

1. Introduction
Isoprenoids represent the largest class of small molecules on earth, 1 so it is not surprising that many of the enzymes that are involved in isoprenoid biosynthesis are drug targets. For example, the most widely prescribed drug, Lipitor, targets cholesterol biosynthesis at an early stage; bisphosphonates such as Fosamax, used to treat bone resorption diseases, target the middle of the isoprenoid biosynthesis pathway, while anti-infectives such as terbinafine (Lamisil) target the later stages of sterol biosynthesis, in fungi/yeasts. The early stages of isoprenoid biosynthesis involve formation of isopentenyl diphosphate (1, IPP) and dimethylallyl diphosphate (2, DMAPP):
 In most pathogenic bacteria, these molecules are produced in the Rohmer or non-mevalonate pathway2, but in humans and in bacteria such as Staphylococcus aureus, they are formed in the mevalonate pathway3. The last two enzymes in the non-mevalonate pathway, IspG and IspH, contain Fe4S4 clusters4,5 and carry out 2H+/2e− reductions, converting 2-C-methyl-D-erythritol 2,4-cyclo-diphosphate (MEcPP, 3) to HMBPP (E-1-hydroxy-2-methyl-but-2-enyl 4- diphosphate,4), and HMBPP to IPP and DMAPP, Scheme I Once formed, IPP and DMAPP condense via a “head-to-tail” mechanism to form geranyl diphosphate (5) and farnesyl diphosphate (6) in reactions catalyzed by the enzyme farnesyl diphosphate synthase (FPPS), and further reaction with IPP catalyzed by the enzyme geranylgeranyl diphosphate synthase (GGPPS) yields the C20 species, geranylgeranyl diphosphate (GGPP, 7)6,7, Scheme II Both FPP and GGPP are used in protein prenylation (of importance in cell survival and signaling pathways), plus, FPP can also condense in a “head-to-head” manner via presqualene diphosphate (PSPP, 8)8 to form triterpenes, Scheme III In humans, this condensation is accompanied by an NADPH reduction step and results in formation of squalene (9)9, but in the bacterium S. aureus, the reduction step is missing and the enzyme CrtM converts FPP to dehydrosqualene (10).10 In many organisms, squalene is epoxidized to form oxidosqualene (11), which is then cyclized to form lanosterol (12) which after several additional steps, is transformed into cholesterol (13) in humans, or ergosterol (14) or episterol (15) in yeasts, fungi, and parasitic protozoa. In S. aureus, 10 is also converted to a carotenoid pigment, staphyloxanthin (16)10, an important virulence factor. The enzymes involved in these reactions are our targets, and I describe here our progress in understanding their structures, mechanism of action, and inhibition, focusing on the use of a less-conventional, knowledge-based approach to inhibitor or drug discovery.
In most pathogenic bacteria, these molecules are produced in the Rohmer or non-mevalonate pathway2, but in humans and in bacteria such as Staphylococcus aureus, they are formed in the mevalonate pathway3. The last two enzymes in the non-mevalonate pathway, IspG and IspH, contain Fe4S4 clusters4,5 and carry out 2H+/2e− reductions, converting 2-C-methyl-D-erythritol 2,4-cyclo-diphosphate (MEcPP, 3) to HMBPP (E-1-hydroxy-2-methyl-but-2-enyl 4- diphosphate,4), and HMBPP to IPP and DMAPP, Scheme I Once formed, IPP and DMAPP condense via a “head-to-tail” mechanism to form geranyl diphosphate (5) and farnesyl diphosphate (6) in reactions catalyzed by the enzyme farnesyl diphosphate synthase (FPPS), and further reaction with IPP catalyzed by the enzyme geranylgeranyl diphosphate synthase (GGPPS) yields the C20 species, geranylgeranyl diphosphate (GGPP, 7)6,7, Scheme II Both FPP and GGPP are used in protein prenylation (of importance in cell survival and signaling pathways), plus, FPP can also condense in a “head-to-head” manner via presqualene diphosphate (PSPP, 8)8 to form triterpenes, Scheme III In humans, this condensation is accompanied by an NADPH reduction step and results in formation of squalene (9)9, but in the bacterium S. aureus, the reduction step is missing and the enzyme CrtM converts FPP to dehydrosqualene (10).10 In many organisms, squalene is epoxidized to form oxidosqualene (11), which is then cyclized to form lanosterol (12) which after several additional steps, is transformed into cholesterol (13) in humans, or ergosterol (14) or episterol (15) in yeasts, fungi, and parasitic protozoa. In S. aureus, 10 is also converted to a carotenoid pigment, staphyloxanthin (16)10, an important virulence factor. The enzymes involved in these reactions are our targets, and I describe here our progress in understanding their structures, mechanism of action, and inhibition, focusing on the use of a less-conventional, knowledge-based approach to inhibitor or drug discovery.
Scheme I.

Formation of Isopentenyl Diphosphate (1) and Dimethyallyl Diphosphate (2) in the Non-Mevalonate Pathway.
Scheme II.

Formation of Farnesyl Diphosphate (6) and Geranylgeranyl Diphosphate (7)
Scheme III.

Formation of Triterpenes from Farnesyl Diphosphate (6)
IspH (LytB), an Fe4S4-cluster containing enzyme
The IspH enzyme is found in the vast majority of pathogenic bacteria11, as well as in malaria parasites12 and, since it is not found in humans and is essential for pathogen survival, it is an important target for anti-infective development. Working with Jomaa and Ermler we reported13 that the enzyme has a unique, trefoil-like structure, Figure 1A,B, with a central Fe3S4 cluster, and a similar structure was then reported by Grawert et al.14 The observation that both proteins contained 3Fe and not 4Fe was inconsistent with the results of EPR5, chemical analysis5,15 and activity5,15 results, which all pointed to an Fe4S4 cluster, so we next used computational methods to construct an Fe4S4 model, with the HMBPP substrate docking to the unique, 4th Fe in oxidized IspH, via its 1-OH group, initially as an alkoxide,13 Figure 1C. Interestingly, very recent x-ray crystallographic results16 have shown that HMBPP does in fact bind to the 4Fe cluster in IspH via O-1 (as we proposed), and the structure of HMBPP bound to the Fe4S4 cluster we deduced13 from computational docking is very similar to that determined by crystallography, Figure 1D (a 0.3 Å ligand rmsd). Apparently then, the 4Fe cluster can be stabilized by ligands binding to the 4th Fe, although the reason for this is not yet known. But how does this Fe4S4 cluster catalyze the 2H+/2e− reduction, the removal of the 1-OH oxygen, to form the IPP and DMAPP products? Based on our crystallographic results and on bioinformatics, we proposed13 that E126 was a key residue in catalysis, providing the H+ needed for activity. The essential nature of E126 was then demonstrated in later work by others14 and we reasoned that by using an inactive IspH mutant (E126A), it might be possible to “trap” a reaction intermediate, which if its structure could be deduced, would give clues as to the catalytic mechanism. To do this, we used EPR and ENDOR spectroscopy17.
FIGURE 1.

Structural results for IspH (LytB). A,B: Crystal structure results for Aquifex aeolicus IspH. C, Initial docking pose for HMBPP to oxidised IspH Fe4S4 cluster obtained by using the “open-form” structure. D, Comparison of HMBPP bound to IspH from X-ray16 (green) and docking13 (red). From Refs. 13, 16, with permission.
Simply adding HMBPP to reduced IspH yielded an EPR spectrum that was essentially the same as that obtained on adding the IPP product (Figure 2A). However, the EPR spectrum obtained when using the E126A mutant was very different, exhibiting g-values of 2.124, 1.999 and 1.958, and had similarities to the EPR spectra of the HMBPP “parent” molecules, ethylene (17) and allyl alcohol (18), when bound to a nitrogenase FeMo cofactor18,19. In nitrogenase, the results of both ENDOR18,19 as well as DFT calculations20 indicated that both of these species (17,18) bind to one of the Fe in the FeMo cofactor cluster, forming π complexes, η2-alkenyl “metallacycles” (19,20), Scheme IV, and it seemed possible that this might occur with the Fe4S4 cluster in IspH as well. A prediction of this binding mode is that there would be substantial hyperfine interactions in the ENDOR spectrum, and as shown in Figure 2B, this is clearly the case with [u-13C]-HMBPP, with hyperfine couplings for 13C being observed, consistent with the idea that HMBPP (4) binds to the [Fe4S4] cluster as the metallacycle 21. This opens up the possibility that this binding mode might in reduced IspH “activate” the molecule such that on protonation (by the E126 CO2H), an η1-allyl complex 22 or the η3 π-allyl complex 23 can form, Figure 3. On reduction and protonation at C2, the IPP product forms, while protonation at C4 would form DMAPP, Figure 3A,B an organometallic as opposed to a purely radical mechanism for catalysis17. These spectroscopic results suggest the likely importance of organometallic intermediates in IspH catalysis, which leads to a new idea for inhibitor design, based on organometallic precedent.
FIGURE 2.

EPR and ENDOR results for IspH. A, EPR spectra of IspH (and an E126A mutant) ±ligands. B, ENDOR spectrum with [u-13C]-26. From Ref. 17, with permission.
Scheme IV.

Schematic Illustration of π/σ Bioorganometallic Species in Nitrogenase and IspH
FIGURE 3.

IspH mechanism proposal. A, deoxygenation steps. B Reductive cleavage forming IPP, DMAPP from allyl species. From Ref. 17, with permission,
In previous work, several groups reported that alkynes could be cis-reduced by “model” Fe4S4 clusters such as [Fe4S4(SPh)4]2-/3- to form olefins21,22, and it was proposed that binding might occur via an η2-alkynyl species, another π or π/σ “metallacycle”. These observations lead to the idea that alkynes might also bind to reduced IspH, and would inhibit catalytic activity. To test this idea, we obtained the EPR and ENDOR spectra of the alkynes 24 and 2517, Scheme V, bound to IspH. Both bound, but were poor inhibitors. However, with propargyl diphosphate 26, there were large changes in the EPR spectra, and the ENDOR spectra of [13C3]-26 bound to IspH17 (e.g. Figure 4A) exhibited large hyperfine couplings (A ~6 MHz for 13C, ~10 MHz for 1H)17. 26 was also a Ki ~970 nM IspH inhibitor (Figure 4B), ~1000x more active than previously reported inhibitors23. A likely explanation of this good inhibition is formation of the π/σ metallacycle 27, in which the alkyne can bind to the unique 4th Fe, Figure 4C, opening up, potentially, a new route to anti-infective development.
Scheme V.
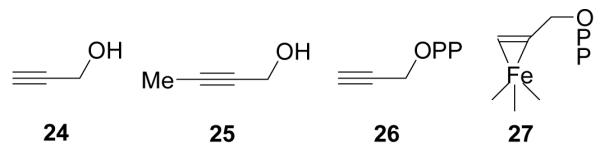
Acetylene Inhibitors of IspH and Proposed Binding Mode
FIGURE 4.

IspH inhibition by the alkyne diphosphate, 26. A, 9GHz ENDOR spectrum of [u-13C]-propargyl diphosphate (26) showing ~6 MHz 13C hyperfine coupling. B, Dose-response curve showing IspH inhibition by 26. C, docking results showing close apposition of the alkyne group to the unique, 4th Fe in IspH. From Ref. 17, with permission.
FPPS (and GGPPS): Structure, Mechanism, and Inhibition by Lipophilic Bisphosphonates
The IPP and DMAPP produced by either the non-mevalonate or mevalonate pathways are next condensed by FPPS and GGPPS to form FPP and GGPP, Scheme II. FPPS is the target of the bisphosphonate class of drugs used to treat bone resorption diseases, but for many years their mechanism of action was unknown. Our interest in these systems arose from several chance observations. First, working with Urbina and Docampo, we found24 that T. cruzi contained very high levels of condensed phosphates, such as diphosphate 28, Scheme VI. This led to the idea that non-hydrolyzable PPi analogs, bisphosphonates such as pamidronate (29) and risedronate (30, Actonel) might inhibit parasite cell growth. This turned out to be the case24,25, but the target was not known! The second chance observation was that we noticed that nitrogen-containing analogs of GPP such as 31, known to be potent, low nM inhibitors of terpene cyclases, looked suspiciously like the bisphosphonate ibandronate, 32, as did their electrostatic potential surfaces φ(r), Figures 5A,B.26 This suggested that cationic bisphosphonates might act as carbocation/diphosphate isosteres, inhibiting isoprenoid biosynthesis, a view supported by the observation that bisphosphonates were reported to act in the mevalonate pathway.27 The third observation was that bisphosphonates such as 33 had been developed by Zeneca as herbicides28, and had been shown to be low nM inhibitors of a daffodil FPPS. 28 Since we noticed that 33 had also been shown29 to be active in bone resorption, we proposed26 that the bone-resorption drugs might act by inhibiting FPPS, mimicking a carbocation reactive intermediate (34), Scheme VII docking into the allylic site in FPPS, Figure 5C26. The FPPS target was soon confirmed30-32 and the allylic binding mode we proposed was later confirmed crystallographically, by Hosfield et al.33 (Figure 5D). In later work, we also showed that pamidronate provided a parasitological cure of cutaneous leishmaniasis in mice, Figure 6A,B34, by blocking FPPS and thus, ergosterol biosynthesis25, opening up the possibility of the clinical use of bisphosphonates as anti-infectives35.
Scheme VI.

Structures of Diphosphate, Several Bisphosphonates and a Terpene Cyclase Inhibitor
FIGURE 5.

Cationic bisphosphonates as FPPS/GGPPS inhibitors. A,B: φ(r) electrostatic potential surfaces for an ammonium diphosphate based terpene cyclase inhibitor (A)and ibandronate, B. C, Early model for bisphosphonate inhibition of FPPS26. D, Crystal structure showing similar pose as in C. E, BPH-715 bound to GGPPS42. From Refs. 26 and 42, with permission.
Scheme VII.

Proposed Carbocation Mechanism for FPPS Catalysis and Similarity Between a Transition State/Reactive Intermediate and the Bisphosphonate Drug, Ibandronate
FIGURE 6.

Effects of the bisphosphonate pamidronate (29) on cutaneous Leishmaniasis (L. mexicana) in mice. A, effects of pamidronate dose on lesion progression. B, cure of infection in Treated mouse (is on the left). From Ref. 34, with permission.
In addition to their activity as bone resorption drugs and anti-parasitics, bisphosphonates kill tumor cells,36 plus, they activate γδ T cells37 to also kill tumor cells.38 There is, therefore, interest in developing bisphosphonates as anti-cancer drugs, and the results of small clinical trials on pamidronate 39 as well as zoledronate (+ interleukin-2)40, have shown promise. More recently, the results of a much larger scale study, of 1803 patients with breast cancer, showed a 30% decrease in the recurrence of disease in patients treated post-surgery with an aromatase inhibitor, plus zoledronate (35)41. Conventional bisphosphonates are, however, rapidly removed from the circulation (in < 1 hour), binding to bone mineral. We reasoned that removing the 1-OH group would reduce bone-binding, and adding more hydrophobic substituents would enhance cell or tissue penetration, so that species such as BPH-715 (36) would be more potent inhibitors of tumor cell growth.
 This turned out to be the case42, with 36 killing tumor cell lines with an IC50 of ~100 nM, at least 100x lower than found with the bisphosphonate zoledronate (35). 36 also blocked tumor cell invasion42, plus, it was a potent activator of γδ T cells43, in addition to having good activity, in vivo42.
This turned out to be the case42, with 36 killing tumor cell lines with an IC50 of ~100 nM, at least 100x lower than found with the bisphosphonate zoledronate (35). 36 also blocked tumor cell invasion42, plus, it was a potent activator of γδ T cells43, in addition to having good activity, in vivo42.
The enhanced activity of 36 is likely due to several factors. First, it inhibits FPPS42, which results in blocking protein (e.g. K-ras) prenylation. Second, since it is lipophilic, it gets into cells more readily than do more polar analogs. Third, when FPPS is inhibited, the substrates IPP and DMAPP build up and these are converted to toxic ATP analogs44 such as ApppI (37)44. Fourth, the buildup of IPP (and DMAPP) in tumor cells on FPPS inhibition leads to activation of γδ T cells, since both IPP and DMAPP are so-called “phosphoantigens”45. Fifth, lipophilic bisphosphonates inhibit GGPPS by docking into the product binding site (Figure 5E)42,46 and the combined effects of FPPS+GGPPS inhibition are likely synergistic (preventing cross-prenylation). When combined with poor bone-binding, this leads to potent in vivo activity.
Dehydrosqualene Synthase (CrtM) and Staphyloxanthin: An Anti-Virulence Approach to Staph Infections
In humans, most FPP is converted via the “head-to-head” triterpene synthase, squalene synthase (SQS), to squalene 9. While involved in some “recreational” reading I noticed an article47 reviewing work48 by Nizet and Liu on the role of the carotenoid virulence factor, staphyloxanthin (16), in S. aureus. This compound is a golden carotenoid pigment found only in S. aureus, the causative agent of staph infections. These workers showed that the pigment acts as a “protective shield”, preventing the organism from being killed by host immune cells that generate reactive oxygen species (such as O2−, ClO·, H2O2), which are thought to be “deactivated” by reacting with the polyene. What caught my attention was that the initial step in staphyloxanthin biosynthesis involved exactly the same first step as in cholesterol/ergosterol biosynthesis: FPP (6) → PSPP (8). I knew from my work with Urbina that many drug leads targeting SQS had been developed by the pharmaceutical industry as cholesterol-lowering agents, and after an examination of the amino acid sequences of the S. aureus dehydrosqualene synthase (called CrtM) and human squalene synthase, it seemed that both enzymes would have similar three dimensional structures. I posited that the bacterial enzyme would be inhibited by the compounds that had already been developed as cholesterol-lowering drugs. As anticipated, we found (with Liu and Wang) that the 3D structure of CrtM49 was very similar to that found with human SQS (Figure 7A), and using a non-reactive, sulfur-containing analog of FPP: S-thiolo-FSPP (38), Scheme VIII, we found two “FPP” binding sites, as hoped (Figure 7B). We then synthesized a range of potential CrtM inhibitors, compounds that had all been developed as SQS inhibitors, and tested them for CrtM inhibition. The most potent inhibitors were bisphosphonates such as 39. However, they did not block staphyloxanthin (16) formation in cells. On the other hand, phosphonosulfonates (40) and phosphonoacetamides (41) inhibited both CrtM activity in vitro, as well as staphyloxanthin biosynthesis in S. aureus, with the crystallographic results showing that they bound to one or the other FPP sites, Figure 7C49. When S. aureus is stripped of its protective carotenoid shield, cells grow normally in vitro since virulence factors are not essential for cell growth. However, the cells are white (Figure 8A) and when exposed to reactive oxygen species, either from H2O2 or by adding neutrophils, cell growth is greatly inhibited (Figures 8B)49-51. Moreover, in mice (Figure 8C), we found a 98% decrease in S. aureus in the kidneys49, on treatment with 40. These results are of interest since they represent a potentially new, highly selective approach to blocking staph infections in which cells are made highly susceptible to killing by the host’s innate immune system. And of course the fact that 40 has already been tested for safety in clinical trials (as a cholesterol lowering agent)52 makes it of particular interest.
FIGURE 7.

CrtM as a target for anti-virulence therapy. A, comparison between CrtM (green) and SQS (yellow) structures. B, FSPP (two molecules) bound to CrtM. C, BPH-652 (40, in blue) bound to CrtM. The two FsPP molecules (green, yellow) are also shown. From Ref. 49 with permission.
Scheme VIII.

Some Inhibitors of the CrtM Enzyme from S. aureus
FIGURE 8.

Effects of BPH-652 (40) on staphyloxanthin biosynthesis and S. aureus infection. A, BPH-652 blocks staphyloxanthin biosynthesis in cells. B, BPH-652 renders staph susceptible to killing by neutrophils in blood and C, reduces infectivity in mice by 98%. From Ref. 49, with permission.
Using the Heart Drug Amiodarone as an Anti-Infective against Chagas Disease and Leishmaniasis
After condensing FPP to squalene, humans, plants, fungi, yeasts as well as the pathogenic protozoa T. cruzi and Leishamania mexicana, carry out an epoxidation to form oxidosqualene (11), which is then cyclized to form lanosterol (12). Again while perusing one of the more populist journals, my attention was drawn to an article53 reporting observations by Courchesne54 and Gupta et al.55 that the class III anti-arrhythmia drug amiodarone (42), Scheme IX, had unexpected activity against bakers’ yeast. An effect on Ca2+ channels was shown, but what was more surprising was that cell growth inhibition activity was synergistic with the azole antibiotics that are commonly used to treat yeast or fungal infections, drugs such as such as fluconazole (43). It seemed likely to me that ergosterol biosynthesis might be involved. I e-mailed Urbina to see if we should try amiodarone in T. cruzi. His response was encouraging: “We are going to pursue vigorously this lead against trypanosomatid parasites, especially because amiodarone is relatively cheap and non-toxic and, most interestingly, is frequently prescribed to Chagas disease patients to control their cardiac arrythmias!!!”
Scheme IX.

Some Sterol Biosynthesis Inhibitors
We screened amiodarone in T. cruzi finding that56: i) It killed T. cruzi. ii) It blocked ergosterol biosynthesis. iii) It acted at the level of oxidosqualene cyclase, OSC. iv) It synergized with the azole posaconazole (44). v) It blocked Ca2+ channels in T. cruzi much more effectively than in host cells. vi) Posaconazole, which blocks ergosterol biosynthesis at the lanosterol 14-α demethylase level, also blocked the parasites’ Ca2+-channels. vii) There were very good parasitological cures of mice treated with the combination therapy of amiodarone + posaconazole. viii) In addition, molecular docking results for lanosterol and a known OSC inhibitor (Ro48-8071; 45) docked to an OSC showed good accord with the known crystallographic structures, and amiodarone bound into the same site.
There was then an apparent lull in activity, but in very recent work, Serrano-Martin et al.57 have reported that amiodarone has similar effects in L. mexicana57, blocking ergosterol biosynthesis and inhibiting cell growth. More importantly, Paniz-Mondolfi et al.58 have begun to report the results of small clinical trials of amiodarone (± itraconazole; 46). In one case, a patient had concurrent Chagas disease and cutaneous leishmaniasis. This is a difficult combination of treat since the standard drugs used to treat leishmaniasis are antimonials, which are problematic in patients with cardiac arrythmias (as in Chagas disease). The patient was treated with amiodarone to stabilize the heart condition, but remarkably, the cutaneous lesions also healed – without use of any specific anti-leishmanial therapy58. In a second study59, the combination amiodarone + itraconazole was used to treat a Chagas disease patient, with a parasitological cure of the T. cruzi infection being reported59. In addition, a 100% cure rate has now been found in 11 patients with cutaneous leishmaniasis.60 This efficacy is very high, and is thought to be due, at least in part, to the very unusual excretion mechanism for amiodarone: through the skin61!
Concluding Remarks and Perspectives
The results described above give a brief summary of the last 10 years work in our laboratory on isoprenoid biosynthesis enzymes, which has focused on discovering new drug targets, mechanisms, and inhibitors. The results with the Fe4S4 cluster-containing protein IspH seem radical, but are simply based on precedent (ethylene, allyl alcohol nitrogenase ENDOR and DFT) and have led to the first μM IspH inhibitors and a new proposal for catalysis, involving organometallic species. With the head-to-tail synthases FPPS (and GGPPS), there are now ~60 crystallographic structures reported, including some with the novel, lipophilic bisphosphonates, which now await more extensive pre-clinical testing. With CrtM, we have the first structure of a head-to-head triterpene synthase containing bound substrate analogs, together with novel inhibitors. These block S. aureus proliferation in vivo, and one has already been tested for safety in humans (in the context of its role as a cholesterol-lowering drug). And finally, we discovered another drug “repurposing”: the use of the anti-arrythmia drug, amiodarone, as an agent against both Chagas disease and cutaneous leishmaniasis. Since Chagas disease affects ~10,000,000 individuals in South America, and there is no cure for the chronic stage of the disease (the leading cause of sudden death on the sub-continent), the combination of amiodarone plus an azole is of considerable interest, as is its use alone in treating some forms of cutaneous leishmaniasis.
In each of the examples described above, we have used a knowledge-based approach, rather than purely screening-based methods, to find new leads in which we use information from one area of research to suggest drug (or inhibitor) leads in another, seemingly un-related area. Since terpenes or isoprenoids are the largest class of small molecules known and their biosynthesis is already the target for many current drugs, it seems likely that many new drugs will be found that target their formation, but as Pasteur famously said: “Chance favors only the prepared mind”.
CONSPECTUS.

The isoprenoid biosynthesis pathways are responsible for the production of the largest class of small molecules on earth: terpenes or isoprenoids. Not surprisingly then, isoprenoid biosynthesis is a target for drug discovery, and many drugs, such as Lipitor, used to lower cholesterol; bisphosphonates such as Fosamax, used to treat osteoporosis; as well as many anti-fungals, target isoprenoid biosynthesis. With the rise in drug resistance in malaria, tuberculosis and in staph infections; the lack of any drugs to treat chronic Chagas disease (the leading cause of sudden death in South America), together with the relatively slow progress in the development of anti-cancer drugs, new approaches and leads are needed. Here, I describe developments in four areas targeting isoprenoid biosynthesis using, in each case, knowledge from one area of Chemistry to guide the development of inhibitors (or drugs/drug leads) in another, seemingly un-related area. First, I describe mechanistic studies of the enzyme IspH that is present in malaria parasites and most pathogenic bacteria, but not in humans. IspH is a 4Fe-4S protein and produces the C5 isoprenoids IPP (isopentenyl diphosphate) and DMAPP (dimethylallyl diphosphate) from HMBPP (E-1-hydroxy-2-methyl-but-2-enyl-4 diphosphate) via a 2H+/2e reduction (of an allyl alcohol to an alkene). The mechanism is unusual in that it involves organometallic species: “metallacycles” (η-alkenes) and η1/η3-allyls. These observations lead to novel alkyne inhibitors, which also form metallacycles. Second, I describe structure/function/inhibition studies of the molecule that condenses IPP and DMAPP to the sesquiterpene, farnesyl diphosphate (FPP) in a “head-to-tail” manner, FPP synthase. This enzyme uses a carbocation mechanism and is potently inhibited by bone resorption drugs, bisphosphonates, which we find are also anti-parasitics which block sterol biosynthesis in protozoa. We also show that “lipophilic” bisphosphonates inhibit protein prenylation and invasiveness in tumor cells, in addition to activating γδ T cells to kill tumor cells. Third, I describe structural and inhibition studies of a “head-to-head” triterpene synthase, dehydrosqualene synthase (CrtM), from S. aureus. CrtM catalyzes the first committed step in biosynthesis of the carotenoid virulence factor staphyloxanthin, the condensation of two FPP molecules to produce the cyclopropane, presqualene diphosphate. The structure of CrtM is similar to that of human squalene synthase (SQS) and some SQS inhibitors (already developed as cholesterol-lowering drugs) block staphyloxanthin biosynthesis. Treated bacteria are white and non-virulent (since they lack the carotenoid shield that protects them from reactive oxygen species produced by neutrophils), rendering them susceptible to innate immune system clearance, a new therapeutic approach. And finally, I show that the heart drug amiodarone, also known to have anti-fungal activity, blocks ergosterol biosynthesis at the level of oxidosqualene cyclase, in Trypanosoma cruzi, work that has led to its use in the clinic as a novel anti-parasitic. In each of these four examples, we use information from one area (organometallic chemistry; bone resorption; cholesterol-lowering; heart disease) to develop drugs or drug leads in an unrelated area, a “knowledge-based” approach.
Acknowledgements
I thank Julio Urbina, Roberto Docampo, Craig Morita, Andrew H-J. Wang, Victor Nizet, Hassan Jomaa and members of their groups, together with Michael Martin, John Sanders, Michael Hudock, Yonghui Zhang, Yongcheng Song, Rong Cao and Weixue Wang, for their contributions. This work was supported by NIH grants GM65307, GM073216 and AI074233, by the World Health Organization, the American Heart Association (Midwest Affiliate) and the Leukemia and Lymphoma Society.
BIOGRAPHICAL INFORMATION
Eric Oldfield was born in London, England, in 1948. He obtained a BSc degree from Bristol University in 1969 and a PhD degree from Sheffield University, in 1972, with Dennis Chapman. After postdoctoral work with Adam Allerhand at Indiana University and with John S. Waugh at MIT, he joined the Chemistry Department at the University of Illinois at Urbana-Champaign in 1975, where he is currently the Alumni Research Scholar Professor of Chemistry. He has been the recipient of ACS’s Award in Pure Chemistry; RSC’s Meldola Medal; the Biochemical Society’s Colworth Medal; the American Heart Association’s Katz Basic Science Research Prize; and the RSC Awards in Spectroscopy, and in Soft Matter and Biophysical Chemistry.
References
- 1.Poulter CD. Bioorganic chemistry. A natural reunion of the physical and life sciences. J Org Chem. 2009;74:2631–45. doi: 10.1021/jo900183c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rohmer M. The discovery of a mevalonate-independent pathway for isoprenoid biosynthesis in bacteria, algae and higher plants. Nat Prod Rep. 1999;16:565–74. doi: 10.1039/a709175c. [DOI] [PubMed] [Google Scholar]
- 3.Goldstein JL, Brown MS. Regulation of the mevalonate pathway. Nature. 1990;343:425–30. doi: 10.1038/343425a0. [DOI] [PubMed] [Google Scholar]
- 4.Seemann M, Bui BT, Wolff M, Tritsch D, Campos N, Boronat A, Marquet A, Rohmer M. Isoprenoid biosynthesis through the methylerythritol phosphate pathway: The (E)-4- hydroxy-3-methylbut-2-enyl diphosphate synthase (GcpE) is a [4Fe-4S] protein. Angew Chem Int Ed Engl. 2002;41:4337–9. doi: 10.1002/1521-3773(20021115)41:22<4337::AID-ANIE4337>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 5.Wolff M, Seemann M, Tse Sum Bui B, Frapart Y, Tritsch D, Estrabot A. Garcia, Rodriguez-Concepcion M, Boronat A, Marquet A, Rohmer M. Isoprenoid biosynthesis via the methylerythritol phosphate pathway: The (E)-4-hydroxy-3-methylbut-2-enyl diphosphate reductase (LytB/IspH) from Escherichia coli is a [4Fe-4S] protein. FEBS Lett. 2003;541:115–20. doi: 10.1016/s0014-5793(03)00317-x. [DOI] [PubMed] [Google Scholar]
- 6.Thulasiram HV, Poulter CD. Farnesyl diphosphate synthase: The art of compromise between substrate selectivity and stereoselectivity. J Am Chem Soc. 2006;128:15819–23. doi: 10.1021/ja065573b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ohnuma S, Hirooka K, Ohto C, Nishino T. Conversion from archaeal geranylgeranyl diphosphate synthase to farnesyl diphosphate synthase. Two amino acids before the first aspartate-rich motif solely determine eukaryotic farnesyl diphosphate synthase activity. J Biol Chem. 1997;272:5192–8. doi: 10.1074/jbc.272.8.5192. [DOI] [PubMed] [Google Scholar]
- 8.Epstein WW, Rilling HC. Studies on the mechanism of squalene biosynthesis. The structure of presqualene pyrophosphate. J Biol Chem. 1970;245:4597–605. [PubMed] [Google Scholar]
- 9.Pandit J, Danley DE, Schulte GK, Mazzalupo S, Pauly TA, Hayward CM, Hamanaka ES, Thompson JF, Harwood HJ., Jr. Crystal structure of human squalene synthase. A key enzyme in cholesterol biosynthesis. J Biol Chem. 2000;275:30610–7. doi: 10.1074/jbc.M004132200. [DOI] [PubMed] [Google Scholar]
- 10.Pelz A, Wieland KP, Putzbach K, Hentschel P, Albert K, Gotz F. Structure and biosynthesis of staphyloxanthin from Staphylococcus aureus. J Biol Chem. 2005;280:32493–8. doi: 10.1074/jbc.M505070200. [DOI] [PubMed] [Google Scholar]
- 11.Eberl M, Hintz M, Reichenberg A, Kollas AK, Wiesner J, Jomaa H. Microbial isoprenoid biosynthesis and human gammadelta T cell activation. FEBS Lett. 2003;544:4–10. doi: 10.1016/s0014-5793(03)00483-6. [DOI] [PubMed] [Google Scholar]
- 12.Rohrich RC, Englert N, Troschke K, Reichenberg A, Hintz M, Seeber F, Balconi E, Aliverti A, Zanetti G, Kohler U, Pfeiffer M, Beck E, Jomaa H, Wiesner J. Reconstitution of an apicoplast-localised electron transfer pathway involved in the isoprenoid biosynthesis of Plasmodium falciparum. FEBS Lett. 2005;579:6433–8. doi: 10.1016/j.febslet.2005.10.037. [DOI] [PubMed] [Google Scholar]
- 13.Rekittke I, Wiesner J, Rohrich R, Demmer U, Warkentin E, Xu W, Troschke K, Hintz M, No JH, Duin EC, Oldfield E, Jomaa H, Ermler U. Structure of (E)-4-hydroxy-3-methyl-but-2-enyl diphosphate reductase, the terminal enzyme of the non-mevalonate pathway. J Am Chem Soc. 2008;130:17206–7. doi: 10.1021/ja806668q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Grawert T, Rohdich F, Span I, Bacher A, Eisenreich W, Eppinger J, Groll M. Structure of active IspH enzyme from escherichia coli provides mechanistic insights into substrate reduction. Angew Chem Int Ed Engl. 2009;48:5756–9. doi: 10.1002/anie.200900548. [DOI] [PubMed] [Google Scholar]
- 15.Xiao Y, Zhao ZK, Liu P. Mechanistic studies of IspH in the deoxyxylulose phosphate pathway: Heterolytic C-O bond cleavage at C4 position. J Am Chem Soc. 2008;130:2164–5. doi: 10.1021/ja710245d. [DOI] [PubMed] [Google Scholar]
- 16.Grawert T, Span I, Eisenreich W, Rohdich F, Eppinger J, Bacher A, Groll M. Probing the reaction mechanism of IspH protein by x-ray structure analysis. Proc Natl Acad Sci U S A. 2010;107:1077–81. doi: 10.1073/pnas.0913045107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang W, Wang K, Liu YL, No JH, Li J, Nilges MJ, Oldfield E. Bioorganometallic mechanism of action, and inhibition, of IspH. Proc Natl Acad Sci U S A. 2010;107:4522–7. doi: 10.1073/pnas.0911087107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee HI, Igarashi RY, Laryukhin M, Doan PE, Dos Santos PC, Dean DR, Seefeldt LC, Hoffman BM. An organometallic intermediate during alkyne reduction by nitrogenase. J Am Chem Soc. 2004;126:9563–9. doi: 10.1021/ja048714n. [DOI] [PubMed] [Google Scholar]
- 19.Lee HI, Sorlie M, Christiansen J, Yang TC, Shao J, Dean DR, Hales BJ, Hoffman BM. Electron inventory, kinetic assignment (En), structure, and bonding of nitrogenase turnover intermediates with C2H2 and CO. J Am Chem Soc. 2005;127:15880–90. doi: 10.1021/ja054078x. [DOI] [PubMed] [Google Scholar]
- 20.Pelmenschikov V, Case DA, Noodleman L. Ligand-bound S = 1/2 FeMo-cofactor of nitrogenase: Hyperfine interaction analysis and implication for the central ligand χ identity. Inorg Chem. 2008;47:6162–72. doi: 10.1021/ic7022743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McMillan RS, Renaud J, Reynolds JG, Holm RH. Biologically related iron-sulfur clusters as reaction centers. Reduction of acetylene to ethylene in systems based on [Fe4S4(SR)4]3. J Inorg Biochem. 1979;11:213–27. doi: 10.1016/s0162-0134(00)80019-7. [DOI] [PubMed] [Google Scholar]
- 22.Itoh T, Nagano T, Hirobe M. The Fe4S4(SR 2−)4] catalytic reduction of diphenylacetylene to cis-stilbene in the presence of NaBH4. Tetrahedron Lett. 1980;21:1343–1346. [Google Scholar]
- 23.Van Hoof S, Lacey CJ, Rohrich RC, Wiesner J, Jomaa H, Van Calenbergh S. Synthesis of analogues of (E)-1-hydroxy-2-methylbut-2-enyl 4-diphosphate, an isoprenoid precursor and human gamma delta T cell activator. J Org Chem. 2008;73:1365–70. doi: 10.1021/jo701873t. [DOI] [PubMed] [Google Scholar]
- 24.Urbina JA, Moreno B, Vierkotter S, Oldfield E, Payares G, Sanoja C, Bailey BN, Yan W, Scott DA, Moreno SN, Docampo R. Trypanosoma cruzi contains major pyrophosphate stores, and its growth in vitro and in vivo is blocked by pyrophosphate analogs. J Biol Chem. 1999;274:33609–15. doi: 10.1074/jbc.274.47.33609. [DOI] [PubMed] [Google Scholar]
- 25.Martin MB, Grimley JS, Lewis JC, Heath HT, 3rd, Bailey BN, Kendrick H, Yardley V, Caldera A, Lira R, Urbina JA, Moreno SN, Docampo R, Croft SL, Oldfield E. Bisphosphonates inhibit the growth of Trypanosoma brucei, Trypanosoma cruzi, Leishmania donovani, Toxoplasma gondii, and Plasmodium falciparum: A potential route to chemotherapy. J Med Chem. 2001;44:909–16. doi: 10.1021/jm0002578. [DOI] [PubMed] [Google Scholar]
- 26.Martin MB, Arnold W, Heath HT, 3rd, Urbina JA, Oldfield E. Nitrogen-containing bisphosphonates as carbocation transition state analogs for isoprenoid biosynthesis. Biochem Biophys Res Commun. 1999;263:754–8. doi: 10.1006/bbrc.1999.1404. [DOI] [PubMed] [Google Scholar]
- 27.Luckman SP, Hughes DE, Coxon FP, Graham R, Russell G, Rogers MJ. Nitrogen-containing bisphosphonates inhibit the mevalonate pathway and prevent post-translational prenylation of GTP-binding proteins, including Ras. J Bone Miner Res. 1998;13:581–9. doi: 10.1359/jbmr.1998.13.4.581. [DOI] [PubMed] [Google Scholar]
- 28.Cromartie TH, Fisher KJ, Grossman JN. The discovery of a novel site of action for herbicidal bisphosphonates. Pesticide Biochem Physiol. 1999;63:114–126. [Google Scholar]
- 29.Sunberg RJ, Ebetino FH, Mosher CT, Roof CF. Designing drugs for stronger bones. Chemtech. 1991;21:304–309. [Google Scholar]
- 30.Grove JE, Brown RJ, Watts DJ. The intracellular target for the antiresorptive aminobisphosphonate drugs in Dictyostelium discoideum is the enzyme farnesyl diphosphate synthase. J Bone Miner Res. 2000;15:971–81. doi: 10.1359/jbmr.2000.15.5.971. [DOI] [PubMed] [Google Scholar]
- 31.Bergstrom JD, Bostedor RG, Masarachia PJ, Reszka AA, Rodan G. Alendronate is a specific, nanomolar inhibitor of farnesyl diphosphate synthase. Arch Biochem Biophys. 2000;373:231–41. doi: 10.1006/abbi.1999.1502. [DOI] [PubMed] [Google Scholar]
- 32.van Beek E, Pieterman E, Cohen L, Lowik C, Papapoulos S. Farnesyl pyrophosphate synthase is the molecular target of nitrogen-containing bisphosphonates. Biochem Biophys Res Commun. 1999;264:108–11. doi: 10.1006/bbrc.1999.1499. [DOI] [PubMed] [Google Scholar]
- 33.Hosfield DJ, Zhang Y, Dougan DR, Broun A, Tari LW, Swanson RV, Finn J. Structural basis for bisphosphonate-mediated inhibition of isoprenoid biosynthesis. J Biol Chem. 2004;279:8526–9. doi: 10.1074/jbc.C300511200. [DOI] [PubMed] [Google Scholar]
- 34.Rodriguez N, Bailey BN, Martin MB, Oldfield E, Urbina JA, Docampo R. Radical cure of experimental cutaneous leishmaniasis by the bisphosphonate pamidronate. J Infect Dis. 2002;186:138–140. doi: 10.1086/341074. [DOI] [PubMed] [Google Scholar]
- 35.Paterson R. Pamidronate next on list as potential cure for leishmaniasis. The Lancet Infectious Diseases. 2002;2:515–515. [Google Scholar]
- 36.Derenne S, Amiot M, Barille S, Collette M, Robillard N, Berthaud P, Harousseau JL, Bataille R. Zoledronate is a potent inhibitor of myeloma cell growth and secretion of IL-6 and MMP-1 by the tumoral environment. J Bone Miner Res. 1999;14:2048–56. doi: 10.1359/jbmr.1999.14.12.2048. [DOI] [PubMed] [Google Scholar]
- 37.Kunzmann V, Bauer E, Wilhelm M. Gamma/delta T-cell stimulation by pamidronate. N Engl J Med. 1999;340:737–8. doi: 10.1056/NEJM199903043400914. [DOI] [PubMed] [Google Scholar]
- 38.Kunzmann V, Bauer E, Feurle J, Weissinger F, Tony HP, Wilhelm M. Stimulation of γδ T cells by aminobisphosphonates and induction of antiplasma cell activity in multiple myeloma. Blood. 2000;96:384–92. [PubMed] [Google Scholar]
- 39.Wilhelm M, Kunzmann V, Eckstein S, Reimer P, Weissinger F, Ruediger T, Tony HP. γδ T cells for immune therapy of patients with lymphoid malignancies. Blood. 2003;102:200–6. doi: 10.1182/blood-2002-12-3665. [DOI] [PubMed] [Google Scholar]
- 40.Dieli F, Vermijlen D, Fulfaro F, Caccamo N, Meraviglia S, Cicero G, Roberts A, Buccheri S, D’Asaro M, Gebbia N, Salerno A, Eberl M, Hayday AC. Targeting human γδ T cells with Zoledronate and interleukin-2 for immunotherapy of hormone-refractory prostate cancer. Cancer Res. 2007;67:7450–7. doi: 10.1158/0008-5472.CAN-07-0199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gnant M, Mlineritsch B, Schippinger W, Luschin-Ebengreuth G, Postlberger S, Menzel C, Jakesz R, Seifert M, Hubalek M, Bjelic-Radisic V, Tausch C, Eidtmann H, Steger G, Kwasny W, Dubsky P, Fridrik M, Fitzal F, Stierer M, Rucklinger E, Greil R, Marth C. Endocrine therapy plus zoledronic acid in premenopausal breast cancer. N Engl J Med. 2009;360:679–91. doi: 10.1056/NEJMoa0806285. [DOI] [PubMed] [Google Scholar]
- 42.Zhang Y, Cao R, Yin F, Hudock MP, Guo RT, Krysiak K, Mukherjee S, Gao YG, Robinson H, Song Y, No JH, Bergan K, Leon A, Cass L, Goddard A, Chang TK, Lin FY, Van Beek E, Papapoulos S, Wang AH, Kubo T, Ochi M, Mukkamala D, Oldfield E. Lipophilic bisphosphonates as dual farnesyl/geranylgeranyl diphosphate synthase inhibitors: An x-ray and NMR investigation. J Am Chem Soc. 2009;131:5153–62. doi: 10.1021/ja808285e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang Y, Cao R, Yin F, Lin FY, Wang H, Krysiak K, No JH, Mukkamala D, Houlihan K, Li J, Morita CT, Oldfield E. Lipophilic pyridinium bisphosphonates: Potent gammadelta T cell stimulators. Angew Chem Int Ed Engl. 2010;49:1136–8. doi: 10.1002/anie.200905933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Monkkonen H, Auriola S, Lehenkari P, Kellinsalmi M, Hassinen IE, Vepsalainen J, Monkkonen J. A new endogenous atp analog (ApppI) inhibits the mitochondrial adenine nucleotide translocase (ANT) and is responsible for the apoptosis induced by nitrogen-containing bisphosphonates. Br J Pharmacol. 2006;147:437–45. doi: 10.1038/sj.bjp.0706628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tanaka Y, Morita CT, Nieves E, Brenner MB, Bloom BR. Natural and synthetic non-peptide antigens recognized by human gamma delta t cells. Nature. 1995;375:155–8. doi: 10.1038/375155a0. [DOI] [PubMed] [Google Scholar]
- 46.Chen CK-M, Hudock MP, Zhang Y, Guo RT, Cao R, No JH, Liang PH, Ko TP, Chang TH, Chang SC, Song Y, Axelson J, Kumar A, Wang AH, Oldfield E. Inhibition of geranylgeranyl diphosphate synthase by bisphosphonates: A crystallographic and computational investigation. J Med Chem. 2008;51:5594–607. doi: 10.1021/jm800325y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Potera C. Pigments of pathogens might provide golden antimicrobial opportunities. ASM News. 2005;71:450–451. [Google Scholar]
- 48.Liu GY, Essex A, Buchanan JT, Datta V, Hoffman HM, Bastian JF, Fierer J, Nizet V. Staphylococcus aureus golden pigment impairs neutrophil killing and promotes virulence through its antioxidant activity. J Exp Med. 2005;202:209–15. doi: 10.1084/jem.20050846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Liu CI, Liu GY, Song Y, Yin F, Hensler ME, Jeng WY, Nizet V, Wang AH, Oldfield E. A cholesterol biosynthesis inhibitor blocks Staphylococcus aureus virulence. Science. 2008;319:1391–4. doi: 10.1126/science.1153018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Song Y, Liu CI, Lin FY, No JH, Hensler M, Liu YL, Jeng WY, Low J, Liu GY, Nizet V, Wang AH, Oldfield E. Inhibition of staphyloxanthin virulence factor biosynthesis in Staphylococcus aureus: In vitro, in vivo, and crystallographic results. J Med Chem. 2009;52:3869–80. doi: 10.1021/jm9001764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Song Y, Lin FY, Yin F, Hensler M, Rodrigues Poveda CA, Mukkamala D, Cao R, Wang H, Morita CT, Pacanowska D. Gonzalez, Nizet V, Oldfield E. Phosphonosulfonates are potent, selective inhibitors of dehydrosqualene synthase and staphyloxanthin biosynthesis in Staphylococcus aureus. J Med Chem. 2009;52:976–88. doi: 10.1021/jm801023u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sharma A, Slugg PH, Hammett JL, Jusko WJ. Clinical pharmacokinetics and pharmacodynamics of a new squalene synthase inhibitor, BMS-188494, in healthy volunteers. J Clin Pharmacol. 1998;38:1116–21. [PubMed] [Google Scholar]
- 53.Ellis C. Doing the two step. Nature Reviews Drug Discovery. 2003;2:605. [Google Scholar]
- 54.Courchesne WE. Characterization of a novel, broad-based fungicidal activity for the antiarrhythmic drug amiodarone. J Pharmacol Exp Ther. 2002;300:195–9. doi: 10.1124/jpet.300.1.195. [DOI] [PubMed] [Google Scholar]
- 55.Gupta SS, Ton VK, Beaudry V, Rulli S, Cunningham K, Rao R. Antifungal activity of amiodarone is mediated by disruption of calcium homeostasis. J Biol Chem. 2003;278:28831–9. doi: 10.1074/jbc.M303300200. [DOI] [PubMed] [Google Scholar]
- 56.Benaim G, Sanders JM, Garcia-Marchan Y, Colina C, Lira R, Caldera AR, Payares G, Sanoja C, Burgos JM, Leon-Rossell A, Concepcion JL, Schijman AG, Levin M, Oldfield E, Urbina JA. Amiodarone has intrinsic anti-Trypanosoma cruzi activity and acts synergistically with posaconazole. J Med Chem. 2006;49:892–9. doi: 10.1021/jm050691f. [DOI] [PubMed] [Google Scholar]
- 57.Serrano-Martin X, Garcia-Marchan Y, Fernandez A, Rodriguez N, Rojas H, Visbal G, Benaim G. Amiodarone destabilizes intracellular Ca2+ homeostasis and biosynthesis of sterols in Leishmania mexicana. Antimicrob Agents Chemother. 2009;53:1403–10. doi: 10.1128/AAC.01215-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Paniz-Mondolfi AE, Perez-Alvarez AM, Reyes-Jaimes O, Socorro G, Zerpa O, Slova D, Concepcion JL. Concurrent Chagas’ disease and borderline disseminated cutaneous leishmaniasis: The role of amiodarone as an antitrypanosomatidae drug. Ther Clin Risk Manag. 2008;4:659–63. doi: 10.2147/tcrm.s2801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Paniz-Mondolfi AE, Perez-Alvarez AM, Lanza G, Marquez E, Concepcion JL. Amiodarone and itraconazole: A rational therapeutic approach for the treatment of chronic Chagas’ disease. Chemotherapy. 2009;55:228–33. doi: 10.1159/000219436. [DOI] [PubMed] [Google Scholar]
- 60.Potera C. Heart drug helps to beat Chagas, Leishmania parasites. Microbe Magazine. 2009 November; [Google Scholar]
- 61.Prystowsky EN. Atrial fibrillation: Dronedarone and amiodarone-the safety versus efficacy debate. Nat Rev Cardiol. 2010;7:5–6. doi: 10.1038/nrcardio.2009.221. [DOI] [PubMed] [Google Scholar]