

Abstract
Importance of the field
The focal adhesion tyrosine kinases FAK and Pyk2 are uniquely situated to act as critical mediators for the activation of signaling pathways that regulate cell migration, proliferation, and survival. By coordinating adhesion and cytoskeletal dynamics with survival and growth signaling, FAK and Pyk2 represent molecular therapeutic targets in cancer as malignant cells often exhibit defects in these processes.
Areas covered in this review
This review examines the structure and function of the focal adhesion kinase Pyk2 and intends to provide a rationale for the employ of modulating strategies that include both catalytic and extra-catalytic approaches that have been developed in the last 3–5 years.
What the reader will gain
Targeting tyrosine kinases in oncology has focused on the ATP binding pocket as means to inhibit catalytic activity and down-regulate pathways involved in tumor invasion. This review will discuss the available catalytic inhibitors and compare them to the alternative approach of targeting protein-protein interactions that regulate kinase activity
Take home message
Development of specific catalytic inhibitors of the focal adhesion kinases has improved but significant challenges remain. Thus, approaches that inhibit the effector function of Pyk2 by targeting regulatory modules can increase specificity and will be a welcome asset to the therapeutic arena.
Keywords: adhesion, focal adhesion kinase, glioma, invasion, inhibitors, migration, Pyk2, tyrosine kinases
1. Introduction
Integrin mediated adhesion to the extracellular matrix plays a central role in the regulation of a number of cellular activities including survival, proliferation, and migration. By linking the extracellular matrix to the actin cytoskeleton, integrins provide a mechanical basis for productive force generation necessary for cell motility. In addition, information from the adhesive interaction is translated into the activation of cellular signaling pathways through the formation of the focal adhesion complex 1. Focal adhesions are multi-protein scaffolds enriched in signaling effectors that mediate signaling inputs. Focal adhesions also serve as a point of convergence for signaling initiated at growth factor and G-protein linked receptors to coordinately regulate cell growth and migration. These complexes exert observable effects on cellular phenotype by sensing not only the local extracellular milieu, but also physical and topographical features (membrane/cytoskeletal flexibility, temporal-spatial dimensionality, and the relationships to extracellular ligands) of the cell within the context of the surrounding environment 2. Coordination and turnover of these contact points involves a series of catalytically-active scaffolding proteins and specific lipids that serve in the transduction of bi-directional (outside-in and inside-out) signaling that ultimately influences cellular behavior 3, 4. The focal adhesion kinases FAK and Pyk2 are uniquely situated to act as critical mediators for the activation of signaling pathways that regulate cell migration, proliferation, and survival. By coordinating adhesion and cytoskeletal dynamics with survival and growth signaling, FAK and Pyk2 represent molecular therapeutic targets in cancer cells as malignant cells often exhibit defects in these processes.
2. The Focal Adhesion Kinases
The prototypical signaling effector coupling integrin-matrix interactions with signaling events at the focal adhesion is the focal adhesion kinase (FAK) 5. FAK is a ubiquitously expressed 125-kDa non-receptor tyrosine kinase that is rapidly phosphorylated following integrin engagement with concomitant increases in its kinase activity 6, 7. Activation of FAK can also occur by non-integrin pathways including a number of growth factors, cytokines, bioactive lipids and neuropeptides 8, 9 Phosphorylation of conserved tyrosine residues provides binding sites for SH2 containing proteins notably Src-family tyrosine kinases (Tyr397) and the Grb2 adaptor protein (Tyr925). Src binding to phosphorylated Tyr397 of FAK leads to full activation of both kinases and the formation of a signaling complex that phosphorylates other focal adhesion proteins including paxillin and p130Cas. A better understanding of the molecular mechanism of FAK activation has been elucidated by structural studies demonstrating that the N-terminal FERM (band Four point one, Ezrin, Radixin, Moesin) domain binds directly to the kinase domain effectively blocking access to the catalytic cleft 10, 11. FAK activation has been proposed to be initiated by displacement of the FERM domain from the kinase domain by the binding of an as yet unidentified activating component to the FERM. This hypothesis is supported by the recent observation that the interaction of the FAK FERM domain with acidic phospholipids induced conformational changes in FAK and is associated with FAK activation 12. Proper subcellular localization is also a critical component of FAK signaling and involves the interaction of the C-terminal focal adhesion targeting (FAT) domain, a four helical bundle, with the integrin associated proteins paxillin and talin 13, 14. Increased expression of FAK has been reported in a number of metastatic cancers and overexpression correlates with invasive phenotypes 15–21. The numerous contributions of FAK to cellular adhesion, motility, cell cycle regulation, and abrogation of programmed cell death have increased its potential as a target for therapeutic intervention 22–25.
A second member of the FAK-family was identified near-contemporaneously by several groups and designated Pyk2 (Proline-rich tyrosine kinase), CAKβ (cell adhesion kinase beta), RAFTK (related adhesion focal tyrosine kinase), or FAK2 26–30. The human Pyk2 gene was found to reside on chromosome 8 (8p21.1; gene reference: PTK2B) 27, 31. These studies demonstrated that Pyk2 shares a similar domain structure with FAK and exhibits ~45% sequence identity over the length of the molecule. The greatest sequence identity, ~60%, is found within the central kinase domain. Despite their structural similarity, Pyk2 and FAK display a number of significant differences. While FAK is ubiquitously expressed, Pyk2 exhibits a more limited tissue distribution. Pyk2 expression has been reported in a number of cell types with expression being highest in cells of hematopoietic lineage and in the CNS 26, 28, 32. Interestingly, knockdown of FAK expression in certain cell types has been accompanied by a compensatory increase of Pyk2 expression 33–35. Notably, the activation of Pyk2 also differs from that of FAK. FAK is primarily activated following integrin mediated adhesion to ECM. In contrast, although Pyk2 can be activated following integrin mediated adhesion, Pyk2 is primarily activated in response to a variety of stimuli that increase intracellular calcium 28, 32, 36, 37. Intracellular distribution of Pyk2 is also different than that of FAK. Although Pyk2 is localized in focal adhesions in certain cell types, it is more commonly found distributed throughout the cell and is often enriched in perinuclear regions 38, 39. Thus, although the FAT regions of FAK and Pyk2 share ~40% identity, these results suggest that they may interact with different proteins resulting in different intracellular localization. Alternatively, Pyk2 may possess an additional targeting sequence that overrides the interactions mediated by the FAT domain in a cell type dependent manner. Similar to FAK, upregulation of Pyk2 expression has been noted in several human tumors. Pyk2 expression is elevated in gliomas and expression is increased with advancing WHO grade 40. Pyk2 overexpression in hepatocellular carcinoma correlated with increased metastasis and reduced survival 41, 42. Similarly, Pyk2 expression is upregulated in non-small cell lung cancer and correlated with higher metastatic potential 43 while knockdown of Pyk2 expression inhibited anchorage independent survival and proliferation of small cell lung cancer cells 44. Pyk2 expression is significantly increased in early and advanced breast cancer and co-overexpressed with ErbB-2 in early stage DCIS and invasive breast cancer 45. Taken in total, Pyk2 is well positioned as a potential target for disease modulation, particularly as it pertains to invasive cancers, osteoporosis, and inflammatory cellular responses. This article reviews the pertinent biological aspects justifying programs for the development of inhibitory strategies that target Pyk2.
3. Pyk2 structure and functional domains
Although a high resolution structure of full length Pyk2 has not been reported, a number of functional domains have been identified and characterized (Figure 1). These functional domains include a N-terminal FERM domain, a central catalytic domain, a number of proline rich sequences, and the C-terminal focal adhesion targeting domain. High resolution structures have been solved for several of these individual functional domains.
Figure 1.

Schematic of Pyk2 functional domains and interacting proteins. Pyk2 contains an N-terminal FERM domain, a central kinase domain, three proline rich motifs (PR1, Pr2, PR3), and a C-terminal focal adhesion targeting domain (FAT). Phosphorylation of Tyr-402 serves as a binding site for Src with subsequent phosphorylation of activation loop residues Tyr-579 and Tyr-580 and Tyr-881 in the FAT domain which promotes binding of the adapter protein Grb2. Proline rich motifs mediate interaction with a variety of SH3 domain containing proteins. Other indicated interactions are Nir1 with the FERM domain, FIP200 interaction with the Pyk2 kinase domain and paxillin and Hic-5 interaction with the FAT domain.
3.1. N-terminal FERM domain
The FERM domain is preceded by 38 amino acids at the amino terminus that have no known function ascribed to them and share very low conservation of sequence with the corresponding residues of FAK. Using hydrophobic cluster analysis, a sequence analysis method based on principles of globular domain folding, Girault and colleagues first proposed that the N-terminal region of Pyk2 contains a divergent FERM domain 46 encompassing ~330 residues. FERM domains are compact cloverleaf shaped structures composed of three structural modules (designated A, B, and C or F1, F2, and F3 respectively) that mediates both protein-membrane targeting as well as protein-protein interactions. The functional activity of the prototypical FERM domain proteins ezrin, radixin, and moesin is regulated by FERM domain mediated intramolecular associations 47, 48. Protein membrane targeting is mediated by basic residues in a cleft between subdomains F1 and F3 that interact with PIP2 49, 50. Interactions with the FERM domains with PIP2 molecules at the membrane has been proposed to induce conformational changes that unmask the full length FERM proteins and stimulate their interaction with the cytoplasmic tails of transmembrane proteins 51. Although a autoregulatory role for the FERM domain of FAK has been described 11, a similar interaction of the Pyk2 FERM domain with the Pyk2 kinase domain has not been reported. Nevertheless, experimental results with chimeric proteins support a substantive role for the Pyk2 FERM domain in the regulation of Pyk2 activity 52. Notably, replacement of the Pyk2 FERM domain with the conserved FAK FERM domain increased Pyk2 catalytic activity and substrate phosphorylation supporting a regulatory role for the Pyk2 FERM domain. Although the molecular mechanism underlying this regulation remains to be determined, Kohno et. al. have recently proposed that the Pyk2 FERM domain regulates Pyk2 activity by mediating a Ca2+/calmodulin dependent Pyk2 homodimer formation and resultant transphosphorylation 53. Other studies have demonstrated that mutations within the Pyk2 FERM domain or expression of an autonomous FERM domain inhibits Pyk2 phosphorylation 54, 55 further supporting a role for the Pyk2 FERM domain in the regulation of Pyk2 activity. These effects could be due to alterations in protein-protein interactions mediated by the FERM domain or changes in cellular localization. Notably, the Pyk2 FERM domain appears to inhibit the focal adhesion targeting of Pyk2 52. Although a number of protein interactions mediated by the FERM domain have been described for the classical ERM proteins and for the FAK FERM domain,56–59 identification of proteins that interact specifically with the Pyk2 FERM domain has been limited. As previously noted, it has been reported that calmodulin binds to the α2-helix of the F2 subdomain of the Pyk2 FERM resulting in the formation of a Pyk2 homodimer and stimulating transphosphorylation 53. A yeast two-hybrid screen was used to identify the Nir family of proteins (Nir1, Nir2, and Nir3) as proteins that interacted with the N-terminal domain of Pyk2 but did not interact with the N-terminal domain of FAK 60. The Nir proteins are calcium binding proteins that possess phosphatidylinosital transfer activity that are phosphorylated by Pyk2 in response to Pyk2 agonists. Given the diverse range of interactions mediated by FERM domains, it is likely that the Pyk2 FERM mediates interactions with other as yet unidentified proteins and that these interactions may contribute to the regulation of Pyk2 function.
3.2. Kinase domain
Pyk2 contains a centrally located kinase domain that is connected to the FERM domain by a short linker segment of ~40 amino acids that contains a Pro-X-X-Pro motif at residues 377–380 and the autophosphorylation site at Tyr402. Phosphorylation at Tyr402 provides a binding site for SH2 containing proteins including Src and p85. Binding of Src leads to phosphorylation of Pyk2 residues Tyr579 and Tyr580 within the kinase domain activation loop and maximal Pyk2 kinase activity. The primary sequence of the catalytic domain of Pyk2 is 60% identical with the catalytic domain of FAK and exhibits high sequence conservation with other protein tyrosine kinases 29. A recently obtained high resolution structure for the Pyk2 kinase domain demonstrates that it exhibits a bi-lobal structure very similar to that of other kinase domains. Interestingly, the kinase domain exhibits unique conformational variability of the canonical Asp-Phe-Gly (DFG) motif in the activation loop that may be of potential use in the design of selective kinase inhibitors 61. A yeast two-hybrid screen was used to identify FIP200 (FAK family kinase-interacting protein of 200 kDa) as a protein that binds to the Pyk2 kinase domain and may function as an potential endogenous inhibitor of Pyk2 62.
3.3. C-terminal domains
The catalytic domain of Pyk2 is followed by two proline rich sequences (713Pro-Pro-Pro-Lys-Pro-Ser-Arg-Pro720 and 855Pro-Pro-Gln-Lys-Pro-Pro-Arg-Leu862) that mediate the interaction of Pyk2 with a number of SH3 domain containing proteins that also interact with FAK including p130Cas, ASAP1, PSGAP, and Graf 36, 63–66. These proline rich sequences also mediate the specific interaction of ASAP2 and PRAP with Pyk2 65, 67. Interestingly, a role for the proline rich sequences in the subcellular localization of Pyk2 has been described. Specifically, mutation of the second proline rich sequence in Pyk2 led to the exclusive nuclear localization of Pyk2 68. Nuclear accumulation of Pyk2 was accompanied by the accumulation of Hic-5 suggesting a potential role for Pyk2 in transcriptional regulation.
The C-terminal domain of Pyk2 also includes a focal adhesion targeting (FAT) domain. This region of Pyk2 is well conserved (~40% identity) with the corresponding FAT domain of FAK that is both necessary and sufficient for efficient localization of FAK to focal adhesions 69, 70. Interestingly, despite this conservation, the FAT domain of Pyk2 does not interact with talin unlike the FAT domain of FAK 71 perhaps contributing to the differential localization of Pyk2 observed in cells. The crystal structure of the FAT domain of Pyk2 demonstrates that it forms an anti-parallel four-helix bundle 72 that exhibits a conserved structure with the FAT domain of FAK 13, 14. The FAT domain of Pyk2 mediates its interaction with the scaffolding protein paxillin whereby two LD4 motifs of paxillin adopt amphipathic helices that interact with hydrophobic patches on opposite sides of the Pyk2 FAT domain creating a six-helix bundle 72. The FAT domain of Pyk2 also interacts with the N-terminus of Hic-5 73. As Hic-5 is closely related to paxillin and contains highly conserved LD motifs it presumably interacts with Pyk2 in a similar molecular manner. The FAT of Pyk2 also interacts with an LD motif in the C-terminus of gelsolin and regulates its activity linking Pyk2 to the regulation of actin cytoskeleton organization 74. The FAT domain also contains Tyr881 which when phosphorylated by Src serves as a binding site for the adaptor Grb2 and linkage to the MAP kinase signaling pathway 75.
3.4. Pyk2 isoforms
At least two novel isoforms have been described for Pyk2. A number of studies had demonstrated that Pyk2 isolated from some cell lines often appeared to run as a doublet on SDS-PAGE gels. Three separate groups isolated and characterized this slightly smaller isoform of Pyk2 66, 76, 77. PCR analysis of Pyk2 indicated this slightly smaller form of Pyk2 resulted from an in frame deletion of 126 base pairs from the C-terminal portion of Pyk2. This isoform arises from alternative mRNA splicing and removes 42 amino acids (residues 738–780) located between the two proline rich SH3 binding domains located in the C-terminal portion of Pyk2 following the kinase domain. This isoform is referred to as Pyk2-H 76 or Pyk2s 66. Pyk2-H is predominately expressed in cells of the hematopeitic lineage including B cells, thymocytes, and NK cells while the unspliced full length Pyk2 is predominately expressed in the brain.
The second isoform described for Pyk2 lacks both the N-terminal FERM domain and the kinase domain. This form of Pyk2 is referred to as PRNK (Pyk2 related nonkinase) and encodes the C-terminal 228 residues of Pyk2 fused to nine unique N-terminal amino acids 66. The structural organization of PRNK is similar to that of FRNK (FAK related non-kinase), the autonomously expressed C-terminal domain of FAK 78, 79 suggesting that it may act as an endogenous regulator of Pyk2 activity in certain tissues. Interestingly, PRNK and Pyk2 exhibit distinct intracellular localization 39, 66. PRNK is localized to focal adhesions while Pyk2 exhibits a predominately cytoplasmic distribution further substantiating the presence of other localization sequences in Pyk2. Similarly, while Pyk2 and PRNK both interact with paxillin, PRNK does not appear to interact with the Pyk2 interacting proteins p130Cas or Graf 66 suggesting that its capacity to act as an endogenous regulator of Pyk2 may be restricted to certain cell types.
4. Functional Investigation of Pyk2 Modulation
Elucidation of the functional role that Pyk2 plays in normal cellular biology is important for gleaning insight into possible derangements seen in human disease states. To this end, several knockout mouse studies added crucial information as to the involvement of Pyk2 in cellular function. One such study which suggested the importance of Pyk2 in cellular adhesion and motility was seen in the hck−/−fgr−/− double knockout mouse 80. Mice deficient in these signaling effectors possessed immune cells with derangements in adhesion and migration with dramatically altered subcellular localization of focal adhesion associated proteins. Macrophages obtained from these knockout mice were observed to have significantly reduced tyrosine phosphorylation of proteins known to be crucial to integrin-mediated function including cortactin, paxillin, and tensin. Moreover, these macrophages had significantly reduced phosphorylation of Pyk2. This work substantiated the importance of Src-family kinases in macrophage migration and implicated Pyk2 as an important proximal contributor to cytoskeletal organization important for the formation of normal filopodia formation and cellular polarity in neutrophils.
Pyk2 involvement in normal bone remodeling was further studied in osteoclasts obtained from the Src−/− mouse. It has been previously shown that engagement of αvβ3 integrins results in activation of Pyk2 and the adaptor protein p130Cas in the sealing zone of actively bone-resorbing osteoclasts 81. Osteoclasts from Src−/− mice displayed defective organization of the microfilament proteins F-actin, vinculin, paxillin, p130cas, and Pyk2 and hyperclustering of αvβ3 integrin complexes. Thus, the formation of functional focal adhesions and the microfilament complex in osteoclasts requires Src and involves Pyk2 82, 83.
Description of the functional role of Pyk2 was further developed by studies using fibroblasts cultured from FAK−/− mice 84. FAK-null fibroblasts exhibit morphological and motility defects despite normal phosphorylation of FAK substrates. Pyk2 expression is elevated in FAK-deficient fibroblasts and Pyk2 activity in these cells is induced by binding to fibronectin 34. Interestingly, Pyk2 activity was not increased upon ECM stimulation in FAK-deficient cells in which FAK was reconstituted. Cell motility could be rescued by reconstitution of FAK, but was only minimally enhanced by Pyk2 overexpression 34. Finally, it was discovered in these fibroblasts that Pyk2 and Src-family members are involved in fibronectin-stimulated signaling through MAPK (Erk2) pathways in the absence of FAK but cannot overcome the loss of FAK regarding cellular motility.
An increase in Pyk2 expression consequent to FAK knockdown has also been recently described in a study investigating the role of FAK in angiogenesis in endothelial cells of adult mice 35. While FAK knockout is an embryonic lethal 84, tamoxifen-inducible conditional knockout of FAK in adult blood vessels did not produce a vascular phenotype and these animals were capable of developing a robust growth factor–induced angiogenic response. Although angiogenesis in wild-type mice was suppressed by pharmacological inhibition of FAK, the inducible FAK-knockout mice were refractory to this treatment due to a compensatory increase in Pyk2 expression. The ability of Pyk2 to assume a compensatory role in integrin-dependent signaling pathways suggests that strategies that selectively target FAK might lack efficacy due to Pyk2 compensation, particularly as it pertains to applications directed towards blocking tumor angiogenesis.
More direct investigation of the functional role of Pyk2 in vivo was obtained by generation of a Pyk2−/− mouse line. Interestingly, targeted deletion of the Pyk2 locus by homologous recombination resulted in a mouse that was viable, fertile, and lacked gross phenotypic aberrancy 85. As the importance of Pyk2 in the biology of immunological cells, particularly macrophages and B-cells had been previously established, lymphoid tissues from the Pyk2−/− mouse were inspected for distributional differences in T- and B-cells, NK cells, macrophages, and monocytes. Notably, Pyk2−/− mice displayed an absence of marginal zone B cells and B cells derived from the Pyk2 deficient mice exhibited decreased motility and decreased responsiveness to chemokines 86. Functional studies with isolated Pyk2 deficient macrophages also identified functional deficits. These macrophages were delayed in the formation of leading edge lamellapodia in response to the chemokine stimulation and failed to detach from substrate at the trailing edge resulting in drastically reduced migration. Moreover, these cells displayed increased spreading and had substantially larger lamellapodia compared to wild type macrophages both in the presence or absence of chemokine. The migratory phenotype could be rescued by reconstitution of Pyk2 and no compensatory overexpression of FAK was observed in the Pyk2−/− macrophages. Further experimentation on the cytoskeletal contractile forces generated by Pyk2−/− macrophages employing optical tweezers to restrain fibronectin-coated beads that were bound by the leading edge lamellapodia of macrophages revealed that rearward displacement of the optically constrained beads was significantly reduced in the Pyk2-null cells compared to the wild type macrophages. The distribution of F-actin in Pyk2-null macrophages in response to chemokine stimulation did not display the typical enrichment at the leading edge lamellapodia, but could be found in various locations in the membrane and non-directional membrane ruffles. In addition, integrin mediated activation of Rho and PI-3 kinase was significantly impaired in Pyk2−/− macrophages. Taken together, Pyk2 appears to be important for the organization of cytoskeletal components in a polarized manner for directional motility in response to chemotactic gradients and in the “flow” of trailing edge components into leading edge structures substantiating a role for Pyk2 in the normal cytoskeletal dynamics of macrophages. Interestingly, Pyk2−/− mice have increased bone mass resulting from an increase in bone formation, as Pyk2 deficiency enhanced the differentiation and activity of osteoprogenitor cells 87. These results indicate that Pyk2 functions as an inhibitor of osteogenic differentiation and suggest that inhibition of Pyk2 activity represents a potential novel approach for the treatment of osteoporosis.
5. Strategies for Pyk2 inhibition
Clinical translation of tyrosine kinase inhibitors has largely focused on competitive inhibition of catalytic domains. Type I inhibitors bind directly to the ATP binding site however, this approach has been challenged by the significant conservation in both sequence and structure of these domains among different protein kinases. This structural conservation often leads to significant undesirable off-target activity. An alternative method includes the use of bipartite inhibitors (Type II inhibitors) that target both the ATP binding site and a less conserved adjacent hydrophobic region formed by an altered conformation of the activation loop containing the conserved DFG motif (“DFG-out”) 88. Both of these approaches have been investigated in the inhibition of Pyk2. A third potential approach for the inhibition of Pyk2 activity is the development of small molecules that target extra-catalytic allosteric sites of protein kinases 89, 90.
5.1. Catalytic inhibitors
Several catalytic inhibitors of the focal adhesion kinases have been developed by the pharmaceutical industry. The bis-anilino pyrimidine compound TAE226 (Novartis Pharma AG, Switzerland) is an ATP competitive inhibitor that inhibits FAK kinase activity in vitro with an IC50 of 5.5 nM 91. TAE226 similarly inhibits Pyk2 kinase activity with an IC50 of 5 nM. The crystal structure of the FAK kinase domain bound to TAE226 demonstrates that TAE226 binding induced a semi-flipped DFG conformation distinct from the “DFG-in” or “DFG-out” conformations observed in other kinases 92. This unique conformation appears to require a glycine residue immediately N-terminal to the DFG motif. This glycine residue is conserved in the Pyk2 kinase domain suggesting that TAE226 likely inhibits Pyk2 in a similar manner. Although the glycine residue preceeding the DFG motif is conserved in both FAK and Pyk2 it is rare among other tyrosine kinases which may contribute to the specificity profile of this and related compounds. Treatment of cultured glioma cell lines 93 or ovarian cancer cell lines 94 with TAE226 inhibited cell proliferation in a dose dependent manner and increased apoptosis in a cell type specific manner. TAE226 inhibition of FAK reduced ovarian tumor growth in vivo and enhanced docetaxel-mediated growth inhibition 94. TAE226 treatment also increased survival in an intracranial glioma xenograft model 91. The contributions of inhibition of FAK and/or Pyk2 to the outcomes in these studies were not defined but given the lack of specificity of TAE226 for FAK or Pyk2 it suggests both kinases were inhibited in these studies.
PF-562,271 (Pfizer, Groton, CN) is a methane sulfonamide diaminopyrimidine ATP competitive inhibitor of FAK and Pyk2 with an IC50 of of 1.5 nM and 13 nM respectively in an in vitro kinase assay 95. Cell based assays indicate a significant selectivity for FAK and Pyk2 over a panel of related kinases. The crystal structure of PF-562,271 bound to the kinase domain of FAK demonstrates that binding of PF-562,271 to FAK induces a variant DFG conformation in FAK very similar to the TAE226 bound conformation 95 suggesting a shared structural basis for their selectivity. Administration of PF-562,271 produced robust antitumor effects across a number of cancer cell types in xenograft tumor models 95 and was well tolerated in patients in a Phase 1 study 96.
Other compounds in this inhibitor series (PF-573,228) exhibit greater selectivity for FAK (IC50=4 nM) over Pyk2 (IC50>1μM). PF-431,396 is a potent dual inhibitor of FAK (IC50=2 nM) and Pyk2 (IC50 30 nM) but lacks stringent specificity 61. Structural studies of PF-431,396 bound to the Pyk2 kinase domain led to the identification and characterization of a novel DFG out conformation for Pyk2 and the development of a Pyk2 inhibitor (PF-4618433) exhibiting increased selectivity for Pyk2 over FAK and a panel of diverse kinases 61. These structural studies suggest that the conformational variability of the DFG motif in Pyk2 (and FAK) can be potentially utilized for the design of potent and selective focal adhesion kinase inhibitors. Thus, the same conformational attributes that enable selectivity for the focal adhesion kinases over other tyrosine kinases also necessitates further synthesis and structure activity relationships to improve selectivity of inhibitors for Pyk2 relative to FAK.
5.2. Non-Catalytic Inhibitors of Pyk2
An alternative approach to the inhibition of kinase activity is to target protein-protein interactions that play a role in the regulation of kinase activity in order to achieve targeting specificity 97, 98. Notably, the last five years have witnessed significant progress in the discovery of small molecule inhibitors that target protein-protein interactions98–100. Similarly, several new ligands have been reported that bind and inhibit kinase function through an allosteric mechanism88, 101, 102. Several studies have implicated the FERM of Pyk2 in the regulation of Pyk2 function52–55 and have demonstrated that the FERM domain of Pyk2 is critical for the Pyk2 stimulated migration of glioma cells55, 103. Thus,targeting discrete, functionally important surface features within the FERM domain could potentially offer a solution for discovery of inhibitors that do not rely upon direct catalytic inhibition. Indeed, as Pyk2 has a scaffolding function in the formation of multi-protein signaling complexes, targeting protein modules that regulate complex assembly may be able to inhibit efficient signal propagation or maintenance even in the presence of continued kinase capacity. Moreover, this approach might employ not only small molecules toward this end, but also highly specific antibody-based biologics.
Determining whether specific FERM domains might be valid targets for molecular therapeutic design and development is an important consideration. Several naturally occurring FERM mutations have been reported that alter protein function and produce pathological conditions. An interesting example of a naturally occurring inactivating mutation highlights the functional importance of the FERM domain of Janus kinase 3. (JAK3). JAKs are FERM domain-containing kinases that are important for transduction of ligand-dependent signals from cytokine receptors104. Unlike the ubiquitously expressed JAK1, the homolog JAK3 is expressed predominantly in hematopoetic cells and naturally occurring mutations result in an autosomal-recessive form of severe-combined immunodeficiency (SCID) in mice and humans105–107. Zhou et. al. studied both naturally occurring and selective mutations within the JAK3 FERM 108. They identified three patient derived FERM domain mutations (Y100C, del58A and D169E) that inhibited cytokine receptor binding and abolished kinase activity. Similarly, substitution of several highly conserved residues in the JAK3 FERM domain inhibited both receptor association and catalytic activity indicating a critical role of the FERM domain in receptor association and maintenance of kinase activity.
The neurofibromatosis 2 (Nf2) gene encodes the protein merlin that functions as a tumor suppressor by negatively regulating cell growth and motility. Merlin is closely related to the classical ERM proteins and mutation in the merlin FERM domain lead to tumor formation in humans 109. Expression of FERM domain mutants of merlin leads to transformation of fibroblasts in culture characterized by lack of contact inhibition and anchorage independent growth and tumors when injected into mice 110. Additionally, merlin expression is significantly reduced in high grade gliomas and re-constitution of merlin inhibits glioma growth in animal models 111. Rac induced phosphorylation of merlin inactivates merlin and potentiates Rac mediated effects on cell motility 112. The FERM domain of merlin binds the phosphatidylinositol 3-kinase enhancer (PIKE-L) preventing its binding to PI3-kinase and blocking the stimulation of cell proliferation 113. The patient derived mutation L64P in the merlin FERM domain abolishes its interaction with PIKE-L allowing PIKE-L to stimulate PI3-kinase activity substantiating a requirement for the FERM domain in merlin function. Together, these studies suggest that selectively perturbing FERM domain structural integrity or disrupting FERM domain interactions with critical effectors, could represent a novel molecularly targeted approach to inhibitor design.
A detailed understanding of how the Pyk2 FERM domain contributes to signal transduction pathway-mediated function is a critical step before embarking upon inhibitor discovery efforts. Despite the availability of many empirically derived FERM domain structures, a high resolution structure is not yet available for the Pyk2 FERM domain. To identify regions and residues within the Pyk2 FERM domain critical for Pyk2 function, a 3-dimensional model of the Pyk2 EFRM domain was developed using threading and multiple molecular dynamic simulations (PDB: 2FO6) 55 and refined following the derivation of the FAK FERM domain structure 10. The model suggests that despite having low overall sequence identity with other classical FERM domains, the Pyk2 FERM domain possesses the characteristic FERM domain fold. With a reasonable structural representation available, the Pyk2 FERM model was compared to several available ligand-bound FERM structures so-as-to develop a “ligand-binding hypothesis” for Pyk2. The available high resolution structures for ligand bound FERM domains indicate that these interactions are mediated by residues in the F3 subdomain. The F3 subdomain exhibits a fold typical of a phosphotyrosine-binding (PTB) or pleckstrin homology (PH) domain consisting of a seven-stranded β sandwich followed by a long a helix at the C-terminus. The binding interactions occur in a long shallow groove on the surface of the F3 subdomain formed by residues from β sheet 5 (β 5C) and the C-terminal α helix (α 1C). Comparison of the binding mode of radixin-ICAM2 (PDB: 1J19) 51, talin-β3 integrin cytoplasmic tail (PDB: 1MK7) 114, and talin-PIPK type 1γ (1Y19) 115 demonstrated that a conserved binding pattern was utilized in each case (Figure 2). Specifically, in all three crystal structures at least three hydrogen bonds were formed between the F3 subdomain and their bound partners anchoring the protein fragments in the binding site. The structural features of the Pyk2 FERM domain 3D homology model (2FO6) and the optimized structure indicates amino acid residues in the β5C-α1C groove that mediate peptide recognition for other FERM domains are conserved in the Pyk2 F3 subdomain (Figure 3). Notably, Pyk2 Ile308 in β5C is exceptionally well conserved among FERM domains and substitution of this residue in Pyk2 significantly inhibited Pyk2 phosphorylation and glioma cell migration55. Moreover, expression of an autonomous Pyk2 FERM domain, inhibits Pyk2 phosphorylation, glioma cell migration in vitro, and increased survival in an intracranial xenograft model 116. In contrast expression of the FERM domain variant Ile308E failed to inhibit Pyk2 phosphorylation and did not increase survival in the xenograft model. These results suggest that the β5C-α1C surface of the Pyk2 FERM domain may be critical for mediating functionally relevant interactions that control Pyk2 activity and that disrupting this interaction represents a novel strategy for inhibiting Pyk2 function.
Figure 2.
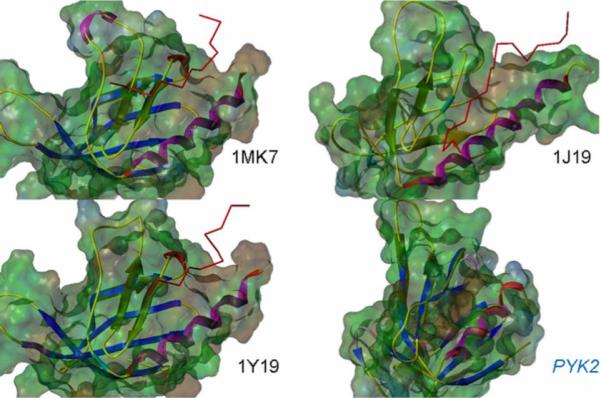
Ribbon representation and overlaid solvent-accessible Connolly surface of ligand-bound FERM domain structures. PDB code 1MK7 - integrin β3 cytoplasmic domain bound to talin FERM; PDB code 1Y19 - talin FERM bound to phosphatidylinositol phosphate kinase type 1-γ); PDB code 1J19 - radixin FERM bound to ICAM-2 cytoplasmic domain peptide PDB ID: 1J19), and the Pyk2 FERM / F3 homology model. Coloring of solvent-accessible surfaces is based on molecular electrostatic potential. Backbone trace of bound partners is represented as a red wire in the experimental structures.
Figure 3.

Refined model for the Pyk2 FERM F3 module. Left: 3D representation of the Pyk2 FERM F3 model highlighting the putative binding site comprised of the α1C-β5C groove. Residues of β5C and α1C are identified are depicted as sticks. Right: Connolly analytic surface with accent on pockets. Green represents surface exposed residues, blue represents hydrophobic residues, and red represents polar residues.
5.2.1 Inhibitors targeting FERM Domains
Targeting of intracellular protein-protein interactions can be contemplated through the use of antibody based biologics 117–119. Intracellular expression of antibodies (“intrabodies”) can inhibit cellular processes in a number of ways including inhibition of intracellular transport or misdirecting intracellular localization 120–122 or by binding to signaling effectors and inhibiting their activity or targeting them for degradation 123, 124. The generally high specificity of the antibody-antigen recognition confers a potential distinct selectivity advantage to this approach over conventional catalytic targeting approcahes. The anti-talin FERM antibody TA205 disrupts actin stress fibers and focal adhesion when microinjected into fibroblasts125 through an allosteric inhibition of the talin FERM domain binding to integrin cytoplasmic domain126 validating that a FERM domain-targeted monoclonal antibody could be used to disrupt FERM-ligand interaction intracellularly. The monoclonal antibody 12A10 specifically targets the Pyk2 FERM domain and recognizes an epitope located on the β5C-α1C surface of the F3 module of the FERM domain that overlaps a site that plays a role in Pyk2 activity 103. Conjugation of 12A10 to a membrane transport peptide led to intracellular accumulation and inhibition of glioma cell migration in a concentration dependent manner. A single chain Fv fragment of 12A10 was stable when expressed in the intracellular environment, interacted directly with Pyk2, reduced Pyk2 phosphorylation, inhibited glioma cell migration in vitro, and extended survival in a glioma xenograft model (Figure 4). As the 12A10 antibody does not react with FAK or the FAK FERM domain 103, these functional effects can be attributed to selective inhibition of Pyk2. Together, these data substantiate a central role for the FERM domain in regulation of Pyk2 activity and identify the F3 module as a novel target to inhibit Pyk2 activity and inhibit glioma progression.
Figure 4.

Inhibition of Pyk2 increased survival of mice bearing intracranial xenografts. Control GBM8 cells or GBM8 cells expressing 12A10 scFv targeting the Pyk2 FERM F3 module were generated by lentiviral transduction and implanted intracranially into immunocompromised mice. Kaplan-Meier curves demonstrate that mice with GBM8-12A10 xenografts exhibited a significant survival benefit relative to mice with control GBM8 xenografts (p = 0.0005).
5.2.2. Small Molecule Inhibitors of the Pyk2 FERM domain
An alternative approach to the use of intrabodies to target the Pyk2 FERM domain mediated protein-protein interactions that regulate kinase activity is through the use of small molecule inhibitors (Figure 5). Indeed, the last five years has witnessed significant progress in the discovery of small molecule inhibitors of protein-protein interaction 97–100. Moreover, several small molecule ligands that inhibit kinase function through an allosteric mechanism have recently been reported 14, 101, 102. There are several reasons to suspect that small molecule inhibitors of Pyk2 might offer significant advantages over biological inhibitors as candidates for therapeutic discovery. First, typical small molecule pharmaceuticals (~450 Da) have less difficulty diffusing through the various extracellular compartments and are generally membrane permeable. These molecules are relatively more amenable to optimization for purposes of enhancing medicinal ADME-Tox properties, such as solubility, binding kinetics, and stability. A protein pharmacophore model for the Pyk2 FERM F3 domain, generated using the structural conservation of ligand bound FERM domains with known 3D structures, was used to identify candidate small molecule inhibitors from the LeadQuest compound library (Tripos International, St. Louis, MO). The highest scoring compound inhibited the binding of the monoclonal antibody 12A10, bound directly to the Pyk2 FERM domain, and inhibited Pyk2 stimulated glioma cell migration 127. Significant optimization work remains to be done before a viable drug candidate is obtained, however it appears clear that extra-catalytic inhibition of Pyk2 (and likely other disease-relevant protein targets) with small molecules is feasible and substantiates the Pyk2 FERM domain as a novel target to inhibit Pyk2 activity.
Figure 5.

Proposed model for non-catalytic inhibition of Pyk2 activity. Pyk2 kinase activity is regulated by FERM domain mediated protein-protein interactions that facilitate phosphorylation, targeting, or scaffolding function that promotes full effector function and activation of signaling pathways mediating growth or migration. Expression of an autonomous FERM domain inhibits Pyk2 activity through competition for FERM domain interacting proteins. Intracellular expression of an anti-Pyk2 F3 monoclonal antibody or interaction of a small molecule with the Pyk2 F3 module inhibits Pyk2 activity by blocking the binding of the interacting protein.
6. Conclusions
This review has focused on the structure and function of the focal adhesion kinase Pyk2. By coordinating adhesion and cytoskeletal dynamics, Pyk2 represents a high-value target for therapeutic discovery efforts due to its prominent position within signaling pathways that regulate cell migration, proliferation, and survival. Clinical translation of tyrosine kinase inhibitors has largely focused on competitive inhibition of catalytic domains. This approach has been challenged by the significant conservation of both the sequence and structure of these domains. An alternative approach to the inhibition of kinase activity is to target protein-protein interactions that play a role in the regulation of kinase activity. Experimental results have demonstrated that non-catalytic modules, such as the N-terminal FERM domain, play a central role in the regulation of Pyk2 activity. Our experience suggests that the identification and characterization of small molecule inhibitors that bind the Pyk2 FERM domain have potential to effectively target protein-protein interactions that regulate Pyk2 kinase activity and to provide mechanistic insights into the molecular interactions that regulate cell motility. In addition to targeting an important mediator of glioma invasion, these small molecules have therapeutic potential in a number of biological processes including inflammation and osteoporosis.
7. Expert opinion
Significant progress in the understanding of intracellular signaling pathways has provided a robust list of potential targets for therapeutic intervention. Approaches to therapeutic discovery of small molecules that prevent protein-protein interactions between key signaling effectors represents an exciting area that will likely lead to agents that work in-concert with inhibitors affecting other points in critical molecular networks. In the nearly 10 years since the approval of the first tyrosine kinase inhibitor for the treatment of cancer (Gleevec for CML), intense discovery and development activity has sought to duplicate the early success. This activity has been met with both disappointments, such as gefitinib treatment for lung cancer, and successes such as sunitinib treatment for renal cell cancer. Clinical experience with kinase inhibitors has demonstrated that the methods through which the function of these proteins are inhibited should not rely exclusively on modulation of catalytic activity due to a set of shortcomings including problems with specificity and the unexpected emergence of resistance to catalytic inhibitors. Certainly, allosteric inhibitors of kinase activity will offer alternatives to inevitable problems of specificity and resistance. However, focusing discovery efforts exclusively on the kinase domain or direct inhibition of catalytic activity is near-sighted. In many cases, Pyk2 serving as an example, inhibition of kinase activity alone may be insufficient to completely abolish cellular signaling as its scaffolding function may be independent of kinase activity. Thus, approaches that inhibit the effector function of disease-relevant proteins without relying on catalytic inhibitory mechanisms will be a welcome asset to the therapeutic arena. Having a variety of agents possessing unique inhibitory properties will provide the clinician with a substantial “tool box” of therapeutic options for patients.
Small molecules have successfully targeted less than 2% of the unique proteins in the human proteome. Therefore, expanding the number of new targets or targeting known proteins including the focal adhesion kinases in new ways will be critical to improve clinical outcome in a number of disease processes. It would be anticipated that the next 5–10 years of inhibitor discovery will likely mirror efforts observed in the field of antibiotic development where beta-lactamase inhibitors are paired with penicillin to escape bacterial penicillin resistance. Protein-protein interactions represent a rich site for potential therapeutic intervention. Indeed, the identification and characterization of discrete functional domains and their interactions with signaling components has opened the door to utilizing a multi-disciplinary approach coupling medicinal chemistry, computational chemistry, and chemoinformatics with biological validation as an approach to exploit to exploit known targets in a heretofore underappreciated manner. With regard to Pyk2, significant experimental data points to an important role for the FERM domain in the regulation of kinase function and as a tractable target for inhibitor development. Perhaps in the future, we will see combination therapies were kinase inhibitors are paired with “FERM domain inhibitors” as a way to boost the therapeutic potency and improve clinical outcomes in complex diseases such as cancer.
Acknowledgements
We acknowledge Natalie Meurice for modeling and graphics of the Pyk2 FERM domain. This work was supported by grants from the National Institutes of Health (CA 103956 and CA 108961 to J.C.L.).
Bibliography
- 1.Schwartz MA, Schaller MD, Ginsberg MH. Integrins: emerging paradigms of signal transduction. Ann Rev Cell Dev Biol. 1995;11:549–99. doi: 10.1146/annurev.cb.11.110195.003001. [DOI] [PubMed] [Google Scholar]
- 2.Geiger B, Spatz JP, Bershadsky AD. Environmental sensing through focal adhesions. Nat Rev Mol Cell Biol. 2009;10:21–33. doi: 10.1038/nrm2593. [DOI] [PubMed] [Google Scholar]
- 3.Berrier AL, Yamada KM. Cell-matrix adhesion. J Cell Physiol. 2007;213:565–73. doi: 10.1002/jcp.21237. [DOI] [PubMed] [Google Scholar]
- 4.Tilghman RW, Parsons JT. Focal adhesion kinase as a regulator of cell tension in the progression of cancer. Semin Cancer Biol. 2008;18:45–52. doi: 10.1016/j.semcancer.2007.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schaller MD, Borgman CA, Cobb BS, Vines RR, et al. pp125FAK: a structurally distinctive protein-tyrosine kinase associated with focal adhesions. Proc Natl Acad Sci U S A. 1992;89:5192–6. doi: 10.1073/pnas.89.11.5192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mitra SK, Hanson DA, Schlaepfer DD. Focal adhesion kinase: in command and control of cell motility. Nat Rev Mol Cell Biol. 2005;6:56–68. doi: 10.1038/nrm1549. [DOI] [PubMed] [Google Scholar]
- 7.Parsons JT. Focal adhesion kinase: the first ten years. J Cell Sci. 2003;116:1409–16. doi: 10.1242/jcs.00373. [DOI] [PubMed] [Google Scholar]
- 8.Schlaepfer DD, Hauck CR, Sieg DJ. Signaling through focal adhesion kinase. Prog Biophys Mol Biol. 1999;71:435–78. doi: 10.1016/s0079-6107(98)00052-2. [DOI] [PubMed] [Google Scholar]
- 9.Schlaepfer DD, Mitra SK. Multiple connections link FAK to cell motility and invasion. Curr Opin Genet Dev. 2004;14:92–101. doi: 10.1016/j.gde.2003.12.002. [DOI] [PubMed] [Google Scholar]
- 10.Ceccarelli DFJ, Song HK, Poy F, Schaller MD, et al. Crystal structure of the FERM domain of focal adhesion kinase. J Biol Chem. 2006;281:252–9. doi: 10.1074/jbc.M509188200. [DOI] [PubMed] [Google Scholar]
- 11.Lietha D, Cai X, Ceccarelli DF, Li Y, et al. Structural basis for the autoinhibition of focal adhesion kinase. Cell. 2007;129:1177–87. doi: 10.1016/j.cell.2007.05.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cai X, Lietha D, Ceccarelli DF, Karginov AV, et al. Spatial and temporal regulation of focal adhesion kinase activity in living cells. Mol Cell Biol. 2008;28:201–14. doi: 10.1128/MCB.01324-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hayashi I, Vuori K, Liddington RC. The focal adhesion targeting (FAT) region of focal adhesion kinase is a four-helix bundle that binds paxillin. Nat Struct Biol. 2002;9:101–6. doi: 10.1038/nsb755. [DOI] [PubMed] [Google Scholar]
- 14.Liu G, Guibao CD, Zheng J. Structural insight into the mechanisms of targeting and signaling of focal adhesion kinase. Mol Cell Biol. 2002;22:2751–60. doi: 10.1128/MCB.22.8.2751-2760.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cance WG, Harris JE, Iacocca MV, Roche E, et al. Immunohistochemical analyses of focal adhesion kinase expression in benign and malignant human breast and colon tissues: Correlation with preinvasive and invasive phenotypes. Clin Cancer Res. 2000;6:2417–23. [PubMed] [Google Scholar]
- 16.Judson PL, He X, Cance WG, L. VL. Overexpression of focal adhesion kinase, a protein tyrosine kinase, in ovarian carcinoma. Cancer. 1999;86:1551–6. doi: 10.1002/(sici)1097-0142(19991015)86:6<1551::aid-cncr23>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- 17.Lark AL, Livasy CA, Calvo B, Caskey L, et al. Overexpression of Focal Adhesion Kinase in Primary Colorectal Carcinomas and Colorectal Liver Metastases: Immunohistochemistry and Real-Time PCR Analyses. Clin Cancer Res. 2003;9:215–22. [PubMed] [Google Scholar]
- 18.Lark AL, Livasy CA, Dressler L, Moore DT, et al. High focal adhesion kinase expression in invasive breast carcinomas is associated with an aggressive phenotype. Mod Pathol. 2005;18:1289–94. doi: 10.1038/modpathol.3800424. [DOI] [PubMed] [Google Scholar]
- 19.Owens L, Xu L, Dent G, Yang X, et al. Focal adhesion kinase as a marker of invasive potential in differentiated human thyroid cancer. Ann Surg Oncol. 1996;3:100–5. doi: 10.1007/BF02409059. [DOI] [PubMed] [Google Scholar]
- 20.Owens LV, Xu L, Craven RJ, Dent GA, et al. Overexpression of the focal adhesion kinase (p125FAK) in invasive human tumors. Cancer Res. 1995;55:2752–5. [PubMed] [Google Scholar]
- 21.Tremblay L, Hauck W, Aprikian AG, R. BL, et al. Focal adhesion kinase (pp125FAK) expression, activation and association with paxillin and p50CSK in human metastatic prostate carcinoma. Int J Cancer. 1996;68:164–71. doi: 10.1002/(sici)1097-0215(19961009)68:2<169::aid-ijc4>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- 22.McLean GW, Carragher NO, Avizienyte E, Evans J, et al. The role of focal-adhesion kinase in cancer - a new therapeutic opportunity. Nat Rev Cancer. 2005;5:505–15. doi: 10.1038/nrc1647. [DOI] [PubMed] [Google Scholar]
- 23.Chatzizacharias NA, Kouraklis GP, Theocharis SE. Focal adhesion kinase: a promising target for anticancer therapy. Expert Opinion on Therapeutic Targets. 2007;11:1315–28. doi: 10.1517/14728222.11.10.1315. [DOI] [PubMed] [Google Scholar]
- 24.Chatzizacharias NA, Kouraklis GP, Theocharis SE. Clinical significance of FAK expression in human neoplasia. Histol Histopathol. 2008;23:629–50. doi: 10.14670/HH-23.629. [DOI] [PubMed] [Google Scholar]
- 25.Parsons JT, Slack-Davis J, Tilghman R, Roberts WG. Focal Adhesion Kinase: Targeting Adhesion Signaling Pathways for Therapeutic Intervention. Clin Cancer Res. 2008;14:627–32. doi: 10.1158/1078-0432.CCR-07-2220. [DOI] [PubMed] [Google Scholar]
- 26.Avraham S, London R, Fu Y, Ota S, et al. Identification and characterization of a novel related adhesion focal tyrosine kinase (RAFTK) from megakaryocytes and brain. J Biol Chem. 1995;270:27742–51. doi: 10.1074/jbc.270.46.27742. [DOI] [PubMed] [Google Scholar]
- 27.Herzog H, Nicholl J, Hort YJ, Sutherland GR, et al. Molecular cloning and assignment of FAK2, a novel human focal adhesion kinase, to 8p11.2–p22 by nonisotopic in situ hybridization. Genomics. 1996;32:484–6. doi: 10.1006/geno.1996.0149. [DOI] [PubMed] [Google Scholar]
- 28.Lev S, Moreno H, Martinez R, Canoll P, et al. Protein tyrosine kinase PYK2 involved in Ca(2+)-induced regulation of ion channel and MAP kinase functions. Nature. 1995;376:737–45. doi: 10.1038/376737a0. [DOI] [PubMed] [Google Scholar]
- 29.Sasaki H, Nagura K, Ishino M, Tobioka H, et al. Cloning and characterization of cell adhesion kinase beta, a novel protein-tyrosine kinase of the focal adhesion kinase subfamily. J Biol Chem. 1995;270:21206–19. doi: 10.1074/jbc.270.36.21206. [DOI] [PubMed] [Google Scholar]
- 30.Yu H, Li X, Marchetto GS, Dy R, et al. Activation of a novel calcium-dependent protein-tyrosine kinase. Correlation with c-Jun N-terminal kinase but not mitogen-activated protein kinase activation. J Biol Chem. 1996;271:29993–8. doi: 10.1074/jbc.271.47.29993. [DOI] [PubMed] [Google Scholar]
- 31.Inazawa J, Sasaki H, Nagura K, Kakazu N, et al. Precise localization of the human gene encoding cell adhesion kinase β (CAKβ/PYK2) to chromosome 8 at p21.1 by fluorescence in situ hybridization. Human Genetics. 1996;98:508–10. doi: 10.1007/s004390050249. [DOI] [PubMed] [Google Scholar]
- 32.Avraham H, Park SY, Schinkmann K, Avraham S. RAFTK/Pyk2-mediated cellular signalling. Cell Signal. 2000;12:123–33. doi: 10.1016/s0898-6568(99)00076-5. [DOI] [PubMed] [Google Scholar]
- 33.Lim Y, Lim S-T, Tomar A, Gardel M, et al. PyK2 and FAK connections to p190Rho guanine nucleotide exchange factor regulate RhoA activity, focal adhesion formation, and cell motility. J Cell Biol. 2008;180:187–203. doi: 10.1083/jcb.200708194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sieg DJ, Ilic D, Jones KC, Damsky CH, et al. Pyk2 and Src-family protein-tyrosine kinases compensate for the loss of FAK in fibronectin-stimulated signaling events but Pyk2 does not fully function to enhance FAK(−) cell migration. EMBO J. 1998;17:5933–47. doi: 10.1093/emboj/17.20.5933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Weis SM, Lim S-T, Lutu-Fuga KM, Barnes LA, et al. Compensatory role for Pyk2 during angiogenesis in adult mice lacking endothelial cell FAK. J Cell Biol. 2008;181:43–50. doi: 10.1083/jcb.200710038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Astier A, Avraham H, Manie SN, Groopman J, et al. The related adhesion focal tyrosine kinase is tyrosine-phosphorylated after beta1-integrin stimulation in B cells and binds to p130cas. J Biol Chem. 1997;272:228–32. doi: 10.1074/jbc.272.1.228. [DOI] [PubMed] [Google Scholar]
- 37.Tokiwa G, Dikic I, Lev S, Schlessinger J. Activation of Pyk2 by stress signals and coupling with JNK signaling pathway. Science. 1996;273:792–4. doi: 10.1126/science.273.5276.792. [DOI] [PubMed] [Google Scholar]
- 38.Klingbeil CK, Hauck CR, Hsia DA, Jones KC, et al. Targeting Pyk2 to β1-integrin-containing focal contacts rescues fibronectin-stimulated signaling and haptotactic motility defects of focal adhesion kinase-null Cells. J Cell Biol. 2001;152:97–110. doi: 10.1083/jcb.152.1.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schaller MD, Sasaki T. Differential signaling by the focal adhesion kinase and cell adhesion kinase beta. J Biol Chem. 1997;272:25319–25. doi: 10.1074/jbc.272.40.25319. [DOI] [PubMed] [Google Scholar]
- 40.Gutenberg A, Bruck W, Buchfelder M, Ludwig HC. Expression of tyrosine kinases FAK and Pyk2 in 331 human astrocytomas. Acta Neuropathol (Berl) 2004;108:224–30. doi: 10.1007/s00401-004-0886-3. [DOI] [PubMed] [Google Scholar]
- 41.Sun CK, Man K, Ng KT, Ho JW, et al. Proline-rich tyrosine kinase 2 (Pyk2) promotes proliferation and invasiveness of hepatocellular carcinoma cells through c-Src/ERK activation. Carcinogenesis. 2008;29:2096–105. doi: 10.1093/carcin/bgn203. [DOI] [PubMed] [Google Scholar]
- 42.Sun CK, Ng KT, Sun BS, Ho JWY, et al. The significance of proline-rich tyrosine kinase2 (Pyk2) on hepatocellular carcinoma progression and recurrence. Br J Cancer. 2007;97:50–7. doi: 10.1038/sj.bjc.6603827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang S, Qiu X, Gu Y, Wang E. Up-regulation of proline-rich tyrosine kinase 2 in non-small cell lung cancer. Lung Cancer. 2008;62:295–301. doi: 10.1016/j.lungcan.2008.05.008. [DOI] [PubMed] [Google Scholar]
- 44.Roelle S, Grosse R, Buech T, Chubanov V, et al. Essential role of Pyk2 and Src kinase activation in neuropeptide-induced proliferation of small cell lung cancer cells. Oncogene. 2007;27:1737–48. doi: 10.1038/sj.onc.1210819. [DOI] [PubMed] [Google Scholar]
- 45.Behmoaram E, Bijian K, Jie S, Xu Y, et al. Focal adhesion kinase-related proline-rich tyrosine kinase 2 and focal adhesion kinase are co-overexpressed in early-stage and invasive ErbB-2-positive breast cancer and cooperate for breast cancer cell tumorigenesis and invasiveness. Am J Pathol. 2008;173:1540–50. doi: 10.2353/ajpath.2008.080292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Girault JA, Labesse G, Mornon JP, Callebaut I. The N-termini of FAK and JAKs contain divergent band 4.1 domains. Trends Biochem Sci. 1999;24:54–7. doi: 10.1016/s0968-0004(98)01331-0. [DOI] [PubMed] [Google Scholar]
- 47.Edwards SD, Keep NH. The 2.7 A crystal structure of the activated FERM domain of moesin: an analysis of structural changes on activation. Biochemistry. 2001;40:7061–8. doi: 10.1021/bi010419h. [DOI] [PubMed] [Google Scholar]
- 48.Pearson MA, Reczek D, Bretscher A, Karplus PA. Structure of the ERM protein moesin reveals the FERM domain fold masked by an extended actin binding tail domain. Cell. 2000;101:259–70. doi: 10.1016/s0092-8674(00)80836-3. [DOI] [PubMed] [Google Scholar]
- 49.Hamada K, Shimizu T, Matsui T, Tsukita S, et al. Structural basis of the membrane-targeting and unmasking mechanisms of the radixin FERM domain. EMBO J. 2000;19:4449–62. doi: 10.1093/emboj/19.17.4449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hirao M, Sato N, Kondo T, Yonemura S, et al. Regulation mechanism of ERM (ezrin/radixin/moesin) protein/plasma membrane association: possible involvement of phosphatidylinositol turnover and Rho-dependent signaling pathway. J Cell Biol. 1996;135:37–51. doi: 10.1083/jcb.135.1.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hamada K, Shimizu T, Yonemura S, Tsukita S, et al. Structural basis of adhesion-molecule recognition by ERM proteins revealed by the crystal structure of the radixin-ICAM-2 complex. EMBO J. 2003;22:502–14. doi: 10.1093/emboj/cdg039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dunty JM, Schaller MD. The N termini of focal adhesion kinase family members regulate substrate phosphorylation, localization, and cell morphology. J Biol Chem. 2002;277:45644–54. doi: 10.1074/jbc.M201779200. [DOI] [PubMed] [Google Scholar]
- 53.Kohno T, Matsuda E, Sasaki H, Sasaki T. Protein-tyrosine kinase CAKβ/PYK2 is activated by binding Ca2+/calmodulin to FERM F2 α2 helix and thus forming its dimer. Biochem J. 2008;410:513–23. doi: 10.1042/BJ20070665. [DOI] [PubMed] [Google Scholar]
- 54.Lipinski CA, Tran NL, Menashi E, Rohl C, et al. The tyrosine kinase pyk2 promotes migration and invasion of glioma cells. Neoplasia. 2005;7:435–45. doi: 10.1593/neo.04712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lipinski CA, Tran NL, Dooley A, Pang YP, et al. Critical role of the FERM domain in Pyk2 stimulated glioma cell migration. Biochem Biophys Res Commun. 2006;349:939–47. doi: 10.1016/j.bbrc.2006.08.134. [DOI] [PubMed] [Google Scholar]
- 56.Chen R, Kim O, Li M, Xiong X, et al. Regulation of the PH-domain-containing tyrosine kinase Etk by focal adhesion kinase through the FERM domain. Nat Cell Biol. 2001;3:439–44. doi: 10.1038/35074500. [DOI] [PubMed] [Google Scholar]
- 57.Medley QG, Buchbinder EG, Tachibana K, Ngo H, et al. Signaling between focal adhesion kinase and Trio. J Biol Chem. 2003;278:13265–70. doi: 10.1074/jbc.M300277200. [DOI] [PubMed] [Google Scholar]
- 58.Poullet P, Gautreau A, Kadare G, Girault J-A, et al. Ezrin Interacts with focal adhesion kinase and induces Its activation independently of cell-matrix adhesion. J Biol Chem. 2001;276:37686–91. doi: 10.1074/jbc.M106175200. [DOI] [PubMed] [Google Scholar]
- 59.Serrels B, Serrels A, Brunton VG, Holt M, et al. Focal adhesion kinase controls actin assembly via a FERM-mediated interaction with the Arp2/3 complex. Nature Cell Biology. 2007;9:1046–56. doi: 10.1038/ncb1626. [DOI] [PubMed] [Google Scholar]
- 60.Lev S, Hernandez J, Martinez R, Chen A, et al. Identification of a novel family of targets of PYK2 related to Drosophila retinal degeneration B (rdgB) protein. Mol Cell Biol. 1999;19:2278–88. doi: 10.1128/mcb.19.3.2278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Han S, Mistry A, Chang JS, Cunningham D, et al. Structural characterization of proline-rich tyrosine kinase 2 (PYK2) reveals a unique (DFG-out) conformation and enables inhibitor design. J Biol Chem. 2009;284:13193–201. doi: 10.1074/jbc.M809038200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ueda H, Abbi S, Zheng C, Guan JL. Suppression of Pyk2 kinase and cellular activities by FIP200. J Cell Biol. 2000;149:423–30. doi: 10.1083/jcb.149.2.423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kruljac-Letunic A, Moelleken J, Kallin A, Wieland F, et al. The tyrosine kinase Pyk2 regulates Arf1 activity by phosphorylation and inhibition of the Arf-GTPase-activating protein ASAP1. J Biol Chem. 2003;278:29560–70. doi: 10.1074/jbc.M302278200. [DOI] [PubMed] [Google Scholar]
- 64.Ren XR, Du QS, Huang YZ, Ao SZ, et al. Regulation of CDC42 GTPase by proline-rich tyrosine kinase 2 interacting with PSGAP, a novel pleckstrin homology and Src homology 3 domain containing rhoGAP protein. J Cell Biol. 2001;152:971–84. doi: 10.1083/jcb.152.5.971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Takahashi T, Yamashita H, Nagano Y, Nakamura T, et al. Identification and characterization of a novel Pyk2/related adhesion focal tyrosine kinase-associated protein that inhibits alpha-synuclein phosphorylation. J Biol Chem. 2003;278:42225–33. doi: 10.1074/jbc.M213217200. [DOI] [PubMed] [Google Scholar]
- 66.Xiong WC, Macklem M, Parsons JT. Expression and characterization of splice variants of PYK2, a focal adhesion kinase-related protein. J Cell Sci. 1998;111:1981–91. doi: 10.1242/jcs.111.14.1981. [DOI] [PubMed] [Google Scholar]
- 67.Andreev J, Simon JP, Sabatini DD, Kam J, et al. Identification of a new Pyk2 target protein with Arf-GAP activity. Mol Cell Biol. 1999;19:2338–50. doi: 10.1128/mcb.19.3.2338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Aoto H, Sasaki H, Ishino M, Sasaki T. Nuclear translocation of cell adhesion kinase beta/proline-rich tyrosine kinase 2. Cell Struct Funct. 2002;27:47–61. doi: 10.1247/csf.27.47. [DOI] [PubMed] [Google Scholar]
- 69.Hildebrand JD, Schaller MD, Parsons JT. Identification of sequences required for the efficient localization of the focal adhesion kinase, pp125FAK, to cellular focal adhesions. J Cell Biol. 1993;123:993–1005. doi: 10.1083/jcb.123.4.993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Shen Y, Schaller MD. Focal adhesion targeting: the critical determinant of FAK regulation and substrate phosphorylation. Mol Biol Cell. 1999;10:2507–18. doi: 10.1091/mbc.10.8.2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zheng C, Xing Z, Bian ZC, Guo C, et al. Differential regulation of Pyk2 and focal adhesion kinase (FAK). The C-terminal domain of FAK confers response to cell adhesion. J Biol Chem. 1998;273:2384–9. doi: 10.1074/jbc.273.4.2384. [DOI] [PubMed] [Google Scholar]
- 72.Lulo J, Yuzawa S, Schlessinger J. Crystal structures of free and ligand-bound focal adhesion targeting domain of Pyk2. Biochem Biophys Res Commun. 2009 doi: 10.1016/j.bbrc.2009.04.011. [DOI] [PubMed] [Google Scholar]
- 73.Matsuya M, Sasaki H, Aoto H, Mitaka T, et al. Cell adhesion kinase beta forms a complex with a new member, Hic-5, of proteins localized at focal adhesions. J Biol Chem. 1998;273:1003–14. doi: 10.1074/jbc.273.2.1003. [DOI] [PubMed] [Google Scholar]
- 74.Wang Q, Xie Y, Du Q-S, Wu X-J, et al. Regulation of the formation of osteoclastic actin rings by proline-rich tyrosine kinase 2 interacting with gelsolin. J Cell Biol. 2003;160:565–75. doi: 10.1083/jcb.200207036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Blaukat A, Ivankovic-Dikic I, Gronroos E, Dolfi F, et al. Adaptor proteins Grb2 and Crk couple Pyk2 with activation of specific mitogen-activated protein kinase cascades. J Biol Chem. 1999;274:14893–901. doi: 10.1074/jbc.274.21.14893. [DOI] [PubMed] [Google Scholar]
- 76.Dikic I, Schlessinger J. Identification of a new Pyk2 isoform implicated in chemokine and antigen receptor signaling. J Biol Chem. 1998;273:14301–8. doi: 10.1074/jbc.273.23.14301. [DOI] [PubMed] [Google Scholar]
- 77.Li X, Hunter D, Morris J, Haskill JS, et al. A calcium-dependent tyrosine kinase splice variant in human monocytes. Activation by a two-stage process involving adherence and a subsequent intracellular signal. J Biol Chem. 1998;273:9361–4. doi: 10.1074/jbc.273.16.9361. [DOI] [PubMed] [Google Scholar]
- 78.Richardson A, Parsons T. A mechanism for regulation of the adhesion-associated proteintyrosine kinase pp125FAK. Nature. 1996;380:538–40. doi: 10.1038/380538a0. [DOI] [PubMed] [Google Scholar]
- 79.Schaller MD, Borgman CA, Parsons JT. Autonomous expression of a noncatalytic domain of the focal adhesion-associated protein tyrosine kinase pp125FAK. Mol Cell Biol. 1993;13:785–91. doi: 10.1128/mcb.13.2.785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Suen PW, Ilic D, Caveggion E, Berton G, et al. Impaired integrin-mediated signal transduction, altered cytoskeletal structure and reduced motility in Hck/Fgr deficient macrophages. J Cell Sci. 1999;112:4067–78. doi: 10.1242/jcs.112.22.4067. [DOI] [PubMed] [Google Scholar]
- 81.Duong LT, Lakkakorpi PT, Nakamura I, Machwate M, et al. PYK2 in osteoclasts is an adhesion kinase, localized in the sealing zone, activated by ligation of αvβ3 integrin, and phosphorylated by Src kinase. J Clin Invest. 1998;102:881–92. doi: 10.1172/JCI3212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lakkakorpi PT, Bett AJ, Lipfert L, Rodan GA, et al. PYK2 autophosphorylation, but not kinase activity, is necessary for adhesion-induced association with c-Src, osteoclast spreading, and bone resorption. J Biol Chem. 2003;278:11502–12. doi: 10.1074/jbc.M206579200. [DOI] [PubMed] [Google Scholar]
- 83.Lakkakorpi PT, Nakamura I, Nagy RM, Parsons JT, et al. Stable association of PYK2 and p130Cas in their co-localization in the sealing zone. J Biol Chem. 1999;274:4900–7. doi: 10.1074/jbc.274.8.4900. [DOI] [PubMed] [Google Scholar]
- 84.Ilic D, Furuta Y, Kanazawa S, Takeda N, et al. Reduced cell motility and enhanced focal adhesion contact formation in cells from FAK-deficient mice. Nature. 1995;377:539–44. doi: 10.1038/377539a0. [DOI] [PubMed] [Google Scholar]
- 85.Okigaki M, Davis C, Falasca M, Harroch S, et al. Pyk2 regulates multiple signaling events crucial for macrophage morphology and migration. Proc Natl Acad Sci U S A. 2003;100:10740–5. doi: 10.1073/pnas.1834348100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Guinamard R, Okigaki M, Schlessinger J, Ravetch JV. Absence of marginal zone B cells in Pyk-2-deficient mice defines their role in the humoral response. Nat Immunol. 2000;1:31–6. doi: 10.1038/76882. [DOI] [PubMed] [Google Scholar]
- 87.Buckbinder L, Crawford DT, Qi H, Ke HZ, et al. Proline-rich tyrosine kinase 2 regulates osteoprogenitor cells and bone formation, and offers an anabolic treatment approach for osteoporosis. Proc Natl Acad Sci USA. 2007;104:10619–24. doi: 10.1073/pnas.0701421104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Liu Y, Gray NS. Rational design of inhibitors that bind to inactive kinase conformations. Nat Chem Biol. 2006;2:358–64. doi: 10.1038/nchembio799. [DOI] [PubMed] [Google Scholar]
- 89.Burke JR, Pattoli MA, Gregor KR, Brassil PJ, et al. BMS-345541 is a highly selective inhibitor of I kappa B kinase that binds at an allosteric site of the enzyme and blocks NF-kappa B-dependent transcription in mice. J Biol Chem. 2003;278:1450–6. doi: 10.1074/jbc.M209677200. [DOI] [PubMed] [Google Scholar]
- 90.Ohren JF, Chen H, Pavlovsky A, Whitehead C, et al. Structures of human MAP kinase kinase 1 (MEK1) and MEK2 describe novel noncompetitive kinase inhibition. Nat Struct Mol Biol. 2004;11:1192–7. doi: 10.1038/nsmb859. [DOI] [PubMed] [Google Scholar]
- 91.Liu TJ, LaFortune T, Honda T, Ohmori O, et al. Inhibition of both focal adhesion kinase and insulin-like growth factor-I receptor kinase suppresses glioma proliferation in vitro and in vivo. Mol Cancer Ther. 2007;6:1357–67. doi: 10.1158/1535-7163.MCT-06-0476. [DOI] [PubMed] [Google Scholar]
- 92.Lietha D, Eck MJ. Crystal structures of the FAK kinase in complex with TAE226 and related bis-anilino pyrimidine inhibitors reveal a helical DFG conformation. PLoS One. 2008;3:e3800. doi: 10.1371/journal.pone.0003800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Shi Q, Hjelmeland AB, Keir ST, Song L, et al. A novel low-molecular weight inhibitor of focal adhesion kinase, TAE226, inhibits glioma growth. Mol Carcinogenesis. 2007;46:488–96. doi: 10.1002/mc.20297. [DOI] [PubMed] [Google Scholar]
- 94.Halder J, Lin YG, Merritt WM, Spannuth WA, et al. Therapeutic efficacy of a novel focal adhesion kinase inhibitor TAE226 in ovarian carcinoma. Cancer Res. 2007;67:10976–83. doi: 10.1158/0008-5472.CAN-07-2667. [DOI] [PubMed] [Google Scholar]
- 95.Roberts WG, Ung E, Whalen P, Cooper B, et al. Antitumor activity and pharmacology of a selective focal adhesion kinase inhibitor, PF-562,271. Cancer Res. 2008;68:1935–44. doi: 10.1158/0008-5472.CAN-07-5155. [DOI] [PubMed] [Google Scholar]
- 96.Siu LL, Burris HA, Mileshkin L, Carnidge DR, et al. Phase I study of a focal adhesion kinase (FAK) inhibitor PF-562,271 in patients with advanced solid tumors. J Clin Oncol. 2007;25:3527. [Google Scholar]
- 97.Sperandio O, Miteva M, Segers K, Nicolaes GAF, et al. Screening outside the catalytic site: Inhibition of macromolecular interactions through structure-based virtual ligand screening experiments. Open Biochem J. 2008;2:29–37. doi: 10.2174/1874091X00802010029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Wells JA, McClendon CL. Reaching for high-hanging fruit in drug discovery at protein-protein interfaces. Nature. 2007;450:1001–9. doi: 10.1038/nature06526. [DOI] [PubMed] [Google Scholar]
- 99.Domling A. Small molecular weight protein-protein interaction antagonists--an insurmountable challenge? Curr Opin Chem Biol. 2008;12:281–91. doi: 10.1016/j.cbpa.2008.04.603. [DOI] [PubMed] [Google Scholar]
- 100.Ruffner H, Bauer A, Bouwmeester T. Human protein-protein interaction networks and the value for drug discovery. Drug DiscovToday. 2007;12:709–16. doi: 10.1016/j.drudis.2007.07.011. [DOI] [PubMed] [Google Scholar]
- 101.Lewis JA, Lebois EP, Lindsley CW. Allosteric modulation of kinases and GPCRs: design principles and structural diversity. Curr OpinChem Biol. 2008;12:269–80. doi: 10.1016/j.cbpa.2008.02.014. [DOI] [PubMed] [Google Scholar]
- 102.Zhao Z, Robinson RG, Barnett SF, Defeo-Jones D, et al. Development of potent, allosteric dual Akt1 and Akt2 inhibitors with improved physical properties and cell activity. Bioorg Med Chem Letts. 2008;18:49–53. doi: 10.1016/j.bmcl.2007.11.015. [DOI] [PubMed] [Google Scholar]
- 103.Loftus JC, Yang Z, Tran NL, Kloss J, et al. The Pyk2 FERM domain as a target to inhibit glioma migration. Mol Can Ther. 2009;8:1505–14. doi: 10.1158/1535-7163.MCT-08-1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Gadina M, Hilton D, Johnston JA, Morinobu A, et al. Signaling by Type I and II cytokine receptors: ten years after. Curr Opin Immunol. 2001;13:363–73. doi: 10.1016/s0952-7915(00)00228-4. [DOI] [PubMed] [Google Scholar]
- 105.Macchi P, Villa A, Giliani S, Sacco MG, et al. Mutations of Jak-3 gene in patients with autosomal severe combined immune deficiency (SCID) Nature. 1995;377:65–8. doi: 10.1038/377065a0. [DOI] [PubMed] [Google Scholar]
- 106.Russell SM, Tayebi N, Nakajima H, Riedy MC, et al. Mutation of Jak3 in a patient with SCID:essential role of Jak3 in lymphoid development. Science. 1995;270:797–800. doi: 10.1126/science.270.5237.797. [DOI] [PubMed] [Google Scholar]
- 107.Thomis DC, Gurniak CB, Tivol E, Sharpe AH, et al. Defects in B lymphocyte maturation and T cell activation in mice lacking Jak3. Science. 1995;270:794–7. doi: 10.1126/science.270.5237.794. [DOI] [PubMed] [Google Scholar]
- 108.Zhou YJ, Chen M, Cusack NA, Kimmel LH, et al. Unexpected effects of FERM domain mutations on catalytic activity of Jak3: structural implication for Janus kinases. Mol Cell. 2001;8:959–69. doi: 10.1016/s1097-2765(01)00398-7. [DOI] [PubMed] [Google Scholar]
- 109.Baser ME. The distribution of constitutional and somatic mutations in the neurofibromatosis 2 gene. Human Mutation. 2006;27:297–306. doi: 10.1002/humu.20317. [DOI] [PubMed] [Google Scholar]
- 110.Johnson KC, Kissil JL, Fry JL, Jacks T. Cellular transformation by a FERM domain mutant of the Nf2 tumor suppressor gene. Oncogene. 2002;21:5990–7. doi: 10.1038/sj.onc.1205693. [DOI] [PubMed] [Google Scholar]
- 111.Lau Y-KI, Murray LB, Houshmandi SS, Xu Y, et al. Merlin is a potent inhibitor of glioma growth. Cancer Res. 2008;68:5733–42. doi: 10.1158/0008-5472.CAN-08-0190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Shaw RJ, Paez JG, Curto M, Yaktine A, et al. The Nf2 tumor suppressor, Merlin, functions in Rac-dependent signaling. Develop Cell. 2001;1:63–72. doi: 10.1016/s1534-5807(01)00009-0. [DOI] [PubMed] [Google Scholar]
- 113.Rong R, Tang X, Gutmann DH, Ye K. Neurofibromatosis 2 (NF2) tumor suppressor merlin inhibits phosphatidylinositol 3-kinase through binding to PIKE-L. Proc Natl Acad Sci USA. 2004;101:18200–5. doi: 10.1073/pnas.0405971102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Garcia-Alvarez B, de Pereda JM, Calderwood DA, Ulmer TS, et al. Structural determinants of integrin recognition by talin. Mol Cell. 2003;11:49–58. doi: 10.1016/s1097-2765(02)00823-7. [DOI] [PubMed] [Google Scholar]
- 115.de Pereda JM, Wegener KL, Santelli E, Bate N, et al. Structural basis for phosphatidylinositol phosphate kinase type I gamma binding to talin at focal adhesions. J Biol Chem. 2005;280:8381–6. doi: 10.1074/jbc.M413180200. [DOI] [PubMed] [Google Scholar]
- 116.Lipinski CA, Tran NL, Viso C, Kloss J, et al. Extended survival of Pyk2 or FAK deficient orthotopic glioma xenografts. J Neuro-Oncol. 2008;90:181–9. doi: 10.1007/s11060-008-9656-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Cao T, Heng BC. Intracellular antibodies (intrabodies) versus RNA interference for therapeutic applications. Ann Clin Lab Sci. 2005;35:227–9. [PubMed] [Google Scholar]
- 118.Manikandan J, Pushparaj PN, Melendez AJ. Protein i: interference at protein level by intrabodies. Front Biosci. 2007;12:1344–52. doi: 10.2741/2152. [DOI] [PubMed] [Google Scholar]
- 119.Williams BR, Zhenping Z. Intrabody-based approaches to cancer therapy: Status and prospects. Curr Med Chem. 2006;13:1473–80. doi: 10.2174/092986706776872899. [DOI] [PubMed] [Google Scholar]
- 120.Boldicke T, Weber H, Mueller PP, Barleon B, et al. Novel highly efficient intrabody mediates complete inhibition of cell surface expression of the human vascular endothelial growth factor receptor-2 (VEGFR-2/KDR) J Immunol Methods. 2005;300:146–59. doi: 10.1016/j.jim.2005.03.007. [DOI] [PubMed] [Google Scholar]
- 121.Wheeler YY, Kute TE, Willingham MC, Chen S-Y, et al. Intrabody-based strategies for inhibition of vascular endothelial growth factor receptor-2: effects on apoptosis, cell growth, and angiogenesis. FASEB J. 2003:1733–5. doi: 10.1096/fj.02-0942fje. [DOI] [PubMed] [Google Scholar]
- 122.Zhu Q, Zeng C, Huhalov A, Yao J, et al. Extended half-life and elevated steady-state level of a single-chain Fv intrabody are critical for specific intracellular retargeting of its antigen, caspase-7. J Immunol Methods. 1999;231:207–22. doi: 10.1016/s0022-1759(99)00158-1. [DOI] [PubMed] [Google Scholar]
- 123.Shin I, Edl J, Biswas S, Lin PC, et al. Proapoptotic activity of cell-permeable anti-Akt single-chain antibodies. Cancer Res. 2005;65:2815–24. doi: 10.1158/0008-5472.CAN-04-2898. [DOI] [PubMed] [Google Scholar]
- 124.Tanaka T, Rabbitts TH. Intrabodies based on intracellular capture frameworks that bind the RAS protein with high affinity and impair oncogenic transformation. EMBO J. 2003;22:1025–35. doi: 10.1093/emboj/cdg106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Bolton SJ, Barry ST, Mosley H, Patel B, et al. Monoclonal antibodies recognizing the N- and C-terminal regions of talin disrupt actin stress fibers when microinjected into human fibroblasts. Cell Motil Cytoskel. 1997;36:363–76. doi: 10.1002/(SICI)1097-0169(1997)36:4<363::AID-CM6>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 126.Xing B, Thuppal S, Jedsadayanmata A, Du X, et al. TA205, an anti-talin monoclonal antibody, inhibits integrin-talin interaction. FEBS Lett. 2006;580:2027–32. doi: 10.1016/j.febslet.2006.02.077. [DOI] [PubMed] [Google Scholar]
- 127.Meurice N, Wang L, Lipinski CA, Yang Z, et al. Structural conservation in FERM domains as a guide to identify inhibitors of the focal adhesion kinase Pyk2. J Med Chem. 2009 doi: 10.1021/jm901247a. [DOI] [PMC free article] [PubMed] [Google Scholar]