

Abstract
Base excision repair (BER) plays a critical role in the repair of bases damaged by oxidative metabolism or alkylating agents, such as those commonly utilized in cancer therapy. Incomplete BER generates intermediates that require activation of homology-dependent DNA repair to resolve. We investigated the effects of lithocholic acid, an inhibitor of the key BER enzyme, DNA polymerase β, in cells deficient in expression of the homology-dependent repair factor, BRCA2. In vitro studies show that lithocholic acid suppresses the DNA polymerase and 5′dRP lyase activities of DNA polymerase β by preventing the formation of a stable pol β-DNA complex, reducing BER effectiveness. Cytotoxicity assays based on colony formation revealed that lithocholic acid exhibits synergism with the alkylating agent, temozolomide, which engages BER through DNA methylation, and that the degree of synergism is increased in cells lacking functional BRCA2. BRCA2-deficient cells also showed heightened susceptibility to both lithocholic acid and temozolomide individually. The potentiation of temozolomide cytotoxicity by lithocholic acid owes to the conversion of single-stranded DNA breaks generated through incomplete BER of methylated nucleotides into double-stranded breaks during DNA replication, as indicated by γH2AX immunofluorescence. Death appears to be induced in co-treated cells through an accumulation of persistent double-stranded DNA breaks. Mutations of the BRCA2 gene have been extensively characterized and are present in various cancers, implying that inhibition of BER may offer a means to augment tumor selectivity in the use of conventional cancer therapies.
Introduction
Lithocholic acid (LCA), a secondary bile acid, has previously been identified as a potent inhibitor of mammalian DNA polymerase β (pol β) (1). In the absence of pol β, the base excision repair (BER) pathway is defective (2). BER is essential to the maintenance of genomic integrity, particularly with respect to oxidative damage and alkylation-induced lesions (3, 4). Pol β has also been demonstrated to catalyze the removal of a 5′-deoxyribose-phosphate (5′-dRP) group during short-patch (single-nucleotide gap) BER when base damage is recognized by a monofunctional DNA glycosylase (5). Inhibition of either the DNA polymerase or 5′-dRP lyase functionalities of pol β can therefore result in termination of short-patch BER.
The oral alkylating agent temozolomide (TMZ) has been extensively studied in the treatment of various tumors, including glioblastoma multiforme, metastatic melanoma, and refractory anaplastic astrocytoma (6–10). The antineoplastic efficacy of TMZ is dependent on its ability to methylate DNA, primarily at the targets O6-guanine, N7-guanine, and N3-adenine (11). Recent work has shown that inhibition of poly(ADP-ribose) polymerase (PARP) activity potentiates the cytotoxic effects of TMZ (12, 13). As the PARP family member PARP-1 functions to detect single-stranded DNA breaks (SSBs) and is required for recruitment of proteins to promote efficient BER (14, 15), the observed potentiation is presumably attributable to an inability to complete BER of deleterious methyl adducts.
Our goal was to determine whether disruption of the BER pathway through the inhibitory effect of LCA on pol β would produce a similar sensitivity to TMZ-induced DNA methylation. We further hypothesized that synergism of LCA with TMZ would be increased in cell lines lacking the ability to efficiently repair double-stranded DNA breaks (DSBs) owing to inactivation of the homology-dependent DNA repair (HDR) pathway. HDR utilizes homologous regions of DNA to repair DSBs and is typically considered to be an error-free pathway, in contrast to more error-prone pathways such as non-homologous end-joining (NHEJ) (16). HDR resolves collapsed replication forks that may arise from SSBs, such as those resulting from failed BER. To this end, we explored the individual and combined effects of LCA and TMZ in cell lines deficient in expression of the critical HDR factor, BRCA2.
The BRCA2 protein functions as a tumor suppressor through its involvement in the process of DSB repair via homologous recombination (17–19). BRCA2 interacts directly with RAD51 and facilitates the formation of helical RAD51–ssDNA filaments, which localize to template DNA to initiate repair (20, 21). Hereditary germline mutations in the BRCA2 gene contribute a significant predisposition to breast and ovarian cancers, arising from increased genetic instability and subsequent cellular transformation as a result of inefficient HDR (22–24). Additionally, BRCA2 silencing and mutational inactivation can contribute to sporadic tumorigenesis (25, 26). Thus, BRCA2-deficient cancers represent an attractive target for synergistic drug therapy.
Materials and Methods
Cell lines
VC-8 (BRCA2-deficient) and VC-8+BRCA2 complemented CHO cells were a gift of Dr. Graeme C. M. Smith of KuDOS Pharmaceuticals Limited (Cambridge, U.K.) and were maintained in DMEM supplemented with 10% FBS. EUFA423 (BRCA2-deficient) and EUFA423+BRCA2 complemented human fibroblasts were a gift of Dr. Simon Powell of the Memorial Sloan-Kettering Cancer Center (New York, NY) and were maintained in DMEM supplemented with 10% FBS, 20 mM HEPES, and 500 mg/mL G418 (complemented only). MEF cell lines 92TAg (WT), and 308TAg (Aag −/−) were a gift of Dr. Leona Samson of the Massachusetts Institute of Technology (Cambridge, MA) and were maintained in DMEM supplemented with 10% FBS and L-glutamine. All cells were cultured at 37°C in a 5% CO2 atmosphere.
Transfection, infection, and expression analysis
To infect the MEFs, the WT pol β and E295K constructs were packaged into retroviruses using the GP2-293 viral packaging cell line. Exponentially growing MEFs at approximately 50% confluence in 60-mm dishes were incubated with 1 mL of virus with 1 μg/mL Polybrene (Sigma) for 2 h. The medium was then removed and replaced with fresh medium containing 1 μg/ml Polybrene. After an overnight incubation, cells were split into 100-mm dishes at different cell densities. All dishes were incubated overnight and then supplemented with 400 μg/ml hygromycin B (Invitrogen) to select clones with stably integrated constructs. Tetracycline (Sigma) at a concentration of 4 μg/ml was also present in the medium to repress expression of the WT or E295K proteins. Individual cell colonies were cloned using cloning cylinders and expanded in the same growth medium.
Chemicals
Lithocholic acid (L6250) and temozolomide (76899) were purchased from Sigma-Aldrich. 100 mM stocks in DMSO were prepared fresh prior to each experiment, diluted as necessary, and added to culture medium at 0.1% v/v. All culture medium was limited to 5% FBS during drug treatment to inhibit LCA precipitation. Ultrapure deoxynucleoside triphosphates, ATP, and [-γ32P] ATP (>6000 Ci/mmol, 150 mCi/mL), cordycepin 5′-triphosphate [-α32P] dATP (>5000 Ci/mmol) were purchased from New England Biolabs, Sigma, and Perkin Elmer, respectively. Uracil DNA glycosylase (UDG) (M0280S), human AP endonuclease I (APE1) (M0282S), terminal transferase (M0252S), T4 polynucleotide kinase (M0201S), and T4 DNA ligase (M0202S) were purchased from New England Biolabs. Sodium borohydride (452882) was purchased from Sigma-Aldrich.
Preparation of DNA substrates
Oligonucleotides were synthesized by the W.M. Keck Facility at Yale University. The substrates used are shown in Table 1. The primer oligonucleotide was labeled at the 5′-end with T4 polynucleotide kinase and γ-32P ATP. Other oligonucleotides were 5′-end-phosphorylated with the kinase and cold ATP. After purification on a Biorad spin column to remove unincorporated dNTPs, annealing was performed by mixing phosphorylated template, radiolabeled primer, and phosphorylated downstream oligos in 50 mM Tris-HCl (pH 8.0) containing 0.25 M NaCl. The mixture was incubated sequentially at 95°C (5 min), slowly cooled to 50°C (for 30 min) and 50°C (for 20 min), and immediately transferred to ice. To verify proper hybridization, the product was analyzed on an 18% native polyacrylamide gel followed by autoradiography to assess the quality of annealing.
Table 1.
DNA substrates employed in primer extension, gel mobility shift, dRP lyase, and base excision repair assays.
| Substrate | Sequence |
|---|---|
| 45AG |
5′GCCTCGCAGCCGTCCAACCAAC CAACCTCGATCCAATGCCGTCC 3′ 3′ CGGAGCGTCGGCAGGTTGGTTGAGTTGGAGCTAGGTTACGGCAGG 5′ |
| LPSD |
5′ CTGCAGCTGATGCGCUGTACGGATCCCCGGGTAC 3′ 3′ GACGTCGACTACGCGGCATGCCTAGGGGCCCATG 5′ |
In vitro primer extension assay
Primer extension experiments were conducted in a solution containing 50 mM Tris-HCl buffer (pH 8.0), 5 mM MgCl2, 2 mM DTT, 20 mM NaCl, 10% glycerol, and 25μM dTTP. Enzyme-1 bp gapped DNA mix (10nM: 5nM) was incubated first with various concentrations of LCA (0–100 μM) and DMSO as control. The enzyme-DNA mixes were incubated with dTTP-MgCl2 and reactions were carried out at 37 °C for 20 min, after which they were stopped by addition of an equal volume of 90% formamide dye and 0.3 M EDTA. Samples were resolved by electrophoresis on 20% polyacrylamide gels containing 8 M urea, then visualized and quantified using a Storm 860 PhosphorImager (Molecular Dynamics, Inc.).
Preparation of 5′-dRP substrate
The 3′ end of DNA substrate LPSD, which contained a single U at position 19 from 3′ end, was radiolabeled using [-α32P] ATP and terminal transferase and annealed with its complementary oligonucleotide. This DNA duplex was treated with UDG (2 units/1 pmol DNA) in 50 mM HEPES (pH 7.5) at 37°C for 10 min, followed by treatment with human AP endonuclease, APE1 (2 units/1 pmol DNA) in buffer R (10 mM MgCl2, 20 mM KCl, and 2 mM DTT) at 37°C for 15 min, which incises the phosphodiester backbone on the 5′-side of the AP site and leaves a 3′-OH and a 5′-dRP residue. Due to the labile nature of the AP site, the 5′-dRP-containing DNA substrate was prepared just before use.
5′-dRP lyase assay
The assay was set up according to Prakash et al. with minor modifications (27). Typically, reaction mixtures (30 μL) contained the DNA polymerases (10 nM) and the 5′-dRP-containing DNA substrate (5 nM) in buffer R (50 mM HEPES (pH 7.5), 10 mM MgCl2, 20 mM KCl, and 2 mM DTT). Reactions were incubated at 37°C for 10 min. Various concentrations of LCA (0–100 μM) were added to a standard dRP lyase assay. The reaction product was stabilized by the addition of 2 M sodium borohydride to a final concentration of 340 mM, followed by incubation on ice for 30 min. Stabilized (reduced) DNA products were ethanol-precipitated in the presence of 0.1 μg/ml of tRNA, resuspended in water, and an equal volume of formamide dye was added. These products were resolved on a 20% polyacrylamide gel and visualized with a Storm 860 PhosphorImager (Molecular Dynamics, Inc.).
Base excision repair assay
5′-end labeled LPSD substrate (Table 1) was used as a BER substrate. For reconstituted BER with purified proteins, we followed the method as described by Prakash with some minor modifications (27). Typically, 100 nM UDG-treated substrate was first incubated for 15 min with commercially available APE1. Approximately 5 nM APE-treated substrate was incubated with purified 10 nM pol β in BER buffer (45 mM HEPES (pH 7.8), 70 mM KCl, 2mM DTT, 7.5 mM MgCl2, 0.5 mM EDTA, 2mM ATP, and 20 μM each of dATP, dTTP, dCTP and dGTP) and T4 DNA ligase with an increasing concentration of lithocholic acid (0–100 μM) for 15 min at 37°C. Finally, EDTA containing formamide dye was added to stop the reaction. The repaired product was resolved on a 20% denaturing polyacrylamide gel followed by visualization on a Storm 860 PhosphorImager (Molecular Dynamics, Inc.).
Gel mobility shift assay
Various concentrations of WT pol β protein (0.1–1000 nM) and 150 μM LCA were incubated with 0.1 nM radiolabeled gapped DNA substrate in buffer containing 50 mM Tris-HCl (pH 8.0), 100 mM NaCl, 10 mM MgCl2, 10% glycerol at room temperature (23°C) for 15 min. Samples were loaded onto a 6% native polyacrylamide gel with the current running at 300 V at 4°C. After the sample was loaded, the voltage was reduced to 150 V. Bound protein was quantified using Imagequant software after scanning the gel using a Storm 860 PhosphorImager (Molecular Dynamics, Inc.). Protein bound to DNA resulted in a shift of the DNA on the gel when compared to DNA without bound protein. Fraction bound is the ratio of the intensity of all shifted species divided by the total. The dissociation constant for DNA (KD) was estimated from fitting the bound protein (Y) versus protein concentration (x) with the equation: Y = [(mx)/(x + KD)] + b, where m is a scaling factor and b is the apparent minimum Y value.
Immunofluorescence
VC-8 and VC-8+BRCA2 complemented cells were seeded onto UV-irradiated coverslips (Corning) and allowed to adhere for 24 h, then co-treated with LCA and TMZ for 3 h. Cells were either prepared immediately or following a 24 h recovery in untreated medium to allow DSB repair. Procedures were performed under reduced light and all washes used 1x PBS. The culture medium was aspirated and cells were washed once and fixed with 4% formaldehyde for 20 min at RT, then washed again. Cells were blocked with 0.5% Triton X-100 buffer containing 3% BSA for 10 min at RT and then incubated with the following antibodies in PBS containing 3% BSA for 20 min at 37°C, with multiple washes after each step: rabbit anti-γH2AX antibody (1:500, Bethyl Laboratories) and FITC-conjugated F(ab′)2 fragment donkey anti-rabbit IgG (H+L) (1:100, Jackson ImmunoResearch). Cells were then stained with 100 ng/mL DAPI (Sigma) for 1 min at RT, washed, and the coverslips mounted on microscope slides using a 90% glycerol/10% PBS solution containing 0.01% p-phenylenediamine. Images were captured using an Axiovert 200 microscope (Carl Zeiss MicroImaging, Inc.).
Clonogenic survival assays
All cell lines were plated in triplicate at 500–1000 cells/well in 6-well plates to provide an optimal counting density. Cells were treated with TMZ for 24 h and with LCA for the entirety of the assay. Cells were cultured for 1–2 weeks until well-defined colonies had formed, replacing culture media every 2–3 days. Cells were briefly permeabilized with 0.9% saline solution and stained with crystal violet in 80% methanol. Colonies of ≥ 50 cells were then counted visually. Data points represent the average of the three values and error bars represent the standard deviation.
Results
LCA reduces BER activity via inhibition of pol β
LCA has been previously demonstrated to inhibit the polymerase activity of pol β. We verified that LCA inhibits pol β-catalyzed DNA polymerization in a dose-dependent manner by assaying the repair of a DNA substrate containing a 1-nt gap (Table 1). Polymerase activity was almost completely absent at an LCA concentration of 60 μM or greater (Fig. 1A). We further demonstrated that LCA inhibits 5′-dRP lyase catalysis by pol β using a prototypical lyase assay (Fig. 1B). A mixture of pol β and 5′-dRP substrate was allowed to react in the presence of increasing concentrations of LCA. In the presence of 60 μM LCA, product formation was reduced to 30% of baseline levels, indicating a similar dose response as for polymerase activity (Fig. 1B).
Fig. 1.

Inhibition of DNA polymerase β by lithocholic acid (A) i. 45AG DNA substrate (Table 1) was incubated with pol β and increasing concentrations of LCA to measure DNA polymerization activity. Formation of n+1 product was analyzed via gel electrophoresis. ii. n+1 polymerization product obtained at each LCA concentration, normalized to untreated sample. (B) i. LPSD DNA substrate (Table 1) was incubated with pol β and increasing concentrations of LCA to measure dRP lyase activity. Formation of 19-nt (no dRP) product was analyzed via gel electrophoresis. ii. 19-nt product obtained at each LCA concentration, normalized to untreated sample. (C) The binding of pol β to a DNA substrate containing a 1-bp gap was analyzed in the presence of DMSO and 150 μM LCA. (D) DNA dissociation constants (KD DNA) of wildtype pol β (33), pol β with DMSO, and pol β with 150 μM LCA, as measured from binding assay. ND: not determinable.
To assess whether this inhibition could be directly attributed to the inability of polβ to form a stable complex with DNA in the presence of LCA, the binding of pol β to a DNA substrate containing a 1-nt gap was assayed (Fig. 1C–D). The DNA dissociation constant (KD DNA) of wild-type pol β has been previously established to be 12±2 nM (28). In the presence of DMSO vehicle alone, the KD DNA increases to 78±14 nM. At 150 μM LCA the binding of pol β to DNA was dramatically reduced, and the KD DNA of pol β could not be determined, suggesting that no stable protein-DNA interaction is occurring.
We hypothesized that the inhibitory action of LCA on both the polymerase and 5′-dRP lyase functionalities of pol β represented an effective mechanism to inhibit the BER pathway. To determine the degree to which BER could be reduced by LCA, the repair of a DNA substrate containing a 1-nt gap was assayed in the presence of pol β, apurinic/apyrimidinic (AP) endonuclease, and T4 DNA ligase (Fig. 2A). The lower bands on the gel represent the starting DNA substrate before (n) and after (n+1) polymerization by pol β, while the highest band represents the completely repaired DNA product. The levels of each are represented by the band intensity. Complete BER of the substrate was reduced by 37.2% upon the addition of 60 μM LCA, relative to DMSO control (Fig. 2B).
Fig. 2.
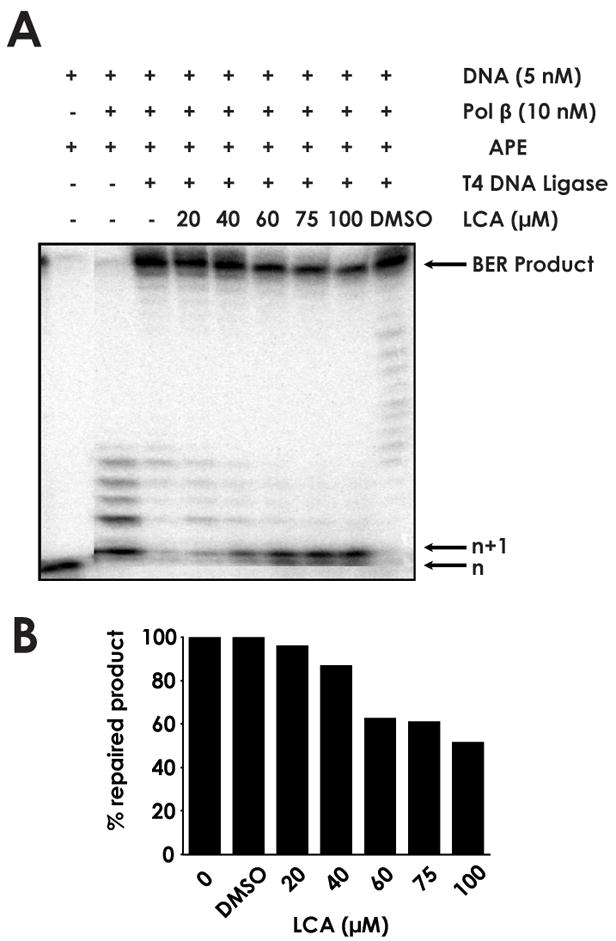
Inhibition of base excision repair by lithocholic acid (A) 45AG DNA substrate was incubated with pol β, apurinic/apyrimidinic (AP) endonuclease, T4 DNA ligase, and increasing concentrations of LCA to measure BER activity. Formation of n+1 product and repaired BER product were analyzed via gel electrophoresis. (B) BER product obtained at each LCA concentration, normalized to untreated sample.
LCA and TMZ synergism is increased in BRCA2-deficient cells
The HDR pathway is the primary cellular mechanism for error-free repair of DSBs and collapsed replication forks. We wished to determine whether inhibition of BER by LCA, with or without engagement of BER by the alkylating agent TMZ, would result in increased toxicity in HDR-deficient cells compared to HDR-proficient cells. We reasoned that blocking BER would yield strand-break intermediates that required HDR for resolution. We therefore treated human and CHO cell lines deficient in BRCA2 with increasing concentrations of LCA and/or TMZ in clonogenic assays. Cells were treated with TMZ for 24 h or with LCA for the duration of the assay.
In the BRCA2-deficient VC-8 CHO and EUFA423 human fibroblast cell lines, both LCA and TMZ individually exhibited increased cytotoxicity over the entire dose range relative to paired cell lines complemented with wild-type BRCA2 (Figs. 3A and 3B, respectively). Differences between the deficient and complemented lines were analyzed statistically via the Wilcoxon signed-rank test and were significant at the p < 0.001 level for LCA and TMZ treatment in VC-8 and TMZ treatment in EUFA423, and at the p < 0.005 level for LCA treatment in EUFA423. The data thus imply that the frequency of endogenous DNA lesions leads to sufficiently high levels of SSBs in the presence of LCA to yield lower cell survival when these breaks are converted to DSBs and remain unrepaired by HDR. Similarly, the potency of TMZ as an alkylating agent allows for sufficient quantities of methyl adducts to be generated and lead to unrepaired DNA lesions even in the presence of a functional BER pathway, conferring an increased sensitivity in BRCA2-deficient cell lines.
Fig. 3.

Cytotoxicity and synergism of lithocholic acid and temozolomide in BRCA2-deficient cells. (A) Clonogenic survival of VC-8 (CHO BRCA2-deficient) and VC-8+BRCA2 cells in response to increasing doses of LCA and TMZ, individually. (B) Clonogenic survival of EUFA423 (human BRCA2-deficient) and EUFA423+BRCA2 cells in response to LCA and TMZ, individually. (C) Synergism of LCA and TMZ in VC-8 and VC-8+BRCA2 cells. Analysis using the Chou-Talalay model yielded CI values of 0.43 and 0.79, respectively, mathematically proving synergism. (D) Synergism of LCA and TMZ in EUFA423 and EUFA423+BRCA2 cells. Analysis using the Chou-Talalay model yielded CI values of 0.58 and 0.71, respectively, mathematically proving synergism.
Having established BRCA2-deficient cell lines as susceptible to both LCA and TMZ individually, we hypothesized that the two agents would exhibit a synergistic effect by creating an inability to complete the repair of any methyl adducts formed. Stalling and subsequent collapse of replication forks as cells containing unrepaired SSBs (owing to ineffective BER) progressed into S phase and began to undergo DNA replication would provide an increased frequency of DSBs. Cells were treated with TMZ for 24 h or LCA for the duration of the assay or both, and clonogenic survival was measured. Synergism was assessed mathematically using the Chou-Talalay mutually nonexclusive model (29). When data are fit using this method, combination index (CI) values < 1, = 1, and > 1 are indicative of synergism, additive effect, and antagonism, respectively. At 10 μM TMZ and 60 μM LCA, VC-8 and VC-8+BRCA2 cells yielded CI values of 0.43 and 0.79, respectively (Fig. 3B). At 20 μM TMZ and 80 μM LCA, EUFA423 and EUFA423+BRCA2 yielded CI values of 0.58 and 0.71, respectively (Fig. 3D). The model thus indicates synergism between LCA and TMZ in each cell line examined and suggests that the synergism was greater in BRCA2-deficient cells relative to BRCA2-complemented cells in both sets of paired lines.
Additional cell lines deficient in the HDR factors FANCA (PD220) and BRCA1 (UWB1.289) were also investigated and were weakly sensitive to TMZ individually but showed no significant increase in LCA/TMZ synergism relative to cell lines complemented with the respective HDR proteins (data not shown).
The in vivo effectiveness of LCA is specifically related to pol β inhibition
To assess whether the synergism of LCA with TMZ in BRCA2-deficient cells is directly attributable to inhibition specifically of pol β rather than another polymerase target, we utilized MEF cell lines exogenously expressing either wild-type pol β or the dominant-negative E295K mutant of pol β, which has previously been shown to render cells deficient in BER (30).
We treated the two cell lines with TMZ in the presence of either 50 μM LCA or DMSO vehicle alone and observed the response in a clonogenic survival assay. In the cells expressing wild-type pol β, there was a significant difference in survival at the p < 0.05 level depending on the treatment or not with LCA (Fig. 4A). However, in the cells also expressing the E295K dominant-negative mutant pol β, no significant difference was observed between LCA- or DMSO-treated cells, indicating that the effect of LCA is eliminated when the BER capacity of pol β is already inhibited by the dominant-negative pol β variant.
Fig. 4.
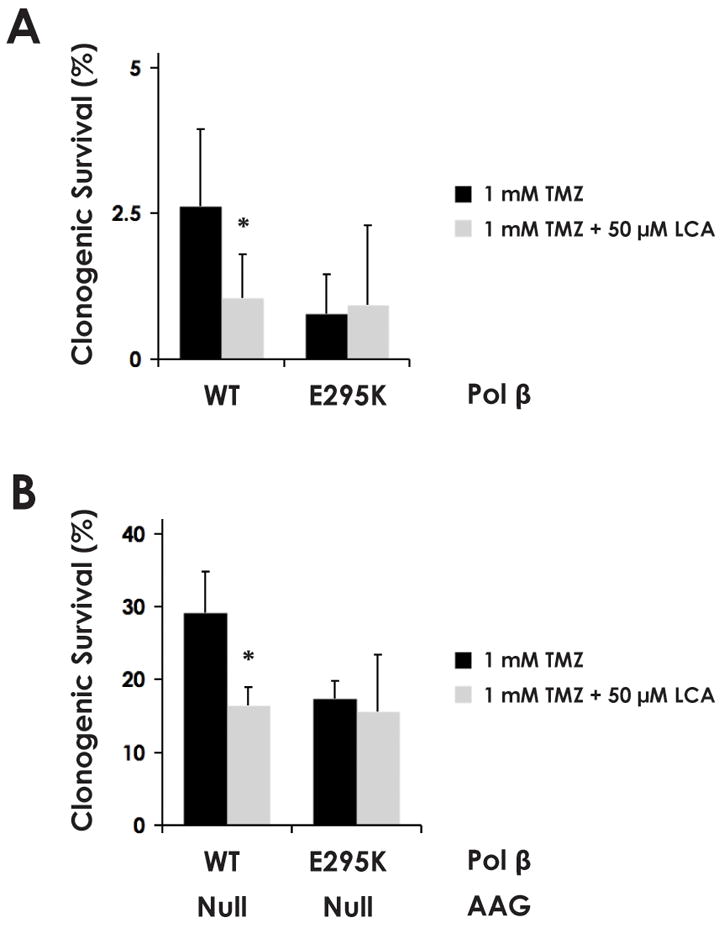
The synergistic effect of LCA on the cytotoxicity of TMZ depends on a functional pol β. Clonogenic survival of AAG-wild-type (A) and AAG-null (B) MEF cell lines modified to stably express exogenous wild-type or dominant negative (E295K-mutant) pol β, as indicated. Statistically significant differences at the p < 0.05 level (*) between treatment with 1 mM TMZ or 1 mM TMZ + 50 μM LCA were observed only in cells expressing wild-type pol β. The BER-deficient E295K mutants exhibited no significant difference in cytotoxicity between the treatments, indicating that LCA exerted no cytotoxic effects in the absence of wild-type pol β function.
Approximately 80% of the lesions induced by TMZ are N-methylated purines (31). Alkyladenine DNA glycosylase (AAG) is the primary DNA glycosylase responsible for recognizing TMZ-induced DNA lesions, and its knockout serves to mitigate TMZ cytotoxicity by preventing the creation of abasic sites. In an AAG-null background (308 cells), the overall levels of survival are higher as compared to the AAG wild-type cells for each experimental condition (Fig. 4B). This is as expected, since, in the absence of AAG, the production of abasic sites as an initial step in BER is substantially reduced, and so survival is improved (even though such cells will likely have increased mutation rates). However, as with the AAG-wild-type cells, in the AAG-null cells expressing wild-type pol β, there is still an effect of LCA on survival to TMZ. In contrast, in the cells expressing the dominant-negative pol β, there is no effect of LCA, again showing that the potentiation of TMZ cytotoxicity by LCA depends specifically on a functional pol β. These data strongly indicate that pol β inhibition is the underlying mechanism for potentiation of TMZ by LCA, rather than an effect on another polymerase or other cellular target.
LCA and TMZ co-treatment produces persistent DSB in the absence of BRCA2
Immunofluorescence analysis of phosphorylated histone H2AX (γH2AX) foci formation was used to measure DSB accumulation in LCA- and TMZ-treated VC-8 (BRCA2-deficient) and VC-8+BRCA2 complemented cells (Fig. 5A). The cells were treated for 3 h and were either analyzed immediately or following a 24 h recovery period to facilitate DNA repair and allow all cells to enter S phase. Relatively low frequencies of γH2AX foci were observable immediately following treatment and occurred at similar levels in both cell lines (Fig. 5B). Treatment with DMSO vehicle or 20 μM LCA induced fewer γH2AX foci than treatment with 20 μM TMZ or 20 μM LCA+20 μM TMZ.
Fig. 5.

Generation and repair of DSB in BRCA2-deficient and complemented cells by LCA and/or TMZ (A) VC-8 (BRCA2-deficient) and VC-8+BRCA2 cells were incubated with 20 μM LCA, 20 μM TMZ, 20 μM LCA+20 μM TMZ, or DMSO vehicle for 3 h and fixed immediately or after 24 h recovery in untreated media for DAPI staining and γH2AX immunofluorescence. Images shown are representative of the majority of cells on coverslip. (B) The fraction of cells exhibiting >15 γH2AX foci was determined. At least 50 cells were analyzed for each treatment condition.
Following the 24 h recovery period, continued DSB generation and persistence was markedly higher in VC-8 than in VC-8+BRCA2. When treated with TMZ alone, foci levels in VC-8+BRCA2 cells fell by 33% after 24 h while foci levels in VC-8 cells rose by 157%. Under combined LCA and TMZ treatment, foci levels in VC-8+BRCA2 cells fell by 48% and foci levels in VC-8 cells rose by 280%. The data agree with our prediction that in the absence of HDR, DSBs produced by LCA and/or TMZ treatment remain unrepaired and the quantity of cells exhibiting γH2AX foci increases as more cells enter S phase and additional breaks are generated. In cells with intact BRCA2 expression, the HDR pathway actively repairs DSBs soon after they occur, reducing the number of cells exhibiting γH2AX foci at the 24 h time point. In addition, the results show that pol β inhibition by LCA leads to even greater generation of DSBs, as would be expected from the higher levels of unrepaired BER intermediates.
Neutral pH comet assays were also employed to assess the generation of LCA-and/or TMZ-induced DSBs in BRCA2-deficient cells and yielded results consistent with γH2AX immunofluorescence (data not shown). Under neutral pH conditions, the DNA is not subjected to alkaline unwinding and the assay does not detect SSBs. Increased comet tail moments therefore indicate a greater quantity of DSBs in cells.
Discussion
The discovery of novel chemotherapeutic adjuvants represents an important step toward improving options for cancer treatment. Here we have shown that lithocholic acid can be used to potentiate the effectiveness of a well-known chemotherapeutic agent in cells exhibiting a prevalent tumorigenic mutation. We observed that LCA reduces BER activity via inhibition of the DNA polymerase and the 5′-dRP lyase activities of pol β. γH2AX immunofluorescence demonstrates that LCA and TMZ co-treatment generates persistent DSBs that can only be repaired effectively when the HDR pathway is functional. To the best of our knowledge, this is also the first study to demonstrate the increased susceptibility of BRCA2-deficient cell lines to treatment with TMZ or LCA.
The base excision repair pathway has previously been identified as a target for temozolomide potentiation using inhibitors of the PARP protein family (12, 13). Our work serves to corroborate molecular targeting of BER as a viable methodology in cancer therapy. Although the PARP enzymes PARP-1 and PARP-2 are known to promote BER activity, their specific roles within the pathway remain uncharacterized. The biochemical function of pol β and the effects of its inactivation on BER have been extensively documented (32–35). We show here that the DNA polymerase and 5′-dRP lyase functionalities of pol β in short-patch BER represent distinct targets of LCA inhibition. The inability of pol β to complex with template DNA in the presence of LCA is consistent with earlier NMR characterization showing that pol β-LCA interaction occurs through the N-terminal 8-kDa template-binding domain of pol β but not the 31-kDa catalytic domain (36).
Temozolomide generates a relatively narrow spectrum of DNA base damage. N7-methylguanine and N3-methyladenine, composing approximately 80% of the induced lesions, are recognized by DNA glycosylases and are therefore substrates for the BER pathway (37, 38). In addition to exogenous damage, an average cell may experience as many as 10,000 spontaneous DNA lesions each day (39). TMZ cytotoxicity in HDR-deficient cells appears to be a result of the inability of BER to efficiently repair the vast number of exogenous and endogenous lesions, leading to the accumulation of BER intermediates and subsequent formation of DSBs. The γH2AX immunofluorescence data presented here demonstrate that TMZ treatment leads to persistent DSBs even in the absence of LCA inhibition, which are likely responsible for cytotoxicity through induction of apoptotic pathways. The toxicity of LCA alone presumably relies on the frequency of endogenous lesions to generate DSBs. BRCA2-deficient cells are more susceptible to LCA than proficient cells, and this sensitivity is increased when also treated with TMZ.
The lowered efficacy of TMZ treatment in some tumors has generated much interest in cellular pathways mediating DNA alkylation resistance. Promoter methylation of the gene encoding O6-methylguanine-DNA methyltransferase (MGMT), which directly repairs the TMZ-induced O6-MeG lesion, is associated with improved prognosis and increased sensitivity to alkylation treatment (40–42). Because the predominant N7-MeG and N3-MeA lesions are BER rather than MGMT substrates, inhibition of BER activity could potentially enhance the effectiveness of TMZ as a chemotherapeutic agent by increasing the number of lesions that progress into DSBs. The use of LCA and other BER inhibitors as adjuvants to treatment with TMZ or similar alkylating agents offers a promising means to increase efficacy in resistant tumors. Besides LCA, we have also tested the chemically related betulinic acid and oleanolic acid. However, neither yielded differential effects on BRCA2-deficient versus proficient cell lines (data not shown).
Defects in homology-dependent DNA repair represent a significant subset of tumorigenic germline mutations. Certain inherited loss-of-function mutations in BRCA1 and BRCA2 have been reported to confer as high as 81% and 85% risks of breast cancer and 54% and 23% risks of ovarian cancer by age 80, respectively, while the lifetime risks of breast and ovarian cancer among women in general have been estimated at 12.5% and 1.8% (43, 44). Evidence is also accumulating for the involvement of BRCA2 mutations in predisposition to numerous other cancers in both males and females (45–47). BRCA2 is also known to play a role in sporadic tumorigenesis, as it has been shown to be silenced by binding of the EMSY protein to exon 3 in sporadic breast and ovarian cancer, in which the EMSY gene is amplified in 13% and 17% of tumors, respectively (26). This amplification leads to increased silencing of the BRCA2 gene and has been correlated with a negative disease prognosis (48). Treatment strategies specifically exploiting inefficient HDR in BRCA2-deficient tumors thus have the potential to improve the specificity and efficacy of cancer therapy while simultaneously minimizing healthy tissue toxicity.
Acknowledgments
We thank S. Rockwell, A. Sartorelli, and D. Kidane for helpful insights. This work was supported by grants from the USPHS (P01CA129186 to PMG and JBS and R01ES05775 to PMG).
This work was supported by grants from the USPHS (P01CA129186 to PMG and JBS and R01ES05775 to PMG).
References
- 1.Ogawa A, Murate T, Suzuki M, Nimura Y, Yoshida S. Lithocholic acid, a putative tumor promoter, inhibits mammalian DNA polymerase beta. Jpn J Cancer Res. 1998;89:1154–9. doi: 10.1111/j.1349-7006.1998.tb00510.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sobol RW, Horton JK, Kuhn R, et al. Requirement of mammalian DNA polymerase-beta in base-excision repair. Nature. 1996;379:183–6. doi: 10.1038/379183a0. [DOI] [PubMed] [Google Scholar]
- 3.Barnes DE, Lindahl T. Repair and genetic consequences of endogenous DNA base damage in mammalian cells. Annual review of genetics. 2004;38:445–76. doi: 10.1146/annurev.genet.38.072902.092448. [DOI] [PubMed] [Google Scholar]
- 4.Sung JS, Demple B. Roles of base excision repair subpathways in correcting oxidized abasic sites in DNA. The FEBS journal. 2006;273:1620–9. doi: 10.1111/j.1742-4658.2006.05192.x. [DOI] [PubMed] [Google Scholar]
- 5.Srivastava DK, Berg BJ, Prasad R, et al. Mammalian abasic site base excision repair. Identification of the reaction sequence and rate-determining steps. The Journal of biological chemistry. 1998;273:21203–9. doi: 10.1074/jbc.273.33.21203. [DOI] [PubMed] [Google Scholar]
- 6.Bleehen NM, Newlands ES, Lee SM, et al. Cancer Research Campaign phase II trial of temozolomide in metastatic melanoma. J Clin Oncol. 1995;13:910–3. doi: 10.1200/JCO.1995.13.4.910. [DOI] [PubMed] [Google Scholar]
- 7.Yung WK. Temozolomide in malignant gliomas. Seminars in oncology. 2000;27:27–34. [PubMed] [Google Scholar]
- 8.Agarwala SS, Kirkwood JM. Temozolomide, a novel alkylating agent with activity in the central nervous system, may improve the treatment of advanced metastatic melanoma. The oncologist. 2000;5:144–51. doi: 10.1634/theoncologist.5-2-144. [DOI] [PubMed] [Google Scholar]
- 9.Middleton MR, Grob JJ, Aaronson N, et al. Randomized phase III study of temozolomide versus dacarbazine in the treatment of patients with advanced metastatic malignant melanoma. J Clin Oncol. 2000;18:158–66. doi: 10.1200/JCO.2000.18.1.158. [DOI] [PubMed] [Google Scholar]
- 10.Biasco G, Pantaleo MA, Casadei S. Treatment of brain metastases of malignant melanoma with temozolomide. The New England journal of medicine. 2001;345:621–2. doi: 10.1056/NEJM200108233450817. [DOI] [PubMed] [Google Scholar]
- 11.D’Atri S, Tentori L, Lacal PM, et al. Involvement of the mismatch repair system in temozolomide-induced apoptosis. Molecular pharmacology. 1998;54:334–41. doi: 10.1124/mol.54.2.334. [DOI] [PubMed] [Google Scholar]
- 12.Palma JP, Rodriguez LE, Bontcheva-Diaz VD, et al. The PARP inhibitor, ABT-888 potentiates temozolomide: correlation with drug levels and reduction in PARP activity in vivo. Anticancer research. 2008;28:2625–35. [PubMed] [Google Scholar]
- 13.Liu X, Shi Y, Guan R, et al. Potentiation of temozolomide cytotoxicity by poly(ADP)ribose polymerase inhibitor ABT-888 requires a conversion of single-stranded DNA damages to double-stranded DNA breaks. Mol Cancer Res. 2008;6:1621–9. doi: 10.1158/1541-7786.MCR-08-0240. [DOI] [PubMed] [Google Scholar]
- 14.de Murcia JM, Niedergang C, Trucco C, et al. Requirement of poly(ADP-ribose) polymerase in recovery from DNA damage in mice and in cells. Proc Natl Acad Sci U S A. 1997;94:7303–7. doi: 10.1073/pnas.94.14.7303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Virag L, Szabo C. The therapeutic potential of poly(ADP-ribose) polymerase inhibitors. Pharmacological reviews. 2002;54:375–429. doi: 10.1124/pr.54.3.375. [DOI] [PubMed] [Google Scholar]
- 16.Wyman C, Kanaar R. DNA double-strand break repair: all’s well that ends well. Annual review of genetics. 2006;40:363–83. doi: 10.1146/annurev.genet.40.110405.090451. [DOI] [PubMed] [Google Scholar]
- 17.Liu Y, West SC. Distinct functions of BRCA1 and BRCA2 in double-strand break repair. Breast Cancer Res. 2002;4:9–13. doi: 10.1186/bcr417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Patel KJ, Yu VP, Lee H, et al. Involvement of Brca2 in DNA repair. Mol Cell. 1998;1:347–57. doi: 10.1016/s1097-2765(00)80035-0. [DOI] [PubMed] [Google Scholar]
- 19.Moynahan ME, Pierce AJ, Jasin M. BRCA2 is required for homology-directed repair of chromosomal breaks. Mol Cell. 2001;7:263–72. doi: 10.1016/s1097-2765(01)00174-5. [DOI] [PubMed] [Google Scholar]
- 20.Lord CJ, Ashworth A. RAD51, BRCA2 and DNA repair: a partial resolution. Nat Struct Mol Biol. 2007;14:461–2. doi: 10.1038/nsmb0607-461. [DOI] [PubMed] [Google Scholar]
- 21.Shivji MK, Venkitaraman AR. DNA recombination, chromosomal stability and carcinogenesis: insights into the role of BRCA2. DNA repair. 2004;3:835–43. doi: 10.1016/j.dnarep.2004.03.008. [DOI] [PubMed] [Google Scholar]
- 22.Krainer M, Silva-Arrieta S, FitzGerald MG, et al. Differential contributions of BRCA1 and BRCA2 to early-onset breast cancer. The New England journal of medicine. 1997;336:1416–21. doi: 10.1056/NEJM199705153362003. [DOI] [PubMed] [Google Scholar]
- 23.Rahman N, Stratton MR. The genetics of breast cancer susceptibility. Annual review of genetics. 1998;32:95–121. doi: 10.1146/annurev.genet.32.1.95. [DOI] [PubMed] [Google Scholar]
- 24.Welcsh PL, King MC. BRCA1 and BRCA2 and the genetics of breast and ovarian cancer. Hum Mol Genet. 2001;10:705–13. doi: 10.1093/hmg/10.7.705. [DOI] [PubMed] [Google Scholar]
- 25.Weber BH, Brohm M, Stec I, Backe J, Caffier H. A somatic truncating mutation in BRCA2 in a sporadic breast tumor. Am J Hum Genet. 1996;59:962–4. [PMC free article] [PubMed] [Google Scholar]
- 26.Hughes-Davies L, Huntsman D, Ruas M, et al. EMSY links the BRCA2 pathway to sporadic breast and ovarian cancer. Cell. 2003;115:523–35. doi: 10.1016/s0092-8674(03)00930-9. [DOI] [PubMed] [Google Scholar]
- 27.Haracska L, Prakash L, Prakash S. A mechanism for the exclusion of low-fidelity human Y-family DNA polymerases from base excision repair. Genes Dev. 2003;17:2777–85. doi: 10.1101/gad.1146103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dalal S, Chikova A, Jaeger J, Sweasy JB. The Leu22Pro tumor-associated variant of DNA polymerase beta is dRP lyase deficient. Nucleic Acids Res. 2008;36:411–22. doi: 10.1093/nar/gkm1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chou TC. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacological reviews. 2006;58:621–81. doi: 10.1124/pr.58.3.10. [DOI] [PubMed] [Google Scholar]
- 30.Lang T, Dalal S, Chikova A, DiMaio D, Sweasy JB. The E295K DNA polymerase beta gastric cancer-associated variant interferes with base excision repair and induces cellular transformation. Mol Cell Biol. 2007;27:5587–96. doi: 10.1128/MCB.01883-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Trivedi RN, Almeida KH, Fornsaglio JL, Schamus S, Sobol RW. The role of base excision repair in the sensitivity and resistance to temozolomide-mediated cell death. Cancer Res. 2005;65:6394–400. doi: 10.1158/0008-5472.CAN-05-0715. [DOI] [PubMed] [Google Scholar]
- 32.Wilson SH, Sobol RW, Beard WA, Horton JK, Prasad R, Vande Berg BJ. DNA polymerase beta and mammalian base excision repair. Cold Spring Harb Symp Quant Biol. 2000;65:143–55. doi: 10.1101/sqb.2000.65.143. [DOI] [PubMed] [Google Scholar]
- 33.Clairmont CA, Sweasy JB. Dominant negative rat DNA polymerase beta mutants interfere with base excision repair in Saccharomyces cerevisiae. J Bacteriol. 1996;178:656–61. doi: 10.1128/jb.178.3.656-661.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cabelof DC, Guo Z, Raffoul JJ, et al. Base excision repair deficiency caused by polymerase beta haploinsufficiency: accelerated DNA damage and increased mutational response to carcinogens. Cancer Res. 2003;63:5799–807. [PubMed] [Google Scholar]
- 35.Dianova, Sleeth KM, Allinson SL, et al. XRCC1-DNA polymerase beta interaction is required for efficient base excision repair. Nucleic Acids Res. 2004;32:2550–5. doi: 10.1093/nar/gkh567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mizushina Y, Ohkubo T, Sugawara F, Sakaguchi K. Structure of lithocholic acid binding to the N-terminal 8-kDa domain of DNA polymerase beta. Biochemistry. 2000;39:12606–13. doi: 10.1021/bi001276m. [DOI] [PubMed] [Google Scholar]
- 37.Tentori L, Graziani G. Pharmacological strategies to increase the antitumor activity of methylating agents. Curr Med Chem. 2002;9:1285–301. doi: 10.2174/0929867023369916. [DOI] [PubMed] [Google Scholar]
- 38.Wood RD, Mitchell M, Lindahl T. Human DNA repair genes, 2005. Mutat Res. 2005;577:275–83. doi: 10.1016/j.mrfmmm.2005.03.007. [DOI] [PubMed] [Google Scholar]
- 39.Sweasy JB, Lang T, Starcevic D, et al. Expression of DNA polymerase {beta} cancer-associated variants in mouse cells results in cellular transformation. Proc Natl Acad Sci U S A. 2005;102:14350–5. doi: 10.1073/pnas.0505166102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Belanich M, Pastor M, Randall T, et al. Retrospective study of the correlation between the DNA repair protein alkyltransferase and survival of brain tumor patients treated with carmustine. Cancer Res. 1996;56:783–8. [PubMed] [Google Scholar]
- 41.Silber JR, Blank A, Bobola MS, Ghatan S, Kolstoe DD, Berger MS. O6-methylguanine-DNA methyltransferase-deficient phenotype in human gliomas: frequency and time to tumor progression after alkylating agent-based chemotherapy. Clin Cancer Res. 1999;5:807–14. [PubMed] [Google Scholar]
- 42.Esteller M, Garcia-Foncillas J, Andion E, et al. Inactivation of the DNA-repair gene MGMT and the clinical response of gliomas to alkylating agents. The New England journal of medicine. 2000;343:1350–4. doi: 10.1056/NEJM200011093431901. [DOI] [PubMed] [Google Scholar]
- 43.King MC, Marks JH, Mandell JB. Breast and ovarian cancer risks due to inherited mutations in BRCA1 and BRCA2. Science. 2003;302:643–6. doi: 10.1126/science.1088759. [DOI] [PubMed] [Google Scholar]
- 44.Feuer EJ, Wun LM, Boring CC, Flanders WD, Timmel MJ, Tong T. The lifetime risk of developing breast cancer. J Natl Cancer Inst. 1993;85:892–7. doi: 10.1093/jnci/85.11.892. [DOI] [PubMed] [Google Scholar]
- 45.Liede A, Karlan BY, Narod SA. Cancer risks for male carriers of germline mutations in BRCA1 or BRCA2: a review of the literature. J Clin Oncol. 2004;22:735–42. doi: 10.1200/JCO.2004.05.055. [DOI] [PubMed] [Google Scholar]
- 46.Levy-Lahad E, Friedman E. Cancer risks among BRCA1 and BRCA2 mutation carriers. Br J Cancer. 2007;96:11–5. doi: 10.1038/sj.bjc.6603535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jakubowska A, Nej K, Huzarski T, Scott RJ, Lubinski J. BRCA2 gene mutations in families with aggregations of breast and stomach cancers. Br J Cancer. 2002;87:888–91. doi: 10.1038/sj.bjc.6600562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rodriguez C, Hughes-Davies L, Valles H, et al. Amplification of the BRCA2 pathway gene EMSY in sporadic breast cancer is related to negative outcome. Clin Cancer Res. 2004;10:5785–91. doi: 10.1158/1078-0432.CCR-03-0410. [DOI] [PubMed] [Google Scholar]