

Abstract
Benzodiazepine withdrawal-anxiety is associated with enhanced AMPA receptor (AMPAR)-mediated glutamatergic transmission in rat hippocampal CA1 synapses due to enhanced synaptic insertion and phosphorylation of GluA1 homomers. Interestingly, attenuation of withdrawal-anxiety is associated with a reduction in NMDA receptor (NMDAR)-mediated currents and subunit expression, secondary to AMPA receptor potentiation. Therefore, in this study ultrastructural evidence for possible reductions in NMDAR GluN1, GluN2A and GluN2B subunits was sought at CA1 stratum radiatum synapses in proximal dendrites using postembedding immunogold labeling of tissues from rats withdrawn for 2-days from 1-week daily oral administration of the benzodiazepine, flurazepam (FZP). GluN1-immunogold density and the percentage of immunopositive synapses were significantly decreased in tissues from FZP-withdrawn rats. Similar decreases were observed for GluN2B subunits, however the relative lateral distribution of GluN2B-immunolabeling within the postsynaptic density did not change after BZ withdrawal. In contrast to the GluN2B subunit, the percentage of synapses labeled with the GluN2A subunit antibody and the density of immunogold labeling for this subunit was unchanged. The spatial localization of immunogold particles asssociated with each NMDAR subunit was consistent with a predominantly postsynaptic localization. The data therefore provide direct evidence for reduced synaptic GluN1/GluN2B receptors and preservation of GluN1/GluN2A receptors in the CA1 stratum radiatum region during BZ withdrawal. Based on collective findings in this benzodiazepine withdrawal-anxiety model, we propose a functional model illustrating the changes in glutamate receptor populations at excitatory synapses during benzodiazepine withdrawal.
Keywords: Electron microscopy, Plasticity, Dependence, Glutamate, LTP, Anxiety
INTRODUCTION
Glutamatergic transmission at hippocampal CA1 synapses is mediated by ionotropic AMPA receptors (AMPAR) assembled from GluA1-3 (GluR1-3) subunits, and NMDA receptors (NMDAR) comprised of the obligatory GluN1 (NR1), and one or more GluN2A (NR2A) or GluN2B (NR2B) subunits (Petralia et al., 2005; Sans et al., 2003; Wenthold et al., 1996). While AMPARs carry most of the fast synaptic current, NMDAR activation triggers synaptic plasticity such as long-term potentiation (LTP), a proposed cellular substrate of learning and memory (Lau and Zukin, 2007; Malenka and Nicoll, 1999). Synaptic plasticity at CA1 synapses occurs largely by NMDA-dependent AMPAR trafficking to the synapse and subsequent phosphorylation. Both are key steps in hippocampal LTP and many other forms of activity-dependent synaptic plasticity (Lee et al., 2009; Malinow and Malenka, 2002; Oh et al., 2006).
While, mechanisms underlying activity-dependent synaptic plasticity, such as GluA1 upregulation and phosphorylation are proposed to underlie drug-induced plasticity (Billa et al., 2009; Boudreau et al., 2007; Kim et al., 2009; Shen et al., 2009, 2010), the role of NMDAR in regulation of AMPARs during drug-dependent plasticity is not well-defined (Kim et al., 2009). As such, alterations in NMDAR expression and function have been reported with various drugs of abuse (Loftis and Janowsky, 2000; 2002; Mao et al., 2009; Obara et al., 2009), including the benzodiazepines (Shen and Tietz, 2008; Van Sickle et al., 2004). Benzodiazepines are widely used in the treatment of anxiety, insomnia and seizures and their clinical effects are mediated by positive allosteric modulation of inhibitory γ-aminobutyric acid type-A (GABAA) receptor function (Wafford, 2005). Although benzodiazepines are highly efficacious, chronic use leads to withdrawal symptoms including heightened anxiety, insomnia and occasionally seizures upon cessation of treatment, symptoms which ultimately compromise their clinical utility and may contribute to misuse and abuse (Chouinard, 2004; Griffiths and Johnson, 2005). Benzodiazepine withdrawal-induced anxiety correlates with CA1 neuron hyperexcitability due to enhancement of glutamatergic AMPAR strength, (Allison and Pratt, 2006; Izzo et al., 2001; Van Sickle et al., 2004). Our group has been analyzing potential similarities between benzodiazepine-induced changes in glutamatergic synapses and other forms of synaptic plasticity. Increased AMPAR function following chronic flurazepam (FZP) administration displayed many similarities to LTP including increased GluA1 mRNA and subunit protein expression, increased synaptic insertion of GluA1-containing AMPAR (Das et al., 2008; Song et al., 2007; Van Sickle et al., 2004), and Ca2+-calmodulin dependent protein kinase II (CaMKII)-mediated phosphorylation of GluA1 homomers leading to enhanced AMPAR conductance (Shen et al., 2010; Song et al., 2007).
While AMPAR potentiation was observed after drug withdrawal and associated with anxiety-like behavior, a reduction in NMDAR function associated with a reduction in GluN1 and GluN2B subunit mRNA and protein expression, averted expression of withdrawal-anxiety (Shen and Tietz, 2008; Van Sickle et al., 2002). Blockade of AMPAR current potentiation in CA1 neurons also prevented the decline in NMDAR function and suggested that the negative regulation of NMDAR was secondary to enhanced AMPAR strength (Xiang and Tietz, 2007). The reduction in NMDAR-mediated whole-cell currents was eliminated by superfusion of the selective GluN2B subunit antagonist, ifenprodil thus implicating GluN2B-containing NMDARs (Shen et al., 2008). Therefore, it was hypothesized that the density of NMDARs at CA1 synapses might be altered during benzodiazepine withdrawal by regulation of the pool of NMDARs containing GluN2B subunits.
NMDAR trafficking via PDZ domain interactions with membrane associated guanylate kinases (MAGUK) is a primary mechanism regulating NMDAR synaptic expression (Prybylowski et al., 2005; Wenthold et al., 2003) and is also critical for modulating surface AMPAR expression (Kim et al., 2005). In particular, NMDAR surface expression is dynamically regulated by mechanisms such as membrane insertion (Barria and Malinow, 2002; Sans et al., 2003), lateral diffusion and internalization (Groc et al., 2004; Nong et al., 2004; Tovar and Westbrook, 2002) and additionally regulated by various kinases including CaMKII (Gardoni et al., 2003; Mayadevi et al., 2002; Strack et al., 2000). Relative to GluN2A, the GluN2B subunit is more mobile and known to be robustly internalized by regulated endocytosis through clathrin-coated pits and to cycle between synaptic and extrasynaptic compartments via lateral diffusion in an activity-dependent manner (Groc et al., 2006; Tovar and Westbrook, 2002). GluN2B subunits also play a very significant role as a CaMKII signaling partner (Robison et al., 2005; Strack et al., 2000).
The goal of the present study was therefore to evaluate the densities of key NMDAR subunits at asymmetric synapses in the hippocampal CA1 region during benzodiazepine withdrawal using a quantitative postembedding immunogold approach. The findings suggest that during benzodiazepine withdrawal, the numbers of NMDAR immunopositive synapses decrease in parallel to a decline in the density of GluN1 and GluN2B, but not GluN2A subunits. Decreased synaptic insertion or enhanced internalization of GluN1/GluN2B receptors during benzodiazepine withdrawal, which occur secondary to AMPAR potentiation, may serve as a protective, negative-feedback mechanism to prevent further AMPAR-mediated neuronal hyperexcitability and limit benzodiazepine withdrawal-anxiety.
MATERIALS AND METHODS
Chronic flurazepam treatment
All procedures involving the use of rats were performed in compliance with the University of Toledo College of Medicine Institutional Animal Care and Use Committee (IACUC) and National Institutes of Health guidelines. Male Sprague-Dawley rats, initially 22–25 postnatal days of age (Harlan, Indianapolis, IN) were housed in individual cages with free access to food. Rats were acclimated for 2–4 days to a 0.02 % saccharin water vehicle as the sole source of drinking water. One-week oral treatment with the water-soluble benzodiazepine, FZP was as described previously (Van Sickle et al., 2004). Briefly, after the acclimation period, rats were offered FZP (100 mg/kg/day) in the saccharin vehicle for the first three days followed by an average dose of 150 mg/kg/day for the next four days. Rats which achieved a weekly average dose greater than 100 mg/kg/day with a goal of > 120 mg/kg/day were used for experiments (Xie and Tietz, 1992, Van Sickle et al., 2004). In the current study, experimental rats drank an average of 127.1 ± 4.2 mg/kg/day (Mean ± SEM, n=5 rats). Due to FZP’s relative potency, oral bioavailability and short half-life in rats (Chouinard, 2004; Lau et al., 1987) this oral dosing regimen results in brain concentrations at the end of treatment (1.2 μM FZP) similar to other chronic benzodiazepine treatment regimens (Xie and Tietz, 1992). While rats do not show any overt behavioral signs during drug treatment or withdrawal, this dosing regimen reliably induces manifestations of both benzodiazepine tolerance and dependence (Tietz et al., 1999; Van Sickle et al., 2004). After discontinuation of this drug regimen, rats consistently show increased anxiety-like behavior the following day that can be measured by a decreased time spent in the open arms of an elevated plus-maze. Rats treated for 1-week show a progressive increase in hippocampal CA1 neuron AMPA mEPSC amplitude after 1-day (~15–30%) and 2-days (~30–50%) of FZP withdrawal and decreased NMDAR function after 2 days (Van Sickle et al., 2002, Van Sickle and Tietz, 2002). The increased AMPAR and the associated anxiety-like behavior and decreased NMDAR function were transient and returned to control levels 4 days after ending treatment (Van Sickle et al., 2004).
At the end of treatment, rats were given saccharin water for an additional 2-days and then prepared for histological processing. Paired control rats were given only saccharin water for the same length of time prior to euthanasia by intracardiac perfusion with fixatives under deep anesthesia.
Tissue fixation and embedding
Control and FZP-withdrawn rats (n = 5/group) were deeply anesthetized with an inhaled isoflurane/O2 mixture (2.5%) and perfused via the aorta with a fixative containing 4% paraformaldehyde and 0.5% glutaraldehyde in 0.1 M phosphate buffer. Coronal sections (200 μm) were processed as described previously (Das et al., 2008). After sections were freeze slammed to a pre-cooled copper mirror (−190°C, Leica EM CPC, Bannockburn, IL), for cryosubstitution and low temperature embedding, tissues were transferred to a Leica EM AFS: 12 hr in 1% uranyl acetate in methanol (−80° C), 4 × 30 min in absolute methanol (this and following at −50° C), 2 hr each in 50%, 75%, 100% lowicryl HM20, then 2 × 8 hr in 100% lowicryl. Sections were flat-embedded using lowicryl between glass coverslips coated with Formen-Trenmittel (Electron Microscopy Sciences, Ft. Washington, PA), and polymerized with UV light (48 hr at −50° C, 72 hr at 0° C, 48 hr at +20° C). After polymerization, the CA1 area was excised and glued to EM blocks. Ultrathin sections (80 nm) were collected on 200-mesh nickel grids coated with a Coat-Quick “G” grid coating pen (Daido Sangyo Co., Ltd., Japan).
Characterization of primary antibodies
The rabbit polyclonal GluN1, GluN2B and GluN2A antibodies were purchased from Frontier Science Co. Ltd. (Hokkaido, Japan). The specificity of immunogold labeling in adult CA1 synapses was tested in GluN2A knockout (KO) mice (see below), but not in GluN1 or GluN2B KO mice because of the lethal postnatal phenotype exhibited by these mice. Fortunately, the specificity of immunogold labeling with these same antibodies has been tested by other authors using conditional knockouts of GluN1 or GluN2B in which these subunits were specifically deleted from hippocampal synapses (Fuyaka et al., 2003; Akashi et al., 2009).
Anti-GluN1 (anti-GluRζ1-C2)
The GluN1 subunit antibody (Code # GluRe2N-Rb-Af660-1) was obtained in a rabbit host by injecting a mouse C-terminal C2 cassette (LQNQKDTVLPRRAIEREEGQLQLCSRHRES) corresponding to amino acids 909-938 of GluN1 carboxy-terminus (Manufacturer’s technical information and Watanabe et al., 1998). Immunoblots of protein extracts from adult mice cerebellum labeled with this antibody showed a single-protein band at 120 kDa with negligible cross-reactivity to other ionotropic glutamate receptors (Yamada et al., 2001). Immunohistochemical analysis also showed intense labeling in various brain regions including the hippocampus and cerebellar cortex with very little background staining (Yamada et al., 2001). Immunoelectron microscopic analysis with this antibody showed that GluN1 was localized in postsynaptic junctions of mossy fiber dendrites in mouse cerebellum (Abe et al., 2004). After conditional deletion of the GluN1 gene targeted to the CA1 region of mouse hippocampus, Fukaya et al. (2003) reported no labeling in CA1 synapses.
Anti-GluN2A (Anti-GluRε1)
The mouse GluN2A C-terminal 1126-1408 amino acid sequence was used as an antigen in a rabbit host to obtain polyclonal antibodies against GluN2A (Code # GluRe1C-Rb-Af542-1). A single protein band of 175 kDa was observed in immunoblot analysis with no cross-reactivity to other glutamate receptor subunits including GluN2B (Manufacturer’s technical information). The specificity of this antibody has been tested previously by immunoblot analysis of mouse brain extracts (Watanabe et al., 1998). In the present study, the specificity of GluN2A antibody was also tested by processing hippocampal sections from one wildtype (WT) and one GluN2A KO mouse (kindly provided by Dr. Stefano Vicini from his colony at Georgetown University, Washington DC) under identical conditions to rat tissues (see Results).
Anti-GluN2B (Anti-GluRε2)
The anti-GluN2B antibody (Code # GluRe2C-Rb-Af264-1) was obtained by injecting a rabbit host with a peptide corresponding to the C-terminal region of the mouse GluN2B (1327-1472 aa) (Manufacturer’s technical information and Watanabe et al., 1998). Immunoblot analysis showed a single band of ~180 kDa without cross reactivity to other iGluR subunits. The specificity of this antibody was tested using immunoblot analysis of GluRε2 (GluN2B) in KO mice expressing a truncated protein (Mori et al., 1998). Western blot analysis of whole brain protein showed the corresponding 180 kDa band in WT and a 130 kDa truncated band in the KO using an N-terminal directed antibody. The difference in size approximated the 459 aa C-terminal deleted region, and the C-terminal directed antibody used herein did not detect any protein in Western blots or in tissue from animals expressing the C-terminal truncated GluN2B protein (Mori et al., 1998). A full description of the production and characterization of this antibody appeared in Watanabe et al (1998). The antibody did not recognize any other NMDAR subunit isoforms in Western blots and the density of labeling was substantially reduced in tissues from a GluN2B+/− heterozygote. A recent study using this antibody in a conditional KO of the GluN2B subunit in the CA3 region reported a reduction in immunoreactivity to background levels in CA3 (Akashi et al., 2009).
Immunogold methods
Post-embedding immunogold labeling was performed on ultrathin sections according to the detailed methods described in Das et al. (2008). In short, after equilibration in Tris-buffered saline with 0.1% Triton X-100 (TBST) pH 7.6, and blocking in normal goat serum (1:10; Cat# 9023, Sigma-Aldrich, St. Louis, MO) containing 0.1 % NaBH4 and 50 mM glycine, grids were incubated at room temperature for 2 hr in rabbit anti-GluN1, GluN2A or GluN2B subunit antibody (1:10), followed by overnight incubation at 4°C, and subsequent incubation for 2 hr in goat anti-rabbit IgG conjugated to 10 nm gold particles (diluted 1:25 in TBST pH 8.2; EMGAR10, BBI, United Kingdom). Grids were counterstained with uranyl acetate and Reynold’s lead citrate for visualization under EM. Grids containing ultrathin sections were also analyzed after omitting the primary antibodies to test for non-specific reaction of the secondary colloidal-gold conjugated antibody with postsynaptic densities (PSD). No significant labeling of PSDs was found after omitting the primary antibody (see Results).
Data collection
Grids containing immunolabeled sections were visualized with a Philips 201 EM electron microscope at 70 kV. Synaptic profiles from individual ultrathin sections were used in our study, rather than fully reconstructed synapses using serial sections. This permitted analyses of a larger sample of synapses. A total of 1,688 synapses were included in the analyses: 42 to 72 synapses per rat and antibody reaction; 10 rats in total, 5 control and 5 FZP-withdrawn. Tissues from each rat were immunolabeled with three different GluN antibodies. Although serial sections were not used, a very large proportion of synapses were identified as positively labeled with all the antibodies used.
All data collection and analyses were performed by an observer blind to experimental group. The cell body layer in the CA1 region of a tissue section was located and then in random surveys of microscopic fields all asymmetric synapses observed in the 100 μm region located just above the cell body layer and towards the stratum radiatum, were digitally photographed at a magnification of × 36,000, whether labeled or unlabeled. Typically a field of view contained more than one synapse. During data analyses, we excluded synapses that were cut in very tangential and oblique planes. Orthogonally cut synapses display a clear synaptic cleft, while in tangentially cut synapses the synaptic cleft is obscured by a superimposed postsynaptic density. Tangentially cut synapses expose a larger and more variable area of the postsynaptic density to antibodies and therefore result in variable degrees of signal overestimation when measuring linear densities of proteins embedded in the postsynaptic density. Thus, for the final data analyses a criterion was set where only synapses demonstrating well-defined pre-and postsynaptic membranes and a clear synaptic cleft were included in the quantitation. Using this criterion, the percentage of synapses omitted from GluN1 analysis was 10% and 7.4% in tissues from controls and FZP-withdrawn groups and did not differ between groups (p>0.05). Similarly, for GluN2A and GluN2B the percentage of synapses omitted from analysis in controls animals was 4.4% and 4.8% and 4.8% and 6.9% in FZP-withdrawn rats, respectively (p>0.05). The total number of synapses included in the analyses for each immunoreaction, animal and experimental group appears in Tables 1 – 3.
TABLE 1.
GluN1 Subunit Immunogold Labeling
| Number | ≥1 Particle | ≥2 Particles | PSD Size | |||||
|---|---|---|---|---|---|---|---|---|
| Rat | Synapses Sampled | Labeled (%) | Density (particles/PSD length) | Labeled (%) | Density (particles/PSD length) | Length (μm) | Width (μm) | Area (μm)2 |
| Control | ||||||||
| 1 | 66 | 100.0 | 33.2 | 91.5 | 29.8 | 0.191 | 0.045 | 0.008 |
| 2 | 71 | 94.3 | 31.1 | 84.5 | 21.0 | 0.182 | 0.034 | 0.006 |
| 3 | 50 | 96.0 | 28.0 | 92.0 | 30.2 | 0.195 | 0.035 | 0.007 |
| 4 | 59 | 98.3 | 27.5 | 87.3 | 25.2 | 0.182 | 0.036 | 0.007 |
| 5 | 58 | 94.5 | 18.3 | 93.9 | 35.7 | 0.192 | 0.042 | 0.008 |
| = 304 | ||||||||
| Mean ± SD/SEM | 96.6 ± 2.4/1.3 | 27.7 ± 5.9/2.6 | 89.8 ± 3.8/1.7 | 30.4 ± 5.9/2.6 | 0.187 ± 0.006/0.003 | 0.038 ± 0.005/0.002 | 0.007 ± 0.001/0.000 | |
| FZP-Withdrawn | ||||||||
| 1 | 69 | 92.8 | 21.8 | 62.1 | 17.4 | 0.190 | 0.037 | 0.007 |
| 2 | 61 | 77.0 | 13.3 | 78.0 | 18.5 | 0.198 | 0.031 | 0.006 |
| 3 | 53 | 84.9 | 20.8 | 73.7 | 27.4 | 0.184 | 0.031 | 0.006 |
| 4 | 50 | 88.0 | 14.8 | 67.2 | 18.9 | 0.215 | 0.040 | 0.009 |
| 5 | 66 | 83.3 | 12.0 | 87.0 | 24.5 | 0.208 | 0.040 | 0.008 |
| = 299 | ||||||||
| Mean ± SD/SEM | 85.2 ± 5.9/2.7 | 16.5 ± 4.5/2.0 | 73.6 ± 9.6/4.3 | 21.4 ± 4.4/2.0 | 0.198 ± 0.011/0.003 | 0.036 ± 0.005/0.002 | 0.007 ± 0.001/0.000 | |
| p Value | 0.004 | 0.009 | 0.008 | 0.026 | 0.099 | 0.406 | ||
TABLE 3.
GluN2A Subunit Immunogold Labeling
| Number | ≥1 Particle | ≥2 Particles | PSD Size | |||||
|---|---|---|---|---|---|---|---|---|
| Rat | Synapses Sampled | Labeled (%) | Density (particles/PSD length) | Labeled (%) | Density (particles/PSD length) | Length (μm) | Width (μm) | Area (μm)2 |
| Control | ||||||||
| 1 | 56 | 71.4 | 12.5 | 62.5 | 19.2 | 0.197 | 0.040 | 0.008 |
| 2 | 60 | 75.0 | 10.6 | 65.0 | 15.4 | 0.191 | 0.045 | 0.009 |
| 3 | 51 | 73.1 | 14.3 | 63.5 | 21.7 | 0.213 | 0.043 | 0.009 |
| 4 | 47 | 83.7 | 17.8 | 75.4 | 23.1 | 0.212 | 0.044 | 0.008 |
| 5 | 70 | 74.3 | 9.8 | 57.6 | 15.6 | 0.201 | 0.042 | 0.009 |
| = 284 | ||||||||
| Mean ± SD/SEM | 75.5 ± 4.8/2.1 | 13.0 ±3.2/1.4 | 64.8 ± 6.6/2.9 | 19.0 ± 3.5/1.6 | 0.201 ± 0.010/0.005 | 0.042 ± 0.003/0.001 | 0.009 ± 0.001/0.000 | |
| FZP-Withdrawn | ||||||||
| 1 | 55 | 67.3 | 7.5 | 41.8 | 14.6 | 0.191 | 0.037 | 0.007 |
| 2 | 72 | 73.6 | 13.2 | 59.7 | 20.9 | 0.198 | 0.049 | 0.010 |
| 3 | 49 | 77.6 | 10.0 | 65.3 | 14.4 | 0.219 | 0.044 | 0.010 |
| 4 | 48 | 64.6 | 7.9 | 45.8 | 15.0 | 0.219 | 0.039 | 0.009 |
| 5 | 57 | 77.2 | 14.2 | 64.9 | 21.8 | 0.192 | 0.041 | 0.008 |
| = 281 | ||||||||
| Mean ± SD/SEM | 72.1 ± 5.9/2.6 | 10.7 ±3.3/1.5 | 55.5 ± 11.0/4.9 | 17.3 ± 3.7/1.6 | 0.204 ± 0.014/0.006 | 0.042 ± 0.005/0.002 | 0.009 ± 0.001/0.000 | |
| p Value | 0.339 | 0.293 | 0.143 | 0.485 | 0.767 | 0.936 | ||
All digitally photographed images included were analyzed using Image Pro Plus software (v. 5.0 Media Cybernetics, Bethesda, MD). As described in detail previously (Das et al., 2008), postsynaptic labeling was defined as immunogold particles inside the PSD, or at a maximum 20 nm from the surrounding edge of the PSD, either towards the cytoplasm or the synaptic cleft. Gold particles falling outside the 20 nm surrounding edge of the PSD were considered non-synaptic. Labeling on the plasma membrane observed within 100 nm lateral to either edge of the PSD, but excluding the 20 nm surrounding zone, was defined as perisynaptic labeling.
Statistical analyses
The number of immunogold particles per synapse between control and FZP-withdrawn groups (0 to ≥ 18) was compared by Mann-Whitney U test. Interval data are reported as mean ± SD/SEM. The density of immunogold labeling was estimated as number of gold particles per linear length (μm) of the PSD. The aggregate mean differences in immunogold density and PSD size (length and width) were based on multiple observations CA1 asymmetric synapses located in the stratum radiatum of control and FZP-withdrawn tissues. PSD area measurements were derived from length and width measurements. Subunit immunogold density and the size of PSDs of all synapses sampled were compared for each antibody reaction using a multivariate general linear model (SPSS 17.0, SPSS, Inc. Chicago, IL). Positively (threshold of either 1 or 2 gold particles) and negatively labeled PSDs were included in the analysis. Between-subjects comparisons were considered significant if the overall F value was <0.05. The percentage of synapses labeled with each antibody were compared between groups be Student’s t-test. The relative position of immunogold particles was determined by measuring the distance between the centers of each gold particle to the outer leaflet of the postsynaptic membrane and analyzed as previously described (Das et al., 2008). For lateral distribution measurements, the distances from the edge of the PSD to the center of each gold particle were measured and particles counted were grouped into 10 bins consisting of the lateral 10% to the medial 40–50% from either edge of the PSD (Park et al., 2008). Excel and GraphPad Prism™ 4.0 software packages were used to analyze data and generate graphs. EM micrographs were composed in Photoshop 5.0 (Adobe Systems, San Jose, CA or CorelDraw 12, Fremont, CA) and only minor adjustments were made to improve the contrast and brightness of images.
RESULTS
Specificity of immunogold labeling and definition of an immunolabeled synapse
A description of the characteristics of GluN antibodies and their specificity was provided in the Materials and Methods. Non-specific labeling was determined in our study using two approaches, immunolabeling without primary antibodies and immunolabeling in CA1 from GluN2A KO mice that do not express detectable levels of the protein (Sakimura et al., 1995). The primary antibodies were omitted to test whether secondary antibodies or spurious gold probes in solution were non-specifically absorbed to PSDs. In rat CA1 tissue sections from control and FZP rats we observed respectively, 2.5% and 3% of synapses labeled with one gold particle (n=78 and 59 synapses analyzed, respectively in section from control and FZP-withdrawn rats). In control tissue we also encountered two synapses with 2 particles (2.5% of all synapses). These results suggested that non-specific absorption of secondary antibodies and/or colloidal gold to the PSD of excitatory synapses in the CA1 region was negligible. Therefore, the observed staining is primarily due to binding of the primary antibody.
We then tested the specificity of GluN2A antibodies in KO mice. We could not test GluN1 and GluN2B antibodies in their respective KO tissues because these mutants do not survive until adulthood. Moreover, the GluN1 and GluN2B antibodies used herein did not label hippocampal tissue sections in which these subunits were conditionally deleted (Fukaya et al., 2003; Akashi et al., 2009). In GluN2A KO mice 9 of 70 synapses (13%) showed some GluN2A immunolabeling; of these all but one displayed a single colloidal gold particle, the other one showed two. In contrast, 65% of 55 CA1 synapses sampled from a WT littermate showed immunolabeling that ranged from 1 to 7 gold particles; two-thirds of the synapses were labeled by more than one colloidal gold particle. Following omission of primary antibodies only one of 41 synapses (2.4%) showed colloidal gold labeling in the GluN2A KO mouse tissue. This suggests that the immunolabeling detected in KO tissue sections is more likely due to non-specific absorption of the primary antibody.
Another possibility that may account for low levels of PSD labeling is that background immunogold particles fall on top of PSDs by chance. To determine the expected number of random background particles over PSDs we used the method detailed in Das et al. (2008). Briefly, twenty-five test rectangles with areas similar to the average PSD area were randomly placed on each image captured from WT and KO mouse tissues or from control and FZP-withdrawn rat tissues for each antibody and animal analyzed. Overall background levels, estimated in random test rectangles placed outside the PSD (see below) were comparable in the WT and KO tissues after primary antibody labeling or after omitting primary antibodies. In each of these three situations the probability of finding one or more background colloidal particles in the test rectangles of similar size and shape to the average PSD was 1%, 0.4% and 0.5%. Background was slightly higher in rat sections, however, the level was comparable in all sections from the 5 control and 5 FZP-withdrawn animals. In rat tissues the probability of finding one or more gold particles in test rectangles by chance was usually above 1%, but always less than 2%, respectively.
In conclusion, colloidal gold labeling due to random placement of particles on top of the PSD or because of non-specific absorption of gold-conjugated secondary antibodies to the PSD was negligible, even when considering labeling by just one particle. However, primary antisera, as in the case of the GluN2A subunit antibody, might non-specifically label some PSDs with only one particle. Accepting synaptic labeling with a threshold of 2 particles per synapse would increase the certainty that all synapses were specifically immunolabeled, but would also discard a large number of weakly labeled, but truly immunolabeled synapses. Since the purpose of the study was to evaluate whether synaptic GluN subunits were downregulated, we believe that the conclusions are strengthened by comparing the quantitative results of all labeled synapses after acceptance of synapses labeled by either 1 or more or 2 or more particles.
GluN1 and GluN2B, but not GluN2A subunits are reduced in hippocampal CA1 synapses during benzodiazepine withdrawal
GluN1, GluN2B and GluN2A receptor subunit immunogold labeling was largely associated with the PSDs of asymmetric synapses in control (Fig. 1A,B, 2A,B, 3A,B) and FZP-withdrawn rats (Fig. 1C,D, 2C,D, 3C,D). Labeling in the postsynaptic cytoplasm was always sparse for all subunits. No labeling was found in symmetric junctions (data not shown).
Figure 1. GluN1 subunit immunogold labeling is decreased in hippocampal CA1 asymmetric synapses during FZP withdrawal.

(A, B) Representative electron micrographs of GluN1-subunit immunogold labeling in hippocampal CA1 stratum radiatum from control tissues. Immunogold particles (10 nm) are located primarily within the postsynaptic density (PSD) and some particles extend into the synaptic cleft. (C, D) Representative images of GluN1-labeled asymmetric synapses from FZP-withdrawn tissues show similar distribution of immunogold labeling. (E) The percentage of synapses with immunogold labeling for GluN1 (containing at least one immunogold particle) was significantly reduced (**p<0.01) in synapses from FZP-withdrawn rats (white dots) compared to controls (black dots). Each dot represents the average obtained from a single animal (n = 50 to 71 synapses per animal, see Table 1). The average for all animals (large horizontal bar) and standard error of the mean (SEM, smaller bars) are superimposed to the dot plots (n=5 animals). (F) GluN1 immunogold density estimated by the number of gold particles per micron synaptic length was also notably reduced in synapses from FZP-withdrawn rats (white dots) compared to controls (black dots)(n=5 rats/group, ***p=0.009). As above, average ± SEM for all five animals is superimposed in the aligned dot plot. Scale bar in C represents 0.25 μm. All images are at the same magnification.
Figure 2. GluN2B subunit immunogold labeling decreased in hippocampal CA1 asymmetric synapses during FZP withdrawal.
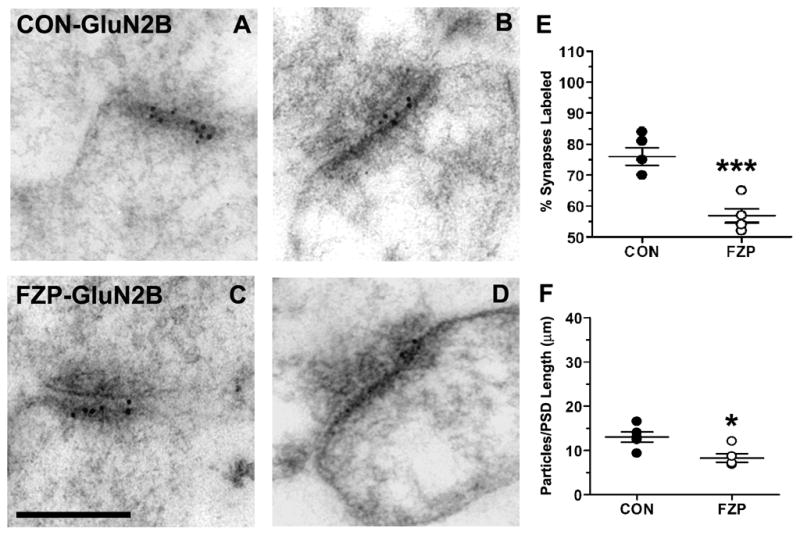
(A, B) Representative electron micrographs of GluN2B-labeled asymmetric synapses from control and (C, D) FZP-withdrawn synapses show 10 nm immunogold particles mainly in the postsynaptic density and extending into the synaptic cleft. (E) FZP-withdrawal caused a significant reduction in the total percentage of synapses labeled with at least one immunogold particle compared to controls (***p<0.001) and (F) in mean immunogold density (n=5 rats/group, *p<0.017). As in Figure 1, each dot represents a single animal average (43 to 59 synapses analyzed in each animal, see Table 2) with the total five animals average and SEM superimposed. Scale bar in C represents 0.25 μm. All images are at the same magnification.
Figure 3. GluN2A subunit immunogold labeling was unaltered in hippocampal CA1 asymmetric synapses during FZP withdrawal.

(A, B) Representative electron micrographs of GluN2A-immunogold labeled synapses from control and (C, D) FZP-withdrawn tissues. In contrast to changes observed in GluN1 and GluN2B, GluN2A immunogold labeling during FZP-withdrawal did not show significant (p>0.017) changes compared to controls in either percentage of synapses labeled with at least one immunogold particle (E) or mean GluN2A immunogold density (F) (n = 5 rats/group). As in Figures 1 and 2 each dot represents a single animal average (47 to 72 synapses analyzed in each animal, see Table 3) with the total five animals average and SEM superimposed. Error bars indicate SEM. Scale bar in C represents 0.25 μm. All images are at the same magnification.
The percentages of asymmetric synapses from control rats that contained 1 or more or 2 or more immunogold particles for GluN1 subunits was 96.6 ± 1.1% and 89.8 ± 1.7%, respectively (± S.E.M.; 50–71 synapses/rat, n = 5 rats) (Fig. 1E, Table 1). The high percentage of immunogold labeled synapses was similar to earlier studies demonstrating a very high level of expression of NR1 subunits in hippocampal CA1 synapses (Nicholson and Geinisman, 2009; Takumi et al., 1999). FZP-withdrawal resulted in a significant decrease in the percentage of GluN1 immunopositive synapses with 1 or more immunogold particles (Fig. 1E; 85.2 ± 2.7%, 50–69 synapses/rat, n = 5 rats, p = 0.004) or 2 or more immunogold particles (73.6 ± 4.3%, p = 0.008). Comparison of resultant labeling suggests that 11.8% of synapses lost all immunolabeling and that in 18.0% of synapses immunolabeling was reduced to less than 2 particles per synapse. Comparing the whole synaptic populations from FZP and control tissues, mean immunogold density (number of gold particles per μm length of PSD) was significantly reduced by 40% in FZP-withdrawn rats (Fig. 1F; 16.5 ± 2.0 particles/μm, p = 0.01) compared to controls (27.7± 2.6 particles/μm). A similar significant 30% reduction was detected when comparing only synapses labeled by 2 or more particles (CON: 30.4 ± 2.6 particles/μm, n = 5 animals with 46–62 synapses labeled by 2 or more particles; FZP: 21.4 ± 2.0 particles/μm, n = 5 animals with 39–60 synapses; p = 0.026). These differences in labeling density are best explained by analysis of the frequency distribution histograms of synapses containing different numbers of GluN1 immunoparticles (Fig. 4A). The fraction of synapses expressing 1, 2 or 3 GluN1 immunogold particles tended to increase in FZP-withdrawn tissues. This was particularly notable for the fraction of synapses containing 2 gold particles (CON: 0.100 ± 0.012; FZP: 0.183 ± 0.016, p = 0.008, Mann-Whitney U test). Additionally, the fraction of GluN1 immunonegative synapses also significantly increased during FZP withdrawal (Fig. 4A; CON: 0.033 ± 0.011; FZP: 0.148 ± 0.026; p= 0.008). In contrast, the fraction of synapses expressing ≥ 6 particles tended to decrease in FZP-withdrawn tissues. A significant decrease in the fraction of synapses containing 8 gold particles was also observed in FZP-withdrawn tissues compared to controls (CON: 0.056 ± 0.020; FZP: 0.003 ± 0.000; p=0.032). Thus, the decreased GluN1 labeling in FZP-withdrawn tissues appeared to be due to an increase in the synaptic fraction expressing lower numbers of immunogold particles and a decrease in the fraction of synapses expressing higher numbers of immunogold particles. This resulted in a significant change in the density of gold particles in those synapses that retained immunolabeling in addition to a reduction of immunolabeled synapses.
Figure 4. Distribution histograms of synapses containing different numbers of (A) GluN1, (B) GluN2B or (C) GluN2A immunogold particles.
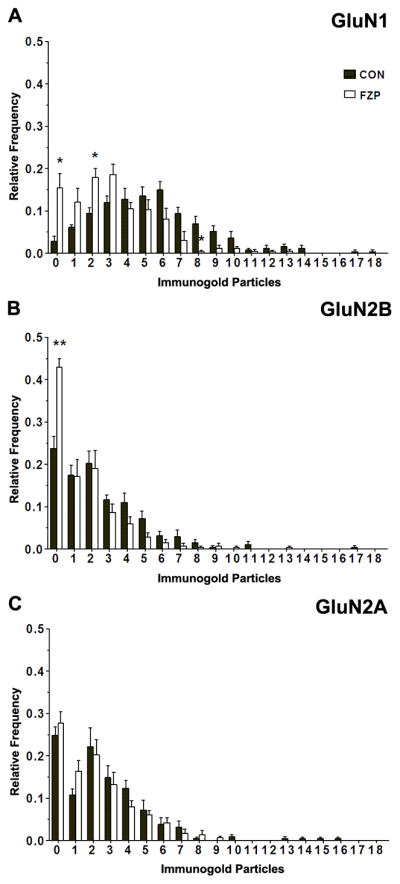
Histograms represent the average relative frequencies of synapses lacking GluN1, GluN2B and GluN2A (0 particles) or containing 1 to ≥ 18 immunogold particles. The fraction of synapses lacking GluN1- or GluN2B-immunogold particles was significantly higher in FZP-withdrawn rats (p<0.05 and p<0.01, respectively, Mann-Whitney U test). A significant decrease was also observed in the fraction of FZP-withdrawn synapses containing 8 GluN1 immunogold particles, that was parallel to increases in the fraction of synapses containing 2 immunogold particles (p<0.05, Mann-Whitney U test). No significant changes in either the fraction of synapses lacking immunogold particles or the fraction of synapses labeled with different numbers of immunogold particles were detected in the case of GluN2A subunit labeling (p>0.05, Mann-Whitney U test).
Fewer synapses exhibited GluN2B-subunit labeling in control tissues (Fig. 2E, Table 2; 76.0 ± 2.8% contained 1 or more immunogold particles and 59.0 ± 1.8% contained 2 or more particles; n=5 rats; 43–57 synapses/rat). The relatively lower immunoreactivity for the GluN2B subunit compared to the GluN1 subunit is consistent with other studies in thalamic synapses (Radley et al., 2007) and the fact that GluN1, but not GluN2B subunits, are obligatory partners of all assembled NMDARs. The percentage of synapses exhibiting GluN2B-immunogold labeling in FZP-withdrawn synapses was significantly reduced compared to controls when considering all immunolabeled synapses (FZP group: 57.0 ± 2.2%, 42–59 synapses/rat, n = 5 rats; 25% reduction from control, p = 0.001, unpaired Student t-test) or only synapses with 2 or more immunogold particles (40.1± 4.2%; 32% reduction from control, p = 0.003) (Fig. 2E, Table 2). Additionally, there was a significant decrease in the density of GluN2B immunogold labeling in FZP-withdrawn synapses (8.3 ± 1.0 particles/μm) compared to controls (Fig. 2F; 13.0 ± 1.2 particles/μm; p = 0.014). This reduction in density was not apparent when considering only synapses labeled by 2 or more particles (CON: 20.4 ± 1.6%, 23–36 synapses/rat, n = 5 rats; FZP: 18.4 ± 1.5%, 15–29 synapses/rat, n = 5 rats, p = 0.395) (Table 2), this is probably because the largest change in FZP-treated tissues was an increase in synapses with no immunogold particles. In the distribution histogram of labeling intensities (Fig. 4B) it was clearly apparent that the fraction of GluN2B immunonegative synapses was significantly higher in FZP-withdrawn animals compared to controls (CON: 0.237 ± 0.039; FZP: 0.429 ± 0.021; p=0.008). While the fraction of synapses labeled with 1 to ≥ 18 GluN2B immunogold particles tended to decrease in FZP-withdrawn tissues, no significant differences were detected in comparison to controls.
TABLE 2.
GluN2B Subunit Immunogold Labeling
| Number | ≥1 Particle | ≥2 Particles | PSD Size | |||||
|---|---|---|---|---|---|---|---|---|
| Rat | Synapses Sampled | Labeled (%) | Density (particles/PSD length) | Labeled (%) | Density (particles/PSD length) | Length (μm) | Width (μm) | Area (μm)2 |
| Control | ||||||||
| 1 | 57 | 75.4 | 16.5 | 63.2 | 25.0 | 0.184 | 0.038 | 0.007 |
| 2 | 55 | 70.3 | 14.1 | 58.2 | 23.0 | 0.215 | 0.049 | 0.011 |
| 3 | 43 | 69.8 | 9.3 | 53.5 | 16.0 | 0.207 | 0.046 | 0.010 |
| 4 | 54 | 81.4 | 12.9 | 57.4 | 20.3 | 0.189 | 0.038 | 0.007 |
| 5 | 51 | 84.3 | 12.4 | 62.7 | 17.8 | 0.187 | 0.038 | 0.007 |
| = 260 | ||||||||
| Mean ± SD/SEM | 76.0 ± 6.4/2.8 | 13.0 ± 2.6/1.2 | 59.0 ± 4.0/1.8 | 20.4 ± 3.7/1.6 | 0.196 ± 0.014/0.006 | 0.042 ± 0.005/0.002 | 0.008 ± 0.002/0.001 | |
| FZP-Withdrawn | ||||||||
| 1 | 51 | 56.9 | 7.1 | 35.3 | 17.2 | 0.198 | 0.043 | 0.009 |
| 2 | 59 | 54.2 | 12.0 | 49.2 | 23.9 | 0.201 | 0.045 | 0.009 |
| 3 | 54 | 57.4 | 6.8 | 27.8 | 19.3 | 0.208 | 0.040 | 0.008 |
| 4 | 42 | 52.4 | 6.8 | 38.0 | 16.0 | 0.198 | 0.038 | 0.008 |
| 5 | 54 | 64.8 | 8.6 | 50.0 | 15.6 | 0.191 | 0.039 | 0.008 |
| = 260 | ||||||||
| Mean ± SD/SEM | 57.0 ± 5.0/2.2 | 8.3 ± 2.2/1.0 | 40.1 ± 9.5/4.2 | 18.4 ± 3.4/1.5 | 0.199 ± 0.006/0.003 | 0.041 ± 0.003/0.001 | 0.008 ± 0.001/0.000 | |
| p Value | 0.001 | 0.014 | 0.003 | 0.394 | 0.688 | 0.775 | ||
GluN2A-subunit immunogold labeling in control tissues was present in a similar number of synapses compared to GluN2B labeling (Tables 2 and 3) and the distribution of synapses with different numbers of immunogold particles was also similar (Fig. 4B,C). Unlike GluN1 and GluN2B-subunits, however, no significant differences were detected between control and FZP-withdrawn rats in the total percentage of GluN2A-labeled synapses labeled (Fig. 3E; CON: 75.2 ± 2.1%, 49–70 synapses/rat; FZP: 72.2 ± 2.6%, 48–72 synapses/rat, n = 5 rats/group, p = 0.396) or overall density of immunogold labeling (Fig. 3F; CON: 13.0 ± 1.4 particles/μm; FZP: 10.7 ± 1.5 particles/μm, p = 0.295). A similar conclusion was reached when considering only synapses labeled by more than 2 gold particles (CON: 64.8 ± 2.9%; FZP: 55.6 ± 4.9%, 48–72; p = 0.143) and no obvious changes were observed in the frequency distribution histograms of the number of GluN2A immunogold particle labeling comparing synapses from control and FZP-treated rats (Fig. 4C). Thus labeling densities at any particular threshold of GluN2A immunogold particle labeling were unchanged.
As illustrated in Figure 4, the frequency distribution histograms of the number of immunogold particles detected per synapse for each NMDAR subunit also indicated that a larger number of immunogold particles were usually found in GluN1-immunolabeled synapses compared to other subunits. This is consistent with the fact that GluN1 antibodies would target the entire population of synaptic NMDARs, whereas GluN2 antibodies would only detect GluN2A and GluN2B subpopulations.
In conclusion, during benzodiazepine withdrawal, GluN1/GluN2B receptor content in a proportion of synapses decreases below detection threshold, leading to a reduction in total NMDAR immunoreactivity in most synapses that is not compensated by parallel increases in GluN1/GluN2A receptors. Thus, decreased CA1 NMDAR-mediated evoked EPSCs and reduced efficacy of the GluN2B selective antagonist ifenprodil on NMDAR-mediated EPSCs during benzodiazepine withdrawal (Shen et al., 2009; Van Sickle et al., 2002) are best explained by the removal of synaptic receptor pools containing GluN1/GluN2B subunits.
PSD sizes of GluN1, GluN2A and GluN2B-labeled asymmetric synapses
As shown in Tables 1–3, the total number of synapses used for PSD size measurements varied from 43 to 71 in control tissues and 42 to 72 in FZP-withdrawn tissues from each of 5 rats. There were no significant differences in average PSD length or width between control and FZP-withdrawn rats for each antibody reaction (GluN1: particle density, F = 11.5, df = 1, p = 0.009; length, F = 3.72, df = 1, p = 0.090; width, F = 0.91, df = 1, p =< 0.369; GluN2A: particle density, F = 1.27, df = 1, p = 0.293; length, F = 0.09, df = 1, p = 0.767; width, F = 0.01, df = 1, p = 0.936; GluN2B: particle density, F = 9.68, df = 1, p = 0.014; length, F = 0.17, df = 1, p = 0.688; width, F = 0.09, df = 1, p = 0.775).
In our previous study (Das et al., 2008), increased insertion of GluA1 subunits in CA1 synapses corresponded with a modest, yet significant increase in mean PSD length in FZP-withdrawn rats compared to controls. This is consistent with several reports of enlargement in synapse size associated with enhanced AMPAR-mediated glutamatergic strength and GluA1 insertion during LTP (Kopec et al., 2007; Matsuzaki et al., 2004; Toni et al., 2001). Further analysis revealed that, in general, GluA1 immunonegative synapses were significantly smaller in length than immunopositive synapses, also in agreement with another study which reported that smaller synapses had low or negligible GluA1 content and were putatively “silent” (Takumi et al., 1999).
In the current study, a similar approach was used to compare mean PSD lengths of GluN immunopositive and immunonegative synapses between control and FZP-withdrawn groups for each antibody reaction. Overall, GluN immunogold labeling appeared to be localized to smaller synapses compared to the GluA1 immunogold labeling (Das et al., 2008) and PSD length of GluN1-immunopositive synapses (0.186 ± 0.003 μm) in control rats was significantly smaller than their immunonegative counterparts (0.232 ± 0.018 μm n = 10 synapses; F= 4.74, df = 3; p = 0.02; p < 0.05). Despite the small sample size of immunonegative synapses, this finding is similar to another report demonstrating that the GluN1-subunit was more concentrated in smaller synapses and that the proportion of synapses lacking GluN1 increased with size (Kharazia and Weinberg, 1999; Shinohara and Hirase, 2009; Sobczyk et al., 2005). Unexpectedly, in FZP-withdrawn tissues, there was no difference in PSD lengths between immunopositive and immunonegative (n = 44) synapses immunolabeled for the GluN1-subunit (0.198 ± 0.005 μm and 0.193 ± 0.011 μm, respectively) or GluN2A (0.203 ± 0.005 μm and 0.197 ± 0.007μm, F=0.876, df = 3; p = 0.87) or GluN2B (0.196 ± 0.006μm, and 0.199 ± 0.009μm F=1.01, df = 3; p = 0.41). A similar conclusion was reached when using a criterion of 2 immunogold particles as the threshold for positive immunolabeling of a synapse. The lack of a significant difference between immunopositive and immunonegative PSD lengths in GluN1-labeled synapses in FZP-withdrawn tissues may reflect the lack of significant changes in synapse size that parallel alterations in NMDA receptor content. In any case, the observed differences in PSD size between immunopositive and immunonegative synapses in this and the earlier study was always rather small and likely related to significant amounts of synapse-to-synapse variability. Perhaps these results arise in part because analyses were performed in random synaptic profiles in single-sections, which does not allow for full reconstruction of the entire size and shape of these synapses.
Spatial localization of NMDAR immunogold labeling in FZP-withdrawn rats
NMDAR synaptic trafficking by insertion, internalization or lateral movement of receptors across the synapse are fundamental mechanisms that regulate synaptic efficacy (Tovar and Westbrook, 2002). To explore whether the possible translocation of NMDARs within the postsynaptic membrane could be correlated with the observed changes in density, we analyzed the localization of NMDAR subunit immunogold particles within PSDs either in a plane orthogonal to the postsynaptic membrane (Fig. 5) or in the membrane plane (see Fig. 5B inset). The location of immunogold particles was first defined by measuring the distance from the center of each gold particle to the outer leaflet of the postsynaptic membrane (Valtschanoff et al., 2000). For GluN1 immunoprocessed sections, one major peak of immunogold particles was observed intracellularly at a distance of 6.2 nm in control and 5.8 nm in FZP-withdrawn tissues (Fig. 5A). In GluN2B labeled sections, two similar peaks were detected at 6.1 and 21.8 nm in controls, whereas in FZP-withdrawn tissues the 6.1 nm peak contained more immunogold particles than the peak at 21.8 nm (Fig. 5B). GluN2A immunolabeled synapses displayed no significant differences in the relative localization of immunogold particles with respect to the plasma membranes (Fig. 5C).
Figure 5. Spatial distribution of GluN1, GluN2A and GluN2B immunogold particles.
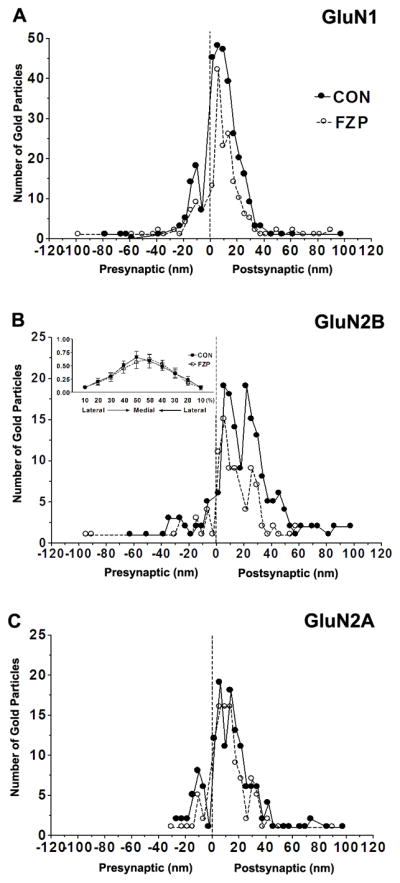
(A–C) Distribution of immunogold particles at different distances within the PSD measured in an orthogonal axis from the center of each 10 nm gold particle to the outer leaflet of postsynaptic membrane (up to 100 nm distance) and grouped into 4 nm wide bins (0 on the abscissa represents external face of postsynaptic membrane). Negative values indicate gold particles located in the direction of the presynaptic bouton and synaptic cleft (average width ~20 nm). Positive values indicate immunogold labeling towards the cytoplasm. (A) GluN1 labeling showed one peak, ~6 nm inside the postsynaptic membrane (image resolution ± 2 nm) in control and FZP-withdrawn tissues. (B) GluN2B immunogold in control synapses was similarly distributed in two peaks located 6 and 22 nm within the PSD, but in FZP-withdrawn rats showed a single peak at 6 nm with the peak at 22 nm much reduced. (B, Inset) Lateral distribution profile of GluN2B subunit immunogold labeling. The distance from the lateral edge of the PSD to the center of GluN2B immunogold particles, parallel to the surface of the postsynaptic membrane was measured. The 0–30 nm PSD bin was subdivided laterally into 10 bins consisting of the lateral 10% to medial 50% from either edge of the PSD. GluN2B subunit distribution was concentrated in the middle of the synapse and was not significantly different between control and FZP-withdrawn rats. (C) The spatial distribution of GluN2A labeling in the PSD did not result in clearly separated peaks within the 40 nm width of the PSD and this pattern was unaltered in FZP-withdrawn rats. Data was obtained from 59, 55 and 56 synapses for GluN1, GluN2A and GluN2B-labeled tissues respectively from controls and 66, 51 and 49 synapses from FZP-withdrawn synapses. In all cases, the labeling was primarily found within the 40 nm width of the PSD and the 20 nm surrounding error zone (spatial resolution of indirect immunogold labeling (Matsubara et al., 1996).
To determine if reductions in synaptic labeling of the more mobile GluN2B subunit was correlated with movement of the receptor towards the lateral edge of PSDs, the distribution of GluN2B-immunolabeling parallel to the postsynaptic membrane, was analyzed using the methodology described in Park et al. (2008). Measurement of distances to the center of gold particles from either edge of the PSD revealed that most gold particles were concentrated in the middle 40–50% of the PSD, with labeling greatly reduced towards the edge (10–20%) of the PSD (Fig 5B, inset). There were no significant differences in immunogold localization within synapses of control and FZP-withdrawn tissues in either middle or lateral bins.
Another possibility is that NMDARs are actually completely removed from the PSD by lateral diffusion and reside in membrane regions adjacent to the PSD. Therefore perisynaptic NMDAR subunit immunogold distribution was also analyzed. Perisynaptic labeling was defined as particles localized 100 nm lateral to the PSD (excluding a 20 nm surrounding edge) (Das et al., 2008). Labeling was very low for all NMDAR subunits in this region. For GluN1, perisynaptic labeling represented only 0.5% and 0.9% of total GluN1 labeling in control and FZP-withdrawn rats, respectively. In GluN2A-labeled tissues, perisynaptic labeling accounted for 1.5% and 0.3% of total GluN2A labeling, respectively and this difference was not statistically significant. Similarly, in GluN2B-labeled tissues, perisynaptic labeling represented 1.6% and 3.5% of total GluN2B labeling in control and FZP-withdrawn rats. These rather small differences did not reach significance given the large variability from synapse to synapse.
In summary, the most significant change observed in the spatial distribution of immunogold particles was a reduction in the number of GluN2B particles relatively deep within the synapse. No significant changes were detected in lateral distribution although there was a definitive trend towards an increase in perisynaptic labeling in a proportion of GluN2B synapses after FZP treatment. GluN1 and GluN2A subunits showed no significant changes in overall distributions within the PSD or adjacent to it.
DISCUSSION
The present study provides ultrastructural evidence for differential regulation of NMDAR GluN1, GluN2A and GluN2B subunits at the synaptic level in the hippocampus during benzodiazepine withdrawal and suggests that the removal of GluN1/GluN2B NMDARs from synapses is a very likely explanation for previous electrophysiological and biochemical findings demonstrating decreased NMDAR-mediated whole-cell currents and subunit protein expression in the CA1 region (Shen et al., 2009; Van Sickle et al., 2002; Van Sickle et al., 2004).
The key findings reported are that GluN1 and GluN2B subunit immunogold content in CA1 neuron synapses was significantly reduced during benzodiazepine withdrawal with a corresponding decrease in the percentage of GluN1 and GluN2B immunopositive synapses, but without significant alterations in GluN2A subunit content. The reduction in GluN2B-containing receptors at the synapse correlates well with prior electrophysiological data that indicated the decreased ability of ifenprodil to antagonize NMDAR-mediated EPSCs in this model (Shen et al., 2009) and suggests that there may be two pools of GluN1 subunits associated with each GluN2 subunit that might be subject to differential regulatory mechanisms; the pool consisting of GluN1/GluN2B subunits may be reduced at the synapse during benzodiazepine withdrawal, whereas the receptor pool consisting of GluN1/GluN2A may remain relatively constant.
Consistent with the reduction in GluN1 and GluN2B mRNA and protein levels (Shen and Tietz, 2008; Van Sickle et al., 2002), the reduction in GluN1/GluN2B receptors might be the result of decreased NMDAR synthesis and synaptic insertion. Enhanced internalization, perhaps via a CaMKII-mediated mechanism, could also contribute to the reduction of synaptic GluN1/GluN2B receptors. GluN2B C-terminal tails preferentially bind CaMKII (Mayadevi et al., 2002) and are stabilized in the synapse by their interaction with MAGUKs and destabilized by increased Ca2+ levels. For instance, an activity-dependent CaMKII-mediated increase in casein kinase 2 activity disrupted the GluN2B-PSD95 interaction by phosphorylation of PDZ-binding domain of the GluN2B subunit (Chung et al., 2004). Since total CaMKII levels were increased in the PSD subfractions derived from the CA1 region of FZP-withdrawn rats (Shen et al., 2010), it is possible that CaMKII-facilitated destabilization of GluN2B-containing receptors led to a reduction in synaptic GluN1/GluN2B receptors during benzodiazepine withdrawal. Internalization may be associated with the mobilization of GluN2B-containing receptors to the lateral edges of the PSD where they are more unstable due to the absence of MAGUK proteins such as PSD-95 (Prybylowski et al., 2005). Though analysis of lateral distribution 2-days after withdrawal suggested that the synaptic reduction of GluN2B subunits was not due to an increased accumulation at the lateral edges of the PSD or in the perisynaptic region, increased trafficking through this compartment during the withdrawal period cannot be completely ruled out because this might occur without a detectable change in the steady-state concentration of the subunits. In the latter case, increased lateral movement of NMDARs must be paralleled by increased internalization from lateral, synaptic and/or perisynaptic regions.
NMDAR subunit labeling was strongly localized just inside the postsynaptic plasma membrane as evidenced by a sharp peak of labeling ~6 nm intracellular from the outer edge of postsynaptic membrane. This finding was consistent with other studies showing that the C-terminus of NMDAR is on the cytoplasmic side of plasma membrane (Bennett and Dingledine, 1995; Kharazia and Weinberg, 1999). Postsynaptic distribution of GluN1 immunogold in olfactory bulb granule cells showed two peaks, proposed to be due to presence of different GluN1 splice variants with different C-terminal tail lengths (Sassoe-Pognetto and Ottersen, 2000). Splice variants of the GluN1 subunit expressing different C-terminal tail lengths were generated dependent on differential splicing of exons encoding C1-or C2-cassettes in the C-terminal domains of the GluN1 subunit (Horak and Wenthold, 2009; Yamada et al., 2001). However, in the current study, two peaks of GluN1 immunogold labeling were not observed suggesting that the C2 cassette-specific GluN1 antibody used may recognize only one GluN1 splice variant in CA1 synapses. Interestingly, in the case of GluN2B subunits, a second major peak more intracellular (~22 nm) relative to the outer edge of postsynaptic plasma membrane was also observed. Since no GluN2 splice variants have been identified, one explanation for the results could be that the C-terminal domains of GluN2 have varied configurations due to their interaction with cytoskeletal proteins (Wyszynski et al., 1997). Alternatively, they may represent pools of receptor GluN2 subunits in different states of trafficking (Park et al., 2008).
NMDARs are considered crucial synaptic elements in mediating induction of hippocampal LTP (Collingridge et al., 1983; Nakazawa et al., 2004; Zalutsky and Nicoll, 1990). Targeted deletion of the GluN1 gene in the CA1 region abolished LTP and impaired spatial memory (Tonegawa et al., 1996; Tsien et al., 1996). Numerous recent studies have documented that GluN2A- and GluN2B-containing NMDAR may differentially govern hippocampal synaptic plasticity. For instance, GluN2A was reported to be required for induction of LTP, but not LTD and GluN2B for LTD, but not LTP (Liu et al., 2004; Massey et al., 2004). Other groups ruled out an exclusive role for GluN2B in LTD (Bellone and Nicoll, 2007; Morishita et al., 2007). GluN2B overexpression enhanced LTP and spatial memory, while GluN2B-antisense knockdown abolished LTP and impaired memory (Berberich et al., 2005; Clayton et al., 2002; Wang et al., 2009). Targeted GluN2B KO in the CA3 region resulted in a near complete loss of NMDAR-mediated EPSCs and abolished LTP (Akashi et al., 2009). GluN2B subunit tyrosine phosphorylation was reported to be critical for LTP maintenance, but not induction, as increased GluN2B phosphorylation was observed 5–15 minutes after LTP induction and sustained for a period of 3 hours (Rostas et al., 1996). Thus, the reduction in GluN2B-containing NMDARs from CA1 synapses from 2-day FZP-withdrawn rats may underlie the modest effect on LTP in CA1 neurons. That is, though LTP induction was unaffected after FZP withdrawal, it could only be maintained for <15 min (Shen et al., 2009). Both GluN2B and phosphoTYR1303 GluN2B subunit levels were significantly decreased in CA1 PSD subfractions from FZP-withdrawn hippocampus, however their ratio was unchanged (Shen and Tietz, 2008). Nonetheless, the absolute numbers of GluN2B subunits available for tyrosine phosphorylation were decreased. Thus, removal of GluN2B, but not GluN2A-containing synaptic NMDARs remains a possible explanation for the lack of LTP maintenance during FZP withdrawal (Shen et al., 2009).
Adaptations of the glutamatergic system, including enhanced GluA1 expression and function, reminiscent of mechanisms underlying LTP, have been reported in different models of drug abuse, including benzodiazepine dependence (Billa et al., 2009; Boudreau et al., 2007; Kim et al., 2009; Das et al., 2008; Song et al., 2007; Shen et al., 2010). Many studies have reported reduced NMDAR subunit expression in a brain region-specific manner and dependent on time of drug withdrawal. As during benzodiazepine withdrawal (Shen et al., 2009; Song et al., 2007), cocaine withdrawal resulted in decreased GluN2B-subunit expression and increased AMPAR GluA1, but not GluA2 subunit levels in rat basolateral amygdala (Lu et al., 2005), reduced GluN1 mRNA expression in nucleus accumbens (Yamaguchi et al., 2002) and reduced GluN1 and GluN2B subunit protein in immunoblots from nucleus accumbens and striatum (Loftis and Janowsky, 2000; 2002). Ethanol withdrawal resulted in decreased GluN2B subunit levels in nucleus accumbens (Obara et al., 2009) and chronic amphetamine significantly lowered GluN2B levels in striatum, but not hippocampus, by a destabilization and degradation of surface GluN2B receptors that resulted in a reduction of ifenprodil-sensitive NMDAR-mediated EPSCs (Mao et al., 2009). Consistent with these reports of drug-induced regulation of glutamatergic function, the present data demonstrated a reduction in CA1 synaptic GluN1/GluN2B receptors at a time-point when AMPAR GluA1 subunit levels and single-channel conductance were increased (Shen et al., 2009). The role of NMDARs in triggering phosphorylation of GluA1 subunits after repeated cocaine injections was suggested by the reduction of phosphoSER831 GluA1 levels following NMDAR blockade (Kim et al., 2009). In contrast, phosphoSER831 GluA1 levels remain upregulated in 2-day FZP withdrawn rats (Shen et al., 2009, 2010) suggesting that GluN1/GluN2B-containing NMDARs are not necessary for the AMPAR potentiation during benzodiazepine withdrawal. Additionally, prior AMPAR antagonist injection prevented the decreased NMDAR function and protein expression (Van Sickle et al., 2004; Xiang and Tietz, 2007). Collectively, the data suggest that changes in synaptic NMDARs during benzodiazepine withdrawal are secondary to enhanced insertion of GluA1 homomeric AMPARs. The proposed functional model of changes in the glutamate receptor population associated with chronic benzodiazepine treatment and withdrawal is summarized in Figure 6.
Figure 6. Benzodiazepine Withdrawal Model.

We propose that 1: persistent benzodiazepine enhancement of the inhibitory GABAA receptor leads to a bicarbonate ion-dependent GABA-mediated depolarization (Zeng and Tietz, 2000) through an as yet undefined mechanism and 2: a doubling of L-type voltage-gated calcium channel (L-VGCC) current density (Xiang et al., 2008). Since both AMPAR potentiation and BZ withdrawal-anxiety were prevented by systemic pre-injection of either an AMPAR or a L-VGCC antagonist, but not an NMDAR antagonist (Van Sickle et al., 2004, Xiang and Tietz, 2007), we postulate that the increased Ca2+ influx through L-VGCCs mediates 3: insertion of AMPAR GluA1 homomers and increased AMPAR current amplitude at CA1 synapses of FZP-withdrawn rats (Song et al., 2007, Shen et al., 2010). Increased Ca2+ influx largely through L-VGCCs, but also possibly through Ca2+-permeable GluA1 homomers leads to 4: CaMKII-mediated Ser831 phosphorylation of GluA1 homomers and increased AMPAR current conductance 2 days after drug withdrawal (Shen et al., 2009, 2010). All dashed arrows represent as yet undefined mechanisms. 5: NMDAR function and expression is decreased secondary to AMPAR potentiation. Data from the current and earlier studies (Van Sickle et al., 2002, Shen et al., 2008) suggest that GluN2B-containing NMDARs in the CA1 synapse are regulated by decreased insertion and/or increased removal from the synapse. Decreased synaptic GluN2B-containing NMDARs may serve as a physiological brake to mitigate CA1 neuron hyperexcitability and BZ withdrawal-anxiety.
In summary, the findings extend prior studies by providing direct evidence for a reduction of GluN1 and GluN2B, but not GluN2A subunits at CA1 neuron synapses, and support the idea that differential regulation of GluN1/GluN2B-containing NMDAR plays a significant role in modulating the expression of benzodiazepine withdrawal-anxiety.
Acknowledgments
Support/Grant Information: This work was supported by NIDA RO1-DA018342 and UT College of Medicine Drug Abuse Research Program (E.I.T.).
The authors thank Krista Pettee, Brian L. Behrle, Maiko Ziegler and Maria Berrocal for excellent technical assistance and Dr. Guofu Shen for assistance with statistical analyses. We gratefully acknowledge Dr. Stefano Vicini for providing the GluN2A KO mice.
Abbreviations
- aa
amino acids
- AMPAR
alpha-Amino-3-hydroxy-5-methylisoxazole-4-propionic acid receptor
- BZ
benzodiazepine
- CNS
central nervous system
- CON
control
- FZP
flurazepam
- GABA
gamma-aminobutyric acid type A
- GluR
glutamate receptor
- KO
knockout
- LTP
long-term potentiation
- mEPSCs
miniature excitatory postsynaptic currents
- MAGUK
Membrane associated guanylate kinase
- NMDAR
N-methyl-D-aspartate receptor
- PSD
postsynaptic density
- WT
wildtype
LITERATURE CITED
- Abe M, Fukaya M, Yagi T, Mishina M, Watanabe M, Sakimura K. NMDA receptor GluRepsilon/NR2 subunits are essential for postsynaptic localization and protein stability of GluRzeta1/NR1 subunit. J Neurosci. 2004;24:7292–7304. doi: 10.1523/JNEUROSCI.1261-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akashi K, Kakizaki T, Kamiya H, Fukaya M, Yamasaki M, Abe M, Natsume R, Watanabe M, Sakimura K. NMDA receptor GluN2B (GluR epsilon 2/NR2B) subunit is crucial for channel function, postsynaptic macromolecular organization, and actin cytoskeleton at hippocampal CA3 synapses. J Neurosci. 2009;29:10869–10882. doi: 10.1523/JNEUROSCI.5531-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allison C, Pratt JA. Differential effects of two chronic diazepam treatment regimes on withdrawal anxiety and AMPA receptor characteristics. Neuropsychopharmacology. 2006;31:602–619. doi: 10.1038/sj.npp.1300800. [DOI] [PubMed] [Google Scholar]
- Barria A, Malinow R. Subunit-specific NMDA receptor trafficking to synapses. Neuron. 2002;35:345–353. doi: 10.1016/s0896-6273(02)00776-6. [DOI] [PubMed] [Google Scholar]
- Bellone C, Nicoll RA. Rapid bidirectional switching of synaptic NMDA receptors. Neuron. 2007;55:779–785. doi: 10.1016/j.neuron.2007.07.035. [DOI] [PubMed] [Google Scholar]
- Bennett JA, Dingledine R. Topology profile for a glutamate receptor: three transmembrane domains and a channel-lining reentrant membrane loop. Neuron. 1995;14:373–384. doi: 10.1016/0896-6273(95)90293-7. [DOI] [PubMed] [Google Scholar]
- Berberich S, Punnakkal P, Jensen V, Pawlak V, Seeburg PH, Hvalby O, Kohr G. Lack of NMDA receptor subtype selectivity for hippocampal long-term potentiation. J Neurosci. 2005;25:6907–6910. doi: 10.1523/JNEUROSCI.1905-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billa SK, Sinha N, Rudrabhatla SR, Moron JA. Extinction of morphine-dependent conditioned behavior is associated with increased phosphorylation of the GluR1 subunit of AMPA receptors at hippocampal synapses. Eur J Neurosci. 2009;29:55–64. doi: 10.1111/j.1460-9568.2008.06560.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boudreau AC, Reimers JM, Milovanovic M, Wolf ME. Cell surface AMPA receptors in the rat nucleus accumbens increase during cocaine withdrawal but internalize after cocaine challenge in association with altered activation of mitogen-activated protein kinases. J Neurosci. 2007;27:10621–10635. doi: 10.1523/JNEUROSCI.2163-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chouinard G. Issues in the clinical use of benzodiazepines: potency, withdrawal, and rebound. J Clin Psychiatry. 2004;65(Suppl 5):7–12. [PubMed] [Google Scholar]
- Chung HJ, Huang YH, Lau LF, Huganir RL. Regulation of the NMDA receptor complex and trafficking by activity-dependent phosphorylation of the NR2B subunit PDZ ligand. J Neurosci. 2004;24:10248–10259. doi: 10.1523/JNEUROSCI.0546-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clayton DA, Mesches MH, Alvarez E, Bickford PC, Browning MD. A hippocampal NR2B deficit can mimic age-related changes in long-term potentiation and spatial learning in the Fischer 344 rat. J Neurosci. 2002;22:3628–3637. doi: 10.1523/JNEUROSCI.22-09-03628.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collingridge GL, Kehl SJ, McLennan H. Excitatory amino acids in synaptic transmission in the Schaffer collateral-commissural pathway of the rat hippocampus. J Physiol. 1983;334:33–46. doi: 10.1113/jphysiol.1983.sp014478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das P, Lilly SM, Zerda R, Gunning WT, 3rd, Alvarez FJ, Tietz EI. Increased AMPA receptor GluR1 subunit incorporation in rat hippocampal CA1 synapses during benzodiazepine withdrawal. J Comp Neurol. 2008;511:832–846. doi: 10.1002/cne.21866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukaya M, Kato A, Lovett C, Tonegawa S, Watanabe M. Retention of NMDA receptor NR2 subunits in the lumen of endoplasmic reticulum in targeted NR1 knockout mice. Proc Natl Acad Sci U S A. 2003;100:4855–4860. doi: 10.1073/pnas.0830996100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardoni F, Mauceri D, Fiorentini C, Bellone C, Missale C, Cattabeni F, Di Luca M. CaMKII-dependent phosphorylation regulates SAP97/NR2A interaction. J Biol Chem. 2003;278:44745–44752. doi: 10.1074/jbc.M303576200. [DOI] [PubMed] [Google Scholar]
- Griffiths RR, Johnson MW. Relative abuse liability of hypnotic drugs: a conceptual framework and algorithm for differentiating among compounds. J Clin Psychiatry. 2005;66(Suppl 9):31–41. [PubMed] [Google Scholar]
- Groc L, Heine M, Cognet L, Brickley K, Stephenson FA, Lounis B, Choquet D. Differential activity-dependent regulation of the lateral mobilities of AMPA and NMDA receptors. Nat Neurosci. 2004;7:695–696. doi: 10.1038/nn1270. [DOI] [PubMed] [Google Scholar]
- Groc L, Heine M, Cousins SL, Stephenson FA, Lounis B, Cognet L, Choquet D. NMDA receptor surface mobility depends on NR2A-2B subunits. Proc Natl Acad Sci U S A. 2006;103:18769–18774. doi: 10.1073/pnas.0605238103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horak M, Wenthold RJ. Different roles of C-terminal cassettes in the trafficking of full-length NR1 subunits to the cell surface. J Biol Chem. 2009;284:9683–9691. doi: 10.1074/jbc.M807050200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izzo E, Auta J, Impagnatiello F, Pesold C, Guidotti A, Costa E. Glutamic acid decarboxylase and glutamate receptor changes during tolerance and dependence to benzodiazepines. Proc Natl Acad Sci U S A. 2001;98:3483–3488. doi: 10.1073/pnas.051628698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kharazia VN, Weinberg RJ. Immunogold localization of AMPA and NMDA receptors in somatic sensory cortex of albino rat. J Comp Neurol. 1999;412:292–302. doi: 10.1002/(sici)1096-9861(19990920)412:2<292::aid-cne8>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- Kim MJ, Dunah AW, Wang YT, Sheng M. Differential roles of NR2A- and NR2B-containing NMDA receptors in Ras-ERK signaling and AMPA receptor trafficking. Neuron. 2005;46:745–760. doi: 10.1016/j.neuron.2005.04.031. [DOI] [PubMed] [Google Scholar]
- Kim SM, Ahn SM, Go BS, Wang JQ, Choe ES. Alterations in AMPA receptor phosphorylation in the rat striatum following acute and repeated cocaine administration. Neuroscience. 2009;163:618–626. doi: 10.1016/j.neuroscience.2009.06.054. [DOI] [PubMed] [Google Scholar]
- Kopec CD, Real E, Kessels HW, Malinow R. GluR1 links structural and functional plasticity at excitatory synapses. J Neurosci. 2007;27:13706–13718. doi: 10.1523/JNEUROSCI.3503-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau CE, Falk JL, Dolan S, Tang M. Simultaneous determination of flurazepam and five metabolites in serum by high-performance liquid chromatography and its application to pharmacokinetic studies in rats. J Chromatogr. 1987;423:251–259. doi: 10.1016/0378-4347(87)80348-1. [DOI] [PubMed] [Google Scholar]
- Lau CG, Zukin RS. NMDA receptor trafficking in synaptic plasticity and neuropsychiatric disorders. Nat Rev Neurosci. 2007;8:413–426. doi: 10.1038/nrn2153. [DOI] [PubMed] [Google Scholar]
- Lee HK, Takamiya K, He K, Song L, Huganir RL. Specific roles of AMPA receptor subunit GluR1 (GluA1) phosphorylation sites in regulating synaptic plasticity in the CA1 region of hippocampus. J Neurophysiol. 2010;103:479–489. doi: 10.1152/jn.00835.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Wong TP, Pozza MF, Lingenhoehl K, Wang Y, Sheng M, Auberson YP, Wang YT. Role of NMDA receptor subtypes in governing the direction of hippocampal synaptic plasticity. Science. 2004;304:1021–1024. doi: 10.1126/science.1096615. [DOI] [PubMed] [Google Scholar]
- Loftis JM, Janowsky A. Regulation of NMDA receptor subunits and nitric oxide synthase expression during cocaine withdrawal. J Neurochem. 2000;75:2040–2050. doi: 10.1046/j.1471-4159.2000.0752040.x. [DOI] [PubMed] [Google Scholar]
- Loftis JM, Janowsky A. Cocaine treatment- and withdrawal-induced alterations in the expression and serine phosphorylation of the NR1 NMDA receptor subunit. Psychopharmacology (Berl) 2002;164:349–359. doi: 10.1007/s00213-002-1209-9. [DOI] [PubMed] [Google Scholar]
- Lu L, Dempsey J, Shaham Y, Hope BT. Differential long-term neuroadaptations of glutamate receptors in the basolateral and central amygdala after withdrawal from cocaine self-administration in rats. J Neurochem. 2005;94:161–168. doi: 10.1111/j.1471-4159.2005.03178.x. [DOI] [PubMed] [Google Scholar]
- Malenka RC, Nicoll RA. Long-term potentiation--a decade of progress? Science. 1999;285:1870–1874. doi: 10.1126/science.285.5435.1870. [DOI] [PubMed] [Google Scholar]
- Malinow R, Malenka RC. AMPA receptor trafficking and synaptic plasticity. Annu Rev Neurosci. 2002;25:103–126. doi: 10.1146/annurev.neuro.25.112701.142758. [DOI] [PubMed] [Google Scholar]
- Mao LM, Wang W, Chu XP, Zhang GC, Liu XY, Yang YJ, Haines M, Papasian CJ, Fibuch EE, Buch S, Chen JG, Wang JQ. Stability of surface NMDA receptors controls synaptic and behavioral adaptations to amphetamine. Nat Neurosci. 2009;12:602–610. doi: 10.1038/nn.2300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massey PV, Johnson BE, Moult PR, Auberson YP, Brown MW, Molnar E, Collingridge GL, Bashir ZI. Differential roles of NR2A and NR2B-containing NMDA receptors in cortical long-term potentiation and long-term depression. J Neurosci. 2004;24:7821–7828. doi: 10.1523/JNEUROSCI.1697-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsubara A, Laake JH, Davanger S, Usami S, Ottersen OP. Organization of AMPA receptor subunits at a glutamate synapse: a quantitative immunogold analysis of hair cell synapses in the rat organ of Corti. J Neurosci. 1996;16:4457–4467. doi: 10.1523/JNEUROSCI.16-14-04457.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuzaki M, Honkura N, Ellis-Davies GC, Kasai H. Structural basis of long-term potentiation in single dendritic spines. Nature. 2004;429:761–766. doi: 10.1038/nature02617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayadevi M, Praseeda M, Kumar KS, Omkumar RV. Sequence determinants on the NR2A and NR2B subunits of NMDA receptor responsible for specificity of phosphorylation by CaMKII. Biochim Biophys Acta. 2002;1598:40–45. doi: 10.1016/s0167-4838(02)00315-1. [DOI] [PubMed] [Google Scholar]
- Mori H, Manabe T, Watanabe M, Satoh Y, Suzuki N, Toki S, Nakamura K, Yagi T, Kushiya E, Takahashi T, Inoue Y, Sakimura K, Mishina M. Role of the carboxy-terminal region of the GluR epsilon2 subunit in synaptic localization of the NMDA receptor channel. Neuron. 1998;21:571–580. doi: 10.1016/s0896-6273(00)80567-x. [DOI] [PubMed] [Google Scholar]
- Morishita W, Lu W, Smith GB, Nicoll RA, Bear MF, Malenka RC. Activation of NR2B-containing NMDA receptors is not required for NMDA receptor-dependent long-term depression. Neuropharmacology. 2007;52:71–76. doi: 10.1016/j.neuropharm.2006.07.005. [DOI] [PubMed] [Google Scholar]
- Nakazawa K, McHugh TJ, Wilson MA, Tonegawa S. NMDA receptors, place cells and hippocampal spatial memory. Nat Rev Neurosci. 2004;5:361–372. doi: 10.1038/nrn1385. [DOI] [PubMed] [Google Scholar]
- Nicholson DA, Geinisman Y. Axospinous synaptic subtype-specific differences in structure, size, ionotropic receptor expression, and connectivity in apical dendritic regions of rat hippocampal CA1 pyramidal neurons. J Comp Neurol. 2009;512:399–418. doi: 10.1002/cne.21896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nong Y, Huang YQ, Salter MW. NMDA receptors are movin’ in. Curr Opin Neurobiol. 2004;14:353–361. doi: 10.1016/j.conb.2004.05.001. [DOI] [PubMed] [Google Scholar]
- Obara I, Bell RL, Goulding SP, Reyes CM, Larson LA, Ary AW, Truitt WA, Szumlinski KK. Differential effects of chronic ethanol consumption and withdrawal on homer/glutamate receptor expression in subregions of the accumbens and amygdala of P rats. Alcohol Clin Exp Res. 2009;33:1924–1934. doi: 10.1111/j.1530-0277.2009.01030.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh MC, Derkach VA, Guire ES, Soderling TR. Extrasynaptic membrane trafficking regulated by GluR1 serine 845 phosphorylation primes AMPA receptors for long-term potentiation. J Biol Chem. 2006;281:752–758. doi: 10.1074/jbc.M509677200. [DOI] [PubMed] [Google Scholar]
- Park CS, Elgersma Y, Grant SG, Morrison JH. alpha-Isoform of calcium-calmodulin-dependent protein kinase II and postsynaptic density protein 95 differentially regulate synaptic expression of NR2A- and NR2B-containing N-methyl-d-aspartate receptors in hippocampus. Neuroscience. 2008;151:43–55. doi: 10.1016/j.neuroscience.2007.09.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petralia RS, Sans N, Wang YX, Wenthold RJ. Ontogeny of postsynaptic density proteins at glutamatergic synapses. Mol Cell Neurosci. 2005;29:436–452. doi: 10.1016/j.mcn.2005.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prybylowski K, Chang K, Sans N, Kan L, Vicini S, Wenthold RJ. The synaptic localization of NR2B-containing NMDA receptors is controlled by interactions with PDZ proteins and AP-2. Neuron. 2005;47:845–857. doi: 10.1016/j.neuron.2005.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radley JJ, Farb CR, He Y, Janssen WG, Rodrigues SM, Johnson LR, Hof PR, LeDoux JE, Morrison JH. Distribution of NMDA and AMPA receptor subunits at thalamo-amygdaloid dendritic spines. Brain Res. 2007;1134:87–94. doi: 10.1016/j.brainres.2006.11.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robison AJ, Bartlett RK, Bass MA, Colbran RJ. Differential modulation of Ca2+/calmodulin-dependent protein kinase II activity by regulated interactions with N-methyl-D-aspartate receptor NR2B subunits and alpha-actinin. J Biol Chem. 2005;280:39316–39323. doi: 10.1074/jbc.M508189200. [DOI] [PubMed] [Google Scholar]
- Rostas JA, Brent VA, Voss K, Errington ML, Bliss TV, Gurd JW. Enhanced tyrosine phosphorylation of the 2B subunit of the N-methyl-D-aspartate receptor in long-term potentiation. Proc Natl Acad Sci U S A. 1996;93:10452–10456. doi: 10.1073/pnas.93.19.10452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakimura K, Kutsuwada T, Ito I, Manabe T, Takayama C, Kushiya E, Yagi T, Alzawa S, Inoue Y, Sugiyama H, Mishina M. Reduced hippocampal LTP and spatial learning in mice lacking NMDA receptorε1 subunit. Nature. 1995;373:151–155. doi: 10.1038/373151a0. [DOI] [PubMed] [Google Scholar]
- Sans N, Prybylowski K, Petralia RS, Chang K, Wang YX, Racca C, Vicini S, Wenthold RJ. NMDA receptor trafficking through an interaction between PDZ proteins and the exocyst complex. Nat Cell Biol. 2003;5:520–530. doi: 10.1038/ncb990. [DOI] [PubMed] [Google Scholar]
- Sassoe-Pognetto M, Ottersen OP. Organization of ionotropic glutamate receptors at dendrodendritic synapses in the rat olfactory bulb. J Neurosci. 2000;20:2192–2201. doi: 10.1523/JNEUROSCI.20-06-02192.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen G, Mohamed MS, Das P, Tietz EI. Positive allosteric activation of GABAA receptors bi-directionally modulates hippocampal glutamate plasticity and behaviour. Biochem Soc Trans. 2009;37:1394–1398. doi: 10.1042/BST0371394. [DOI] [PubMed] [Google Scholar]
- Shen G, Tietz EI. Regulation of synaptic NR2B subunit-containing NMDA receptors: one component of glutamatergic plasticity in benzodiazepine withdrawal. Soc Neurosci Abstr, 2008 2008 [Google Scholar]
- Shen G, Van Sickle BJ, Tietz EI. Calcium/calmodulin dependent protein kinase II mediates hippocampal AMPAR-mediated synaptic plasticity during benzodiazepine withdrawal. Neuropsychopharmacology. 2010 doi: 10.1038/npp.2010.61. in press. [2010 May 5. Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song J, Shen G, Greenfield LJ, Jr, Tietz EI. Benzodiazepine withdrawal-induced glutamatergic plasticity involves up-regulation of GluR1-containing alpha-amino-3-hydroxy-5-methylisoxazole-4-propionic acid receptors in Hippocampal CA1 neurons. J Pharmacol Exp Ther. 2007;322:569–581. doi: 10.1124/jpet.107.121798. [DOI] [PubMed] [Google Scholar]
- Strack S, McNeill RB, Colbran RJ. Mechanism and regulation of calcium/calmodulin-dependent protein kinase II targeting to the NR2B subunit of the N-methyl-D-aspartate receptor. J Biol Chem. 2000;275:23798–23806. doi: 10.1074/jbc.M001471200. [DOI] [PubMed] [Google Scholar]
- Takumi Y, Matsubara A, Rinvik E, Ottersen OP. The arrangement of glutamate receptors in excitatory synapses. Ann N Y Acad Sci. 1999;868:474–482. doi: 10.1111/j.1749-6632.1999.tb11316.x. [DOI] [PubMed] [Google Scholar]
- Tonegawa S, Tsien JZ, McHugh TJ, Huerta P, Blum KI, Wilson MA. Hippocampal CA1-region-restricted knockout of NMDAR1 gene disrupts synaptic plasticity, place fields, and spatial learning. Cold Spring Harb Symp Quant Biol. 1996;61:225–238. [PubMed] [Google Scholar]
- Toni N, Buchs PA, Nikonenko I, Povilaitite P, Parisi L, Muller D. Remodeling of synaptic membranes after induction of long-term potentiation. J Neurosci. 2001;21:6245–6251. doi: 10.1523/JNEUROSCI.21-16-06245.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tovar KR, Westbrook GL. Mobile NMDA receptors at hippocampal synapses. Neuron. 2002;34:255–264. doi: 10.1016/s0896-6273(02)00658-x. [DOI] [PubMed] [Google Scholar]
- Tietz EI, Zeng XJ, Chen S, Lilly SM, Rosenberg HC, Kometiani P. Antagonist-induced reversal of functional and structural measures of hippocampal benzodiazepine tolerance. J Pharmacol Exp Ther. 1999;291:932–942. [PubMed] [Google Scholar]
- Tsien JZ, Huerta PT, Tonegawa S. The essential role of hippocampal CA1 NMDA receptor-dependent synaptic plasticity in spatial memory. Cell. 1996;87:1327–1338. doi: 10.1016/s0092-8674(00)81827-9. [DOI] [PubMed] [Google Scholar]
- Valtschanoff JG, Burette A, Davare MA, Leonard AS, Hell JW, Weinberg RJ. SAP97 concentrates at the postsynaptic density in cerebral cortex. Eur J Neurosci. 2000;12:3605–3614. doi: 10.1046/j.1460-9568.2000.00256.x. [DOI] [PubMed] [Google Scholar]
- Van Sickle BJ, Cox AS, Schak K, Greenfield LJ, Jr, Tietz EI. Chronic benzodiazepine administration alters hippocampal CA1 neuron excitability: NMDA receptor function and expression(1) Neuropharmacology. 2002;43:595–606. doi: 10.1016/s0028-3908(02)00152-1. [DOI] [PubMed] [Google Scholar]
- Van Sickle BJ, Tietz EI. Selective enhancement of AMPA receptor-mediated function in hippocampal CA1 neurons from chronic benzodiazepine-treated rats. Neuropharmacology. 2002;43:11–27. doi: 10.1016/s0028-3908(02)00065-5. [DOI] [PubMed] [Google Scholar]
- Van Sickle BJ, Xiang K, Tietz EI. Transient plasticity of hippocampal CA1 neuron glutamate receptors contributes to benzodiazepine withdrawal-anxiety. Neuropsychopharmacology. 2004;29:1994–2006. doi: 10.1038/sj.npp.1300531. [DOI] [PubMed] [Google Scholar]
- Wafford KA. GABAA receptor subtypes: any clues to the mechanism of benzodiazepine dependence? Curr Opin Pharmacol. 2005;5:47–52. doi: 10.1016/j.coph.2004.08.006. [DOI] [PubMed] [Google Scholar]
- Wang D, Cui Z, Zeng Q, Kuang H, Wang LP, Tsien JZ, Cao X. Genetic enhancement of memory and long-term potentiation but not CA1 long-term depression in NR2B transgenic rats. PLoS One. 2009;4:e7486. doi: 10.1371/journal.pone.0007486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe M, Fukaya M, Sakimura K, Manabe T, Mishina M, Inoue Y. Selective scarcity of NMDA receptor channel subunits in the stratum lucidum (mossy fibre-recipient layer) of the mouse hippocampal CA3 subfield. Eur J Neurosci. 1998;10:478–487. doi: 10.1046/j.1460-9568.1998.00063.x. [DOI] [PubMed] [Google Scholar]
- Wenthold RJ, Petralia RS, Blahos J, II, Niedzielski AS. Evidence for multiple AMPA receptor complexes in hippocampal CA1/CA2 neurons. J Neurosci. 1996;16:1982–1989. doi: 10.1523/JNEUROSCI.16-06-01982.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenthold RJ, Prybylowski K, Standley S, Sans N, Petralia RS. Trafficking of NMDA receptors. Annu Rev Pharmacol Toxicol. 2003;43:335–358. doi: 10.1146/annurev.pharmtox.43.100901.135803. [DOI] [PubMed] [Google Scholar]
- Wyszynski M, Lin J, Rao A, Nigh E, Beggs AH, Craig AM, Sheng M. Competitive binding of alpha-actinin and calmodulin to the NMDA receptor. Nature. 1997;385:439–442. doi: 10.1038/385439a0. [DOI] [PubMed] [Google Scholar]
- Xiang K, Tietz EI. Benzodiazepine-induced hippocampal CA1 neuron alpha-amino-3-hydroxy-5-methylisoxasole-4-propionic acid (AMPA) receptor plasticity linked to severity of withdrawal anxiety: differential role of voltage-gated calcium channels and N-methyl-D-aspartic acid receptors. Behav Pharmacol. 2007;18:447–460. doi: 10.1097/FBP.0b013e3282d28f2b. [DOI] [PubMed] [Google Scholar]
- Xiang K, Earl DE, Davis KM, Giovannucci DR, Greenfield LJ, Jr, Tietz EI. Chronic benzodiazepine administration potentiates high-voltage activated calcium currents in hippocampal neurons. J Pharmacol Exp Ther. 2008;327:872–883. doi: 10.1124/jpet.108.144444. [DOI] [PubMed] [Google Scholar]
- Xie XH, Tietz EI. Reduction in potency of selective gamma-aminobutyric acidA agonists and diazepam in CA1 region of in vitro hippocampal slices from chronic flurazepam-treated rats. J Pharmacol Exp Ther. 1992;262:204–211. [PubMed] [Google Scholar]
- Yamada K, Fukaya M, Shimizu H, Sakimura K, Watanabe M. NMDA receptor subunits GluRepsilon1, GluRepsilon3 and GluRzeta1 are enriched at the mossy fibre-granule cell synapse in the adult mouse cerebellum. Eur J Neurosci. 2001;13:2025–2036. doi: 10.1046/j.0953-816x.2001.01580.x. [DOI] [PubMed] [Google Scholar]
- Yamaguchi M, Suzuki T, Abe S, Hori T, Kurita H, Asada T, Okado N, Arai H. Repeated cocaine administration differentially affects NMDA receptor subunit (NR1, NR2A-C) mRNAs in rat brain. Synapse. 2002;46:157–169. doi: 10.1002/syn.10132. [DOI] [PubMed] [Google Scholar]
- Zalutsky RA, Nicoll RA. Comparison of two forms of long-term potentiation in single hippocampal neurons. Science. 1990;248:1619–1624. doi: 10.1126/science.2114039. [DOI] [PubMed] [Google Scholar]
- Zeng X, Tietz EI. Role of bicarbonate ion in mediating decreased synaptic conductance in benzodiazepine tolerant hippocampal CA1 pyramidal neurons. Brain Res. 2000;868:202–214. doi: 10.1016/s0006-8993(00)02330-1. [DOI] [PubMed] [Google Scholar]