal cord injury in dogs: a Phase I clinical trial. J. Neurotrauma. 1991;8:103–119. doi: 10.1089/neu.1991.8.103. [DOI] [PubMed] [Google Scholar]
- Carlson G.D. Gorden C.D. Oliff H.S. Pillai J.J. LaManna J.C. Sustained spinal cord compression: part I: time-dependent effect on long-term pathophysiology. J. Bone Joint Surg. Am. 2003;85-A:86–94. [PubMed] [Google Scholar]
- Casha S. Yu W.R. Fehlings M.G. Oligodendroglial apoptosis occurs along degenerating axons and is associated with FAS and p75 expression following spinal cord injury in the rat. Neuroscience. 2001;103:203–218. doi: 10.1016/s0306-4522(00)00538-8. [DOI] [PubMed] [Google Scholar]
- Fehlings M.G. Nashmi R. A new model of acute compressive spinal cord injury in vitro. J. Neurosci. Methods. 1997;71:215–224. doi: 10.1016/s0165-0270(96)00147-1. [DOI] [PubMed] [Google Scholar]
- Fehlings M.G. Perrin R.G. The role and timing of early decompression for cervical spinal cord injury: update with a review of recent clinical evidence. Injury. 2005;36(Suppl. 2):B13–B26. doi: 10.1016/j.injury.2005.06.011. [DOI] [PubMed] [Google Scholar]
- Fehlings M.G. Tator C.H. An evidence-based review of decompressive surgery in acute spinal cord injury: rationale, indications, and timing based on experimental and clinical studies. J. Neurosurg. 1999;91:1–11. doi: 10.3171/spi.1999.91.1.0001. [DOI] [PubMed] [Google Scholar]
- Fehlings M.G. Sekhon L.H. Tator C. The role and timing of decompression in acute spinal cord injury: what do we know? What should we do? Spine. 2001;26:S101–S110. doi: 10.1097/00007632-200112151-00017. [DOI] [PubMed] [Google Scholar]
- Fu Y. Huff T.B. Wang H.W. Wang H. Cheng J.X. Ex vivo and in vivo imaging of myelin fibers in mouse brain by coherent anti-Stokes Raman scattering microscopy. Opt. Express. 2008;16:19396–19409. doi: 10.1364/oe.16.019396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y. Wang H. Huff T.B. Shi R. Cheng J.X. Coherent anti-Stokes Raman scattering imaging of myelin degradation reveals a calcium-dependent pathway in lyso-PtdCho-induced demyelination. J. Neurosci. Res. 2007;85:2870–2881. doi: 10.1002/jnr.21403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghasemlou N. Kerr B.J. David S. Tissue displacement and impact force are important contributors to outcome after spinal cord contusion injury. Exp. Neurol. 2005;196:9–17. doi: 10.1016/j.expneurol.2005.05.017. [DOI] [PubMed] [Google Scholar]
- Haghighi S.S. Pugh S.L. Perez-Espejo M.A. Oro J.J. Effect of 4-aminopyridine in acute spinal cord injury. Surg. Neurol. 1995;43:443–447. doi: 10.1016/0090-3019(95)80087-w. [DOI] [PubMed] [Google Scholar]
- Hayes K.C. Potter P.J. Wolfe D.L. Hsieh J.T. Delaney G.A. Blight A.R. 4-Aminopyridine-sensitive neurologic deficits in patients with spinal cord injury. J. Neurotrauma. 1994;11:433–446. doi: 10.1089/neu.1994.11.433. [DOI] [PubMed] [Google Scholar]
- Jensen J.M. Shi R. Effects of 4-aminopyridine on stretched mammalian spinal cord: the role of potassium channels in axonal conduction. J. Neurophysiol. 2003;90:2334–2340. doi: 10.1152/jn.00868.2002. [DOI] [PubMed] [Google Scholar]
- Kamencic H. Griebel R.W. Lyon A.W. Paterson P.G. Juurlink B.H. Promoting glutathione synthesis after spinal cord trauma decreases secondary damage and promotes retention of function. FASEB J. 2001;15:243–250. doi: 10.1096/fj.00-0228com. [DOI] [PubMed] [Google Scholar]
- Lim P.A. Tow A.M. Recovery and regeneration after spinal cord injury: a review and summary of recent literature. Ann. Acad. Med. Singapore. 2007;36:49–57. [PubMed] [Google Scholar]
- Li S. Mealing G.A. Morley P. Stys P.K. Novel injury mechanism in anoxia and trauma of spinal cord white matter: glutamate release via reverse Na+-dependent glutamate transport. J. Neurosci. 1999;19:RC16. doi: 10.1523/JNEUROSCI.19-14-j0002.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma M. Basso D.M. Walters P. Stokes B.T. Jakeman L.B. Behavioral and histological outcomes following graded spinal cord contusion injury in the C57Bl/6 mouse. Exp. Neurol. 2001;169:239–254. doi: 10.1006/exnr.2001.7679. [DOI] [PubMed] [Google Scholar]
- McBride J.M. Smith D.T. Byrn S.R. Borgens R.B. Shi R. 4-Aminopyridine derivatives enhance impulse conduction in guinea-pig spinal cord following traumatic injury. Neuroscience. 2007;148:44–52. doi: 10.1016/j.neuroscience.2007.05.039. [DOI] [PubMed] [Google Scholar]
- Morell P. Quarles R.H. Myelin formation, structure, and biochemistry. In: Siegel G., editor; Agranoff B.W., editor; Alberts R.W., editor; Molinoff P.B., editor. Basic Neurochemistry: Molecular, Cellular, and Medical Aspects. Lippincott Williams & Wilkins; Philadelphia: 1999. [Google Scholar]
- Morino T. Ogata T. Horiuchi H. Takeba J. Okumura H. Miyazaki T. Yamamoto H. Delayed neuronal damage related to microglia proliferation after mild spinal cord compression injury. Neurosci. Res. 2003;46:309–318. doi: 10.1016/s0168-0102(03)00095-6. [DOI] [PubMed] [Google Scholar]
- Nashmi R. Fehlings M.G. Changes in axonal physiology and morphology after chronic compressive injury of the rat thoracic spinal cord. Neuroscience. 2001;104:235–251. doi: 10.1016/s0306-4522(01)00009-4. [DOI] [PubMed] [Google Scholar]
- Ng W.P. Fehlings M.G. Cuddy B. Dickman C. Fazl M. Green B. Hitchon P. Northrup B. Sonntag V. Wagner F. Tator C.H. Surgical treatment for acute spinal cord injury study pilot study #2: evaluation of protocol for decompressive surgery within 8 hours of injury. Neurosurg. Focus. 1999;6:e3. doi: 10.3171/foc.1999.6.1.4. [DOI] [PubMed] [Google Scholar]
- Ouyang H. Galle B. Li J. Nauman E. Shi R. Biomechanics of spinal cord injury: a multimodal investigation using ex vivo guinea pig spinal cord white matter. J. Neurotrauma. 2008;25:19–29. doi: 10.1089/neu.2007.0340. [DOI] [PubMed] [Google Scholar]
- Ouyang H. Galle B. Li J. Nauman E. Shi R. Critical roles of decompression in functional recovery of ex vivo spinal cord white matter. J. Neurosurg. Spine. 2009;10:161–170. doi: 10.3171/2008.10.SPI08495. [DOI] [PubMed] [Google Scholar]
- Poliak S. Peles E. The local differentiation of myelinated axons at nodes of Ranvier. Nat. Rev. Neurosci. 2003;4:968–980. doi: 10.1038/nrn1253. [DOI] [PubMed] [Google Scholar]
- Poliak S. Salomon D. Elhanany H. Sabanay H. Kiernan B. Pevny L. Stewart C.L. Xu X. Chiu S.Y. Shrager P. Furley A.J. Peles E. Juxtaparanodal clustering of Shaker-like K+ channels in myelinated axons depends on Caspr2 and TAG-1. J. Cell Biol. 2003;162:1149–1160. doi: 10.1083/jcb.200305018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie J.M. Chiu S.Y. Distribution of sodium and potassium channels in mammalian myelinated nerve. Adv. Neurol. 1981;31:329–342. [PubMed] [Google Scholar]
- Scheff S.W. Rabchevsky A.G. Fugaccia I. Main J.A. Lumpp J.E., Jr. Experimental modeling of spinal cord injury: characterization of a force-defined injury device. J. Neurotrauma. 2003;20:179–193. doi: 10.1089/08977150360547099. [DOI] [PubMed] [Google Scholar]
- Sherman D.L. Brophy P.J. Mechanisms of axon ensheathment and myelin growth. Nat. Rev. Neurosci. 2005;6:683–690. doi: 10.1038/nrn1743. [DOI] [PubMed] [Google Scholar]
- Shi R. Blight A.R. Compression injury of mammalian spinal cord in vitro and the dynamics of action potential conduction failure. J. Neurophysiol. 1996;76:1572–1580. doi: 10.1152/jn.1996.76.3.1572. [DOI] [PubMed] [Google Scholar]
- Shi R. Borgens R.B. Acute repair of crushed guinea pig spinal cord by polyethylene glycol. J. Neurophysiol. 1999;81:2406–2414. doi: 10.1152/jn.1999.81.5.2406. [DOI] [PubMed] [Google Scholar]
- Shi R. Whitebone J. Conduction deficits and membrane disruption of spinal cord axons as a function of magnitude and rate of strain. J. Neurophysiol. 2006;95:3384–3390. doi: 10.1152/jn.00350.2005. [DOI] [PubMed] [Google Scholar]
- Shi R. Asano T. Vining N.C. Blight A.R. Control of membrane sealing in injured mammalian spinal cord axons. J. Neurophysiol. 2000;84:1763–1769. doi: 10.1152/jn.2000.84.4.1763. [DOI] [PubMed] [Google Scholar]
- Shields C.B. Zhang Y.P. Shields L.B. Han Y. Burke D.A. Mayer N.W. The therapeutic window for spinal cord decompression in a rat spinal cord injury model. J. Neurosurg. Spine. 2005;3:302–307. doi: 10.3171/spi.2005.3.4.0302. [DOI] [PubMed] [Google Scholar]
- Takahashi T. Suto Y. Kato S. Ohama E. Experimental acute dorsal compression of cat spinal cord: correlation of magnetic resonance signal intensity with spinal cord evoked potentials and morphology. Spine (Phila. Pa. 1976) 1996;21:166–173. doi: 10.1097/00007632-199601150-00002. [DOI] [PubMed] [Google Scholar]
- Totoiu M.O. Keirstead H.S. Spinal cord injury is accompanied by chronic progressive demyelination. J. Comp. Neurol. 2005;486:373–383. doi: 10.1002/cne.20517. [DOI] [PubMed] [Google Scholar]
- Tysseling-Mattiace V.M. Sahni V. Niece K.L. Birch D. Czeisler C. Fehlings M.G. Stupp S.I. Kessler J.A. Self-assembling nanofibers inhibit glial scar formation and promote axon elongation after spinal cord injury. J. Neurosci. 2008;28:3814–3823. doi: 10.1523/JNEUROSCI.0143-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waxman S.G. Black J.A. Stys P.K. Ransom B.R. Ultrastructural concomitants of anoxic injury and early post-anoxic recovery in rat optic nerve. Brain Res. 1992;574:105–119. doi: 10.1016/0006-8993(92)90806-k. [DOI] [PubMed] [Google Scholar]
- Waxman S.G. Membranes, myelin, and the pathophysiology of multiple sclerosis. N. Engl. J. Med. 1982;306:1529–1533. doi: 10.1056/NEJM198206243062505. [DOI] [PubMed] [Google Scholar]
- Young W. Spinal cord contusion models. Prog. Brain Res. 2002;137:231–255. doi: 10.1016/s0079-6123(02)37019-5. [DOI] [PubMed] [Google Scholar]