


Abstract
The intrinsic flexibility of DNA and the difficulty of identifying its interaction surface have long been challenges that prevented the development of efficient protein–DNA docking methods. We have demonstrated the ability our flexible data-driven docking method HADDOCK to deal with these before, by using custom-built DNA structural models. Here we put our method to the test on a set of 47 complexes from the protein–DNA docking benchmark. We show that HADDOCK is able to predict many of the specific DNA conformational changes required to assemble the interface(s). Our DNA analysis and modelling procedure captures the bend and twist motions occurring upon complex formation and uses these to generate custom-built DNA structural models, more closely resembling the bound form, for use in a second docking round. We achieve throughout the benchmark an overall success rate of 94% of one-star solutions or higher (interface root mean square deviation ≤4 Å and fraction of native contacts >10%) according to CAPRI criteria. Our improved protocol successfully predicts even the challenging protein–DNA complexes in the benchmark. Finally, our method is the first to readily dock multiple molecules (N > 2) simultaneously, pushing the limits of what is currently achievable in the field of protein–DNA docking.
INTRODUCTION
The computational docking field is proceeding ever faster to become an integral part of the research workflow in life sciences. Most of the developments in docking methodology were pioneered in the fields of small molecule docking and protein–protein docking (1–3). Docking has become a valuable tool in drug design, molecular interaction studies, NMR and X-ray structural studies, biochemical experiment design and validation (4–6). While docking is flourishing in these fields, less progress has been made in the development of successful protein–DNA docking algorithms. This is in part due to two system-dependent problems: (i) identifying the location of the interaction interface(s) on the DNA and (ii) modelling DNA conformational changes while maintaining a correct representation of the DNA double-helix during a simulation. The field of protein–DNA docking is, however, receiving renewed interest as the vital role of protein–DNA interactions in regulating gene expression and guarding genome integrity has become apparent (7). As a consequence, new protein–DNA docking methods are put forward and proven protein–protein docking concepts are extended to deal with these systems (8–17).
We have in the past adapted our data driven docking method HADDOCK, to deal with protein–DNA systems (18) and showed that it is able to deal with the two main challenges mentioned above. The ability of HADDOCK to use experimental data to drive the docking greatly facilitates the identification and positioning of the interaction interfaces during the docking (19,20). The incorporation of flexibility, both explicitly during the docking and implicitly by the use of custom-built DNA structural models, has proven to facilitate the conformational changes in the protein and DNA needed to establish the complex. The protocol was initially tested by docking the unbound structures of three monomeric transcription factors to their respective operator half-sites [phage 434 Cro (21), phage λ Arc (22) and Escherichia coli Lac (23)]. The resulting near native docking solutions reproduced many of the contacts observed in the experimental structures as well as specific conformational changes in the DNA. Our initial protein–DNA docking protocol has been successfully used in a number of practical applications by various laboratories worldwide (24–28). Driven by this success we have worked on improving the method’s performance and user friendliness by facilitating the generation of custom DNA structural models (29) as well as establishing a protein–DNA docking benchmark as a test bed for future developments (30). Next to that, HADDOCK has been made available to the community as a web server (http://www.haddocking.org; http://haddock.chem.uu.nl).
Here we bring all these elements together and challenge our method using the 47 test cases from the protein–DNA benchmark to define the limits of our current approach. We focus on the same two questions addressed in the previous work (18): how successful is the method in dealing with conformational changes upon complex formation and how well is it able to identify the correct interaction interfaces? Compared to the three test cases used previously, the 47 test cases in the benchmark pose some considerable challenges. The initial test cases were all major groove interacting transcription factors in their monomeric form, targeting one operator half-side that effectively spans one helical turn of DNA. The DNA-interacting domain of these transcription factors changes only conformation with respect to the side-chains of the DNA-interacting residues. The global conformational changes in the DNA were expressed as a uniform bend and change in groove width. In contrast, among the 47 test cases of the benchmark, not only transcription factors but also enzymes and structural proteins are present. These interact using a variation of structural domains, often involving multiple proteins, targeted to one or multiple sites on the DNA. Furthermore, the DNA length is often more than one helical turn. As a consequence, conformational changes can no longer be expressed in a smooth and uniform way but rather as an accumulation of local DNA bending and twisting events. To cope with these challenges we have improved our method for the generation of custom DNA structural models by extending its ability to capture the main bend and twist motions occurring in the DNA upon complex formation, and by subsequently using this information for the generation of custom DNA models.
The new results, again, show that the use of explicit flexibility in combination with implicit flexibility by means of an ensemble of custom-built DNA structural models, greatly improves the protein–DNA docking efficiency with respect to rigid-body docking. This is especially clear for the intermediate and difficult categories of the benchmark where DNA conformational changes readily occur. The use of experimental information for the docking of a representative subset of the benchmark, demonstrates the ability of our method to identify the correct interfaces and assemble the complex under ‘real life’ docking conditions. Furthermore, our method is the first to dock multiple molecules simultaneously, a valuable feature in a benchmark containing 40% of multi-component complexes. Top ranking docking solutions throughout the benchmark readily score one and two stars according to the CAPRI quality criteria (31) and three-star predictions are getting within reach for ‘easy’ test cases.
To our knowledge this is the first time a protein–DNA docking study of such a magnitude has been performed. Our results stress the importance of conformational adaptation in the docking of protein–DNA complexes and show the potential of HADDOCK to deal with them. We hope that they will stimulate the docking community to put their methods to the test on the same benchmark and foster further developments.
MATERIALS AND METHODS
Protein–DNA docking benchmark
The performance of HADDOCK was evaluated using the coordinate files for the bound and unbound proteins of 47 protein–DNA complexes available in the protein–DNA benchmark version 1.2 [http://haddock.chem.uu.nl/dna/benchmark.html (30)]. Canonical B-DNA 3D structural models were built using the 3D-DART web server [http://haddock.chem.uu.nl/dna (29)]. Their conformation was of BII type with the sugar pucker in the C2′-endo conformation [sugar pseudo-rotation phase angle (P) = 155°, DNA backbone torsion angles: α=309°, β=159°, γ=37°, δ=146°, ε=218°, ζ=191° and χ=260°].
Restraints used in the docking
Ambiguous interaction restraints, based on the true interface
Ideal ambiguous interaction restraints (AIR) restraint sets were generated based on the true interface(s) of the reference complexes as follows: (i) retrieval of all intermolecular atom–atom contacts below a cutoff of 5.0 Å; (ii) transformation of the atom–atom contacts to their respective residue–residue counterparts distinguishing between three categories: amino-acid to nucleotide base contacts, amino-acid to nucleotide sugar–phosphate backbone contacts or amino-acid to full nucleotide contacts. Contacts that originated from amino-acid residues having a relative main- or side-chain solvent accessibility of <30% as measured by NACCESS (32) where discarded.
All residues used in creating the interaction restraint file were defined as ‘active’. In effect we used the same procedure to generate AIRs as in the case of experimental information with the difference that they are only defined between the residues that are known to be in close vicinity in the reference complex.
AIRs based on experimental information
To evaluate the performance of HADDOCK in docking protein–DNA complexes using experimental information, we selected six representative tested cases from the ‘easy’ (3cro, 1by4), ‘intermediate’ (1azp, 1jj4) and ‘difficult’ (1a74, 1zme) category of the benchmark. For these we collected biochemical and biophysical information from literature sources. Only residues that are solvent accessible in the unbound proteins, using the same criteria as described above, were considered. For those DNA bases shown to be involved in specific interactions with the protein, only atoms able to interact by hydrogen-bond or non-bonded interactions were defined. This selection was further subdivided into atoms facing either the major or minor groove in case information about the protein-binding mode was available (Table 1). In case of non-specific interactions with the DNA, only the atoms of the sugar–phosphate backbone that are able to interact via hydrogen bonds or non-bonded interactions were defined (Table 1). Solvent accessible residues located in the predicted interaction interface, for which no experimental information was available, were defined as ‘passive’. Residues for which experimental information was available were defined as ‘active’. An overview of the data used is listed in Table 2.
Table 1.
Nucleotide atom subsets used in the definition of AIRs
| DNA base | Minor groove atoms | Major groove atoms |
|---|---|---|
| Thy | H3, O2, C2′ | H3, O4, C4, C5, C6, C7′ |
| Ade | N1, N3, C2, C4′ | H61, H62, N1, N7, C5, C6, C8′ |
| Gua | H1, H21, H22, N3, C2, C4′ | H1, H21, N7, O6, C5, C6, C8′ |
| Cyt | N3, O2, C2′ | H41, H42, N3, C4, C5, C6′ |
| Non-specific backbone atoms | ||
| Sugar–phosphate backbone | C1′, C2′, O3′, O5′, P, O1P, O2P | |
Subsets are defined for atoms capable of interacting using non-bonded or hydrogen bonded interactions. Individual subsets are defined for those atoms facing the DNA major and minor groove for the four bases and for the sugar–phosphate backbone atoms.
Table 2.
Definition of the AIRs based on experimental data for the six selected test-cases
| Protein | DNA | References | |
|---|---|---|---|
| ‘Easy’ | |||
| 1by4 (37) | Act: (K31,R32)a,b ⇒ T5,C6,G25,A26 (E24,K27)a,b ⇒ G3/4,C27/28 (K72,K73,R80)b ⇒ A2,G3/4 | Act: (T5,C6,A26,C27,C28,T29)a (G3,G4)a,c,d, (A2,T24)a,c T23c,G25a,d | (38–46) |
| Pas: V34,A75,V76,Q77, R55,N56,Q59,R62 | |||
| 3cro (21) | Act: (K29,Q31,S32,K42-P44)a L35b⇒C14,T15/T23,33 | Act: (C6,A7,T16-T18,C24,A25,T34- T36)a,(T32,T33)a,b,c | |
| Pas: K9,T18-T20,G27,V28,Q30,Q34, I36,E37,V40,T41,R45,F46 | (T4,A5,T13,C14,T15,T22,A23, G31)a,c | (47–50) | |
| ‘Intermediate’ | |||
| 1azp (51) | Act: W24e ⇒ G3,G15 V26b,M29b,S31e,V45e ⇒ C2-A4, T13-G15 (K22,T33,R42)e ⇒ T5-G7,C10-A12 | Act: C2,G3f,A4,T5,C6,G7, C10,G11,A12,T13,C14,G15f | (52–55) |
| Pas: K21,R25,G27,K28,K39,T40,A44, S46,E47 | |||
| 1jj4 (56) | Act: (N13,K16,C17,R19-R21)a | Act: (A3,C4,T30)a,(C5,G28,G29)a,d (T25-C27)c | (57,58) |
| Pas: S34,T35,H37 ⇒ T26-C27 | |||
| ‘Difficult’ | |||
| 1a74 (59) | Act: (H97,N122)a,b ⇒ A35,G36 (A54-N56,T59,R60,R65,R73, G75)a ⇒ T1-C7 | Act: (T1-C7)a,b,d,(A35,G36)b, G40d | (60–65) |
| Pas: V51,G57,P58,T66,V71,H77, H100,K119 | |||
| 1zme (66) | Act: (R9,R11,H12,R80,R82,H83)a Pas: A4,K14,K39-S43, A75,K85,K100-S114 | Act: (C2,G3,G4,C15,C17,G18, C20,G21,G22,C33,C34,G35)a (T26-C32,C9-T14) | (67–72) |
Active residues (Act) are grouped according to the available information. Continuous stretches of residues are separated by a dash. Arrows indicate active restraints for specific pairs of residues. Passive residues (Pas) are only defined for the protein. Since 1by4, 1jj4 and 1a74 are symmetrical dimers only the restraints for one subunit are shown. Base-specific restraints for 3cro, 1by4, 1jj4, 1a74 and 1zme are targeted to the atoms of the nucleotides facing the major groove and those of 1azp to those facing the minor groove (Table 1).
aConserved residues.
bMutagenesis data.
cEthylation interference data.
dMethylation interference data.
eNMR native state amide hydrogen exchange.
fRaman spectroscopy.
DNA restraints
In order to preserve the helical conformation during the flexible stages of the docking the DNA was restrained as described before (18). For the docking of the unbound protein(s) to a canonical B-DNA structural model, the dihedral angles of the sugar–phosphate backbone of the input structure (inp) were measured and used as restraints (restricted to α=αinp ± 10°, β=βinp ± 40°, γ=γinp ± 20°, δ=δinp ±50°, ε=εinp ± 10° and ζ=ζinp ± 50°). For the docking of the unbound protein(s) to the ensemble of custom-built DNA structural models, the same protocol for sugar–phosphate backbone restraints was used but the restraint error values were reduced to half of those in the canonical B-DNA case.
Docking protocol
The default protein–DNA docking protocol as described before (18) and implemented in HADDOCK version 2.0 (33) was used for all the docking runs. This protocol includes the random removal of 50% of the ambiguous interaction restraints for each docking trial. Several docking-specific modifications were made as follows.
Bound–bound docking
Only rigid body docking generating 2000 solutions. Protein and DNA structures were used in the bound conformation obtained from the reference complex.
Unbound–unbound docking using a canonical B-DNA structural model
A single component HADDOCK run was performed using the unbound proteins to yield a better sampling of side-chains and loop conformations. The residues of the interface either defined based on the reference complex or on experimental information were allowed to sample additional conformations during the semi-flexible refinement stage. Here, semi-flexible refinement signifies the combination of the semi-flexible simulated annealing stage in torsion angle space and the final water refinement stage in Cartesian space. Four protein models and the original unbound protein structure were used together with the canonical B-DNA model as an input ensemble for unbound–unbound docking. A total of 4000 docking solutions (every combination of models is sampled 800 times) were generated in the rigid body docking stage and the top 10% based on the HADDOCK score were used in the subsequent semi-flexible refinement stage. During the semi-flexible simulated annealing stage, the full DNA excluding the terminal base pairs was treated as semi-flexible. The amino-acid residues within 5.0 Å of any partner molecule were automatically defined as semi-flexible.
Unbound–unbound docking using five custom-built DNA structural models
The same protocol as for unbound–unbound docking starting from canonical B-DNA was used with as difference; five custom-built DNA structural models were used instead of canonical B-DNA; the conformational freedom of the DNA in the semi-flexible simulated annealing stage was limited by automatically defining both the amino-acid residues and nucleotides within 5.0 Å of any partner molecule as semi-flexible; the error range for the sugar–phosphate backbone dihedral angles as described above were reduced by half. Every combination of protein–DNA input models is sampled 160 times in the rigid body docking stage. The procedure for generating custom DNA structural models used as input for this docking run is described below.
Generation of custom DNA structural models
The generation of five custom DNA structural models is based on an analysis and a modelling step.
Analysis
The 10 best solutions from the top ranking cluster, both according to the HADDOCK score, were selected. The DNA structures in these solutions were analyzed using 3DNA (34,35) and the DNA bend analysis algorithm used in the 3D-DART server (29). This resulted in average parameter values for the six base pair (step) parameters (36) for every base pair (step) in the structure. These describe the conformation of the DNA. The average global bend vector with respect to a common reference frame between every successive base pair in the structures was calculated by 3D-DART. This information was used in the modelling stage.
Modelling
The modelling of custom DNA structures is based on the progressive introduction of global and local DNA conformational changes to a canonical B-DNA starting model.
A default set of base pair (step) parameters representing a canonical B-DNA conformation with the same sequence as the reference structure is generated by 3D-DART using the ‘fiber’ utility of the 3DNA software suite.
The Roll and Tilt values in the default set are updated by 3D-DART to reflect the average global bend vector for every base pair step in the sequence. The central base pair is used as origin of the global reference frame and default Twist values are used for correcting the vectors direction relative to the reference frame. The introduced bend vector between base pairs is scaled, enabling sampling of conformation change beyond the limits of the values defined by the average ± the standard deviation determined in the analysis stage. The scaling factor is set between 2.0 and 3.0 for those ensembles that show little deviation from a canonical helix and between 4.0 and 6.0 for the remaining test cases. For the docking of 1a74 using experimentally derived restraints the scaling factor was set to 10.0 to match the amount of DNA bend to the curved interaction surface of the protein (see ‘Results’ section).
-
All base pair step parameters are updated to reflect the average values as determined by the analysis stage resulting in a new weighted parameter
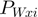 at base pair step i defined as follows:
at base pair step i defined as follows:
where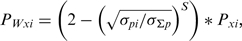
(1) 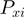 is the average value for the given parameter at base pair step i obtained from the analysis stage,
is the average value for the given parameter at base pair step i obtained from the analysis stage, 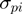 defines the standard deviation for the given parameter at base pair step i and
defines the standard deviation for the given parameter at base pair step i and 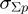 is the standard deviation for the given parameter for all base pair steps. S is a parameter-specific scaling factor that compensates for the over- or under-estimation of a given parameter as a result of the HADDOCK semi-flexible refinement stages. S was set to: twist: 0.8, roll: 0.8, tilt: 0.8, rise: 0.0, slide: 0.2 and shift: 0.8.The new value
is the standard deviation for the given parameter for all base pair steps. S is a parameter-specific scaling factor that compensates for the over- or under-estimation of a given parameter as a result of the HADDOCK semi-flexible refinement stages. S was set to: twist: 0.8, roll: 0.8, tilt: 0.8, rise: 0.0, slide: 0.2 and shift: 0.8.The new value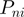 for the parameter at base pair step i is now calculated as follows:
for the parameter at base pair step i is now calculated as follows:
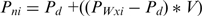
(2) Here Pd is the default value from canonical B-DNA for the given parameter at base pair step i and V is a variance value used to sample the parameter above or below its adjusted average (set to 0.8 by default).
The default base pair parameters are updated in the same way as for the base pair step parameters. The base pair parameter-specific scaling factors (S) used are: shear: 1.0, stretch: 1.0, stagger: 1.0, buckle: −1.0 and propeller twist: −1.0. The variance parameter V is set to 0.8 by default.
The updated list of base pair (step) parameters is used to build a 3D DNA structure using the same parameters for the sugar pucker and phosphate backbone dihedral angles as in the case of canonical B-DNA.
Analysis
The quality of the generated solutions was evaluated using the CAPRI criteria expressed as stars; three stars (high quality): Fnat > 0.5, l- or i-r.m.s.d < 1.0 Å; two stars (medium quality): Fnat > 0.3, l-r.m.s.d < 5.0 Å or i-r.m.s.d < 2.0 Å; one star (acceptable quality): Fnat > 0.1, l-r.m.s.d < 10.0 Å or i-r.m.s.d < 4.0 Å. Fnat is the fraction of native contacts within a 5 Å cutoff, i-r.m.s.d is the interface backbone (Cα,P) r.m.s.d and l-r.m.s.d is the ligand backbone r.m.s.d calculated by superimposition on all phosphate atoms of the reference DNA and subsequently on all Cα atoms of the reference protein. For the results in Figure 4 and the docking using experimentally derived restraints, the reported r.m.s.d values were calculated after superimposition of all heavy atoms of the reference belonging to either the DNA, the protein, the interface or the full complex. The r.m.s.d values were calculated using ProFit (A.C.R. Martin, http://www.bioinf.org.uk/software/profit)
Figure 4.

Best solutions from unbound flexible docking using an ensemble of custom-built DNA structural models (blue) superimposed on to the reference structure (yellow). The complexes are grouped according to their docking difficulty (‘easy’, ‘intermediate’ and ‘difficult’) as indicated in the benchmark. The CAPRI score for each solution is indicated as one or two stars after the PDB code as well as the fraction of native contacts (a), the interface (b) and DNA r.m.s.d (c) from the reference structure. r.m.s.d values (Å) were calculated after superimposition on all heavy atoms of the selected regions of the reference complex. The figures were generated using Pymol (DeLano Scientific LLC, www.pymol.org).
Hardware
HADDOCK docking runs were performed on a Transtec (Transtec AG, Tubingen, Germany) computer cluster operating with 48, 2.0 GHz, 64 bit Opteron processors. As a measure of CPU requirements, one complete run starting with 4000 structures in the rigid-body docking stage could be performed in 4 h on 48 processors.
RESULTS
The power of HADDOCK as a method relies among others on its use of AIRs and explicit flexibility. An AIR defines that a residue on the surface of a biomolecule should be in close vicinity to another residue or group of residues on the partner biomolecule when they form the complex. By default this is described as an ambiguous distance restraint between all atoms of the source residue to all atoms of all reference residue(s) that are assumed to be in the interface in the complex. The effective distance between all those atoms, 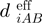 is calculated as follows:
is calculated as follows:
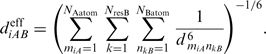 |
(3) |
Here NAatom indicates all atoms of the source residue on molecule A, NresB the residues defined to be at the interface of the reference molecule B, and NBatom all atoms of a residue on molecule B. The 1/r6 summation somewhat mimics the attractive part of the Lennard–Jones potential and ensures that the AIRs are satisfied as soon as any two atoms of the biomolecules are in contact. The AIRs are incorporated as an additional energy term to the energy function that is minimized during the docking. The ambiguous nature of these restraints easily allows experimental data that often provide evidence for a residue making contacts to be used as driving force for the docking. As such the AIRs define a network of restraints between the possible interaction interface(s) of the molecules to be docked without defining the relative orientation of the molecules, minimizing the necessary search through conformational space needed to assemble the interfaces. Because the AIRs are part of the energy function they might also contribute to induce the conformational changes during the flexible stage of the docking.
To objectively answer the question: ‘how successful is HADDOCK in dealing with conformational changes upon complex formation?’ the effects of the quality and quantity of AIRs on complex formation and conformational change should be kept to a minimum. This was realized by constructing ideal AIR restraint sets based on the true interface(s) of the reference complexes (see ‘Materials and Methods’ section). Using these restraints we first evaluated the ability of HADDOCK to reconstruct the complex from its components in their bound conformation. Challenges in reconstruction due to structural characteristics, the inability of the restraints to drive correct complex formation or selection of top ranking solutions due to scoring problems can be identified at this stage. Next we used the same restraints to drive the docking between the unbound protein and a canonical B-DNA 3D structural model using our two-stage protein–DNA docking approach. We focused on the two stages individually, first evaluating the effects of explicit flexibility on the docking by comparing the docking solutions from rigid body refinement with those after semi-flexible refinement. Subsequently we analyzed the conformation of the DNA in the final docking solutions. Here, the focus was on the ability of HADDOCK to introduce those specific DNA conformational changes in terms of DNA bending and twisting that can lead to the final conformation of the DNA in the complex. With this information an ensemble of custom DNA structural models was generated using a modified protocol of our 3D-DART DNA modelling web server (see ‘Materials and Methods’ section). The resulting models were used as input for a second, ‘refinement’, docking run. The results were compared with those of the previous run starting from a canonical B-DNA structural model to analyze the effect of this implicit treatment of flexibility. Finally, the same two-stage docking protocol was applied to a subset of six test cases from the benchmark using AIR restraints based on experimental information obtained from literature sources.
Bound–bound docking
A bound–bound docking experiment is essentially an exercise of separating the reference complex into its individual biomolecules and reconstructing it again. As the different components are already in their bound conformation flexibility is not required and only rigid-body docking needs to be performed. The ability of HADDOCK to sample conformational space in search of the correct interaction interface(s) using ideal AIR restraints was evaluated using the CAPRI star ranking as a quality measure commonly used in protein–protein docking (31). These criteria define one-star predictions as ‘acceptable’, two-star as ‘medium’ and three-star as ‘high’ quality with respect to their reference structure (see ‘Materials and Methods’ section).
The results illustrate that for 75% of the test cases three-star solutions are generated (Figure 1, dark-grey bars). For the first half of the test cases (left half of Figure 1) more than 95% of the solutions ranked one-star or higher but for the remaining, a sharp decline in the total number of star-ranked solutions was observed. The latter group of test cases corresponds mostly with the ‘intermediate’ and ‘difficult’ categories of the benchmark. They are characterized by larger and more segmented interface(s). Many of them require rearrangements of protein domains, loops and secondary structure elements at the interfaces upon interaction to generate a well-packed complex. These, for instance, involve enzymes that perform their catalytic function on single nucleotides that are flipped out of the helix into a catalytic pocket of the protein (1emh, 7mht), restriction enzymes clamping themselves around the DNA (3bam,1rva) or proteins with complex dimerization interfaces (1tro, 1f4k). Effective docking of the bound conformation of these cases is hindered by non-bonded repulsions associated with interface penetration and the correct alignment of the segmented interfaces during the rotation and translation stages of the rigid body refinement. This limits the efficiency of the rigid-body bound-bound docking and in part explains the lower the total number of star-ranked solutions for these cases.
Figure 1.

Cumulative bar graph expressing the quality of the docking solutions according to the CAPRI star rating for all 2000 bound–bound rigid-body docking solutions. Complexes are sorted according to the total number of obtained stars. CAPRI criteria are defined as; three stars (high quality): Fnat > 0.5, l-r.m.s.d or i-r.m.s.d < 1.0 Å; two stars (medium quality): Fnat > 0.3, l-r.m.s.d < 5.0 Å or i-r.m.s.d < 2.0 Å; one star (acceptable quality): Fnat > 0.1, l-r.m.s.d < 10.0 Å or i-r.m.s.d < 4.0 Å. Fnat is the fraction of native contacts within a 5 Å cutoff.
Despite the differences in total number of star-ranked solutions, the 10 best solutions were selected based on the HADDOCK score in all cases coincided with the best solutions based on the CAPRI criteria. This indicates that the HADDOCK scoring function at this stage is sufficient to retrieve the best solutions.
Unbound–unbound docking starting from a canonical B-DNA structural model
We proceeded with the docking of the unbound conformation of the proteins with canonical B-DNA models using ideal AIRs. To increase the sampling of conformational space for the proteins, especially those that use flexible loops to interact with DNA grooves, we first performed a simulated annealing on the interface residues followed by a refinement in explicit water. This procedure resulted in an ensemble of five structures, including the original unbound protein, sampling different conformations of the interface. In 66% of the cases, conformations closer to the bound conformation then the unbound reference protein were sampled. The protein–DNA docking protocol, at this stage, effectively incorporates two modes of flexibility: implicit sampling by means of the ensemble of protein starting structures and explicit sampling of protein and DNA conformational space during semi-flexible refinement.
Figure 2 illustrates the docking results using only rigid-body docking (A) and the effect of a subsequent semi-flexible refinement (B). Here, the cumulative bar graphs show the percentage of CAPRI one-star (white bars) and two-star solutions (grey bars) over all rigid-body (4000) and refined (400) solutions. Overall, 96% of the cases improve due to explicit flexibility. For a number of complexes, one- and two-star solutions were already obtained after rigid-body docking. In all cases, except for 1dfm, the number of one- or two-star solutions increased significantly after semi-flexible refinement. The number of star ranking solutions obtained after rigid-body docking and there subsequent improvement due to explicit flexibility, clearly divides the complexes into three groups that coincide reasonably well with the ‘easy’, ‘intermediate’ and ‘difficult’ categories of the benchmark. For the ‘easy’ category the inclusion of explicit flexibility readily results in a shift from one- to two-star solutions, for the ‘intermediate’ category the number of one-star solutions greatly improves and for the ‘difficult’ category one-star solutions are often only achieved because of explicit flexibility.
Figure 2.

Cumulative bar graphs expressing the quality of the best 400 docking solutions according to the HADDOCK score in terms of CAPRI one-star (grey) and two-star (white) results, for the two-stage unbound–unbound protein–DNA docking using true interface derived restraints. Results are presented for; the rigid-body docking starting from a canonical B-DNA model (A); after the semi-flexible refinement (B) and after semi-flexible refinement using an ensemble of custom DNA 3D structural models (C). Complexes are sorted according to the total number of obtained stars in (B), reclassifying the benchmark into ‘easy’, ‘intermediate’ and ‘difficult’ categories. See caption of Figure 1 for the definition of the CAPRI criteria.
Unbound–unbound docking starting from custom-built B-DNA structural models
The previous docking results show the improvements that can be obtained when using explicit flexibility versus rigid-body docking. In all cases, the DNA and the proteins could adapt their conformation to better interact with each other. For the DNA, these conformational changes range from small local changes in helical bend and groove width, while maintaining a relative straight helix, to larger global changes that effectively bend and twist the DNA structure. However, the amount of conformational space that can be sampled during the semi-flexible refinement stage is limited. Starting from a canonical B-DNA structural model, the semi-flexible refinement stage improved the DNA model on average by 0.84 ± 0.36 Å all heavy atom r.m.s.d with respect to the reference. This clearly cannot account for the often large DNA conformational changes observed in the benchmark (ranging from 3 up to 10 Å).
The amount and consistency of the DNA conformational changes that did occur during semi-flexible refinement, can however provide an indication of the extent of conformational change to be expected in the final complex as we have shown before (18). By analyzing the conformational changes in the top 10 solutions of the best cluster, both selected based on the HADDOCK score, we generated five new DNA structural models with custom conformations reflecting the conformational changes that took place in the DNA during the first docking round for every test case (see ‘Materials and methods’ section).
The effects of using a custom-built DNA structural ensemble on the docking results obtained after semi-flexible refinement is illustrated in Figure 2C. Again, the cumulative bar graph shows the percentage of CAPRI one-star (white bars) and two-star solutions (grey bars) among all (400) refined docking solutions according to the HADDOCK score.
In a number of cases there is a marked increase in one- and/or two-star solutions due to the use of the ensemble, while in other cases there is no improvement or even a reduction. However, because the ensemble contains custom built DNA structures in different conformations, it is possible that one or several of these are less successful in sampling relevant conformational space than the canonical B-DNA model used in the first run. However, if even only one of the five models is significantly better that canonical B-DNA, and the scoring and clustering stage select solutions obtained from this model then an improvement is achieved compared to only semi-flexible refinement. Figure 3 better illustrates the results by individual graphs showing for every test case the various r.m.s.d values and fraction of native contacts for the 10 best solutions of the top-ranking cluster, both selected based on the HADDOCK score. The figure shows statistics for the corresponding solutions after semi-flexible refinement, the solutions from the rigid-body stage starting from canonical B-DNA, and the solutions after semi-flexible refinement using an ensemble of custom-built DNA starting structures (source data can be found in Supplementary Tables S1–S3 of the Supplementary Data). With respect to the best 10 solutions, our two-stage docking protocol improved the results in 91% of the cases relative to rigid-body docking. The use of an ensemble of custom-built DNA structural models (the second stage of the docking) further improved the results in 72% of the cases compared to the first stage only. For most complexes there is a marked improvement in terms of r.m.s.d from the reference complex, when progressing from rigid-body docking to the use of an ensemble of custom built DNA structural models. The improvement in DNA, interface and all heavy-atom r.m.s.d becomes more significant with the increasing difficulty of the test cases. This trend is to be expected as the conformational changes between unbound and bound structures are small in the ‘easy’ category and become more pronounced in the ‘intermediate’ and ‘difficult’ categories of the benchmark. These results show the efficiency of the DNA modelling procedure in capturing the essential motions that occur in the DNA upon complex formation. The fraction of native contacts improves significantly throughout the benchmark even when the solutions improve little in terms of r.m.s.d. Apart from this, the convergence in the 10 best solutions in general improves, which is apparent in the smaller standard deviations (Figure 3) and an improved clustering (Supplementary Table S3, Supplementary Data).
Figure 3.

All heavy atom r.m.s.d values from the reference complex [(A) DNA only, (B) full complex, (C) interface] and fraction of native contacts [Fnat, (D)] for the 10 best solutions of the best cluster, both selected based on the HADDOCK score, after rigid-body docking (open squares) and semi-flexible refinement (closed circles) starting from a canonical B-DNA structural model and after semi-flexible refinement (open triangle) starting from an ensemble of custom-built DNA models.
Unbound–unbound docking using experimental derived restraints
In a ‘real-life’ docking situation, AIRs are typically defined based on experimental data or interface predictions (19,20). The quality and quantity of available data can influence the correct assembly of the interaction interface(s) and the conformational changes brought about in the flexible stages of the docking. To evaluate the performance of our two-stage protein–DNA docking protocol under these circumstances we selected six representative test cases from the ‘easy’, ‘intermediate’ and ‘difficult’ categories of the benchmark (two of each). These are, respectively, the protein–DNA complexes formed by the phage 434 Cro (3cro) transcription factor and retinoid X receptor (1by4), the hyperthermophile chromosomal protein SAC7D (1azp) and papillomavirus type 18 E2 (1jj4) protein, the homing endonuclease I-PpoI (1a74) and the proline utilization transcription activator PUT3 (1zme). For these we defined AIRs based on experimental data collected from literature sources (see ‘Material and Methods’ section). Docking the protein and DNA in their bound conformation (Table 3, bound-rigid) using rigid-body energy minimization only illustrates that the AIRs defined based on experimental data are also able to reconstruct the correct interaction interface(s) in all cases resulting in high quality predictions. The overall results for the unbound docking again show a significant improvement in terms of r.m.s.d from the reference complexes and fraction of native contacts when progressing from rigid body docking to semi-flexible refinement and finally a second docking round starting from an ensemble of custom-built DNA structural models (Table 3). The best docking solutions superimposed onto their reference structures are presented in Figure 4.
Table 3.
Performance of the two-stage docking protocol when using AIRs based on experimental information: the r.m.s.d values from the reference and fraction of native contacts for the top ten docking solutions of the top ranking cluster both selected based on the HADDOCK score
| r.m.s.d (Å) |
Fnate | CAPRIf *,**,*** | ||||
|---|---|---|---|---|---|---|
| Totala | Interfaceb | DNAc | Proteind | |||
| ‘Easy’ | ||||||
| 1by4 | ||||||
| Bound rigid | 0.410.08 | 0.340.07 | 0.000.00 | 0.380.07 | 0.890.02 | 0,0,10 |
| Unbound rigid | 4.330.72 | 4.010.53 | 1.410.00 | 4.660.73 | 0.110.04 | 4,0,0 |
| Unbound flex | 6.722.10 | 5.871.71 | 1.900.19 | 6.982.21 | 0.170.05 | 5,0,0 |
| DNA lib | 5.522.43 | 4.912.32 | 1.610.14 | 5.852.46 | 0.270.09 | 4,3,0 |
| 3cro | ||||||
| Bound rigid | 0.320.16 | 0.380.19 | 0.000.00 | 0.440.22 | 0.850.09 | 0,0,10 |
| Unbound rigid | 3.790.60 | 3.510.63 | 3.700.00 | 3.500.83 | 0.150.05 | 10,0,0 |
| Unbound flex | 3.570.63 | 3.290.68 | 2.860.30 | 3.190.68 | 0.270.07 | 6,2,0 |
| DNA lib | 2.890.40 | 2.620.73 | 2.080.21 | 2.960.43 | 0.400.06 | 3,7,0 |
| ‘Intermediate’ | ||||||
| 1azp | ||||||
| Bound rigid | 0.330.07 | 0.310.07 | 0.000.00 | 0.110.00 | 0.920.03 | 0,0,10 |
| Unbound rigid | 7.122.06 | 7.092.25 | 3.250.00 | 3.580.02 | 0.020.02 | 0,0,0 |
| Unbound flex | 6.902.00 | 6.682.26 | 2.870.32 | 3.640.13 | 0.040.04 | 0,0,0 |
| DNA lib | 4.560.79 | 4.000.45 | 1.830.26 | 3.760.16 | 0.100.04 | 5,0,0 |
| 1jj4 | ||||||
| Bound rigid | 0.390.10 | 0.400.09 | 0.000.00 | 0.100.03 | 0.820.07 | 0,0,10 |
| Unbound rigid | 4.230.37 | 4.760.48 | 3.190.00 | 1.470.05 | 0.090.02 | 3,0,0 |
| Unbound flex | 4.250.43 | 4.550.58 | 3.190.21 | 2.400.02 | 0.160.07 | 6,0,0 |
| DNA lib | 3.220.30 | 3.620.38 | 2.380.14 | 2.370.05 | 0.210.07 | 9,1,0 |
| ‘Difficult’ | ||||||
| 1a74 | ||||||
| Bound rigid | 0.060.01 | 0.070.01 | 0.000.00 | 0.010.00 | 0.840.01 | 0,0,10 |
| Unbound rigid | 5.430.99 | 6.880.97 | 7.440.00 | 1.680.14 | 0.040.02 | 0,0,0 |
| Unbound flex | 4.950.38 | 6.300.46 | 7.120.32 | 1.840.14 | 0.140.04 | 8,0,0 |
| DNA lib | 2.720.25 | 3.370.32 | 3.760.19 | 1.780.12 | 0.240.05 | 9,1,0 |
| 1zme | ||||||
| Bound rigid | 0.480.11 | 0.460.08 | 0.000.00 | 0.010.00 | 0.790.06 | 0,0,10 |
| Unbound rigid | 6.290.64 | 5.490.68 | 4.280.00 | 5.670.61 | 0.060.03 | 0,0,0 |
| Unbound flex | 6.150.62 | 5.290.59 | 4.680.33 | 5.880.27 | 0.120.06 | 4,0,0 |
| DNA lib | 5.270.62 | 4.630.80 | 3.350.13 | 5.550.48 | 0.150.04 | 8,0,0 |
Average all heavy atom r.m.s.d values from the reference structure (Å, standard deviation in subscript) calculated over:
aThe entire complex.
bThe interface.
cThe DNA only for the 10 top ranking solutions.
dThe protein only for the 10 top ranking solutions.
The r.m.s.d values are reported for; bound rigid-body docking (bound rigid); unbound rigid-body docking (unbound rigid), semi-flexible refinement (unbound flex.) starting from canonical B-DNA; unbound semi-flexible docking using a library of custom-built DNA structural models as input (DNA library).
eFnat is the fraction of native contacts.
fNumber of one-, two- and three-star CAPRI ranked solutions obtained in the top 10 solutions.
Although the overall results improved for all six test cases, differences were observed. The bound and unbound components of the retinoid X receptor–DNA complex (1by4) differ little from each other in terms of r.m.s.d from the reference and rigid body docking readily generates one-star solutions. The complex is composed of two proteins that interact with the DNA major groove but not with each other. Independent movement of both proteins resulted in a relative large variation in the 10 best solutions after semi-flexible refinement when starting from a canonical B-DNA model. The use of a custom built DNA library does not reduce this variation but does significantly improve the fraction of native contacts and medium quality solutions. The phage 434 Cro–DNA complex (3cro) is a similar case with the exception that the proteins dimerize. This results in far less variation in the 10 best solutions after the flexible stages and a sequential improvement of the r.m.s.d values and fraction of native contacts at each step of the docking. The hyperthermophile chromosomal protein SAC7D–DNA complex (1azp) binds in a non-specific manner to the DNA minor groove. The experimental data available for this complex are less well defined than for the other test cases. Despite this, the two-stage docking protocol did reproduce the characteristic minor groove widening observed for this system resulting in a significant improvement in r.m.s.d when using an ensemble of custom built DNA structural models. The specific kink in the DNA structure observed at the second C–G base pair (61°) in the reference complex was, however, predicted at the third G–A base pair step (∼25°) in the docking solutions. The potential of our two-stage docking protocol to deal with large DNA conformational changes is best illustrated in the case of the homing endonuclease I-PpoI–DNA complex (1a74). Here, the overall bend of ∼38° is reproduced in the best solutions (∼45°). The information available for this complex results in a well defined, curved, interaction interface on the protein and indicates that there is little conformational difference of the protein in its bound and unbound state. As such, the sharp bend introduced in the DNA by the analysis and modelling step could be sampled up to 10 times the standard deviation from the average to match the protein surface (see ‘Materials and methods’ section). The proline utilization transcription activator PUT3 (1zme) is a difficult case from both protein and DNA perspectives. The protein contains two globular DNA binding domains connected to a core domain with a long flexible linker. The NMR ensemble of the unbound protein contains the DNA binding domains in many different orientations that prevent effective docking in the rigid body stage. Therefore, we cut the protein at the flexible linkers, resulting in three parts that were docked as separated bodies. Peptide linker restraints were defined between the amino acids at the scission sites. After semi-flexible refinement, we reconnected the different parts in the 10 best solutions and used the resulting protein ensemble for the second docking stage starting from an ensemble of custom built DNA structural models.
DISCUSSION
The use of AIRs is essential to the success of the HADDOCK docking methodology in general. These are used to position the protein at the interaction interface of the DNA and, together with the flexible stages of the docking, to facilitate conformational changes. We have shown previously the importance of AIRs in protein–DNA docking (18) using three monomeric transcription factor DNA complexes as test cases. In the current study we refined our initial method and evaluated its performance on a benchmark of 47 protein–DNA complexes (30). Compared with the initial three test cases the benchmark contains complexes from various structural functional classes in which one or multiple proteins interact with the DNA using various binding modes. Because of the presence of multiple proteins or DNA-binding domains, 40% of the benchmark required docking following a multi-body (N > 2) approach. This challenging benchmark offers a good platform to evaluate the capabilities of our docking method. We will discuss in the following the two questions that were the focus of both this study as well as the previous work describing the initial protein–DNA docking method.
How well is the method able to identify the correct interaction interface(s)?
The assembly of the interaction interface(s) is a process driven by AIRs. In ‘real-life’ docking settings the AIRs are typically defined based on experimental data and/or interface predictions. The quality of the docking solutions is therefore closely related to the amount and quality of available data in terms of their accuracy and information content. We started from an ideal situation in which the restraints were derived from the intermolecular contacts in the reference complex. Bound docking resulted for 75% of cases in three-star (high quality) predictions among the top 10 solutions based on the HADDOCK score (Figure 1). The percentage of generated high-quality solutions and the total number of star-ranked solutions, however, declined for the ‘intermediate’ and ‘difficult’ cases due to interface topology features such as segmentation and rearrangement of structure elements. Such rearrangements occur in protein domains, loops and secondary-structure elements at the interfaces during the process of complex formation; they are required to form a well-packed complex. The difference between the bound and unbound conformation of the protein and DNA interfaces in the benchmark (30) further illustrates this. Consequently, in a bound–bound docking setting, the docking efficiency is hindered by non-bonded repulsions associated with interface penetration and by the correct alignment of the segmented interfaces during the rotation and translation stages of the rigid body refinement. The increase in the total number of star-ranked solutions for many of the ‘difficult’ test cases in unbound–unbound docking relative to bound–bound docking further illustrates this process as rearrangements are allowed to take place. Still there are a number of test cases such as 1tro and 1f4k in which non-bonded repulsions hamper the docking. Given that these cases can be identified beforehand, the docking efficiency could be improved by scaling down the non-bonded energy terms (inter_rigid term to 0.001 or lower in HADDOCK); this allows penetration to occur during the docking. An initial test with a scaled down non-bonded energy term for the above-mentioned two test cases resulted in a significant increase in the number of one- and two-star solutions (Supplementary Table S4, Supplementary Data). This shows that the AIRs are not the limiting factor but also raises the question whether a change in the non-bonded energy term scaling factor could be beneficial throughout the benchmark. Our experience in protein–protein docking however indicates that the scoring becomes more challenging, which might be detrimental at the end.
The unbound two-stage flexible docking using the same restraints (Figures 2 and 3) resulted in the prediction of one- to two-star solutions depending on the level of difficulty of the test cases. Although these results are significantly better than unbound rigid-body docking only, they still indicate that conformational changes are the limiting factor in protein–DNA docking.
The same series of docking experiments were performed with a representative selection of six test cases using AIRs defined based on experimental information (Table 3, Figure 4). The results were comparable to the use of ideal restraints in terms of the CAPRI quality criteria. This clearly illustrates that readily available non-structural experimental data are sufficient to assemble the correct interaction interface(s) in these challenging, often multi-component, protein–DNA systems. Still, the quality of the generated solutions is directly related to the quality of the used experimental information. Sparse- and/or low-quality information will likely result in poor-quality docking solutions, especially for multi-component systems. The AIRs can, however, be defined based on a wider variety of information sources than used in the current work. For instance, NMR data or even statistical protein–DNA interaction potentials, are promising means of improving the results either by driving the docking or filtering solutions afterwards. With respect to the latter we should note that the many different solutions generated in this benchmark docking effort, provide a compelling set of decoy structures that can be useful for the development and validation of scoring functions.
How successful is the method in dealing with conformational changes upon complex formation?
The correct treatment of conformational changes upon complex formation is likely the most challenging aspect of protein–DNA docking. Both protein(s) and DNA readily change their conformation upon complex formation. The extent of this change forms the basis of the protein–DNA benchmark categorization. Our two-stage protein–DNA docking method was designed to deal with this challenge and its performance is best illustrated in the docking of unbound proteins with canonical B-DNA using ideal AIRs. While a single docking run was sufficient to generate two-star solutions for the ‘easy’ cases, the two-stage protocol was often required to generate one–two-star solution for the ‘intermediate’ and ‘difficult’ cases. Altogether, this approach was successful in generating at least one-star solutions for 96% of the complete benchmark. This illustrates that the explicit flexibility implemented in HADDOCK is sufficient to generate two-star solution in the ‘easy’ cases where conformational changes are limited but that this approach fails for cases where such changes are more pronounced such as in the ‘intermediate’ and ‘difficult’ cases. For the latter, our DNA analysis and modelling procedure is capable of extracting the main bend and twist motions that occur in the DNA upon complex formation and use these for the benefit of DNA modelling. In that way, a larger part of the relevant DNA conformational space can be sampled than what is feasible within a single round of semi-flexible refinement. Even results of the ‘easy’ test cases with limited conformational changes are improved by this two-stage procedure. Finally, the use of experimentally-derived AIRs on a subset of six test cases showed that our method also significantly improved the docking results under real-life conditions when less ideal AIR restraints are available.
Although the semi-flexible refinement stage of HADDOCK is able to introduce many of the DNA conformational changes required for correct complex formation it has difficulties predicting DNA groove expansion facilitated by negative base pair step sliding (for example in 1a74 and 1g9z). Consequently, this mode of conformational change is not detected by our DNA analysis procedure and not introduced in the custom-built DNA ensemble. Although the improvements in r.m.s.d to the reference complex and fraction of native contacts clearly illustrate that our method outperforms rigid-body docking it does raise questions on the quality of the DNA in the generated solutions. This however, remains a difficult issue due to the lack of DNA structure validation procedures. Furthermore, our method predominantly focuses on the conformational changes in the DNA, but also proteins can often change their conformation upon complex formation, sometimes quite drastically as, for example, in the restriction endonuclease MvaI (2oaa). While accounting for small conformational changes by means of flexible refinement and the use of protein ensembles that sample different interface conformations, large conformational changes such as loop and domain rearrangements or disordered to order transitions remain a challenge. Such events are present in some of the test cases where the use of an ensemble of custom-built DNA structural models did not improve the results significantly. This still leaves plenty of opportunities for improvements, for instance in those cases where protein domain rearrangements are facilitated by flexible ‘hinges’ connecting them. Such domains can be docked as separate bodies, enabling them to sample conformational space individually. This procedure has been successfully used for the proline utilization transcription activator PUT3 (1zme) in this study.
The flexible protein–DNA docking approach described in this article can benefit protein–DNA interaction studies at several levels. It can be used to generate models of protein–DNA complexes from the structures of the unbound proteins and a canonical B-DNA in the presence of suitable experimental data without any prior knowledge of the DNA conformational changes required to establish the complex. It should also be useful for studying the effects of mutations or different operator sequences on complex formation. In addition, it can assist in experimental structural studies by, for instance, providing initial DNA structural models to guide and speed up the NMR analysis and assignment process.
In summary, by allowing the inclusion of a large variety of experimental and/or prediction data, together with a flexible description of the DNA, the proposed docking approach should be a useful tool in structural studies of protein–DNA complexes.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
European Community (FP6 I3 project ‘EU-NMR’, contract no. RII3-026145 and FP7 I3 project ‘eNMR’, contract no. 213010-e-NMR) and VICI grant from the Netherlands Organization for Scientific Research (NWO) to A.M.J.J.B. (grant no. 700.96.442). Funding for open access charge: VICI grant from the Netherlands Organization for Scientific Research (NWO) (grant no. 700.96.442 to A.M.J.J.B.).
Conflict of interest statement. None declared.
Supplementary Material
REFERENCES
- 1.Halperin I, Ma B, Wolfson H, Nussinov R. Principles of docking: An overview of search algorithms and a guide to scoring functions. Proteins. 2002;47:409–443. doi: 10.1002/prot.10115. [DOI] [PubMed] [Google Scholar]
- 2.Schneidman-Duhovny D, Nussinov R, Wolfson HJ. Predicting molecular interactions in silico: II. Protein-protein and protein-drug docking. Curr. Med. Chem. 2004;11:91–107. doi: 10.2174/0929867043456223. [DOI] [PubMed] [Google Scholar]
- 3.Ritchie DW. Recent progress and future directions in protein-protein docking. Curr. Protein Pept. Sci. 2008;9:1–15. doi: 10.2174/138920308783565741. [DOI] [PubMed] [Google Scholar]
- 4.Gane PJ, Dean PM. Recent advances in structure-based rational drug design. Curr. Opin. Struct. Biol. 2000;10:401–404. doi: 10.1016/s0959-440x(00)00105-6. [DOI] [PubMed] [Google Scholar]
- 5.Joseph-McCarthy D. Computational approaches to structure-based ligand design. Pharmacol. Ther. 1999;84:179–191. doi: 10.1016/s0163-7258(99)00031-5. [DOI] [PubMed] [Google Scholar]
- 6.Kuntz ID. Structure-based strategies for drug design and discovery. Science. 1992;257:1078–1082. doi: 10.1126/science.257.5073.1078. [DOI] [PubMed] [Google Scholar]
- 7.Dunn RK, Kingston RE. Gene regulation in the postgenomic era: technology takes the wheel. Mol. Cell. 2007;28:708–714. doi: 10.1016/j.molcel.2007.11.022. [DOI] [PubMed] [Google Scholar]
- 8.Adesokan AA, Roberts VA, Lee KW, Lins RD, Briggs JM. Prediction of HIV-1 integrase/viral DNA interactions in the catalytic domain by fast molecular docking. J. Med. Chem. 2003;47:821–828. doi: 10.1021/jm0301890. [DOI] [PubMed] [Google Scholar]
- 9.Aloy P, Moont G, Gabb HA, Querol E, Aviles FX, Sternberg MJ. Modelling repressor proteins docking to DNA. Proteins. 1998;33:535–549. [PubMed] [Google Scholar]
- 10.Bastard K, Thureau A, Lavery R, Prevost C. Docking macromolecules with flexible segments. J. Comput. Chem. 2003;24:1910–1920. doi: 10.1002/jcc.10329. [DOI] [PubMed] [Google Scholar]
- 11.Fan L, Roberts VA. Complex of linker histone H5 with the nucleosome and its implications for chromatin packing. Proc. Natl Acad. Sci. USA. 2006;103:8384–8389. doi: 10.1073/pnas.0508951103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fanelli F, Ferrari S. Prediction of MEF2A-DNA interface by rigid body docking: a tool for fast estimation of protein mutational effects on DNA binding. J. Struct. Biol. 2006;153:278–283. doi: 10.1016/j.jsb.2005.12.002. [DOI] [PubMed] [Google Scholar]
- 13.Knegtel RM, Boelens R, Kaptein R. Monte Carlo docking of protein-DNA complexes: incorporation of DNA flexibility and experimental data. Protein Eng. 1994;7:761–767. doi: 10.1093/protein/7.6.761. [DOI] [PubMed] [Google Scholar]
- 14.Liu Z, Guo JT, Li T, Xu Y. Structure-based prediction of transcription factor binding sites using a protein-DNA docking approach. Proteins. 2008;72:1114–1124. doi: 10.1002/prot.22002. [DOI] [PubMed] [Google Scholar]
- 15.Poulain P, Saladin A, Hartmann B, Prevost C. Insights on protein-DNA recognition by coarse grain modelling. J. Comput. Chem. 2008;29:2582–2592. doi: 10.1002/jcc.21014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Roberts VA, Case DA, Tsui V. Predicting interactions of winged-helix transcription factors with DNA. Proteins. 2004;57:172–187. doi: 10.1002/prot.20193. [DOI] [PubMed] [Google Scholar]
- 17.Sandmann C, Cordes F, Saenger W. Structure model of a complex between the factor for inversion stimulation (FIS) and DNA: modeling protein-DNA complexes with dyad symmetry and known protein structures. Proteins. 1996;25:486–500. doi: 10.1002/prot.8. [DOI] [PubMed] [Google Scholar]
- 18.van Dijk M, van Dijk AD, Hsu V, Boelens R, Bonvin AM. Information-driven protein-DNA docking using HADDOCK: it is a matter of flexibility. Nucleic Acids Res. 2006;34:3317–3325. doi: 10.1093/nar/gkl412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Melquiond ASJ, Bonvin AMJJ. Experimental Constraint-Driven Docking. In: Zacharias M, editor. Protein-protein Complexes: Analysis, Modelling and Drug Design. London: Imperial College Press; 2009. pp. 183–209. [Google Scholar]
- 20.van Dijk AD, Boelens R, Bonvin AM. Data-driven docking for the study of biomolecular complexes. FEBS J. 2005;272:293–312. doi: 10.1111/j.1742-4658.2004.04473.x. [DOI] [PubMed] [Google Scholar]
- 21.Mondragon A, Harrison SC. The phage 434 Cro/OR1 complex at 2.5 A resolution. J. Mol. Biol. 1991;219:321–334. doi: 10.1016/0022-2836(91)90568-q. [DOI] [PubMed] [Google Scholar]
- 22.Raumann BE, Rould MA, Pabo CO, Sauer RT. DNA recognition by beta-sheets in the Arc repressor-operator crystal structure. Nature. 1994;367:754–757. doi: 10.1038/367754a0. [DOI] [PubMed] [Google Scholar]
- 23.Chuprina VP, Rullmann JA, Lamerichs RM, van Boom JH, Boelens R, Kaptein R. Structure of the complex of lac repressor headpiece and an 11 base-pair half-operator determined by nuclear magnetic resonance spectroscopy and restrained molecular dynamics. J. Mol. Biol. 1993;234:446–462. doi: 10.1006/jmbi.1993.1598. [DOI] [PubMed] [Google Scholar]
- 24.Bessiere D, Lacroix C, Campagne S, Ecochard V, Guillet V, Mourey L, Lopez F, Czaplicki J, Demange P, Milon A, et al. Structure-function analysis of the THAP zinc finger of THAP1, a large C2CH DNA-binding module linked to Rb/E2F pathways. J. Biol. Chem. 2008;283:4352–4363. doi: 10.1074/jbc.M707537200. [DOI] [PubMed] [Google Scholar]
- 25.Cai S, Zhu L, Zhang Z, Chen Y. Determination of the three-dimensional structure of the Mrf2-DNA complex using paramagnetic spin labeling. Biochemistry. 2007;46:4943–4950. doi: 10.1021/bi061738h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gamsjaeger R, Swanton MK, Kobus FJ, Lehtomaki E, Lowry JA, Kwan AH, Matthews JM, Mackay JP. Structural and biophysical analysis of the DNA binding properties of myelin transcription factor 1. J. Biol. Chem. 2008;283:5158–5167. doi: 10.1074/jbc.M703772200. [DOI] [PubMed] [Google Scholar]
- 27.Liu W, Vierke G, Wenke AK, Thomm M, Ladenstein R. Crystal structure of the archaeal heat shock regulator from Pyrococcus furiosus: a molecular chimera representing eukaryal and bacterial features. J. Mol. Biol. 2007;369:474–488. doi: 10.1016/j.jmb.2007.03.044. [DOI] [PubMed] [Google Scholar]
- 28.Singh S, Hager MH, Zhang C, Griffith BR, Lee MS, Hallenga K, Markley JL, Thorson JS. Structural insight into the self-sacrifice mechanism of enediyne resistance. ACS Chem. Biol. 2006;1:451–460. doi: 10.1021/cb6002898. [DOI] [PubMed] [Google Scholar]
- 29.van Dijk M, Bonvin AM. 3D-DART: a DNA structure modelling server. Nucleic Acids Res. 2009;37:W235–W239. doi: 10.1093/nar/gkp287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.van Dijk M, Bonvin AM. A protein-DNA docking benchmark. Nucleic Acids Res. 2008;36:e88. doi: 10.1093/nar/gkn386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Janin J. Assessing predictions of protein-protein interaction: the CAPRI experiment. Protein Sci. 2005;14:278–283. doi: 10.1110/ps.041081905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hubbard SJ, Thornton JM. University College London; 1993. ‘NACCESS’, computer program, Department of Biochemistry and Molecular Biology. [Google Scholar]
- 33.de Vries SJ, van Dijk AD, Krzeminski M, van Dijk M, Thureau A, Hsu V, Wassenaar T, Bonvin AM. HADDOCK versus HADDOCK: new features and performance of HADDOCK2.0 on the CAPRI targets. Proteins. 2007;69:726–733. doi: 10.1002/prot.21723. [DOI] [PubMed] [Google Scholar]
- 34.Lu XJ, Olson WK. 3DNA: a software package for the analysis, rebuilding and visualization of three-dimensional nucleic acid structures. Nucleic Acids Res. 2003;31:5108–5121. doi: 10.1093/nar/gkg680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lu XJ, Olson WK. 3DNA: a versatile, integrated software system for the analysis, rebuilding and visualization of three-dimensional nucleic-acid structures. Nat. Protoc. 2008;3:1213–1227. doi: 10.1038/nprot.2008.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dickerson RE. Definitions and nomenclature of nucleic acid structure parameters. J. Biomol. Struct. Dyn. 1989;6:627–34. doi: 10.1080/07391102.1989.10507726. [DOI] [PubMed] [Google Scholar]
- 37.Zhao Q, Chasse SA, Devarakonda S, Sierk ML, Ahvazi B, Rastinejad F. Structural basis of RXR-DNA interactions. J. Mol. Biol. 2000;296:509–520. doi: 10.1006/jmbi.1999.3457. [DOI] [PubMed] [Google Scholar]
- 38.Danielsen M, Hinck L, Ringold GM. Two amino acids within the knuckle of the first zinc finger specify DNA response element activation by the glucocorticoid receptor. Cell. 1989;57:1131–1138. doi: 10.1016/0092-8674(89)90050-0. [DOI] [PubMed] [Google Scholar]
- 39.Glass CK. Differential recognition of target genes by nuclear receptor monomers, dimers, and heterodimers. Endocr. Rev. 1994;15:391–407. doi: 10.1210/edrv-15-3-391. [DOI] [PubMed] [Google Scholar]
- 40.Haussler MR, Whitfield GK, Haussler CA, Hsieh JC, Thompson PD, Selznick SH, Dominguez CE, Jurutka PW. The nuclear vitamin D receptor: biological and molecular regulatory properties revealed. J. Bone Miner. Res. 1998;13:325–349. doi: 10.1359/jbmr.1998.13.3.325. [DOI] [PubMed] [Google Scholar]
- 41.Koszewski NJ, Reinhardt TA, Horst RL. Vitamin D receptor interactions with the murine osteopontin response element. J. Steroid Biochem. Mol. Biol. 1996;59:377–388. doi: 10.1016/s0960-0760(96)00127-6. [DOI] [PubMed] [Google Scholar]
- 42.Lee MS, Kliewer SA, Provencal J, Wright PE, Evans RM. Structure of the retinoid X receptor alpha DNA binding domain: a helix required for homodimeric DNA binding. Science. 1993;260:1117–1121. doi: 10.1126/science.8388124. [DOI] [PubMed] [Google Scholar]
- 43.Mader S, Kumar V, de Verneuil H, Chambon P. Three amino acids of the oestrogen receptor are essential to its ability to distinguish an oestrogen from a glucocorticoid-responsive element. Nature. 1989;338:271–274. doi: 10.1038/338271a0. [DOI] [PubMed] [Google Scholar]
- 44.Nelson CC, Hendy SC, Faris JS, Romaniuk PJ. Retinoid X receptor alters the determination of DNA binding specificity by the P-box amino acids of the thyroid hormone receptor. J. Biol. Chem. 1996;271:19464–19474. doi: 10.1074/jbc.271.32.19464. [DOI] [PubMed] [Google Scholar]
- 45.Rastinejad F, Perlmann T, Evans RM, Sigler PB. Structural determinants of nuclear receptor assembly on DNA direct repeats. Nature. 1995;375:203–211. doi: 10.1038/375203a0. [DOI] [PubMed] [Google Scholar]
- 46.Umesono K, Evans RM. Determinants of target gene specificity for steroid/thyroid hormone receptors. Cell. 1989;57:1139–1146. doi: 10.1016/0092-8674(89)90051-2. [DOI] [PubMed] [Google Scholar]
- 47.Harrison SC, Anderson JE, Koudelka GB, Mondragon A, Subbiah S, Wharton RP, Wolberger C, Ptashne M. Recognition of DNA sequences by the repressor of bacteriophage 434. Biophys. Chem. 1988;29:31–37. doi: 10.1016/0301-4622(88)87022-4. [DOI] [PubMed] [Google Scholar]
- 48.Koudelka GB. Recognition of DNA structure by 434 repressor. Nucleic Acids Res. 1998;26:669–675. doi: 10.1093/nar/26.2.669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Koudelka GB, Lam CY. Differential recognition of OR1 and OR3 by bacteriophage 434 repressor and Cro. J. Biol. Chem. 1993;268:23812–23817. [PubMed] [Google Scholar]
- 50.Wharton RP, Brown EL, Ptashne M. Substituting an alpha-helix switches the sequence-specific DNA interactions of a repressor. Cell. 1984;38:361–369. doi: 10.1016/0092-8674(84)90491-4. [DOI] [PubMed] [Google Scholar]
- 51.Robinson H, Gao YG, McCrary BS, Edmondson SP, Shriver JW, Wang AH. The hyperthermophile chromosomal protein Sac7d sharply kinks DNA. Nature. 1998;392:202–205. doi: 10.1038/32455. [DOI] [PubMed] [Google Scholar]
- 52.Clark AT, Smith K, Muhandiram R, Edmondson SP, Shriver JW. Carboxyl pK(a) values, ion pairs, hydrogen bonding, and the pH-dependence of folding the hyperthermophile proteins Sac7d and Sso7d. J. Mol. Biol. 2007;372:992–1008. doi: 10.1016/j.jmb.2007.06.089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dostal L, Chen CY, Wang AH, Welfle H. Partial B-to-A DNA transition upon minor groove binding of protein Sac7d monitored by Raman spectroscopy. Biochemistry. 2004;43:9600–9609. doi: 10.1021/bi049192r. [DOI] [PubMed] [Google Scholar]
- 54.Kahsai MA, Martin E, Edmondson SP, Shriver JW. Stability and flexibility in the structure of the hyperthermophile DNA-binding protein Sac7d. Biochemistry. 2005;44:13500–13509. doi: 10.1021/bi051167d. [DOI] [PubMed] [Google Scholar]
- 55.Peters WB, Edmondson SP, Shriver JW. Effect of mutation of the Sac7d intercalating residues on the temperature dependence of DNA distortion and binding thermodynamics. Biochemistry. 2005;44:4794–4804. doi: 10.1021/bi047382w. [DOI] [PubMed] [Google Scholar]
- 56.Kim SS, Tam JK, Wang AF, Hegde RS. The structural basis of DNA target discrimination by papillomavirus E2 proteins. J. Biol. Chem. 2000;275:31245–31254. doi: 10.1074/jbc.M004541200. [DOI] [PubMed] [Google Scholar]
- 57.Bedrosian CL, Bastia D. The DNA-binding domain of HPV-16 E2 protein interaction with the viral enhancer: protein-induced DNA bending and role of the nonconserved core sequence in binding site affinity. Virology. 1990;174:557–575. doi: 10.1016/0042-6822(90)90109-5. [DOI] [PubMed] [Google Scholar]
- 58.Sanchez IE, Dellarole M, Gaston K, de Prat Gay G. Comprehensive comparison of the interaction of the E2 master regulator with its cognate target DNA sites in 73 human papillomavirus types by sequence statistics. Nucleic Acids Res. 2008;36:756–769. doi: 10.1093/nar/gkm1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Flick KE, Jurica MS, Monnat R.J., Jr, Stoddard BL. DNA binding and cleavage by the nuclear intron-encoded homing endonuclease I-PpoI. Nature. 1998;394:96–101. doi: 10.1038/27952. [DOI] [PubMed] [Google Scholar]
- 60.Argast GM, Stephens KM, Emond MJ, Monnat R.J., Jr I-PpoI and I-CreI homing site sequence degeneracy determined by random mutagenesis and sequential in vitro enrichment. J. Mol. Biol. 1998;280:345–353. doi: 10.1006/jmbi.1998.1886. [DOI] [PubMed] [Google Scholar]
- 61.Eklund JL, Ulge UY, Eastberg J, Monnat R.J., Jr Altered target site specificity variants of the I-PpoI His-Cys box homing endonuclease. Nucleic Acids Res. 2007;35:5839–5850. doi: 10.1093/nar/gkm624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ellison EL, Vogt VM. Interaction of the intron-encoded mobility endonuclease I-PpoI with its target site. Mol. Cell Biol. 1993;13:7531–7539. doi: 10.1128/mcb.13.12.7531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Galburt EA, Chadsey MS, Jurica MS, Chevalier BS, Erho D, Tang W, Monnat R.J., Jr, Stoddard BL. Conformational changes and cleavage by the homing endonuclease I-PpoI: a critical role for a leucine residue in the active site. J. Mol. Biol. 2000;300:877–887. doi: 10.1006/jmbi.2000.3874. [DOI] [PubMed] [Google Scholar]
- 64.Muscarella DE, Ellison EL, Ruoff BM, Vogt VM. Characterization of I-Ppo, an intron-encoded endonuclease that mediates homing of a group I intron in the ribosomal DNA of Physarum polycephalum. Mol. Cell Biol. 1990;10:3386–3396. doi: 10.1128/mcb.10.7.3386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wittmayer PK, McKenzie JL, Raines RT. Degenerate DNA recognition by I-PpoI endonuclease. Gene. 1998;206:11–21. doi: 10.1016/s0378-1119(97)00563-5. [DOI] [PubMed] [Google Scholar]
- 66.Swaminathan K, Flynn P, Reece RJ, Marmorstein R. Crystal structure of a PUT3-DNA complex reveals a novel mechanism for DNA recognition by a protein containing a Zn2Cys6 binuclear cluster. Nat. Struct. Biol. 1997;4:751–759. doi: 10.1038/nsb0997-751. [DOI] [PubMed] [Google Scholar]
- 67.Axelrod JD, Majors J, Brandriss MC. Proline-independent binding of PUT3 transcriptional activator protein detected by footprinting in vivo. Mol. Cell Biol. 1991;11:564–567. doi: 10.1128/mcb.11.1.564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Brandriss MC. Evidence for positive regulation of the proline utilization pathway in Saccharomyces cerevisiae. Genetics. 1987;117:429–435. doi: 10.1093/genetics/117.3.429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Marczak JE, Brandriss MC. Isolation of constitutive mutations affecting the proline utilization pathway in Saccharomyces cerevisiae and molecular analysis of the PUT3 transcriptional activator. Mol. Cell Biol. 1989;9:4696–4705. doi: 10.1128/mcb.9.11.4696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Marczak JE, Brandriss MC. Analysis of constitutive and noninducible mutations of the PUT3 transcriptional activator. Mol. Cell Biol. 1991;11:2609–2619. doi: 10.1128/mcb.11.5.2609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Siddiqui AH, Brandriss MC. The Saccharomyces cerevisiae PUT3 activator protein associates with proline-specific upstream activation sequences. Mol. Cell Biol. 1989;9:4706–4712. doi: 10.1128/mcb.9.11.4706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Walters KJ, Dayie KT, Reece RJ, Ptashne M, Wagner G. Structure and mobility of the PUT3 dimer. Nat. Struct. Biol. 1997;4:744–750. doi: 10.1038/nsb0997-744. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.