


Abstract
Bacteria expand their genetic diversity, spread antibiotic resistance genes, and obtain virulence factors through the highly coordinated process of conjugative plasmid transfer (CPT). A plasmid-encoded relaxase enzyme initiates and terminates CPT by nicking and religating the transferred plasmid in a sequence-specific manner. We solved the 2.3 Å crystal structure of the relaxase responsible for the spread of the resistance plasmid pCU1 and determined its DNA binding and nicking capabilities. The overall fold of the pCU1 relaxase is similar to that of the F plasmid and plasmid R388 relaxases. However, in the pCU1 structure, the conserved tyrosine residues (Y18,19,26,27) that are required for DNA nicking and religation were displaced up to 14 Å out of the relaxase active site, revealing a high degree of mobility in this region of the enzyme. In spite of this flexibility, the tyrosines still cleaved the nic site of the plasmid’s origin of transfer, and did so in a sequence-specific, metal-dependent manner. Unexpectedly, the pCU1 relaxase lacked the sequence-specific DNA binding previously reported for the homologous F and R388 relaxase enzymes, despite its high sequence and structural similarity with both proteins. In summary, our work outlines novel structural and functional aspects of the relaxase-mediated conjugative transfer of plasmid pCU1.
INTRODUCTION
Antibiotics have effectively treated a broad range of bacterial infections for years. However, with the emergence of antibiotic resistance, the treatment of these same bacterial infections is becoming increasingly difficult (1) (http://www.cdc.gov/hicpac/mdro/mdro_toc.html). Currently, 70% of all hospital acquired infections are resistant to at least one antibiotic, and antibiotic resistance to at least one drug has been reported for every major strain of pathogenic bacteria (2). As a result, the Centers for Disease Control has declared antibiotic resistance a primary threat to human health, and multiple efforts have been made to understand and inhibit the mechanisms responsible for its spread (2–8).
Conjugative plasmid transfer (CPT) is a primary method by which bacteria acquire antibiotic resistance genes. During CPT, one bacterium (the donor) transfers genetic material, in the form of a conjugative DNA plasmid, to a neighboring cell (the recipient) via physical cell–cell contact. Each conjugative plasmid encodes the majority of proteins found within the relaxosome, a multi-protein complex necessary to promote plasmid transfer in a sequence-specific manner. The relaxosome works in concert with a type IV secretion system (T4SS), and these two complexes are linked by a type IV coupling protein (T4CP), believed to form the pore through which the T (transferred) strand of the plasmid travels into the recipient bacterium (9–13).
One component of the relaxosome, the relaxase, is responsible for the initiation and termination of T-strand transfer (14,15). A generalized mechanism describing the role of the relaxase was first determined through the study of the F (fertility) plasmid system (14–27), and further details were contributed by the study of other conjugative systems, including plasmids R388 (28–37), RP4 (38–40), R1162 (41–46) and R100 (47,48). To initiate transfer, the relaxase binds the origin of transfer (oriT) of the plasmid’s T-strand and cleaves the strand at the nic site. After the T-strand has been unwound from the parent strand, the relaxase terminates transfer by religating the nicked strand (14).
The structures of the F TraI relaxase, the R388 TrwC relaxase and the R1162 MobA relaxase have been reported previously (3,23,24,33,34,44). In addition, the F TraI and R388 TrwC relaxase structures have been resolved in complex with their respective DNA substrates, revealing that the relaxase binds the T-strand oriT as a partial DNA hairpin (33,34). The 3′ end of the oriT extends beyond the hairpin as a single strand and forms a U-turn around the protein N-terminus, before entering the active site of the enzyme (24,33,34). The active site is located on two strands of a central beta sheet and contains three histidines coordinating a metal cation, the identity of which has been debated (3,24,34,46). The scissile phosphate of the DNA substrate’s nic site is positioned above the chelated metal. As a result, the metal cation withdraws electron density from the phosphate center to promote nucleophilic attack on the scissile phosphate by one of the enzyme’s conserved tyrosine residues (3,24,34). All three relaxase enzymes bind and cleave their specific DNA substrate with high affinity and sequence-specificity (22,33,41).
We have examined the atomic structure and the DNA binding and nicking activities of the relaxase enzyme responsible for the transfer of the resistance plasmid pCU1, originally isolated from Salmonella typhimurium (49–52). pCU1 is a derivative of resistance plasmid R46 and confers on its host resistance to the antibiotics ampicillin, streptomycin and spectinomycin (52,53). The relaxase activity encoded by pCU1 is located within the N-terminal 299 residues of a multifunctional TraI enzyme, and productive, plasmid-specific transfer is dependent upon TraI (51,52). Our analysis of the pCU1 TraI relaxase has identified structural and functional characteristics unique to the pCU1 relaxase. These results advance our understanding of both the general mechanism of relaxase-mediated DNA binding and cleavage, as well as the unique nature of each CPT system.
MATERIALS AND METHODS
Relaxase constructs
The relaxase of plasmid pCU1 is located within the pCU1-encoded protein TraI (51,52). To determine the extent of the relaxase domain within pCU1 TraI, we aligned the pCU1 TraI amino acid sequence (GenBank: AAD27542) with those of R388 TrwC (GenBank: CAA44853) and F TraI (GenBank: BAA97974). Additional details describing how the alignments were performed can be found in the Supplementary Data. Residues 1 to ∼300 of pCU1 TraI corresponded to the relaxase domains of F TraI and R388 TrwC. Secondary-structure predictors (Jpred3, version 2.2, http://www.compbio.dundee.ac.uk/www-jpred/) identified loop regions near residue 300, and the pCU1 TraI relaxase construct extending from residue 1 to 299 was chosen as the final construct. Primers were designed to isolate residues 1–299 of pCU1 TraI using the Vector NTI Express 10.0.1 suite of programs (Invitrogen, 2005) and were then commercially synthesized [Integrated DNA Technologies (IDT), 2009]. All cloning and mutagenesis was verified by sequencing at the UNC-CH Genome Analysis Facility. Residues 1–299 of pCU1 TraI were cloned into the vector pTYB2 (IMPACT system, New England Biolabs), between NdeI and SmaI restriction sites in order to C-terminally fuse the relaxase to an intein and a chitin binding domain (CBD) affinity tag and thus generate the construct WT_299. Several additional relaxase constructs containing tyrosine to phenylalanine mutations were generated by Quick Change site directed mutagenesis (Stratagene) of WT_299. Using ligation independent cloning (LIC), residues 1–299 of the relaxase domain were inserted into vector pMCSG9 (54) to create the mutant construct Nterm_299. In vector pMCSG9, the relaxase was N-terminally fused to a maltose binding protein (MBP) affinity tag and a 6-His (MBP-HIS) affinity tag with a TEV protease cleavage site located between the protein sequence and affinity tags (54). During purification, these affinity tags were removed by the tobacco etch virus (TEV) protease, leaving three non-native residues (Ser–Asn–Ala) at the N-terminus of Nterm_299.
Protein expression and purification
All proteins were over-expressed in BL21 Escherichia coli, and the cells were harvested by centrifugation and resuspended in buffer C (WT_299) (500 mM NaCl, 20 mM Tris–HCl pH 7.5, 10% glycerol, 0–5 mM EDTA, 0.01% azide) or buffer A (Nterm_299) [500 mM NaCl, 20 mM Na2H(PO4) pH 8.0, 10% glycerol, 5 mM imidazole, 0–1 mM EDTA, 0.01% azide]. Resuspended cells were stored at −80°C. Additional details can be found in the Supplementary Data.
Prior to protein purification, cells were thawed in the presence of lysozyme and a cocktail of protease inhibitors, lysed on ice using a Sonic Dismembrator, Model 500 (Fisher Scientific), and centrifuged to isolate the soluble fraction. WT_299 was purified on Chitin Resin (NEB) using a batch bind method followed by an extended wash step, as described by the manufacturer. Cleavage of the intein and CBD tags, and subsequent release of WT_299 from the chitin resin, was induced by incubating the Chitin Resin with 50 mM d,l -dithiothreitol for 16 h. WT_299 was then eluted from the Chitin Resin. Buffer C was used throughout the purification. Nterm_299 was purified over a 5 ml HisTrap HP column (GE Healthcare) on an Aktaxpress FPLC (GE Healthcare). Fusion protein was eluted with a high imidazole buffer B [500 mM NaCl, 20 mM Na2H(PO4) pH 8.0, 10% glycerol, 500 mM imidazole, 0–1 mM EDTA, 0.01% azide] after washing the column to baseline with buffer A. The affinity tags were removed by cleavage with 4% (w/w) TEV protease during dialysis overnight into buffer A at 4°C. The protein was then concentrated and further purified over two sequential 5 ml HisTrap HP gravity columns. The cleaved protein that flowed through the final column was collected. All proteins were visualized by SDS–PAGE and found to be >95% pure (Supplementary Figure S1).
For crystallization, WT_299 and Nterm_299 were dialyzed into 250 mM ammonium acetate while concentrating to 5–6 mg/ml (Nterm_299) or ∼14 mg/ml (WT_299), then used immediately for crystallization. For analysis by circular dichroism (CD) spectroscopy, WT_299 and Nterm_299 were dialyzed into buffer CD (100 mM NaF, 20 mM Tris–HCl pH 7.5, 5% glycerol, 0.01% azide) and then used immediately. For DNA binding, nicking and cross-over assays with WT_299, an additional size exclusion chromatography purification step was performed. During this step, WT_299 was loaded onto a HiLoad 16/60 Superdex 200 column (GE Healthcare) pre-equilibrated with buffer S (100 mM NaCl, 20 mM Tris–HCl pH 7.5, 5% glycerol, 0–5 mM EDTA, 0.01% azide) on an Atkaxpress FPLC (GE Healthcare). The protein was eluted from the column in buffer S. When necessary, any EDTA present was removed by dialysis of WT_299 and Nterm_299 into buffer D (100 mM NaCl, 20 mM Tris–HCl pH 7.5, 5% glycerol, 0.01% azide). For inductively coupled plasma mass spectrometry (ICP-MS) analysis, a separate preparation of WT_299 and Nterm_299 was performed in order to prevent contaminating the protein sample with metals from outside sources. All water was obtained from the laboratory Barnstead E-pure water filtration system, at >17 megaohm-cm (ddH2O). Following purification by size-exclusion chromatography, the sample was then dialyzed into buffer ICP (20 mM NaCl, 10 mM Tris–HCl pH 7.5, 0.01% azide).
Protein crystallization
Crystals of WT_299 and Nterm_299 were grown at 20°C by the hanging-drop vapor diffusion method. Equal volumes of purified protein and well solution (250 mM triNa citrate, 22% PEG 3350) were mixed and crystals grew over a course of 4–5 weeks. Crystals were initially small and hexagonal, but following micro-seeding and the addition of 5 mM MgCl2 to the purified protein sample, larger tetragonal crystals were obtained. The crystals were cryoprotected in 250 mM triNa citrate, 26% PEG 3350 and 10% ethylene glycol and flash frozen in liquid nitrogen for data collection at 100 K.
Data collection, structure determination and refinement
Crystals were initially screened at the UNC Biomolecular X-ray Crystallography Facility with a Rigaku R-Axis IV++ and CCD, while full data sets were collected at either the Southeast Regional Collaborative Access Team (SER-CAT) Sector 22-ID beam-line at the Advanced Photon Source (APS) at Argonne National Laboratory or the Stanford Synchrotron Radiation Laboratory (SSRL) using remote access of beam-line 9-2 ID. Data from the best diffracting WT_299 crystals were indexed and scaled with X-ray Detector Software (XDS, MPI for Medical Research, Heidelberg, 2009, http://xds.mpimf-heidelberg.mpg.de/). Data from the best diffracting Nterm_299 crystals were indexed and scaled with HKL2000 (HKL Research Inc., 2005, http://www.hkl-xray.com). Initial phases were determined by molecular replacement in Phaser (http://www-structmed.cimr.cam.ac.uk/phaser), a component of the Collaborative Computing Project No. 4 (CCP4) (http://www.ccp4.ac.uk, http://ccp4wiki.org) using the relaxase domain of TrwC as the search model (PDB code 1omh). Data were refined using refmac (http://www.ysbl.york.ac.uk/∼garib/refmac/), a component of CCP4, CNS (version 1.2, http://cns.csb.yale.edu/v1.2), and Phenix (version 1.6, http://www.phenix-online.org), and model building was performed in Coot (http://biop.ox.ac.uk/coot). Ligands were found in the standard monomer library in CCP4, on the HIC-Up server (release 12.1, http://xray.bmc.uu.se/hicup/), or were constructed using the Dundee PRODRG2 server (http://davapc1.bioch.dundee.ac.uk/prodrg/). Data were verified using MolProbity (http://molprobity.biochem.duke.edu). Figures were constructed in PyMOL (DeLano Scientific LLC, San Carlos, CA, USA, http://www.pymol.org, 2009). AREAIMOL, a component of CCP4, was used to calculate the solvent exposed surface area of each monomer within the asymmetric unit of the crystal. The character of the dimer interface was analyzed using the Protein Interfaces, Surfaces and Assemblies service (PISA) (version 1.18, http://www.ebi.ac.uk/msd-srv/prot_int/pistart.html). Structural alignments were performed with TM-align (http://zhanglab.ccmb.med.umich.edu/TM-align), using the ProCKSI-Server (version procksi-8.7, www.procksi.net), to generate the root mean square deviation (RMSD) of Cα positions of the proteins analyzed. Within Coot, the OptAlign command, using the Kabsch algorithm (http://pymolwiki.org/index.php/Kabsch), was used to generate the RMSD of atoms within the histidine triad of the proteins analyzed. The coordinates of WT_299 and Nterm_299 have been deposited in the PDB (accession codes 3L57, 3L6T).
Inductively coupled plasma mass spectrometry (ICP-MS)
WT_299 and Nterm_299 were analyzed by ICP-MS to determine the identity and concentration of metals present in the samples. Data were collected by the UNC-CH Chemistry Department Mass Spectrometry Core Facility on a Varian 820-MS (Palo Alto, CA). Protein was first digested in 70% HNO3. Each sample was then diluted in 10 ml of 1.4% HNO3. A control consisting of each buffer used during protein purification was analyzed to determine the concentration of metal in the buffers used. Each experimental sample, as well as the buffer control, was monitored for Mg24, Mn55, Ca44, Fe57, Ni60 and Zn66. Data were collected as part per billion (ppb) and converted to µM. The original concentration of each metal in each sample was then determined in Excel 2007 (Microsoft, 2006). Data were then presented as fold increase in metal concentration over background levels, where background values represented the signal due to 1.4% HNO3 alone.
DNA binding assays
The affinity of WT_299 and Nterm_299 for a panel of DNA substrates was determined by fluorescence anisotropy-based DNA binding assays. 5′ fluorescein-labeled DNA substrates were commercially synthesized and HPLC purified (IDT). Table 1 provides the nucleic acid sequence of each substrate investigated. Each substrate was resuspended in buffer R (50 mM NaCl, 10 mM Tris–HCl pH 7.5, 0.05 mM EDTA), heated to 95°C for 10 min, and then allowed to slow cool to room temperature. Each stock was then diluted to 0.1 µM in ddH2O prior to use. Additional details on the assay protocol and data analysis can be found in the Supplementary Data.
Table 1.
DNA binding assay substrates
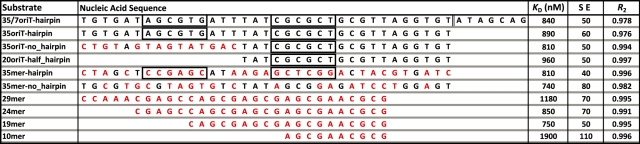 |
The nucleic acid sequence, secondary structure and pCU1 TraI relaxase binding affinity, as determined by fluorescence anisotropy-based binding assays, for each substrate are provided. The names of substrates encoding a wild-type nic site and minimal oriT include the phrase ‘oriT’. The names of mutant substrates include the phrase ‘mer’. Mutated bases are in red font. Inverted repeats predicted to form a hairpin are boxed in black. The predicted nic site for each substrate (if present) is indicated by vertical double black lines. The DNA binding affinity of WT_299 for each substrate is presented as the dissociation constant (KD), as calculated by Equation 1, and the error represents the standard error of the KD when calculated by Equation 1. Additional details can be found in the Materials and Methods section and the Supplementary Data.
DNA nicking and DNA cross-over assays
The DNA nicking and religation activity of WT_299 and Nterm_299 for a panel of DNA substrates was determined by DNA nicking and DNA cross-over assays. 5′ fluorescein-labeled and unlabeled DNA substrates were commercially synthesized (IDT); 5′ fluorescein-labeled DNA substrates were HPLC purified. Table 2 provides the nucleic acid sequence of each substrate investigated. Each substrate was resuspended in buffer R, heated to 95°C for 10 min, and then either allowed to cool passively to room temperature or snap cooled on ice. Both preparations yielded similar results. Each stock was then diluted to 10 µM in ddH2O prior to use. Additional details on the assay protocol can be found in the Supplementary Data.
Table 2.
DNA nicking and DNA cross-over assay substrates
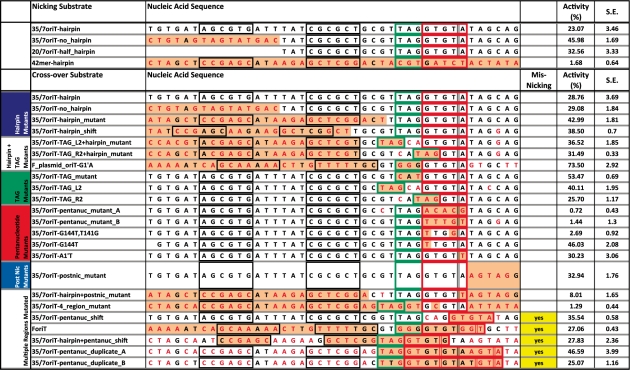 |
The nucleic acid sequence, secondary structure and nicking or religation activity of WT_299, as measured by DNA nicking and cross-over assays, for each substrate are provided. Cross-over assay substrates are listed according to the region of the pCU1 oriT which is mutated (see Figure 6 for a definition of these regions). Mutated bases are in red font. Light orange shading emphasizes the key areas of each mutant substrate relative to the wild-type substrate. Inverted repeats that are predicted to form a hairpin are boxed in black. The TAG region of each substrate is boxed in green. The pentanucleotide region of each substrate is boxed in red. The predicted nic site for each substrate is indicated by vertical double black lines, and non-canonical nic sites, as observed in DNA cross-over assays, are indicated by vertical double red lines. To determine the DNA nicking or religation activity of the wild-type pCU1 TraI relaxase, 5 μM enzyme was incubated with 1 μM labeled substrate and, for cross-over assays, 1 μM unlabeled substrate for 1 h at 37°C; products were then separated on denaturing polyacrylamide gels. ‘Misnicking’ activity is reported for each substrate that generated a product during cross-over assays that was larger or smaller than anticipated, indicating that the initial nicking reaction most likely occurred at a non-canonical nic site. Enzyme activity is represented as percent product generated. Each data point is the average of at least three experiments, and the error is the standard error of these replicates. Additional details can be found in the Materials and Methods section and the Supplementary Data.
To test the metal dependence of the pCU1 relaxase, WT_299 (10 μM) was treated with 10 mM EDTA for 24 h. The protein was then diluted either 2- or 10-fold upon the addition of 1 µM 35/7oriT-hairpin, and 0–15 mM metal was added to each reaction to generate an excess of 0–10 mM metal. Stocks (10×) of all metals investigated were made by dissolving the corresponding metal salt [MgCl2, MnCl2, CaCl2, NiCl2, Cu(SO4), Zn(acetate)2 and ZnCl2] in 1 M Tris–HCl pH 7.5.
DNA nicking activity and DNA religation activity were reported as percent product generated (the intensity of the product band divided by the sum of product and substrates band intensities).
Each data point represents the average of three or more reactions and error bars represent the standard error of these data points. All data processing was performed in Excel 2007 and plots were generated in SigmaPlot 8.02a (Systat, 2004) or Graphpad PRISM v5.03 (Graphpad, 2010).
RESULTS
Crystal structure of the pCU1 TraI relaxase domain
The 2.3 Å structure of WT_299 was determined by X-ray crystallography. Crystals were grown by hanging-drop vapor diffusion, and the resulting structure was determined to 2.3 Å by molecular replacement using the R388 TrwC relaxase [PDB code 1omh (33)] as the search model (Table 3). The protein crystallized as a dimer in the asymmetric unit (asu) in the space group P21. The two monomers were determined to have a combined 20 600 Å2 of solvent accessible surface area of which 1670 Å2 was buried upon dimerization. While this analysis could suggest a physiologically relevant dimer (55), only 18 potential hydrogens bonds and four potential salt bridges could be formed at the dimer interface, and the structure’s complexation significance score (CSS) of 0.000 indicated that the dimer was most likely to be a result of crystal packing (56). Indeed, the protein was determined to be a monomer by size exclusion chromatography (Supplementary Figure S2). The electron density of monomer A was consistently higher in quality than that of monomer B. As a result, out of the protein’s 299 residues, 223 were confidently modeled into the electron density of monomer A, while only 203 were placed into the electron density of the B monomer (Table 3).
Table 3.
Crystallographic statistics
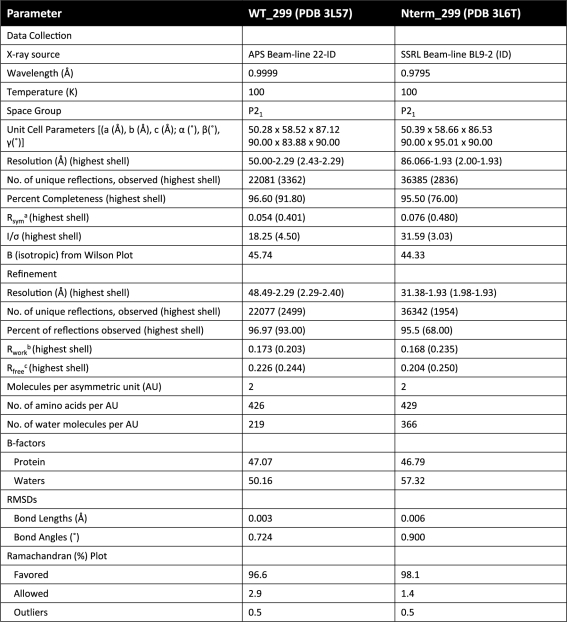 |
aRsym = ∑|I – Imean|/∑ I, where I is the observed intensity and Imean is the average intensity of several symmetry-relate observations.
bRwork = ∑|Fo – Fc|/∑ Fo, where Fo and Fc are the observed and calculated structure factors, respectively.
cRfree = Calculated as above for 5% of data not used in any step of refinement.
The overall fold of the pCU1 TraI relaxase domain (Figure 1A) resembles that of a human hand, as seen in previously solved relaxase structures (3,23,24,33,34,44). The palm consists of five central anti-parallel β-strands, and the surrounding α-helices and β-sheets form the fingers, back of the hand and bottom edge of the palm. The N-terminus is located at the beginning of the first β-strand (β1), and the remaining strands that compose the palm are in the order β1–β3–β7–β6–β2, when looking at the ‘palm side’ of the protein. The C-terminal 44 (monomer A) and 46 (monomer B) residues of WT_299 are disordered, but are predicted to be α-helical (Figure 2). Many of these residues have been placed in structures of homologous proteins, but the entire region has only been observed when the relaxase is in complex with its DNA substrate (24,34).
Figure 1.

The 2.3 Å resolution crystal structure of the pCU1 TraI relaxase. (A) The overall structure of the pCU1 relaxase (WT_299) with secondary-structures colored as represented in Figure 2 (β-strands are represented by yellow arrows, α-helices by red helices and loop regions by gray loops). The histidine triad of the active site and the tyrosines implicated in DNA nicking are shown as sticks. The manganese ion bound by the histidine triad is shown as a grey sphere. (B) The active site of the pCU1 relaxase (WT_299) with secondary structures represented as in (A). At the center is the histidine triad and bound manganese ion. In the upper right is loop A and the four tyrosines implicated in DNA nicking, and to the left is the N-terminus, represented as a black sphere. (C) The active site of the pCU1 relaxase [WT_299, colored as described in (A)] aligned with the active sites of the F plasmid TraI relaxase (in blue) and the relaxase of plasmid R388 TrwC (in magenta). The histidine triad and the primary DNA nicking tyrosine of each protein are shown as sticks. The distances between the tyrosines are indicated. Note that the in the structure of the plasmid R388 TrwC, Tyr18 was mutated to Phe18.
Figure 2.

Amino acid sequence alignment and secondary structure of the pCU1 TraI relaxase. The initial 337 amino acids of pCU1 TraI (GenBank ID AAD27542) were aligned with R388 TrwC (GenBank ID CAA44853) and F TraI (GenBank ID BAA97974) using the Clustal X program in BioEdit. Identical residues are shaded green, similar residues are shaded blue. Orange and yellow boxes indicate the first and last residues of the relaxase domains of each protein. Two constructs of the F TraI relaxase domain have been crystallized (3,23), therefore the terminal residues of both are boxed. Red boxes indicate the location of the conserved tyrosine residues and the active site histidine triad. Red lines underline the residues forming the ‘thumb’ regions of each protein. Since this region is disordered in the pCU1 TraI relaxase crystal, the extent of this region is estimated. Above the three sequences is the secondary structure of the pCU1 relaxase. β-strands are represented by yellow arrows; α-helices by red cylinders; and loop regions by black wavy lines. The secondary structure of residues 1–225 reflects that observed in the crystal structure of WT_299; the secondary structure of residues 226–337 reflects that predicted by Jpred 3.
The active site of WT_299 is found within the palm of the hand, and consists of three histidines, two on β7 (His160, 162) and one on β6 (His149) (Figure 1B). Together, they chelate a divalent metal cation, occupying three of the bound metal’s coordination sites. The remaining coordination sites surrounding the divalent cation can be satisfied by a variety of compounds, such as water molecules or components from the cryoprotectant or well solution. In the structure of WT_299, these remaining sites are occupied by the small molecule citrate (monomer A), or a water molecule and ethylene glycol (monomer B). Initially, several divalent metals were sequentially modeled into the active site of WT_299 to fulfill the electron density chelated by the histidines. Mn2+, Fe2+, Ni2+, Cu2+ and Zn2+ all satisfied the electron density as assessed by difference density and R-factors. Mn2+ was later determined to be the most likely metal in the active site based on a series of experiments described below.
The four tyrosines of WT_299 implicated in DNA nicking (Tyr18,19,26,27) are located on loop A, adjacent to the active site (Figure 1B). In monomer B, only the first few residues of the loop were sufficiently ordered to be modeled into the structure. In monomer A, however, the entire loop was observed, and it was located in a unique orientation relative to previous relaxase structures. Tyr18 and Tyr19 were found displaced from the active site, flipped out and pointing away from the bound metal ion (Figure 1C). In the F TraI relaxase structure, the equivalent tyrosine residues are oriented towards the active site, with Tyr16 (equivalent to pCU1 relaxase Tyr18) positioned to attack the scissile phosphate of the DNA substrate (3,23,24). The equivalent tyrosines (Tyr18,19) in the R388 TrwC relaxase are also directed towards the active site, with Tyr18 shifted slightly behind the scissile phosphate, relative to Tyr16 of the F TraI (33,34). When the structure of WT_299 monomer A was overlaid with the R388 TrwC relaxase [PDB ID 2cdm (34)] and F TraI relaxase [PDB ID 1p4d (23)] (Figure 1C), Tyr18 of WT_299 was displaced 14.2 Å away from the histidine triad when compared to Tyr16 of F TraI and 12.4 Å when compared to Tyr18 of R388 TrwC.
DNA binding by the pCU1 TraI relaxase
The relaxase binds the oriT of its DNA plasmid’s T-strand prior to cleaving the T-strand’s nic site (22,25,33). We determined the binding affinity and specificity of the pCU1 TraI relaxase for a panel of DNA substrates (Table 1) using fluorescence anisotropy-based DNA binding assays. The effect of DNA substrate length, secondary structure and sequence on relaxase binding affinity was investigated. For all assays, a hyperbolic binding curve was generated by measuring the change in fluorescence anisotropy of the labeled DNA substrate as a function of enzyme concentration. By fitting these curves with Equation 1 (see Supplementary Data) using non-linear regression, we calculated dissociation constants (KD) for each experiment and discovered that both WT_299 and Nterm_299 bound DNA substrates in an oriT- and hairpin-independent manner. The binding affinity of both WT_299 and Nterm_299 was similar for all substrates investigated, and varied between 700 nM and 1.2 μM (Table 1, Nterm_299 data not shown).
This low affinity, sequence-independent DNA binding was unexpected, given the highly sequence-specific and high affinity DNA binding reported for similar relaxase enzymes (22,25,33,41). WT_299 was confirmed to be folded and stable in solution by circular dichromism spectroscopy (Supplementary Figures S3 and S4), and the design of the WT_299 construct incorporated the full extent of the relaxase domain of pCU1 TraI (Figure 2). Therefore, the design of the DNA binding assay was modified to determine if the experimental conditions for WT_299 DNA binding could be further optimized. When incubating the Y18,19,26,27F mutant of the pCU1 relaxase with a DNA substrate that extended beyond the nic site (35/7oriT-hairpin), no improvement in DNA binding affinity or selectively was observed, and when supplementing the reaction conditions with 4 mM MnCl2 there was only a 2- to 3-fold increase in DNA binding affinity (Figure 3A and B). To determine if the best equation had been used to fit the experimental data, data were also fit by two additional equations using the Graphpad PRISM software (two-sites specific binding and one-site specific binding with Hill Slope). The data were ambiguously fit when assuming two binding sites. When fitting the data with the Hill parameter, the calculated KD did not improve, while the goodness-of-fit parameters (dependency and R2) showed slight or no improvement. These results did not justify the use of an additional parameter. Therefore, the data indicate that WT_299 binds DNA in a weak, sequence-independent manner, and may require an additional protein partner to initiate sequence-specific DNA binding.
Figure 3.

DNA binding by the pCU1 TraI relaxase. DNA binding curves were generated by monitoring the change in fluorescence anisotropy (FA) of a fluorescein-labeled DNA substrate as a function of protein concentration and then plotting the normalized FA as a function of total protein concentration. Each data point is the average of at least three replicates, with error bars representing the standard error of these replicates. The DNA binding affinity (KD, in nM) was calculated by fitting the data with Equation 1 using non-linear regression. The error represents the standard error of the KD when calculated by Equation 1. (A) Binding of the substrates 35oriT-hairpin and 35/7oriT-hairpin by WT_299 and the pCU1 relaxase mutant Y18,19,26,27F_299, respectively. The substrate 35/7oriT-hairpin extends 7 nt beyond the oriT nic site, while 35oriT-hairpin is truncated at the nic site. (B) Binding of the substrate 35/7oriT-hairpin by Y18,19,26,27F_299 in the absence or presence of 4 mM MnCl2.
Tyrosine-dependent DNA nicking by the pCU1 TraI relaxase
In the crystal structure of WT_299, the tyrosine residues predicted to nick DNA (Y18,19,26,27) were displaced up to 14 Å from the active site (Figure 1C). To determine if this significant displacement impacted the activity of the tyrosines when nicking DNA, we assessed the enzyme’s DNA nicking activity by incubating WT_299 with 5′ fluorescently labeled synthetic oligonucleotides and then monitored the amount of nicked product generated (Figure 4A). WT_299 efficiently nicked DNA containing the pCU1 oriT sequence and nic site. We then cloned, expressed and purified mutants of WT_299 in which one (Y18F and Y26F), two (Y18,26F) or four (Y18,19,26,27F) of the tyrosine residues were mutated to the catalytically inactive phenylalanine residue. The nicking activity of each mutant enzyme was then compared to that of WT_299. Single and double tyrosine mutants suffered an ∼50% decrease in activity, while the mutation of all four tyrosines effectively eliminated all nicking activity (Figure 4A). Thus, the catalytic tyrosines of the pCU1 relaxase were shown to be structurally mobile and functionally redundant.
Figure 4.

DNA nicking and religation by the pCU1 TraI relaxase domain. (A) To determine the DNA nicking activity of the wild-type pCU1 TraI relaxase, as well as several relaxase mutants, 5 μM enzyme was incubated with 1 μM substrate (35/7oriT- or 20/7oriT-hairpin) for 1 h at 37°C, and products were then separated on denaturing polyacrylamide gels. Nicking and religation activity is represented as percent product generated. Each experiment was performed in triplicate; the error is the standard error of these replicates. For each experiment, a representative gel is provided. Below each gel, a reaction scheme is provided, where a fluorescein label is indicated by a green star and the substrate nic site by a red triangle. Additional experimental details can be found in the Materials and Methods section and the Supplementary Data. (B) A reaction scheme illustrating a DNA cross-over experiment, where a fluorescein label is indicated by a green star and the substrate nic site by a red triangle.
Metal-dependent DNA nicking by the pCU1 TraI relaxase
Both the F TraI and R388 TrwC relaxase enzymes exhibit metal-dependent DNA nicking, though the optimal metal for each is under debate (3,24,26,34,46). To determine the metals most likely to be used by WT_299 during DNA nicking, the nicking activity of the enzyme was measured in the presence of EDTA and several metals (Mg2+, Mn2+, Ca2+, Ni2+, Cu2+ and Zn2+) (Figure 5). In the presence of EDTA, the DNA nicking activity of WT_299 was inhibited, but this activity could be rescued by then supplementing the reaction with an increasing concentration of a variety of metals. Mg2+ provided the most significant enhancement in nicking activity, as compared to the EDTA-treated sample, but Mg2+, Mn2+, Ca2+ and Ni2+ all supported DNA nicking in a concentration dependent manner. As was also observed by Boer et al. (34), the relaxase maintained residual DNA nicking activity in the presence of EDTA. Despite controlling the pH of Cu(SO4), ZnCl2 and Zn(acetate)2 solutions, relaxase activity was inhibited as the concentration of these metals increased, and relaxase activity was limited even at low concentrations of both Zn-containing compounds. We were unable to determine relaxase nicking activity in the presence of iron due to the metal’s insolubility within the pH range of the assay. Using inductively coupled plasma mass spectrometry (ICP-MS), we then determined the concentrations of Mg24, Mn55, Ca44, Fe57, Ni60 and Zn66 in samples of WT_299 (Table 4). When ranked according to fold increase over background levels, Zn66 was by far the most prevalent metal in the WT_299 sample, followed by Ni60 and Mn55. Taken together, these data indicate that while the relaxase can function in the presence of many metals, its nicking activity is best supported by Mg2+, Mn2+, Ca2+ or Ni2+, despite the overabundance of Zn66 bound by the protein sample.
Figure 5.

DNA nicking by the pCU1 TraI relaxase in the presence of metals. To determine the DNA nicking activity of the pCU1 TraI relaxase (WT_299) after treatment with EDTA and in the presence of the indicated concentration of metal, 5 μM enzyme was incubated with 1 μM substrate (35/7oriT-hairpin) for 1 h at 37°C, and products were then separated on denaturing polyacrylamide gels. Nicking and religation activity is represented as percent product generated. Each experiment was performed in triplicate; the error is the standard error of these replicates. Additional details can be found in the Materials and Methods section and the Supplementary Data.
Table 4.
Summary of metal data
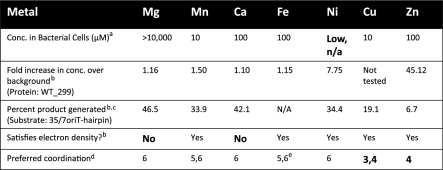 |
aSource: Finney et al. (59).
bSource: This article.
cDNA nicking data is the average of three replicates and the activity reported is the percent product generated at the optimal concentration for each metal; Cu2+ becomes inhibitory as its concentration increases.
dSource: Harding et al. (61).
eThis analysis data includes both ferric and ferrous groups.
Sequence-dependent DNA nicking by the pCU1 TraI relaxase
DNA binding assays demonstrated the sequence-independent DNA binding of WT_299. To determine if WT_299 also nicked and religated DNA in a sequence-independent manner, the DNA nicking and religation activities of WT_299 were tested using a series of DNA nicking and cross-over assays. First, to examine the effect of DNA secondary structure on pCU1 TraI relaxase DNA nicking activity, 5′ fluorescently labeled substrates containing the pCU1 oriT sequence, nic site and either a full hairpin-forming inverted repeat (35/7oriT-hairpin), a mutated inverted repeat unable to form a hairpin (35/7-no_hairpin), or a truncated hairpin (20/7oriT-half_hairpin) were incubated with WT_299 (Table 2). Each substrate was cleaved, but the percent product generated increased upon truncation of the inverted repeat. A similar trend was observed when studying the nicking activity of both the F TraI and R388 TrwC relaxase enzymes (25,37).
Second, to examine the effect of nucleic acid sequence on pCU1 TraI relaxase-mediated DNA nicking, WT_299 was incubated with either an oriT-containing substrate or a control substrate that did not contain the pCU1 oriT sequence. WT_299 only cleaved the oriT-containing substrate (Table 2). In the same manner, during DNA cross-over assays, WT_299 could only successful religate oriT-containing substrates. In fact, both donor and recipient substrates had to contain the pCU1 oriT sequence and nic site. If either donor or recipient did not encode the oriT, or were truncated prior to the nic site, no cross-over products were observed (Supplementary Figure S5).
To determine the specific bases within the pCU1 oriT sequence required for relaxase-mediated DNA nicking, a series of 42 nt-long unlabeled DNA substrates were designed in which mutations were made within one or multiple regions of the wild-type pCU1 oriT sequence (Table 2, Figure 6). These regions were designated ‘hairpin’, ‘TAG’, ‘pentanucleotide’, or ‘post nic’, and corresponded to sections of the oriT previously shown by work with the F and R388 plasmids to influence DNA nicking, binding, or plasmid transfer (22,25,31,37). With these data in mind, the TAG and pentanucleotide regions were predicted to strongly influence DNA nicking, while the hairpin and post nic regions were expected to have only a limited effect. WT_299 was then incubated with each of these unlabeled mutant DNA substrates (the donor substrate) and a fluorescently labeled 20 nt-long recipient substrate (20-oriT_half-hairpin) that encoded the pCU1 oriT and terminated at the nic site. An experiment was thus performed to measure the ability of the relaxase to nick the donor substrate and generate a 27 nt-long cross-over product (Figure 4B). The percent cross-over product generated from each mutant substrate was compared to that detected during a DNA cross-over experiment between a wild-type donor substrate (35/7oriT-hairpin) and the same recipient substrate. Mutations within the hairpin, post nic or TAG region alone did not decrease overall relaxase activity. Mutation of the two flanking nucleotides within the pentanucleotide region also had no inhibitory effect on enzyme activity. However, the mutation of bases within the core pentanucleotide region eliminated enzyme activity, as did the mutation of a flanking nucleotide within the pentanucleotide region along with a mutation within the hairpin region or the post nic region (Table 2).
Figure 6.

Nucleic acid sequence and sub-divisions of the pCU1 oriT. The nucleic acid sequence of the plasmid pCU1 oriT is divided into four regions (hairpin in dark blue, TAG in green, pentanucleotide in red and post nic in light blue). The bases forming the inverted repeat are shaded black and the location of the predicted hairpin is indicated.
A number of the substrates were apparently nicked at erroneous nic sites by WT_299 prior to religation onto 20oriT-half_hairpin, thus creating products either longer or shorter than the expected 27mer (Table 2). In particular, these misnicked products were observed if the mutant substrate contained a duplicated or shifted pentanucleotide region. If the mutant substrate also contained a mutant TAG region, multiple misnicked products were observed. If the mutant substrate contained a TAG and pentanucleotide region that were shifted in concert, only one alternative product was generated. However, mutation or shifting of the TAG region alone did not consistently affect nicking activity. The likelihood of erroneous nicking was also not affected by the presence, absence, or location of the hairpin forming inverted repeat.
The pentanucleotide region of the pCU1 oriT is expected to wrap around the N-terminus of the pCU1 TraI relaxase, forming an intra-strand U-turn that has been previously described for both the F and R388 oriT (27,37). To illustrate the importance of the interaction between the pentanucleotide region and the pCU1 TraI N-terminus, we compared the structure and function of WT_299 to that of a mutant pCU1 TraI relaxase, Nterm_299, in which three residues (Ser–Asn–Ala) were appended to the protein N-terminus. We measured the DNA nicking activity of Nterm_299 and determined its structure to 1.9 Å by X-ray crystallography. The nicking activity of Nterm_299 was in fact inhibited relative to WT_299 (Figure 4A). The structures of WT_299, Nterm_299 and the DNA-bound relaxase domain of the R388 TrwC relaxase [PDB code 2cdm (34)] were overlaid (Supplementary Figure S6). In the TrwC relaxase structure, its DNA substrate, a 27mer oligonucleotide containing the R388 oriT, is bound by the relaxase as a partial hairpin. The overall folds of the three proteins are similar (maximum rmsd over equivalent Cα positions = 2.05 Å) (57,58), and the histidine triad-dominated active sites are practically identical (averaged rmsd of these residues between each of the three structures = 0.076 Å). However, the three non-native N-terminal residues of Nterm_299 clearly clash with the bound DNA substrate of TrwC. Therefore, Nterm_299 should be incapable of nicking oriT DNA without either physically moving its N-terminus out of the way, or by binding the DNA in a novel orientation. Both options destroy all sequence-specific protein–DNA interactions formed between the enzyme N-terminus and the oriT DNA. Thus, this structural clash clearly explains the functional inhibition observed during DNA nicking assays.
DISCUSSION
The role of tyrosine mobility during pCU1 relaxase-mediated DNA nicking
CPT with concomitant replication is proposed to proceed through two sequential DNA nicking steps that are accomplished by catalytic tyrosine residues contributed by relaxase enzymes (3,32,35). These reactions can be completed using either single tyrosines on two relaxases acting sequentially, or by one multi-tyrosine relaxase, through a ‘flip-flop’ mechanism of DNA nicking and religation described previously (3,14,32,35). While the ‘flip-flop’ mechanism requires significant rearrangement of tyrosine residues within the active site of the multi-tyrosine relaxase, it abrogates the need for multiple enzymes to accomplish the transfer of one conjugative plasmid.
Our structural and function data have emphasized the flexible nature of the tyrosines within multi-tyrosine systems. In particular, the crystal structure of WT_299 demonstrated the mobility of loop A and its accompanying four catalytic tyrosines. As compared to homologous structures, loop A of WT_299 monomer A is rotated away from the active site and its first two catalytic tyrosines are displaced from the metal-bound histidine triad (Figure 1C). In monomer B, loop A was too mobile to be modeled into the structure. Our DNA nicking data also highlighted the flexibility of the four tyrosines. Mutation of up to two tyrosine residues only eliminated ∼50% of nicking activity, indicating that tyrosines other than the predicted primary DNA nicking residue, Tyr18, were also able to enter the active site in a productive orientation (Figure 4A). These data support the conclusion that the active site residues of the pCU1 relaxase, and perhaps additional multi-tyrosine systems, are sufficiently mobile to allow for the rearrangement necessary to accommodate the ‘flip-flop’ mechanism of plasmid transfer.
Metal ions in the pCU1 relaxase active site
Productive CPT requires that the relaxase nick the T-strand DNA within the enzyme’s metal-chelating active site (3,32,35). However, the identity of this essential metal ion is debated (3,26,34,46). Therefore, we used enzyme activity assays to establish that Mg2+, Mn2+, Ca2+ and Ni2+ all supported pCU1 relaxase-mediated DNA nicking (Figure 5). In contrast, both Zn2+ and Cu2+ supported only limited DNA nicking by WT_299 at low metal concentrations and were actually inhibitory as the concentration of metal increased. We then measured concentration of each metal in a purified sample of WT_299 using ICP mass spectrometry (Table 4). Based on these experimental data, as well as the low concentration of Ni2+ within the bacterial cytoplasm (59,60) and the inability of Ca2+ and Mg2+ to satisfy the electron density within the active site of the crystal structure of WT_299, we concluded that the pCU1 relaxase most likely employed Mn2+ during DNA nicking and religation reactions.
Our results mirror those reported by Xia et al. who found that the DNA nicking activity of the R1162 MobA relaxase was supported by Mn2+ but inhibited by Zn2+. In addition, Mn2+ was shown to stabilize MobA and was bound by the active site of the crystallized MobA relaxase domain (44,46). Larkin et al. demonstrated by isothermal titration calorimetry (ITC) that the affinity of F TraI relaxase was ∼25 times stronger for Mn2+ than for Ca2+ and Mg2+ (24), and while the relaxase domain of R388 TrwC has been crystallized in complex with Zn2+, the DNA nicking activity of this enzyme was also inhibited at high Zn2+ concentrations (34).
A comparison of the chemical properties of these metals may provide insight into the inhibitory nature of Cu2+ and Zn2+. While Mg2+, Mn2+, Ca2+ and Ni2+ are usually surrounded by five to six ligands in an trigonal bipyramidal or octahedral arrangement, Zn2+ and Cu2+ prefer three or four coordinating ligands (61). Within the relaxase active site, an octahedral or trigonal bipyramidal arrangement of the three histidine side chains, the bound DNA ligand, and the attacking tyrosine hydroxyl about the metal ion could ideally position the DNA’s scissile phosphate for attack by the catalytic tyrosine. Lujan et al. (3) and Datta et al. (23) both observed an octahedral arrangement of ligands surrounding the bound Mg2+ in the active site of the F TraI relaxase, and Lujan and colleagues noted that this octahedral arrangement would allow for the ‘flip-flop’ mechanism proposed for the relaxase (3). In summary, while the pCU1 relaxase can utilize a variety of metal catalysts, Mn2+ appears to be the most likely candidate to occupy the active site during productive DNA nicking due to its availability and preferred coordination number.
Low affinity and sequence-independent DNA binding by the pCU1 relaxase
In contrast to the high affinity, sequence-specific DNA binding by the F TraI and R388 TrwC relaxase enzymes (22,25,33), the pCU1 TraI relaxase bound DNA in a non-specific manner and with low affinity (Table 1, Figure 3). We propose that the pCU1 enzyme relies upon a second DNA binding protein to bind selectively to the pCU1 oriT. In fact, a pCU1-encoded protein, TraK, has been identified, for which the efficiency of pCU1 plasmid transfer decreases 100-fold upon deletion (51). Homologs of TraK have been identified within the F, R388 and RP4 plasmid sequences; all are known to be DNA binding proteins that enhance relaxase function (13,21,40,62). In particular, studies of RP4 have shown that its TraK homolog, TraJ, enhances sequence-specific binding and nicking by the RP4 TraI enzyme (13,40). In addition, the F plasmid-encoded TraK homolog, TraY, has been shown necessary for relaxase binding and nicking in vivo (13). Therefore, pCU1 sequence-specific binding may be accomplished by a two-component system. pCU1 TraK could bind selectively to the pCU1 oriT, to then promote high affinity DNA binding by TraI.
Sequence-specific DNA cleavage by the pCU1 relaxase
In order to cleave the T-strand DNA at the scissile phosphate of the plasmid nic site, the relaxase must position the oriT DNA within its active site such that the nic site is located directly above the chelated metal ion (24). However, due to the pCU1 relaxase’s low affinity and non-specific DNA binding, it was unclear whether or not this enzyme would be able to nick DNA in the anticipated sequence-specific manner. In crystal structures of the DNA-bound F TraI and R388 TrwC relaxase enzymes, the DNA substrate is observed to form a U-turn around the enzyme N-terminus immediately prior to entering the active site. As a result, the DNA bases immediately upstream of the substrate’s nic site form several contacts with the relaxase N-terminus that have been shown to regulate relaxase-mediated DNA nicking activity (24,27,33,34). Therefore, we determined the DNA nicking activity of the pCU1 relaxase against a library of DNA substrates designed to test the sequence selectivity of the enzyme for the pCU1 oriT, with a particular emphasis on the regions of the pCU1 oriT corresponding to the bases of the F and R388 oriT known to form this U-turn structure (Table 2, Figure 6).
The results of the DNA nicking assays indicated that the bases of the pCU1 oriT, in particular those within the pentanucleotide region, cooperate with the N-terminus of WT_299 to position the nic site within the enzyme active site to promote sequence-specific DNA nicking (Table 2). Depending on the location of the mutation(s) within the pentanucleotide region, WT_299 failed to cleave the substrate’s nic site entirely, cleaved the site at a reduced level, or cleaved the substrate at non-canonical nic sites. In particular, by shifting the location of the pentanucleotide region relative to the nic site, the location of DNA nicking could be shifted correspondingly. DNA nicking was also inhibited by mutation of the enzyme N-terminus, as illustrated by the failure of mutant enzyme Nterm_299 to nick oriT-containing DNA substrates (Figure 4A). Of particular interest, the bases within the pentanucleotide region had previously been shown to be necessary for pCU1 plasmid transfer during cell-based plasmid transfer assays (52). This correlation emphasizes the pivotal role of the relaxase enzyme within the relaxosome during sequence-specific CPT.
ACCESSION NUMBERS
PDB accession code 3L57, 3L6T.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Institutes of Health [grant numbers AI078924 to M.R.R., ES016488 to R.P.N., GM008719 to UNC-CH]. Funding for open access charge: National Institutes of Health (grant number AI078924).
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Suzanne Paterson for the generous gift of the TraI gene. We thank Jeff Keenan for assistance with the initial cloning of the TraI gene. We thank Sarah Kennedy for collecting data sets leading to PDB accession code 3L57. We thank Eric Ortlund, Bret Wallace, Jillian Orans and Laurie Betts for guidance when analyzing the crystallographic data leading to PBD accession codes 3L57 and 3L6T. We thank Joseph V. Lomino for assistance when analyzing CD data. We thank the Matson laboratory for instruction when performing non-denaturing DNA gels. We thank Dan McNamara, Andrew Hemmert and members of the Redinbo lab for assistance with manuscript preparation. Use of the Advanced Photon Source (APS) was supported by the US Department of Energy, Office of Science, Office of Basic Energy Sciences, under contract No W-31-109-Eng-38. We thank SER-CAT for access to beam-line 22-ID at the APS. Portions of this research were carried out at the Stanford Synchrotron Radiation Laboratory (SSRL), a national user facility operated by Stanford University on behalf of the US Department of Energy, Office of Basic Energy Sciences. The SSRL Structural Molecular Biology Program is supported by the Department of Energy, Office of Biological and Environmental Research, and by the National Institutes of Health, National Center for Research Resources, Biomedical Technology Program, and the National Institute of General Medical Sciences. We thank SSRL for access to beam-line BL9-2.
REFERENCES
- 1.Siegel JD, Rhinehart E, Jackson M, Chiarello L. The Centers for Disease Control and Prevention. Healthcare Infection Control Practices Advisory Committee; 2006. Management of Multidrug-Resistant Organisms in Healthcare Settings. [DOI] [PubMed] [Google Scholar]
- 2.Clatworthy AE, Pierson E, Hung DT. Targeting virulence: a new paradigm for antimicrobial therapy. Nat. Chem. Biol. 2007;3:541–548. doi: 10.1038/nchembio.2007.24. [DOI] [PubMed] [Google Scholar]
- 3.Lujan SA, Guogas LM, Ragonese H, Matson SW, Redinbo MR. Disrupting antibiotic resistance propagation by inhibiting the conjugative DNA relaxase. Proc. Natl Acad. Sci. USA. 2007;104:12282–12287. doi: 10.1073/pnas.0702760104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fernandez-Lopez R, Machon C, Longshaw CM, Martin S, Molin S, Zechner EL, Espinosa M, Lanka E, de la Cruz F. Unsaturated fatty acids are inhibitors of bacterial conjugation. Microbiology. 2005;151:3517–3626. doi: 10.1099/mic.0.28216-0. [DOI] [PubMed] [Google Scholar]
- 5.Garcillan-Barcia MP, Jurado P, Gonzalez-Perez B, Moncalian G, Fernandez LA, de la Cruz F. Conjugative transfer can be inhibited by blocking relaxase activity within recipient cells with intrabodies. Mol. Microbiol. 2007;63:404–416. doi: 10.1111/j.1365-2958.2006.05523.x. [DOI] [PubMed] [Google Scholar]
- 6.Williams JJ, Hergenrother PJ. Exposing plasmids as the Achilles' heel of drug-resistant bacteria. Curr. Opin. Chem. Biol. 2008;12:389–399. doi: 10.1016/j.cbpa.2008.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Qiu X-Q, Wang H, Lu X-F, Zhang J, Li S-F, Cheng G, Wan L, Yang L, Zuo J-Y, Zhou Y-Q, et al. An engineered multidomain bactericidal peptide as a model for targeted antibiotics against specific bacteria. Nat. Biotechnol. 2003;21:1480–1485. doi: 10.1038/nbt913. [DOI] [PubMed] [Google Scholar]
- 8.Potts RG, Lujan SA, Redinbo MR. Winning the asymmetric war: new strategies for combating antibacterial resistance. Future Microbiol. 2008;3:119–123. doi: 10.2217/17460913.3.2.119. [DOI] [PubMed] [Google Scholar]
- 9.Willetts N, Wilkins B. Processing of plasmid DNA during bacterial conjugation. Microbiol. Rev. 1984;48:24–41. doi: 10.1128/mr.48.1.24-41.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lanka E, Wilkins BM. DNA processing reactions in bacterial conjugation. Annu. Rev. Biochem. 1995;64:141–169. doi: 10.1146/annurev.bi.64.070195.001041. [DOI] [PubMed] [Google Scholar]
- 11.de la Cruz F, Davies J. Horizontal gene transfer and the origin of species: lessons from bacteria. Trends Microbiol. 2000;8:128–133. doi: 10.1016/s0966-842x(00)01703-0. [DOI] [PubMed] [Google Scholar]
- 12.Llosa M, Gomis-Ruth FX, Coll M, de la Cruz F. Bacterial conjugation: a two-step mechanism for DNA transport. Mol. Microbiol. 2002;45:1–8. doi: 10.1046/j.1365-2958.2002.03014.x. [DOI] [PubMed] [Google Scholar]
- 13.de la Cruz F, Frost LS, Meyer RJ, Zechner EL. Conjugative DNA Metabolism in Gram-negative bacteria. FEMS Microbiol. Rev. 2009;34:18–30. doi: 10.1111/j.1574-6976.2009.00195.x. [DOI] [PubMed] [Google Scholar]
- 14.Byrd DRN, Matson SW. Nicking by transesterification: the reaction catalysed by a relaxase. Mol. Microbiol. 1997;25:1011–1022. doi: 10.1046/j.1365-2958.1997.5241885.x. [DOI] [PubMed] [Google Scholar]
- 15.Matson SW, Sampson JK, Byrd D.RN. F plasmid conjugative DNA transfer: the TraI helicase activity is essential for DNA strand transfer. J. Biol. Chem. 2001;276:2372–2379. doi: 10.1074/jbc.M008728200. [DOI] [PubMed] [Google Scholar]
- 16.Tatum EL, Lederberg J. Gene Recombination in the bacterium Escherichia coli. J. Bacteriol. 1947;53:673–684. doi: 10.1128/jb.53.6.673-684.1947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Matson SW, Nelson WC, Morton BS. Characterization of the reaction product of the oriT nicking reaction catalyzed by Escherichia coli DNA helicase I. J. Bacteriol. 1993;175:2599–2606. doi: 10.1128/jb.175.9.2599-2606.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sherman JA, Matson SW. Escherichia coli DNA helicase I catalyzes a sequence-specific cleavage/ligation reaction at the F plasmid origin of transfer. J. Biol. Chem. 1994;269:26220–26226. [PubMed] [Google Scholar]
- 19.Howard MT, Nelson WC, Matson SW. Stepwise assembly of a relaxosome at the F plasmid origin of transfer. J. Biol. Chem. 1995;270:28381–28386. [PubMed] [Google Scholar]
- 20.Matson SW, Ragonese H. The F-plasmid TraI protein contains three functional domains required for conjugative DNA strand transfer. J. Bacteriol. 2005;187:697–706. doi: 10.1128/JB.187.2.697-706.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nelson WC, Howard MT, Sherman JA, Matson SW. The traY gene product and integration host factor stimulate Escherichia coli DNA helicase I-catalyzed nicking at the F plasmid oriT. J. Biol. Chem. 1995;270:28374–28380. [PubMed] [Google Scholar]
- 22.Stern JC, Schildbach JF. DNA recognition by F factor TraI36: highly sequence-specific binding of single-stranded DNA. Biochemistry. 2001;40:11586–11595. doi: 10.1021/bi010877q. [DOI] [PubMed] [Google Scholar]
- 23.Datta S, Larkin C, Schildbach JF. Structural insights into single-stranded DNA binding and cleavage by F factor TraI. Structure. 2003;11:1369–1379. doi: 10.1016/j.str.2003.10.001. [DOI] [PubMed] [Google Scholar]
- 24.Larkin C, Datta S, Harley MJ, Anderson BJ, Ebie A, Hargreaves V, Schildbach JF. Inter- and intramolecular determinants of the specificity of single-stranded DNA binding and cleavage by the F factor relaxase. Structure. 2005;13:1533–1544. doi: 10.1016/j.str.2005.06.013. [DOI] [PubMed] [Google Scholar]
- 25.Williams SL, Schildbach JF. Examination of an inverted repeat within the F factor origin of transfer: context dependence of F TraI relaxase DNA specificity. Nucleic Acids Res. 2006;34:426–435. doi: 10.1093/nar/gkj444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Larkin C, Haft R.JF, Harley MJ, Traxler B, Schildbach JF. Roles of active site residues and the HUH motif of the F plasmid TraI relaxase. J. Biol. Chem. 2007;282:33707–33713. doi: 10.1074/jbc.M703210200. [DOI] [PubMed] [Google Scholar]
- 27.Hekman K, Guja K, Larkin C, Schildbach JF. An intrastrand three-DNA-base interaction is a key specificity determinant of F transfer initiation and of F TraI relaxase DNA recognition and cleavage. Nucleic Acids Res. 2008;36:4565–4572. doi: 10.1093/nar/gkn422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Llosa M, Bolland S, de la Cruz F. Structural and functional analysis of the origin of conjugal transfer of the broad-host-range IncW plasmid R388 and comparison with the related IncN plasmid R46. Mol. General Genet. 1991;226:473–483. doi: 10.1007/BF00260661. [DOI] [PubMed] [Google Scholar]
- 29.Grandoso G, Llosa M, Zabala JC, de la Cruz F. Purification and biochemical characterization of TrwC, the helicase involved in plasmid R388 conjugal DNA transfer. Eur. J. Biochem. 1994;226:403–412. doi: 10.1111/j.1432-1033.1994.tb20065.x. [DOI] [PubMed] [Google Scholar]
- 30.Llosa M, Grandoso G, de la Cruz F. Nicking activity of TrwC directed against the origin of transfer of the IncW plasmid R388. J. Mol. Biol. 1995;246:54–62. doi: 10.1006/jmbi.1994.0065. [DOI] [PubMed] [Google Scholar]
- 31.Llosa M, Grandoso G, Hernando MA, de la Cruz F. Functional domains in protein TrwC of plasmid R388: dissected DNA strand transferase and DNA helicase activities reconstitute protein function. J. Mol. Biol. 1996;264:56–67. doi: 10.1006/jmbi.1996.0623. [DOI] [PubMed] [Google Scholar]
- 32.Grandoso G, Avila P, Cayon A, Hernando MA, Llosa M, de la Cruz F. Two active-site tyrosyl residues of protein TrwC act sequentially at the origin of transfer during plasmid R388 conjugation. J. Mol. Biol. 2000;295:1163–1172. doi: 10.1006/jmbi.1999.3425. [DOI] [PubMed] [Google Scholar]
- 33.Guasch A, Lucas M, Moncalian G, Cabezas M, Perez-Luque R, Gomis-Ruth FX, de la Cruz F, Coll M. Recognition and processing of the origin of transfer DNA by conjugative relaxase TrwC. Nature Struct. Biol. 2003;10:1002–1010. doi: 10.1038/nsb1017. [DOI] [PubMed] [Google Scholar]
- 34.Boer R, Russi S, Guasch A, Lucas M, Blanco AG, Perez-Luque R, Coll M, de la Cruz F. Unveiling the molecular mechanism of a conjugative relaxase: the structure of TrwC complexed with a 27-mer DNA comprising the recognition hairpin and the cleavage site. J. Mol. Biol. 2006;358:857–869. doi: 10.1016/j.jmb.2006.02.018. [DOI] [PubMed] [Google Scholar]
- 35.Gonzalez-Perez B, Lucas M, Cooke LA, Vyle JS, de la Cruz F, Moncalian G. Analysis of DNA processing reactions in bacterial conjugation by using suicide oligonucleotides. EMBO J. 2007;26:3847–3857. doi: 10.1038/sj.emboj.7601806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gonzalez-Perez B, Carballeira JD, Moncalian G, de la Cruz F. Changing the recognition site of a conjugative relaxase by rational design. Biotechnol. J. 2008;4:554–557. doi: 10.1002/biot.200800184. [DOI] [PubMed] [Google Scholar]
- 37.Lucas M, Gonzalez-Perez B, Cabezas M, Moncalian G, Rivas G, de la Cruz F. Relaxase DNA binding and cleavage are two distinguishable steps in conjugative DNA processing that involve different sequence elements of the nic site. J. Biol. Chem. 2010 doi: 10.1074/jbc.M109.057539. doi: 10.1074/jbc.M109.057539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pansegrau W, Schroder W, Lanka E. Relaxase (TraI) of IncPa plasmid RP4 catalyzes a site-specific cleaving-joining reaction of single-stranded DNA. Proc. Natl Acad. Sci. USA. 1993;90:2925–2929. doi: 10.1073/pnas.90.7.2925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Balzer D, Pansegrau W, Lanka E. Essential motifs of relaxase (TraI) and TraG proteins involved in conjugative trasfer of plasmid RP4. J. Bacteriol. 1994;176:4285–4295. doi: 10.1128/jb.176.14.4285-4295.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pansegrau W, Lanka E. Mechanisms of initiation and termination reactions in conjugative DNA processing. J. Biol. Chem. 1996;271:13068–13076. doi: 10.1074/jbc.271.22.13068. [DOI] [PubMed] [Google Scholar]
- 41.Bhattacharjee M, Meyer RJ. Specific binding of MobA, a plasmid-encoded protein involved in the initiation and termination of conjugal DNA transfer, to single-stranded oriT DNA. Nucleic Acids Res. 1993;21:4563–4568. doi: 10.1093/nar/21.19.4563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Becker EC, Meyer RJ. Recognition of oriT for DNA processing at termination of a round of conjugal transfer. J. Mol. Biol. 2000;300:1067–1077. doi: 10.1006/jmbi.2000.3902. [DOI] [PubMed] [Google Scholar]
- 43.Becker EC, Meyer RJ. Relaxed specificity of the R1162 nickase: a model for evolution of a system for conjugative mobilization of plasmids. J. Bacteriol. 2003;185:3538–3546. doi: 10.1128/JB.185.12.3538-3546.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Monzingo AF, Ozburn A, Xia S, Meyer RJ, Robertus JD. The structure of the minimal relaxase domain of MobA at 2.1 Å resolution. J. Mol. Biol. 2007;366:165–178. doi: 10.1016/j.jmb.2006.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Meyer RJ. The R1162 Mob proteins can promote conjugative transfer from cryptic origins in the bacterial chromosome. J. Bacteriol. 2008;191:1574–1580. doi: 10.1128/JB.01471-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Xia S, Robertus JD. Effect of divalent ions on the minimal relaxase domain of MobA. Arch. Biochem. Biophys. 2009;488:42–47. doi: 10.1016/j.abb.2009.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Harley MJ, Schildbach JF. Swapping single-stranded DNA sequence specificities of relaxases from conjugative plasmids F and R100. Proc. Natl Acad. Sci. USA. 2003;100:11243–11248. doi: 10.1073/pnas.2035001100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fekete RA, Frost LS. Mobilization of chimeric oriT plasmids by F and R100-1: role of relaxosome formation in defining plasmid specificity. J. Bacteriol. 2000;182:4022–4027. doi: 10.1128/jb.182.14.4022-4027.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Khatoon H, Iyer RV, Iyer VN. A new filamentous bacteriophage with sex-factor specificity. Virology. 1972;48:145–155. doi: 10.1016/0042-6822(72)90122-5. [DOI] [PubMed] [Google Scholar]
- 50.Konarska-Kozlowska M, Iyer VN. Physical and genetic organization of the IncN-group plasmid pCU1. Gene. 1981;14:195–204. doi: 10.1016/0378-1119(81)90115-3. [DOI] [PubMed] [Google Scholar]
- 51.Paterson ES, More MI, Pillay G, Cellini C, Woodgate R, Walker GC, Iyer VN, Winans SC. Genetic analysis of the mobilization and leading regions of the IncN plasmids pKM101 and pCU1. J. Bacteriol. 1999;181:2572–2583. doi: 10.1128/jb.181.8.2572-2583.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Paterson ES, Iyer VN. Localization of the nic site of IncN conjugative plasmid pCU1 through formation of a hybrid oriT. J. Bacteriol. 1997;179:5768–5776. doi: 10.1128/jb.179.18.5768-5776.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Paterson ES, Iyer VN. The oriT region of the conjugative transfer system of plasmid pCU1 and specificity between it and the mob region of other N tra plasmids. J. Bacteriol. 1992;174:499–507. doi: 10.1128/jb.174.2.499-507.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Donnelly MI, Zhou M, Millard CS, Clancy S, Stols L, Eschenfeldt WH, Collart FR, Joachimiak A. An expression vector tailored for large-scale, high throughput purification of recombinant proteins. Protein Expression Purif. 2006;47:446–454. doi: 10.1016/j.pep.2005.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lo Conte L, Chothia C, Janin J. The atomic structure of protein-protein recognition sites. J. Mol. Biol. 1999;285:2177–2198. doi: 10.1006/jmbi.1998.2439. [DOI] [PubMed] [Google Scholar]
- 56.Krissinel E, Henrick K. Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 2007;372:774–797. doi: 10.1016/j.jmb.2007.05.022. [DOI] [PubMed] [Google Scholar]
- 57.Barthel D, Hirst J, Blazewicz J, Burke E, Krasnogor N. ProCKSI: a decision support system for protein (structure) comparison, knowledge, similarity and information. BMC Bioinformatics. 2007;8:416. doi: 10.1186/1471-2105-8-416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhang Y, Skolnick J. TM-align: a protein structure alignment algorithm based on TM-score. Nucleic Acids Res. 2005;33:2302–2309. doi: 10.1093/nar/gki524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Finney LA, O'Halloran TV. Transition metal speciation in the cell: insights from the chemistry of metal ion receptors. Science. 2003;300:931–936. doi: 10.1126/science.1085049. [DOI] [PubMed] [Google Scholar]
- 60.Outten C, O'Halloran T. Femtomolar sensitivity of metalloregulatory proteins controlling zinc homeostasis. Science. 2001;292:2488–2492. doi: 10.1126/science.1060331. [DOI] [PubMed] [Google Scholar]
- 61.Harding MM. Geometry of metal-ligand interactions in proteins. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2001;57:401–411. doi: 10.1107/s0907444900019168. [DOI] [PubMed] [Google Scholar]
- 62.Moncalian G, de la Cruz F. DNA binding properties of protein TrwA, a possible structural variant of the Arc repressor superfamily. Biochim. Biophys. Acta. 2004;1701:15–23. doi: 10.1016/j.bbapap.2004.05.009. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.