

Abstract
Assessment of abuse potential of opioid analgesics has a long history in both laboratory animals and humans. This article reviews the methods used in animals and in humans and then presents the data collected in the evaluation of tramadol, an atypical centrally acting opioid analgesic approved for marketing in the United States in 1998. Finally, data on the abuse of tramadol from postmarketing surveillance and case reports are presented. The consistency between animal and human study results and the predictive value of both are discussed. Overall, there was substantial agreement between animal and human data, with each having predictive value. Nonetheless, it is suggested that abuse-potential screening of new medications would benefit from an organized, integrated cross-species program.
Introduction
The increase in abuse of marketed medications in recent years has highlighted the need for abuse-liability assessment (SAMHSA, 2003b; Zacny et al., 2003). The numbers of new nonmedical users of four major classes of prescription-type drugs (pain relievers, tranquilizers, stimulants, and sedatives) increased between 1991 and 2001, with the largest increase in pain relievers, which increased from 628,000 initiates in 1990 to 2.4 million in 2001 (SAMHSA, 2003a, 2003c). The number of primary treatment admissions for narcotic-analgesic abuse increased 76 percent between 1997 and 2000 (SAMHSA, 2003a). Data from abuse-liability studies are used to determine whether a medication needs to be controlled under the United States (US) Controlled Substances Act (CDA). Under this act, a substance deemed to have some liability for abuse can be scheduled at one of five levels reflecting how stringently its manufacture and distribution will be regulated. Compounds with high abuse liability and no medical use recognized by the Food and Drug Administration (FDA) are placed in Schedule I, while those whose medical use is recognized are placed in one of the other four schedules (II–V), with higher numbers reflecting less stringent control. The primary purpose of scheduling is to deter diversion and to provide a mechanism for detection if diversion should occur. Scheduling also serves as a warning to physicians and patients that a particular medication has the potential to be abused.
Among the criteria for a drug’s scheduling, or exemption from scheduling, as outlined in Section 811 of the Controlled Substances Act (DEA, 2003), are the drug’s actual or relative potential for abuse, scientific evidence of its pharmacological effect (if known), the state of current scientific knowledge regarding the substance, risk to the public health, and psychological or physiological dependence liability. Standardized experimental procedures have been developed to evaluate pharmacological entities according to actual or relative potential for abuse, pharmacological effects, and psychological or physiological dependence liability.
In the past, abuse-liability data on analgesics were collected as part of a screening program that was carried out in parallel in laboratory animals and in humans, though this has become increasingly rare. An extensive testing program was coordinated by the Committee (now College) on Problems of Drug Dependence of the National Academy of Sciences, with human testing conducted at the Addiction Research Center (ARC) in Lexington, Kentucky, and animal testing at centers at the ARC as well the University of Michigan and later Virginia Commonwealth University. The organized testing program in humans is no longer in place, and pharmaceutical companies generally contract for human studies on a drug-by-drug basis. As a consequence, less-extensive testing is conducted in humans, primarily because assessment for physical-dependence potential is now done almost exclusively in laboratory-animal models. In this paper, we review the procedures used to evaluate the behavioral pharmacology of opioids in laboratory animals and humans and then examine the predictive value of the two sources of information, using postmarketing data for tramadol, an atypical, centrally acting opioid analgesic.
It should be noted that the research that formed the basis for our current knowledge and methods predates the identification of opioid receptors and endogenous opioid ligands as well as modern receptor and second-messenger assays. Furthermore, the understanding of the roles of some components of drug effects in drug addiction has changed over time. Although terminology has also evolved over time, some terms from earlier published studies have been perpetuated, sometimes because no other, more suitable names have been identified. One example is the “direct addiction” studies of physical dependence, so named because physical-dependence potential was once considered to be synonymous with addiction potential; the procedure involved testing whether physical dependence could be produced de novo by administration of a test drug. For the purposes of this review, we have used the names of tests as they appear in the literature, to avoid creating confusion by renaming them. We have tried to indicate where terms may be misleading or outdated.
Behavioral-Pharmacology Paradigms for Opioids
Abuse Potential
One important indicator of abuse potential is the extent to which a drug produces reinforcing effects. This is typically estimated in self-administration studies by use of operant-conditioning paradigms that measure the ability of the drug to act as a positive reinforcer. In self-administration studies, research subjects, usually laboratory animals, are given access to a drug under controlled experimental conditions, and their drug-taking behavior is evaluated. A drug is considered to be reinforcing if the frequency of a designated behavioral response (e.g., a lever press) is increased when drug delivery is contingent on the performance of that response in comparison to the frequency of responses in the absence of the drug. The capacity of a drug to reinforce behavior and, thus, maintain self-administration under experimental conditions is associated with a significant likelihood of abuse by human drug abusers (Willner, 1997). The number of self-administration studies in laboratory animals exceeds by far the number in humans.
Numerous variations of the self-administration paradigm have been used. Drug can be introduced de novo to naïve animals or substituted for another drug in animals experienced in self-administration; the outcome measure in these studies is usually response rate. In choice procedures, the test drug is made available along with another drug, and the animal is given the opportunity to choose which drug to self-administer; the proportion of choices made for the test drug is usually the outcome variable. In the progressive-ratio paradigm, the response requirement for acquisition of each dose increases until the animal no longer responds for drug; the highest response requirement resulting in drug delivery, referred to as the breakpoint, becomes the outcome measure. The type of test used depends on the objectives of study, a complete description of which is beyond the scope of this article; for reviews, see Collins et al., 1984 and Woolverton & Nader, 1990.
Because self-administration procedures are quite complex and can be technically difficult, conditioned place preference is sometimes used as an alternative. In conditioned place preference, a laboratory animal is repeatedly placed in a novel environment (containing distinctive sensory cues) while experiencing the effects of a test drug, and, in separate sessions, is placed in a different novel environment while not experiencing the effects of the drug. In a subsequent test session with no drug administered, the animal is allowed to move freely between the two environments. A preponderance of time spent in the drug-associated environment is taken as an index of drug reward. (For reviews, see Bardo & Bevins, 2000 and Tzschentke, 1998.)
Another approach to abuse-liability assessment is to estimate the extent to which a novel compound is pharmacologically similar to compounds that are already scheduled. This is typically done with drug-discrimination studies and subjective-effects studies. In drug-discrimination studies, laboratory animals or human subjects are trained to discriminate the presence or absence of a prototypic drug, and novel drugs are tested for their ability to substitute for the prototypic drug. Drug discrimination is not itself a measure of reinforcing efficacy. Although virtually all abused drugs can be trained as discriminative stimuli, most psychoactive drugs, including those that are not self-administered, are also discriminable from vehicle or no drug (Overton, 1987). Thus, the indicator of abuse liability is not discriminability per se, but rather the relative similarity of a drug’s stimulus effects to those of a known drug of abuse. Nevertheless, there is substantial concordance between discriminative stimulus and subjective effects in humans (Preston & Bigelow, 1991) and self-administration behavior in laboratory animals (Young, 1991). As noted above, pharmacologic similarity is a primary factor considered in federal scheduling of psychoactive substances. Thus, the utility of drug-discrimination studies in evaluating abuse liability is primarily due to their capacity to classify drugs according their similarity or dissimilarity to a standard or prototypic drug. Pharmacological specificity is a major feature of drug discrimination: drugs that have similar effects in other physiological or behavioral assays (often drugs within the same pharmacological class) usually share discriminative-stimulus effects, while drugs in different classes usually do not (Young, 1991).
Subjective-effects studies are conducted with human participants (Jasinski, 1977). A novel drug is administered over a range of doses, and its subjective effects are measured on a battery of questionnaires and compared to those of prototypic drugs of abuse. Each study usually incorporates multiple questionnaires in order to gather a complete profile of a drug’s subjective effects. The questionnaires typically contain elements designed to measure the presence or absence of drug effects and a qualitative and quantitative characterization of those effects as well as drug-induced mood changes. A critical element is the assessment of whether a subject likes the effects of the drug and possibly whether the drug is a euphoriant. For opioids, the measures most commonly used are scales of global drug effects (especially ratings of “liking”), subscales of the Addiction Research Center Inventory (ARCI) (Haertzen, 1966) (especially the MBG scale, reflecting effects common to morphine and Benzedrine [amphetamine]), the Adjective Rating Scales, and the Drug-Class Questionnaire (Fraser, VanHorn, Martin, Wolbach, & Isbell, 1961).
Physical-Dependence Capacity
Physical-dependence capacity alone is neither necessary nor sufficient for abuse potential, but it can increase the likelihood of abuse and the extent of adverse consequences of abuse. Historically, physical-dependence capacity, the nature of any withdrawal syndrome, and the ability to suppress opioid withdrawal had further significance as bioassays for opioid activity because opioid research substantially predates modern receptor and second messenger assay technology. While physical-dependence capacity is no longer a major component of abuse-liability assessment or opioid bioassays, it continues to have practical importance. For example, when the partial agonist buprenorphine was developed as a maintenance medication for opioid dependence, physical-dependence studies were needed to determine its ability to suppress withdrawal from full agonists as well as the circumstances under which it would induce withdrawal (for example, Greenwald et al., 2003; Breen et al., 2003; Eissenberg et al., 1997; Schuh et al., 1996).
Physical-dependence capacity can be determined by “direct addiction,” substitution, or suppression studies with similar procedures in both laboratory animals and humans (Howes, 1981; Jasinski, 1977; Martin & Jasinski, 1977). (“Direct addiction” is the term used historically for induction of physical dependence by administration of the test drug; the term is used here because it is found in the literature, but is not meant to suggest that physical dependence is equivalent to addiction.) In direct-addiction studies, a test drug is administered repeatedly over time; doses are initially low and then gradually increased as tolerance develops to toxic effects. After subjects have been stabilized at a specified dose, tolerance and other effects of chronic drug administration are evaluated; methods for evaluating tolerance have been described in detail elsewhere (Brady & Lukas, 1984; Branch, 1993). Physical dependence can then be documented by abrupt cessation of drug administration or by administration of a selective antagonist for the appropriate receptor type (e.g., naloxone for opioid dependence). Subjects are observed for signs of an abstinence syndrome. Self-report measures can also be included in studies with human subjects to gain additional qualitative information about the character of the abstinence syndrome, including dysphoric effects. Drug-seeking behavior, such as requests for test drug or other medication during the withdrawal period, can be used as indicators of psychological or physical dependence (for example, see Jasinski & Mansky, 1972). Alternatively, self-administration procedures can be used to evaluate drug-seeking during and after chronic drug administration (Mello & Mendelson, 1980; Preston, Bigelow, & Liebson, 1985).
Variations on “direct addiction” procedures include substitution and suppression procedures, each of which has been used to evaluate the physical-dependence capacity of opioids (Howes, 1981; Jasinski, 1977). In suppression studies, subjects are initially made physically dependent and maintained on a prototypic opioid agonist, for example, morphine administered in four injections or infusions per day. In test sessions, agonist administration is abruptly discontinued, and at the peak of withdrawal, test medications are administered and evaluated for their ability to suppress withdrawal. This methodology was used to document the ability of opioid agonists to suppress the abstinence syndrome. In substitution studies, the maintenance drug is replaced with a test drug or placebo. The ability of the test drug to suppress the onset of the withdrawal syndrome is assessed over a specified period of time (for example, 24 hours). This procedure enables crossover studies, testing multiple drugs or multiple dose levels of a test drug; subjects are restabilized on the maintenance medication between experimental test sessions.
Opioid-Receptor Antagonism
Opioid-receptor antagonism reduces the degree to which a substance produces opioid-agonist-like effects and thus affects abuse potential. Substances with antagonist activity have less than full efficacy as agonists; in addition, the effects of full agonists can be decreased in the presence of substances with lower efficacy. In vitro assays have been developed to assess opioid antagonism in isolated tissues (Hayes, Birch, Rogers, & Sheehan, 1990), cell preparations (Clark et al., 1997), and spinal animal models (Dewey, Snyder, Harris, & Howes, 1969). In vitro assays do not always reflect in vivo activity (Hayes et al., 1990); therefore, antagonism is also investigated in vivo.
In both laboratory animals and humans, similar procedures are used to assess blockade of opioid-agonist effects in individuals with or without physical dependence; sophisticated methods have been developed for this purpose (Aceto, McKean, & Pearl, 1969; Broadbear et al., 2000; Kishioka, Paronis, Lewis, & Woods, 2000). In non-dependent laboratory animals, the most frequently used outcome measures are blockade of analgesic effects, respiratory effects, or other species-specific opioid effects. In non-dependent humans, the most frequently used outcome measures are blockade of agonist-induced respiratory depression, subjective effects, and pupillary effects (Glass, Jhaveri, & Smith, 1994; Jones, Johnson, Fudala, Henningfield, & Heishman, 2000; Walsh, Sullivan, Preston, Garner, & Bigelow, 1996). In physically dependent laboratory animals or humans, administration of an opioid antagonist produces the signs and symptoms of abstinence; methods for quantifying these signs and symptoms include comparison of the effects of the test medication versus placebo and a drug with known antagonist activity (such as naloxone or naltrexone) (Jasinski, 1977; Martin & Jasinski, 1977).
Tramadol – assessment in laboratory animals, humans, and in the marketplace
Tramadol [(±)-trans-2-(dimethylaminomethyl)-1-(m-methoxyphenyl)-cyclohexanol hydrochloride] was introduced as a non-scheduled drug on the market in the US in 1995 for treatment of moderate to moderately severe pain. It had been available in Germany since 1977. As a centrally acting analgesic, tramadol is atypical because it produces analgesia through both mu opioid and monoamine actions (Bamigbade, Davidson, Langford, & Stamford, 1997; Driessen & Reimann, 1992; Driessen, Reimann, & Giertz, 1993; Raffa et al., 1992). The pharmacology of tramadol is further complicated by the fact that it is administered as a racemic mixture with two active enantiomers (Raffa et al., 1993), each of which is biotransformed to an active metabolite: (+)-O-desmethyltramadol or (−)-O-desmethyltramadol (Valle, Garrido, Pavon, Calvo, & Troconiz, 2000). These metabolites are believed to be responsible for most of tramadol’s mu-agonist properties (Gillen, Haurand, Kobelt, & Wnendt, 2000).
Clinically, the recommended dose of tramadol for mild to moderate pain is 50–100 mg every 6 hours p.o. or parenterally. Based on analgesic studies, tramadol is thought to be approximately one-tenth as potent as morphine when each is administered parenterally, and approximately one-third as potent when each is administered orally (Gutstein & Akil, 2001; Lehmann, Kratzenberg, Schroeder-Bark, & Horrichs-Haermeyer, 1990).
Results of Animal Studies
Only a few self-administration studies with tramadol have been published. Yanagita (1978) conducted three tramadol self-administration experiments in rhesus monkeys. In one study, four monkeys were trained to self-administer lefetamine, an amphetamine with opioid-agonist activity. After lefetamine self-administration had stabilized at approximately 100 injections per 4-hr session (0.1 mg/kg/injection), saline was substituted until responding extinguished. Lefetamine and saline availability were switched repeatedly until reinstatement of responding and extinction occurred reliably within 24 hours. When tramadol (0.25 mg/kg/injection) was substituted for lefetamine, the number of injections fell to 41.0% (range 14.7–69.6%; SD 22.5%) of those taken when lefetamine was available, not significantly higher than the proportion taken when saline was available (14.1; range 7.6–23.1; SD 6.5%). In a second study, four rhesus monkeys, two naïve and two self-administration experienced, were given unrestricted access to tramadol. In all four monkeys, rates of operant responding for drug increased above saline levels when the dose/injection of tramadol was increased to 1.0 mg/kg. Two of the monkeys also participated in a third study with tramadol 1.0 mg/kg injection available on a progressive-ratio schedule. The maximum response ratios that the monkeys completed for injections were 64:1 and 32:1; these values were less than the 128:1 ratio expected for other known analgesics such as morphine. The author concluded that tramadol had reinforcing effects but that these effects were less than those of pentazocine (Yanagita, 1978).
More recently, however, tramadol was shown to produce conditioned place preference in rats (Sprague et al., 2002; Tzschentke, Bruckmann, & Friderichs, 2002). The effects of tramadol on place preference were similar in magnitude to those of morphine, which served as a positive control in each study. Preference for the tramadol-associated compartment of the conditioning box produced by 10 mg/kg (i.p.) was prevented by pretreatment with naloxone 0.215 mg/kg (s.c.) (Tzschentke et al., 2002).
In the drug-discrimination paradigm, tramadol was tested in rats trained to discriminate 4.0 mg/kg morphine from saline and in rats trained to discriminate 0.5 mg/kg methamphetamine from saline (Ren & Zheng, 2000). Tramadol fully substituted for morphine, with approximately 96% of responses on the morphine-appropriate lever when tramadol doses reached or exceeded 32 mg/kg. This effect was completely blocked by concomitant administration of naltrexone. Tramadol did not substitute for methamphetamine at any dose tested (3.2 – 56 mg/kg). Tramadol also failed in rats to substitute for the flupirtine, a novel analgesic with probable alpha-2 adrenergic actions, or for anpirtoline, a drug with serotonergic actions (Swedberg, Shannon, Nickel, & Goldberg, 1988, 1992).
The capacity of tramadol to produce physical dependence has been tested in mice, rats, and rhesus monkeys. The results of these studies have been mixed. Miranda & Pinardi (1998) administered tramadol (39.1or 100 mg/kg; sc) three times daily for five days to mice and then tested them for tolerance in an experimental pain model (the acetic acid writhing test) and for physical dependence by injection with naloxone (1 mg/kg; i.p.). There was no evidence of tolerance to the antinociceptive response to the ED50 dose (7.82 mg/kg) of tramadol, and there were few or no signs of withdrawal after administration of naloxone at either dose of tramadol. In contrast, a control group that received an identical regimen of morphine (1.05 or 100 mg/kg) injections showed significant tolerance to the morphine ED50 dose (0.21 mg/kg) and showed opiate-withdrawal signs on administration of naloxone. Almost no cross-tolerance was demonstrated: the antinociceptive response to tramadol was unchanged in the morphine-treated group, and there was only a trend for decreased response to morphine in the tramadol-treated group. Thus, tramadol produced neither tolerance nor physical dependence in mice. Similarly, Murano et al. (1978) evaluated tolerance and physical dependence in rats treated with up to 160 mg/kg/day in four divided s.c. injections. Tolerance to tramadol’s antinociceptive effects was observed, but there was no evidence of physical dependence as indicated by weight loss following abrupt discontinuation of tramadol administration or following administration of levallorphan. However, some evidence of physical dependence was detected in rats receiving tramadol orally (50 mg/kg/d) and subjected to 24-hr withdrawal with or without injection of naloxone (Nickel & Aledter, 1987). Similarly, there was some evidence of withdrawal in eight rhesus monkeys receiving tramadol four times a day (32 to 96 mg/kg/day; s.c.) for 59 days: although few or no withdrawal signs were seen when naloxone (1.0 mg/kg; s.c.) was administered on four occasions during the administration period, withdrawal signs did emerge in the five days after tramadol was discontinued. These signs were graded as only mild (or, after the highest dose regimen, intermediate), not progressing to such severe signs as vomiting or diarrhea (Yanagita, 1978). In a concurrently run experiment, four rhesus monkeys self-administering tramadol for four to six weeks and administered naloxone (1.0 mg/kg; s.c.) at weeks 2 and 4 showed only mild-to-moderate withdrawal signs. The author concluded that the physical-dependence potential of tramadol is lower than that of pentazocine (Yanagita, 1978).
No antagonist activity has been demonstrated for tramadol in laboratory animals. Tramadol had only additive effects in an analgesic assay when combined with low doses of morphine and did not precipitate withdrawal jumping in morphine-dependent mice (Friderichs, Felgenhauer, Jongschaap, & Osterloh, 1978).
In summary, results of animal studies suggested that tramadol is an atypical opioid analgesic. It has some abuse potential, but, based on the self-administration studies in monkeys, less than that of prototypic opioids such as morphine. The evidence for physical-dependence capacity is mixed; withdrawal was not detected in mice, withdrawal was not detected consistently in rats, and only mild-to-moderate withdrawal was detected in rhesus monkeys.
Results of Human studies
There are no published human studies on self-administration or discrimination of tramadol; however, it has been investigated in a series of single-dose subjective-effects studies by several routes of administration.
The first abuse-liability study assessed the subjective, behavioral, and miotic effects of intramuscularly administered tramadol (75, 150 and 300 mg) compared to those of morphine (15 and 30 mg; i.m.), and placebo in 12 non-dependent opiate abusers. The results suggested that tramadol had a low abuse potential, at least when administered by the intramuscular route (Preston, Jasinski, & Testa, 1991). On subjective measures of opiate-like and positive mood effects, tramadol 75 and 150 mg were not different from placebo. Tramadol 300 mg was identified as an opiate but did not produce other morphine-like effects. Consistent with its not being a typical opioid and having inhibitory activity at catecholamine-reuptake sites, tramadol initially induced mydriasis (an increase in pupil diameter). In contrast, morphine induced miosis (a decrease in pupil diameter), as well as typical opioid-agonist-like subjective effects and identification as an opiate. Tramadol was expected to be one-tenth as potent as morphine based on its analgesic effects in other studies (Gutstein & Akil, 2001); however, in terms of opiate-like subjective effects, it was estimated to be only one twentieth as potent as those of morphine. Thus, the results showed a dissociation between tramadol’s potency as an analgesic and as an inducer of opioid-like subjective effects, consistent with its having a low potential for abuse when administered intramuscularly.
Abuse of prescription medications is more likely to occur by the oral route or by intravenous injection of crushed, dissolved oral formulations. Therefore, our laboratory has conducted evaluations of intravenously and orally administered tramadol in experienced opioid abusers. These studies have been reported only in abstract form (Jasinski, Preston, Sullivan, & Testa, 1993) or as a conference presentation (Jasinski, Sullivan, & Testa, 1994). The studies were approved by the appropriate Institutional Review Board for human research, and participants gave informed consent and were paid for their participation.
In an initial intravenous dose-ranging study, tramadol (700 mg; i.v.) administered over 1 min produced a seizure, as did a lower dose (300 mg; i.v.) delivered over 2.5 min. Thus, toxicity is likely to limit abuse of high doses of IV tramadol. At a lower dose (200 mg; i.v) administered over 5 min, no seizures occurred.
In a follow-up crossover study of intravenous tramadol, 10 experienced opioid abusers were tested with placebo, morphine (10 and 20 mg; i.v.), and tramadol (100 and 200 mg; i.v.) administered over 5 minutes according to two 5×5 balanced Latin squares under double-blind conditions. Both tramadol and morphine significantly increased ratings of “feel drug effect” compared to placebo. Morphine 10 and 20 mg significantly increased ratings of “liking,” and morphine 20 mg increased ratings on the ARCI MBG scale (Figure 1). In contrast, neither dose of tramadol increased ratings on the liking or MBG scales (Figure 1) or on any other subjective measure of opiate-like effects.
Figure 1.

Mean area under the curve scores produced by intravenously administered tramadol, morphine, and saline placebo on “Liking” and MBG scale scores in 10 experienced opioid abusers. Brackets indicate one half of Fisher’s Least Significant Difference.
A very different pattern of effects was produced when tramadol was administered orally. Tramadol (175, 350, and 700 mg; p.o.) was compared to placebo and oxycodone (20 and 40 mg; p.o.) in 12 experienced opioid abusers in two 6×6 balanced Latin squares under double-blind conditions. Tramadol and oxycodone both increased ratings on the MBG scale of the ARCI and were identified by participants as opiate-like on a questionnaire that listed various drug classes (data not shown). Tramadol and oxycodone also decreased pupil diameter and increased ratings on the “feel drug” and “liking” scales (Figure 2). However, the maximum responses to tramadol occurred much later than the maximum responses to oxycodone (Figure 2, right panels). These findings are consistent with the observation that tramadol’s mu-agonist properties require its biotransformation to an active metabolite.
Figure 2.
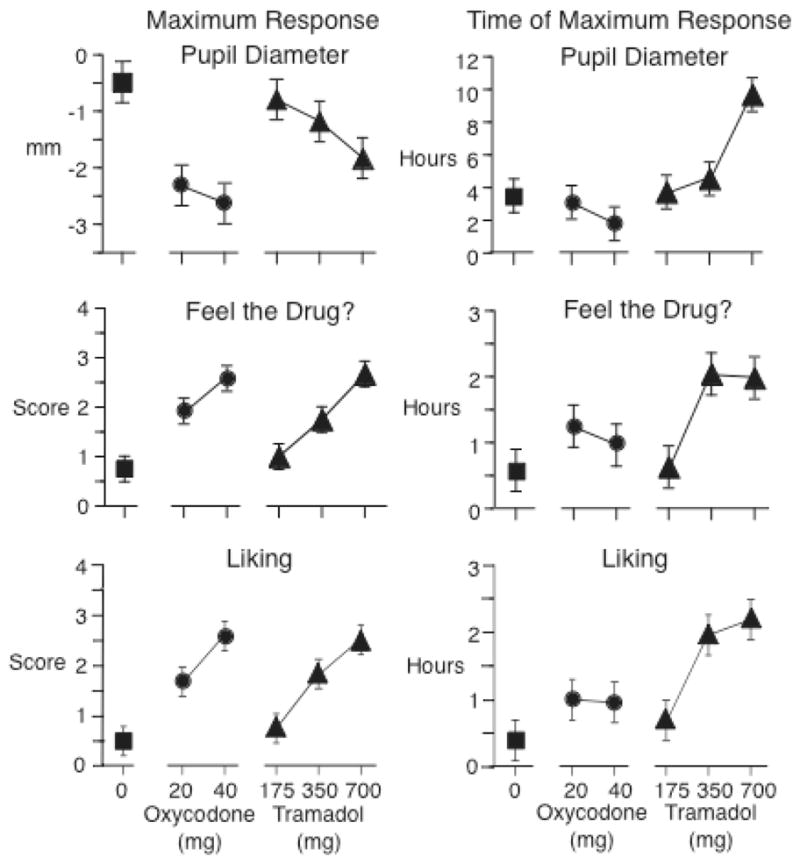
Maximum effects (left panel) and time to maximum effect (right panel) of orally administered tramadol, oxycodone, and placebo on pupil diameter, and “Feel drug,” and “Liking” scores in 12 experienced opioid abusers. Brackets indicate one half of Fisher’s Least Significant Difference.
The physical-dependence capacity of tramadol has not been evaluated in human laboratory studies as it has for buprenorphine and nalbuphine (Jasinski & Mansky, 1972; Jasinski, Pevnick, & Griffith, 1978). However, development of dependence was evaluated during a clinical trial of tramadol for treatment of severe pain (Richter, Barth, Flohe, & Giertz, 1985). Patients were treated orally with tramadol capsules up to a maximum of 400 mg/day for three weeks (mean dose approximately 250 mg/day); there was no evidence that tolerance developed to the analgesic effects. At the end of the three-week treatment, patients were randomized to receive naloxone (1.6 mg; i.m.) or placebo under double-blind conditions three hours after the last tramadol ingestion. Three of 54 participants showed marginal or slight elevations in opiate-withdrawal scores following naloxone, while 1 of 55 participants showed a marginal elevation in opiate-withdrawal scores following placebo. This difference was not statistically significant. Thus, under conditions of intermediate duration of tramadol administration within the recommended oral dose range, there was no strong evidence for development of physical dependence.
The opioid antagonism of tramadol was assessed in 6 male opioid-dependent volunteers who had been maintained on methadone (30 mg/day; p.o.) for at 10 days prior to the study (Cami, Lamas, & Farre, 1994). The subjective, behavioral, and physiological effects of tramadol (100 and 300 mg; i.m.) were not significantly different from those of placebo. Tramadol neither produced morphine-like effects nor precipitated a withdrawal syndrome. Thus, there was no evidence that tramadol has antagonist activity.
In summary, opiate-like effects were produced by oral but not parenteral administration of tramadol. There was minimal evidence for physical dependence and no evidence for opioid antagonism. Unlike other opioids, tramadol’s abuse-potential indices appeared low relative to its analgesic potency, at least by parenteral routes.
Postmarketing surveillance
Tramadol was approved by the FDA in 1998 for the management of moderate to moderately severe pain under the brand name Ultram without being scheduled under the Controlled Substances Act. The FDA, however, required that the sponsoring company conduct postmarketing surveillance of abuse and diversion. The tramadol surveillance program included: spontaneous reports to the manufacturer; adverse-event data from the FDA’s MedWatch; a key-informant network of treatment researchers who completed quarterly questionnaires; and monitoring of tramadol use in a population of impaired professionals (Cicero et al., 1999; Knisely, Campbell, Dawson, & Schnoll, 2002; Woody et al., 2003). This combination of elements greatly increased the program’s sensitivity relative to that of MedWatch alone. MedWatch is a program maintained by the FDA to collect adverse-event reports on marketed medications from health-care providers, drug companies, and individual consumers, and to provide safety information for health-care professionals and the public (Corrigan, 2002; Woody et al., 2003); it does not specifically target abuse or dependence as adverse events, nor does it target specific medications. In contrast, the tramadol key-informant-network program solicited reports of tramadol abuse at three-month intervals from drug-abuse experts (including researchers, clinicians, treatment counselors, and methadone-program directors) and conducted Internet searches for information on tramadol abuse (Cicero et al., 1999). The impaired-professionals surveillance recruited participants from monitoring programs in four states (Florida, Illinois, Pennsylvania, and Washington) (Knisely et al., 2002). Urine specimens collected as part of the monitoring programs were tested for tramadol. Participants were asked to report all drug use at each urine collection but were not aware that tramadol was the drug of interest. Findings from all of these programs are summarized in the Table.
Table.
Summary of abuse and dependence liability studies of tramadol
| Laboratory/Clinical Trial | Epidemiological | |||
|---|---|---|---|---|
| rodents | monkeys | humans | ||
| Physical dependence | (+) | (+) | (+) | (+)/− |
| Postmarking data | (+) | |||
| Internet mentions | − | |||
| Case reports | (+) | |||
| Abuse potential | ||||
| Conditioned place preference | + | |||
| Discrimination (similarity to morphine) | + | |||
| Self-administration | (+) | |||
| Positive subjective effects | (+) | |||
| Reports of actual abuse | ||||
| Postmarking data | (+) | |||
| Internet mentions | (+) | |||
| Case reports | (+) | |||
| Antagonist activity | − | − | ? | |
+ = positive finding
(+) = mildly or equivocally positive finding
− = negative finding
? = not clear
Reports from MedWatch and the key-informant program showed a low rate of abuse of tramadol, initially rising and then falling during the first three years of its availability in the US (Cicero et al., 1999). The rate of abuse peaked at two to three cases per month per 100,000 patients exposed during the first two years, then fell at three years to approximately one case per month per 100,000 patients exposed. In contrast, during the same period, the number of patients who were prescribed tramadol increased from 700,000 in the first year to about 900,000 in the third year. Internet searches identified more than 150 mentions over six months of the mood effects of tramadol. Most of the mentions indicated that tramadol did not produce euphorigenic effects, though a small number did report mood alteration or enhancement of the effects of other drugs.
Results from the postmarketing surveillance in impaired health-care professionals showed a similarly low rate of abuse among at-risk individuals (Knisely et al., 2002). The incidence of tramadol use, including legitimate prescription use, was 69 per 1000 individuals per year in 1601 impaired professionals participating in state monitoring programs between November 1995 and August 1998. Approximately one third (560) were primary opioid abusers. The incidence of tramadol abuse was approximately one tenth of that rate, 6.9 per 1000 individuals per year. Within the entire sample of 1601 individuals, 140 ever used tramadol and 15 met the study’s criteria for abuse/dependence. The authors considered this rate very low, given that the population studied was at high risk for relapse.
The MedWatch reports and postmarketing surveillance were also used to investigate whether tramadol produces physical dependence and a withdrawal syndrome (Senay et al., 2003; Woody et al., 2003). During the first three years of surveillance, 1248 adverse events were reviewed (Senay et al., 2003). Approximately one third (N=422) were rated as withdrawal, with most (N=367) indicating typical opioid withdrawal-like signs and symptoms, but with a small proportion (N=55) identified as atypical (not opioid withdrawal-like). The typical opioid withdrawal signs and symptoms included abdominal cramps, anxiety, bone pain, depression, diarrhea, goose flesh, insomnia, lacrimation, nausea, restlessness, rhinorrhea, and sweating. The atypical-withdrawal reports fell into four categories: severe anxiety and panic attacks, unusual CNS symptoms, unusual sensory phenomena, and hallucinations. CNS symptoms included confusion, depersonalization, derealization, and paranoia. Sensory phenomena reported were numbness, tingling, paresthesia, and tinnitus. The signs and symptoms of atypical withdrawal are similar to the discontinuation syndrome reported for selective serotonin reuptake inhibitors (SSRIs) (Bogetto, Bellino, Revello, & Patria, 2002; Rosenbaum, Fava, Hoog, Ascroft, & Krebs, 1998).
Clinical experiences with abuse and dependence can also be gleaned from published case reports. The case reports on tramadol have generally been consistent with the results of the formal postmarketing studies, describing incidents of abuse with and without prior history of other substance abuse (Ehrenreich & Poser, 1993; Lange-Asschenfeldt, Weigmann, Hiemke, & Mann, 2002; Reeves & Liberto, 2001), or describing physical dependence with a discontinuation syndrome in both abusers and in non-abusing patients (Barsotti, Mycyk, & Reyes, 2003; Freye & Levy, 2000; Leo, Narendran, & DeGuiseppe, 2000; Thomas & Suresh, 2000; Yates, Nguyen, & Warnock, 2001).
Comparison between laboratory-animal data and human data
The Table summarizes findings from laboratory animals and humans, and contrasts those findings with epidemiological findings.
Abuse potential
The results of laboratory tests have generally been consistent across primate species: in rhesus monkeys, tramadol is self-administered only to a modest degree, and in humans, tramadol elicits only modestly positive subjective effects. Each of these findings seems to have been appropriately predictive of the clinical and epidemiological experience with tramadol; postmarking data, case reports, and Internet mentions all suggest that tramadol is rarely perceived as a highly desirable euphoriant.
In light of this, data from rat models of abuse potential seem to represent false positives. In rats, tramadol produces fairly robust conditioned place preference and fully substitutes for the morphine discriminative stimulus. There are at least two possible explanations for this cross-species discrepancy. First, the biotransformation of tramadol to its main active metabolites is known to be much more rapid and complete in rats than in humans; 72 hours after administration of tramadol, humans excrete 25–32% of the dose unmetabolized, while rats excrete only 1% unmetabolized (Lintz, Erlacin, Frankus, & Uragg, 1981). The metabolites are believed to be responsible for most of tramadol’s mu-agonist properties (Gillen et al., 2000), so more rapid and complete biotransformation is consistent with greater abuse potential. Second, direct cross-species comparison is hampered by the use of different paradigms in difference species. The monkey and human studies used self-administration and subjective-effects measures, respectively; the rat studies used conditioned place preference and drug discrimination. Therefore, the apparently superior predictive value of the monkey and human data cannot be decisively attributed to species differences. An organized, integrated program of cross-species assessment, like the one formerly in place, would be desirable.
Physical dependence
Data on tramadol’s capacity to produce physical dependence appear more consistent across species, with the exception of one negative finding in mice. Rats and rhesus monkeys showed evidence of mild withdrawal symptoms; the occurrence of such symptoms in humans has been borne out by postmarketing surveillance and case reports, as has the symptoms’ relative mildness. Still, there are gaps in our knowledge. It has not been directly demonstrated that the withdrawal syndrome seen in humans is mediated primarily through opioid receptors; in fact, as discussed above, some of the withdrawal symptoms seen in humans resemble those seen with discontinuation of SSRIs. These so-called “atypical” withdrawal symptoms have not been reported in other species, but this may be due to their not having been systematically assessed. Again, these gaps in our knowledge suggest benefits that would accrue from an organized, integrated program of cross-species assessment.
Concluding remarks
As summarized in the Table, the tramadol experience in the US seems to represent a successful case of premarketing assessment for abuse and dependence potential that led to appropriate scheduling of a medication. While data from rodents suggests that some restrictions on availability would be necessary, data from primates and humans suggested that these restrictions could be minimal; postmarketing data suggest that the decision not to schedule tramadol was correct.
Nevertheless, the tramadol data also point to areas where premarketing screening could be improved. First, the use of different paradigms in different species precludes direct comparison of interspecies data. Most of the paradigms reviewed in this article can be used in rodents, monkeys, and humans; investigators should make more concerted efforts to see that this is done. Second, assuming for the moment that differences in paradigms can be overlooked, the pattern of premarketing results with tramadol suggest that rodent studies are useful but not sufficient for predicting abuse and dependence potential. Although it could be argued that tramadol is a special case because of its complex pharmacology (in particular, being both a drug and a prodrug), species differences in metabolic pathways and receptor profiles are likely to affect findings with other drugs as well. The current reliance on rodent models, while expedient in terms of drug throughput, increases the likelihood that a useful medication might be abandoned or too highly restricted due to exaggerated indices of abusability (or not sufficiently restricted due to insufficient detection of abusability). Rodent, monkey, and human studies should again be conducted in an integrated manner.
Acknowledgments
The authors thank John T. Sullivan, M.D., for his role in the human behavioral pharmacology studies of intravenous and oral tramadol. Dr. Epstein and Dr. Preston were supported by the Intramural Research Program of the NIH, National Institute on Drug Abuse.
Contributor Information
David H. Epstein, NIDA Intramural Research Program, 5500 Nathan Shock Drive, Baltimore, MD 21224
Kenzie L. Preston, NIDA Intramural Research Program, 5500 Nathan Shock Drive, Baltimore, MD 21224
Donald R. Jasinski, Johns Hopkins School of Medicine, Department of Medicine, 4940 Eastern Ave, Baltimore, MD 21224
References
- Aceto MD, McKean DB, Pearl J. Effects of opiates and opiate antagonists on the Straub tail reaction in mice. British Journal of Pharmacology. 1969;36(2):225–239. doi: 10.1111/j.1476-5381.1969.tb09500.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bamigbade TA, Davidson C, Langford RM, Stamford JA. Actions of tramadol, its enantiomers and principal metabolite, O-desmethyltramadol, on serotonin (5-HT) efflux and uptake in the rat dorsal raphe nucleus. British Journal of Anaesthesia. 1997;79(3):352–356. doi: 10.1093/bja/79.3.352. [DOI] [PubMed] [Google Scholar]
- Bardo MT, Bevins RA. Conditioned place preference: what does it add to our preclinical understanding of drug reward? Psychopharmacology. 2000;153(1):31–43. doi: 10.1007/s002130000569. [DOI] [PubMed] [Google Scholar]
- Barsotti CE, Mycyk MB, Reyes J. Withdrawal syndrome from tramadol hydrochloride. American Journal of Emergency Medicine. 2003;21(1):87–88. doi: 10.1053/ajem.2003.50039. [DOI] [PubMed] [Google Scholar]
- Bogetto F, Bellino S, Revello RB, Patria L. Discontinuation syndrome in dysthymic patients treated with selective serotonin reuptake inhibitors: a clinical investigation. CNS Drugs. 2002;16(4):273–283. doi: 10.2165/00023210-200216040-00006. [DOI] [PubMed] [Google Scholar]
- Brady JV, Lukas SE. Testing Drugs for Physical Dependence Potential and Abuse Liability. NIDA Research Monograph. 1984;52 [PubMed] [Google Scholar]
- Branch MN. Behavioral factors in drug tolerance. In: van Haaren F, editor. Methods in Behavioral Pharmacology. Amsterdam: Elsevier; 1993. [Google Scholar]
- Breen CL, Harris SJ, Lintzeris N, Mattick RP, Hawken L, Bell J, et al. Cessation of methadone maintenance treatment using buprenorphine: transfer from methadone to buprenorphine and subsequent buprenorphine reductions. Drug and Alcohol Dependence. 2003;71(1):49–55. doi: 10.1016/s0376-8716(03)00071-1. [DOI] [PubMed] [Google Scholar]
- Broadbear JH, Sumpter TL, Burke TF, Husbands SM, Lewis JW, Woods JH, et al. Methocinnamox is a potent, long-lasting, and selective antagonist of morphine-mediated antinociception in the mouse: comparison with clocinnamox, beta-funaltrexamine, and beta-chlornaltrexamine. Journal of Pharmacology and Experimental Therapeutics. 2000;294(3):933–940. [PubMed] [Google Scholar]
- Cami J, Lamas X, Farre M. Acute effects if tramadol in methadone-maintained volunteers. Drugs. 1994;47:39–43. doi: 10.2165/00003495-199400471-00007. [DOI] [PubMed] [Google Scholar]
- Cicero T, Adams E, Geller A, Inciardi J, Munoz A, Schnoll S, et al. A postmarketing surveillance program to monitor Ultram (tramadol hydrochloride) abuse in the United States. Drug and Alcohol Dependence. 1999;57:7–22. doi: 10.1016/s0376-8716(99)00041-1. [DOI] [PubMed] [Google Scholar]
- Clark MJ, Emmerson PJ, Mansour A, Akil H, Woods JH, Portoghese PS, et al. Opioid efficacy in a C6 glioma cell line stably expressing the delta opioid receptor. Journal of Pharmacology and Experimental Therapeutics. 1997;283(2):501–510. [PubMed] [Google Scholar]
- Collins R, Weeks J, Cooper M, Good P, Russell R. Prediction of abuse liability of drugs using IV self-administration by rats. Psychopharmacology. 1984;82:6–13. doi: 10.1007/BF00426372. [DOI] [PubMed] [Google Scholar]
- Corrigan OP. A risky business: the detection of adverse drug reactions in clinical trials and post-marketing exercises. Social Science & Medicine. 2002;55(3):497–507. doi: 10.1016/s0277-9536(01)00183-6. [DOI] [PubMed] [Google Scholar]
- DEA. 21 USC, Section 811. 2003 Retrieved January 8, 2004, from http://www.deadiversion.usdoj.gov/21cfr/21usc/811.htm.
- Dewey WL, Snyder JW, Harris LS, Howes JF. The effect of narcotics and narcotic antagonists on the tail-flick response in spinal mice. Journal of Pharmacy and Pharmacology. 1969;21(8):548–550. doi: 10.1111/j.2042-7158.1969.tb08312.x. [DOI] [PubMed] [Google Scholar]
- Driessen B, Reimann W. Interaction of the central analgesic, tramadol, with the uptake and release of 5-hydroxytryptamine in the rat brain in vitro. British Journal of Pharmacology. 1992;105(1):147–151. doi: 10.1111/j.1476-5381.1992.tb14226.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Driessen B, Reimann W, Giertz H. Effects of the central analgesic tramadol on the uptake and release of noradrenaline and dopamine in vitro. British Journal of Pharmacology. 1993;108(3):806–811. doi: 10.1111/j.1476-5381.1993.tb12882.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrenreich H, Poser W. Dependence on tramadol. Clinical Investigator. 1993;72(1):76. doi: 10.1007/BF00231123. [DOI] [PubMed] [Google Scholar]
- Eissenberg T, Johnson RE, Bigelow GE, Walsh SL, Liebson IA, Strain EC, et al. Controlled opioid withdrawal evaluation during 72 h dose omission in buprenorphine-maintained patients. Drug and Alcohol Dependence. 1997;45:81–91. doi: 10.1016/s0376-8716(97)01347-1. [DOI] [PubMed] [Google Scholar]
- Fraser H, VanHorn G, Martin W, Wolbach A, Isbell H. Methods for evaluating addiction liability.(A) “attitude” of opiate addicts toward opiate-like drugs,(B) a short-term “direct” addiction test. Journal of Pharmacology and Experimental Therapeutics. 1961;133(3):371–387. [PubMed] [Google Scholar]
- Freye E, Levy J. Acute abstinence syndrome following abrupt cessation of long-term use of tramadol (Ultram): a case study. European Journal of Pain. 2000;4(3):307–311. doi: 10.1053/eujp.2000.0187. [DOI] [PubMed] [Google Scholar]
- Friderichs E, Felgenhauer F, Jongschaap P, Osterloh G. Pharmacological studies on analgesia, dependence on and tolerance of tramadol, a potent analgetic drug (author’s transl) Arzneimittelforschung. 1978;28(1a):122–134. [PubMed] [Google Scholar]
- Gillen C, Haurand M, Kobelt DJ, Wnendt S. Affinity, potency and efficacy of tramadol and its metabolites at the cloned human mu-opioid receptor. Naunyn-Schmiedebergs Archives of Pharmacology. 2000;362(2):116–121. doi: 10.1007/s002100000266. [DOI] [PubMed] [Google Scholar]
- Glass PS, Jhaveri RM, Smith LR. Comparison of potency and duration of action of nalmefene and naloxone. Anesthesia and Analgesia. 1994;78:536–541. doi: 10.1213/00000539-199403000-00021. [DOI] [PubMed] [Google Scholar]
- Greenwald MK, Schuh KJ, Stine SM. Transferring methadone-maintained outpatients to the buprenorphine sublingual tablet: a preliminary study. American Journal on Addictions. 2003;12(4):365–374. [PubMed] [Google Scholar]
- Gutstein HB, Akil H. Opioid analgesics. In: Hardman JG, Limbird LE, editors. Goodman & Gilman’s The Pharmacological Basis of Therapeutics. New York, NY: McGraw-Hill; 2001. pp. 569–619. [Google Scholar]
- Haertzen C. Development of scales based on patterns of drug effects, using the addiction research center inventory (ARCI) Psychological Reports. 1966;18:163–194. doi: 10.2466/pr0.1966.18.1.163. [DOI] [PubMed] [Google Scholar]
- Hayes AG, Birch PJ, Rogers H, Sheehan MJ. Use of isolated tissue preparations to evaluate opioid drugs of abuse. In: Adler MW, Cowan A, editors. Testing and Evaluation of Drugs of Abuse. Vol. 6. New York: Wiley-Liss; 1990. pp. 19–32. [Google Scholar]
- Howes JF. A simple, reliable method for predicting the physical dependence liability of narcotic antagonist analgesics in the rat. Pharmacology Biochemistry & Behavior. 1981;14(5):689–692. doi: 10.1016/0091-3057(81)90132-5. [DOI] [PubMed] [Google Scholar]
- Jasinski DR. Assessment of the abuse potentiality of morphinelike drugs (methods used in man) In: Martin WR, editor. Drug Addiction 1: Morphine, sedative/hypnotic and alcohol dependence. Berlin: Springer-Verlag; 1977. [Google Scholar]
- Jasinski DR, Mansky PA. Evaluation of nalbuphine for abuse potential. Clinical Pharmacology and Therapeutics. 1972;13(1):78–90. doi: 10.1002/cpt197213178. [DOI] [PubMed] [Google Scholar]
- Jasinski DR, Pevnick JS, Griffith JD. Human pharmacology and abuse potential of the analgesic buprenorphine: a potential agent for treating narcotic addiction. Archives of General Psychiatry. 1978;35(4):501–516. doi: 10.1001/archpsyc.1978.01770280111012. [DOI] [PubMed] [Google Scholar]
- Jasinski DR, Preston K, Sullivan JT, Testa MP. Abuse potential of oral tramadol. NIDA Research Monograph. 1993;132:103. [PubMed] [Google Scholar]
- Jasinski DR, Sullivan JT, Testa M. Abuse potential of IV tramadol. Paper presented at the 55th Annual Scientific Meeting of the College on Problems of Drug Dependence.1994. [Google Scholar]
- Jones HE, Johnson RE, Fudala PJ, Henningfield JE, Heishman SJ. Nalmefene: blockade of intravenous morphine challenge effects in opioid abusing humans. Drug and Alcohol Dependence. 2000;60(1):29–37. doi: 10.1016/s0376-8716(99)00138-6. [DOI] [PubMed] [Google Scholar]
- Kishioka S, Paronis CA, Lewis JW, Woods JH. Buprenorphine and methoclocinnamox: agonist and antagonist effects on respiratory function in rhesus monkeys. European Journal of Pharmacology. 2000;391(3):289–297. doi: 10.1016/s0014-2999(00)00039-x. [DOI] [PubMed] [Google Scholar]
- Knisely JS, Campbell ED, Dawson KS, Schnoll SH. Tramadol post-marketing surveillance in health care professionals. Drug and Alcohol Dependence. 2002;68(1):15–22. doi: 10.1016/s0376-8716(02)00107-2. [DOI] [PubMed] [Google Scholar]
- Lange-Asschenfeldt C, Weigmann H, Hiemke C, Mann K. Serotonin syndrome as a result of fluoxetine in a patient with tramadol abuse: plasma level-correlated symptomatology. Journal of Clinical Psychopharmacology. 2002;22(4):440–441. doi: 10.1097/00004714-200208000-00022. [DOI] [PubMed] [Google Scholar]
- Lehmann KA, Kratzenberg U, Schroeder-Bark B, Horrichs-Haermeyer G. Postoperative patient-controlled analgesia with tramadol: analgesic efficacy and minimum effective concentrations. Clinical Journal of Pain. 1990;6(3):212–220. doi: 10.1097/00002508-199009000-00008. [DOI] [PubMed] [Google Scholar]
- Leo RJ, Narendran R, DeGuiseppe B. Methadone detoxification of tramadol dependence. Journal of Substance Abuse Treatment. 2000;19(3):297–299. doi: 10.1016/s0740-5472(00)00098-2. [DOI] [PubMed] [Google Scholar]
- Lintz W, Erlacin S, Frankus E, Uragg H. Biotransformation of tramadol in man and animal (author’s transl) Arzneimittelforschung. 1981;31(11):1932–1943. [PubMed] [Google Scholar]
- Martin W, Jasinski DR. Assessment of the abuse potential of narcotic analgesics in animals. In: Born G, Eichler O, Farah A, Herken H, Welch A, editors. Handbook of Experimental Pharmacology. 45. Berlin: Springer-Verlag; 1977. pp. 159–196. [Google Scholar]
- Mello NK, Mendelson JH. Buprenorphine suppresses heroin use by heroin addicts. Science. 1980;207(8):657–659. doi: 10.1126/science.7352279. [DOI] [PubMed] [Google Scholar]
- Miranda HF, Pinardi G. Antinociception, tolerance, and physical dependence comparison between morphine and tramadol. Pharmacology Biochemistry & Behavior. 1998;61(4):357–360. doi: 10.1016/s0091-3057(98)00123-3. [DOI] [PubMed] [Google Scholar]
- Murano T, Yamamoto H, Endo N, Kudo Y, Okada N, Masuda Y, et al. Studies on dependence on tramadol in rats. Arzneimittelforschung. 1978;28(1a):152–158. [PubMed] [Google Scholar]
- Nickel B, Aledter A. Comparative physical dependence studies in rats with flupirtine and opiate receptor stimulating analgesics. Postgraduate Medical Journal. 1987;63(Suppl 3):41–43. [PubMed] [Google Scholar]
- Overton DA. Applications and limitations of the drug discrimination method for the study of drug abuse. In: Bozarth MA, editor. Methods of assessing the reinforcing properties of abused drugs. New York: Springer-Verlag; 1987. pp. 291–340. [Google Scholar]
- Preston KL, Bigelow GE. Subjective and discriminative effects of drugs. Behavioural Pharmacology. 1991;2(4–5):293–313. [PubMed] [Google Scholar]
- Preston KL, Bigelow GE, Liebson IA. Self-administration of clonidine, oxazepam, and hydromorphone by patients undergoing methadone detoxification. Clin Pharmacol Ther. 1985;38(2):219–227. doi: 10.1038/clpt.1985.162. [DOI] [PubMed] [Google Scholar]
- Preston KL, Jasinski DR, Testa M. Abuse potential and pharmacological comparison of tramadol and morphine. Drug and Alcohol Dependence. 1991;27(1):7–17. doi: 10.1016/0376-8716(91)90081-9. [DOI] [PubMed] [Google Scholar]
- Raffa RB, Friderichs E, Reimann W, Shank RP, Codd EE, Vaught JL. Opioid and nonopioid components independently contribute to the mechanism of action of tramadol, an “atypical” opioid analgesic. Journal of Pharmacology and Experimental Therapeutics. 1992;260(1):275–285. [PubMed] [Google Scholar]
- Raffa RB, Friderichs E, Reimann W, Shank RP, Codd EE, Vaught JL, et al. Complementary and synergistic antinociceptive interaction between the enantiomers of tramadol. Journal of Pharmacology and Experimental Therapeutics. 1993;267(1):331–340. [PubMed] [Google Scholar]
- Reeves RR, Liberto V. Abuse of combinations of carisoprodol and tramadol. Southern Medical Journal. 2001;94(5):512–514. [PubMed] [Google Scholar]
- Ren YH, Zheng JW. Influence of tramadol on morphine discriminative behavior in rats. Acta Pharmacologica Sinica. 2000;21(10):924–926. [PubMed] [Google Scholar]
- Richter W, Barth H, Flohe L, Giertz H. Clinical investigation on the development of dependence during oral therapy with tramadol. Arzneimittelforschung. 1985;35(11):1742–1744. [PubMed] [Google Scholar]
- Rosenbaum JF, Fava M, Hoog SL, Ascroft RC, Krebs WB. Selective serotonin reuptake inhibitor discontinuation syndrome: a randomized clinical trial. Biological Psychiatry. 1998;44(2):77–87. doi: 10.1016/s0006-3223(98)00126-7. [DOI] [PubMed] [Google Scholar]
- SAMHSA. The DASIS report: Treatment admissions involving narcotic painkillers. 2003a Retrieved January 9, 2004, from http://www.samhsa.gov/oas/2k3/painTX/painTX.htm.
- SAMHSA. Nonmedical use of prescription–type drugs among youths and young adults. 2003b Retrieved January 9, 2004, from http://www.samhsa.gov/oas/2k3/prescription/prescription.htm.
- SAMHSA. Overview of Findings from the 2002 National Survey on Drug Use and Health. 2003c Retrieved January 9, 2004, from http://www.samhsa.gov/oas/NHSDA/2k2NSDUH/Results/2k2results.htm.
- Schuh KJ, Walsh SL, Bigelow GE, Preston KL, Stitzer ML. Buprenorphine, morphine and naloxone effects during ascending morphine maintenance in humans. Journal of Pharmacology and Experimental Therapeutics. 1996;278(2):836–846. [PubMed] [Google Scholar]
- Senay EC, Adams EH, Geller A, Inciardi JA, Munoz A, Schnoll SH, et al. Physical dependence on Ultram (tramadol hydrochloride): both opioid-like and atypical withdrawal symptoms occur. Drug and Alcohol Dependence. 2003;69(3):233–241. doi: 10.1016/s0376-8716(02)00321-6. [DOI] [PubMed] [Google Scholar]
- Sprague JE, Leifheit M, Selken J, Milks MM, Kinder DH, Nichols DE. In vivo microdialysis and conditioned place preference studies in rats are consistent with abuse potential of tramadol. Synapse. 2002;43(2):118–121. doi: 10.1002/syn.10025. [DOI] [PubMed] [Google Scholar]
- Swedberg MD, Shannon HE, Nickel B, Goldberg SR. Pharmacological mechanisms of action of flupirtine: a novel, centrally acting, nonopioid analgesic evaluated by its discriminative effects in the rat. Journal of Pharmacology and Experimental Therapeutics. 1988;246(3):1067–1074. [PubMed] [Google Scholar]
- Swedberg MD, Shannon HE, Nickel B, Goldberg SR. D-16949 (anpirtoline): a novel serotonergic (5-HT1B) psychotherapeutic agent assessed by its discriminative effects in the rat. Journal of Pharmacology and Experimental Therapeutics. 1992;263(3):1015–1022. [PubMed] [Google Scholar]
- Thomas AN, Suresh M. Opiate withdrawal after tramadol and patient-controlled analgesia. Anaesthesia. 2000;55(8):826–827. doi: 10.1046/j.1365-2044.2000.01629-30.x. [DOI] [PubMed] [Google Scholar]
- Tzschentke TM. Measuring reward with the conditioned place preference paradigm: a comprehensive review of drug effects, recent progress and new issues. Progress in Neurobiology. 1998;56(6):613–672. doi: 10.1016/s0301-0082(98)00060-4. [DOI] [PubMed] [Google Scholar]
- Tzschentke TM, Bruckmann W, Friderichs E. Lack of sensitization during place conditioning in rats is consistent with the low abuse potential of tramadol. Neuroscience Letters. 2002;329(1):25–28. doi: 10.1016/s0304-3940(02)00571-2. [DOI] [PubMed] [Google Scholar]
- Valle M, Garrido MJ, Pavon JM, Calvo R, Troconiz IF. Pharmacokinetic-pharmacodynamic modeling of the antinociceptive effects of main active metabolites of tramadol, (+)-O-desmethyltramadol and (−)-O-desmethyltramadol, in rats. Journal of Pharmacology and Experimental Therapeutics. 2000;293(2):646–653. [PubMed] [Google Scholar]
- Walsh SL, Sullivan JT, Preston KL, Garner JE, Bigelow GE. Effects of naltrexone on response to intravenous cocaine, hydromorphone and their combination in humans. Journal of Pharmacology and Experimental Therapeutics. 1996;279:524–538. [PubMed] [Google Scholar]
- Willner P. Animal models of addiction. Human Psychopharmacology. 1997;12:59–68. [Google Scholar]
- Woody GE, Senay EC, Geller A, Adams EH, Inciardi JA, Schnoll S, et al. An independent assessment of MEDWatch reporting for abuse/dependence and withdrawal from Ultram (tramadol hydrochloride) Drug and Alcohol Dependence. 2003;72(2):163–168. doi: 10.1016/s0376-8716(03)00198-4. [DOI] [PubMed] [Google Scholar]
- Woolverton W, Nader M. Experimental evaluation of the reinforcing effects of drugs. Modern Methods in Pharmacology. 1990;6:165–192. [Google Scholar]
- Yanagita T. Drug dependence potential of 1-(m-methoxyphenyl)-2-dimethylaminomethyl)-cyclohexan-1-ol hydrochloride (tramadol) tested in monkeys. Arzneimittelforschung. 1978;28(1a):158–163. [PubMed] [Google Scholar]
- Yates WR, Nguyen MH, Warnock JK. Tramadol dependence with no history of substance abuse. American Journal of Psychiatry. 2001;158(6):964. doi: 10.1176/appi.ajp.158.6.964. [DOI] [PubMed] [Google Scholar]
- Young A. Discriminative stimulus profiles of psychoactive drugs. In: Mello NK, editor. Advances in Substance Abuse. 4. London: Jessica Kingsley Publishers; 1991. pp. 140–203. [Google Scholar]
- Zacny J, Bigelow G, Compton P, Foley K, Iguchi M, Sannerud C. College on Problems of Drug Dependence taskforce on prescription opioid non-medical use and abuse: position statement. Drug and Alcohol Dependence. 2003;69(3):215–232. doi: 10.1016/s0376-8716(03)00003-6. [DOI] [PubMed] [Google Scholar]