

Abstract
A ruthenium-catalyzed, redox neutral C-O bond cleavage of 2-aryloxy-1-arylethanols was developed that yields cleavage products in 62-98% isolated yield. This reaction is applicable to breaking the key ethereal bond found in lignin-related polymers. The bond transformation proceeds by a tandem dehydrogenation/reductive ether cleavage. Initial mechanistic investigations indicate that the ether cleavage is most likely an organometallic C-O activation. A catalytic depolymerization of a lignin-related polymer quantitatively yields the corresponding monomer with no added reagent.
Lignin depolymerization is one of the most significant barriers to reaching the full potential of lignocellulosic bio-fuels as fossil fuel replacements.1 The problem stems from the fact that lignin comprises 15-25% of the mass found in lignocellulose and up to 40% of the energy content.2 For lignocellulosic bio-fuels to be sustainable, chemistry must be developed to convert lignin into small molecules that can be upgraded into a fuel stream.3 Homogeneous and heterogeneous catalytic processes for both oxidative and reductive lignin depolymerizations are known.4 Here we describe a tandem catalytic dehydrogenation/C-O bond cleavage that enables a redox neutral approach for lignin depolymerization. This approach is noteworthy both as a novel C-O bond cleavage reaction and as a process for depolymerization that requires no added reagent.
The chemical structure of lignin is highly variable therefore choosing a small-molecule system and strategy for reaction development is challenging.5 The β-[O]-4′-glycerolaryl ether linkages (1) are ubiquitous in lignin found across many species including Miscanthus giganteus, a species of particular interest for industrial lignocellulosic bio-fuels production (Figure 1).6 2-Aryloxy-1-arylethanols (2), or “β-[O]-4′–ethanolaryl ethers”, are often used to model the β-[O]-4′-glycerolaryl ether linkages and were chosen for the purposes of reaction development.7
Figure 1.
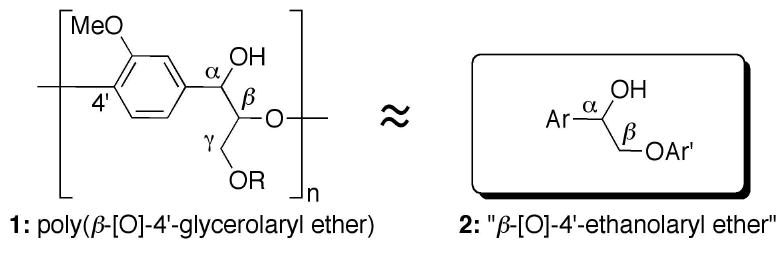
2-Aryloxy-1-arylethanols approximate the functionality in β-[O]-4′-glycerolaryl ethers
Structural analysis of polymer 1 suggested a redox neutral depolymerization strategy for reaction development. In the desired reaction, catalytic dehydrogenation of the α-carbinol provides the reducing equivalents needed to cleave the β-arylether C-O bond. To be successful, however, this strategy requires a catalyst that can perform the requisite hydrogen shuttling and the novel C-O bond activation processes in tandem. The ruthenium complex RuH2(CO)(PPh3)3 is known to be competent for both dehydrogenation8 and C-O activation chemistry and was chosen as the starting point for catalyst development.9
Table 1 summarizes a screen of phosphine ligands using catalytic amounts of RuH2(CO)(PPh3)3 with 2-phenoxy-1-phenethanol (2a). In the absence of added ligand, little product was obtained (entry 1). Adding monodentate phosphines resulted in lower conversion (entries 2 & 3). Bidentate ligands (entries 4-7) with a range of phosphine bite-angles effected dehydrogenation to yield α-phenoxyacetophenone, but were not able to promote the reductive ether cleavage. However, a quantitative yield was attained with the wide-bite angle ligand, 9,9-dimethyl-bis(diphenylphosphino)-xanthene (Ph-xantphos), providing proof-of-principle for the tandem catalysis strategy (entry 8).
Table 1.
Ligand screening for C-O bond cleavage of 2a
 | ||||
|---|---|---|---|---|
| Entrya | Ligand | Conv.c,d (%) | Yield (%)c,d | |
| PhCOMe | PhOH | |||
| 1 | none | 45 | 5 | 6 |
| 2 | PPh3b | 39 | <1 | 5 |
| 3 | PCy3b | 27 | 4 | 5 |
| 4 | dppm | 11 | 0 | 0 |
| 5 | dppp | 32 | 0 | <1 |
| 6 | dppbz | 32 | 0 | 0 |
| 7 | dppf | 37 | 5 | 6 |
| 8 | Ph-xantphos | >99 | >99 | >99 |
Reactions were run in sealed tubes under nitrogen.
2 equivalents relative to Ru.
Yields and conversions were determined by GC/MS relative to an internal standard.
Average of two duplicate experiments.
Additional reaction optimization yielded a set of general conditions for 2-aryloxy-1-arylethanols. The substrates in Table 2 model the substitution patterns found in lignin. Increased methoxyl substitution of the O-terminus aryl ring resulted in diminished yields (2a-c), while methoxylation of the C-terminus aryl ring had comparatively little effect on the reaction outcome (2d). The conditions also worked well for the highly substituted substrate 2e.
Table 2.
C-O bond cleavage of various 2-aryloxy-1-arylethanols
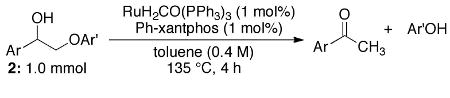 | |||
|---|---|---|---|
| Substrate | Ar | Ar′ | Yield (%)a |
| 2a | C6H5 | C6H5 | 98 |
| 2b | C6H5 | 2-(CH3O)-C6H4 | 88 |
| 2c | C6H5 | 2,6-(CH3O)2-C6H3 | 62 |
| 2d | 4-(CH3O)-C6H4 | C6H5 | 98 |
| 2e | 3,4-(CH3O) -C6H3 | 2-(CH3O)-C6H4 | 89 |
Average isolated yield of the corresponding ketone based on two duplicate experiments.
The next challenge came in applying the small-molecule chemistry to an actual polymer of 2-aryloxy-1-arylethanol. Scheme 1 shows the quantitative depolymerization of poly(4′-hydroxy-1-phenethanol)10 (3) to 4′-hydroxyacetophenone. Solvent, temperature, and catalyst loading were modified to ameliorate the poor solubility and reactivity of 3 under the conditions developed for the 2-aryloxy-1-arylethanols (Scheme 1). Complete conversion of polymer to monomer in 99% isolated yield demonstrates the utility of the catalytic system for depolymerizing polyethers that are in the molecular weight range of isolated lignin.
Scheme 1.
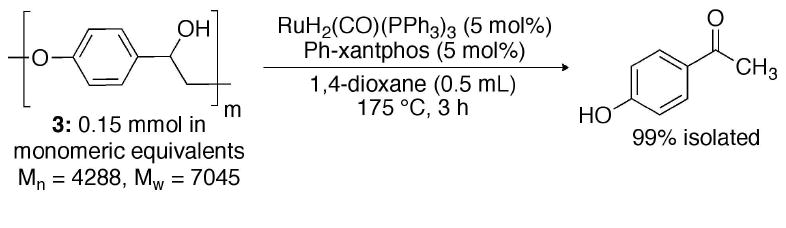
Depolymerization of lignin related polymer
The proposed mechanism for the transformation is shown in Figure 2. It begins with a well-known Ru-catalyzed dehydrogenative equilibrium between a benzylic alcohol and the corresponding aryl ketone.10 This is followed by loss of HX from the catalyst precursor and formation of Ru(0) complex 5. C-O activation in 5 leads to Ru-enolate (6). Hydrogenation of 6 yields a Ru-alkoxide (7) followed by reductive elimination of phenol and association with 4 to close the cycle.
Figure 2.
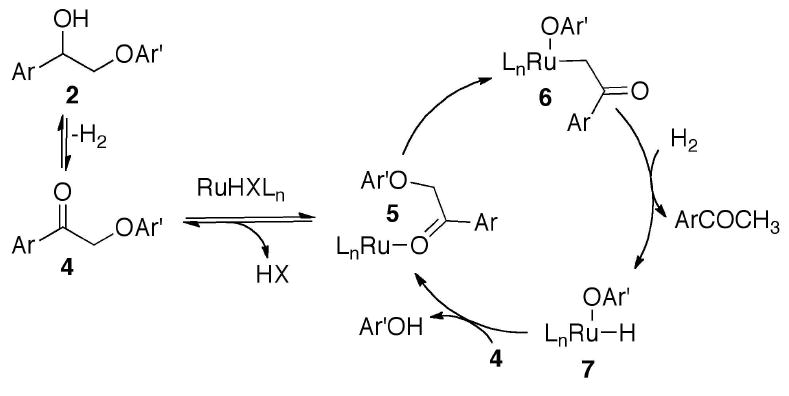
Mechanistic rationale for C-O bond cleavage
In situ monitoring of the reaction time course for 2-phenoxyphenethanol (2a) suggests that 2-phenoxyacetophenone (4a) is an intermediate. Blocking the dehydrogenation process that forms intermediate 4a prevents the C-O cleavage reaction (Scheme 2a) under conditions in which the C-O bond cleavage functions. Using silane as a surrogate for molecular hydrogen, intermediate 4a is reduced to yield the corresponding C-O bond cleavage products in 89% yield (Scheme 2b). Together, these experiments support the intermediacy of α-aryloxy ketones (4).
Scheme 2.
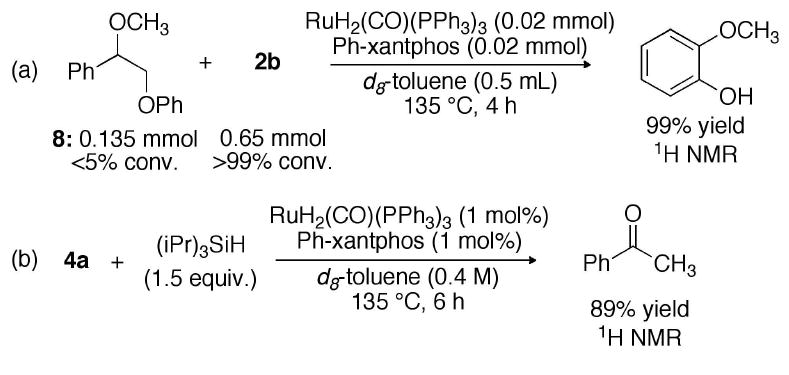
(a) Blocking formation of 4a prevents C-O cleavage; (b) Hydrosilation of 4a yields C-O cleavage
Alternate mechanisms that do not proceed through the α-aryloxy ketone are not consistent with the experimental observations summarized in Scheme 2. These mechanisms include a free-radical mechanism initiated by the formation of a benzyl radical,11 or the elimination of phenol to yield styrylenolethers. Compound 8 is theoretically able to participate in both mechanisms and yet is not converted to product under the reaction conditions. Moreover, the radical inhibitor 2,6-di-(tert-butyl)-4-methylphenol (BHT) has no effect on the apparent half-life, or yield, for the disproportionation of 2b (t1/2 = 2 h, 135 °C, [2b] = 0.1 M). Finally, hydrosilation of ketone 4a with Et3SiD forms acetophenone with 55% deuterium incorporation at the α-keto position as confirmed by 2H NMR. The selective deuteration result is consistent with a mechanism in which a ruthenium enolate (6) is formed and trapped by the deuterated silane.13 These experiments indicate that radical and elimination mechanisms are not likely responsible for the observed transformation and support the proposed model outlined in Figure 2.7b, 12
Future work will be directed toward the investigation of the elementary steps of the organometallic C-O activation process as well as applications to natural lignin and other model systems.
Supplementary Material
Acknowledgments
The authors gratefully acknowledge the Energy Biosciences Institute (Grant No. OO7J37) for financial support. J.M.N. thanks the National Institutes of Health for support through the Ruth L. Kirschstein Postdoctoral Fellowship (F32GM084597).
Footnotes
Supporting Information Available: Substrate syntheses and experimental procedures (23 pages). This material is available free of charge via the Internet at http://pubs.acs.org.
Contributor Information
Robert G. Bergman, Email: rbergman@berkeley.edu.
Jonathan A. Ellman, Email: jonathan.ellman@yale.edu.
References
- 1.a) Ragauskas A, Williams C, Davison B, Britovsek G, Cairney J, Eckert C, Frederick W, Hallett J, Leak D, Liotta C, Mielenz J, Murphy R, Templer R, Tschaplinski T. Science. 2006;311:484. doi: 10.1126/science.1114736. [DOI] [PubMed] [Google Scholar]; b) Chang MCY. Curr Opin Chem Biol. 2007;11:677. doi: 10.1016/j.cbpa.2007.08.039. [DOI] [PubMed] [Google Scholar]
- 2.Regalbuto JR. Science. 2009;325:822. doi: 10.1126/science.1174581. [DOI] [PubMed] [Google Scholar]
- 3.Ohlrogge J, Allen D, Berguson B, DellaPenna D, Shachar-Hill Y, Stymne S. Science. 2009;324:1019. doi: 10.1126/science.1171740. [DOI] [PubMed] [Google Scholar]
- 4.Zakzeski J, Bruijnincx PCA, Jongerius AL, Weckhuysen BM. Chem Rev. 2010;110:3552. doi: 10.1021/cr900354u. [DOI] [PubMed] [Google Scholar]
- 5.For variations in lignin structure, see:; a) Bunzel M, Ralph JJ. Ag Food Chem. 2006;54:8352. doi: 10.1021/jf061525z. [DOI] [PubMed] [Google Scholar]; b) del Rio JC, Marques G, Rencoret J, Martinez AT, Gutierrez A. J Ag Food Chem. 2007;55:5461. doi: 10.1021/jf0705264. [DOI] [PubMed] [Google Scholar]; c) Ibarra D, Isabel Chavez M, Rencoret J, Del Rio JC, Gutierrez A, Romero J, Camarero S, Martinez MJ, Jimenez-Barbero J, Martinez AT. J Ag Food Chem. 2007;55:3477. doi: 10.1021/jf063728t. [DOI] [PubMed] [Google Scholar]
- 6.Detailed characterization of native milled-wood lignin isolated from M. giganteus indicates that 93% of the polymeric linkages are β-[O]-4′–glycerolaryl ethers.; Villaverde JJ, Li J, Ek M, Ligero P, de Vega A. J Ag Food Chem. 2009;57:6262. doi: 10.1021/jf900483t. [DOI] [PubMed] [Google Scholar]
- 7.a) Cyr A, Chiltz F, Jeanson P, Martel A, Brossard L, Lessard J, Menard H. Can J Chem. 2000;78:307. [Google Scholar]; b) Kandanarachchi P, Autrey T, Franz J. J Org Chem. 2002;67:7937. doi: 10.1021/jo025581k. [DOI] [PubMed] [Google Scholar]; c) Kim YS, Chang Hm, Kadla JF. Holzforschung. 2008;62:38. [Google Scholar]
- 8.For some recent examples, see:; a) Owston NA, Parker AJ, Williams JMJ. Chem Commun. 2008:624–625. doi: 10.1039/b717073d. [DOI] [PubMed] [Google Scholar]; b) Shibahara F, Bower JF, Krische MJ. J Am Chem Soc. 2008;180:14120. doi: 10.1021/ja805356j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kakiuchi F, Usui M, Ueno S, Chatani N, Murai S. J Am Chem Soc. 2004;126:2706. doi: 10.1021/ja0393170. [DOI] [PubMed] [Google Scholar]
- 10.Kishimoto T, Uraki Y, Ubukata M. Org Biomol Chem. 2005;3:1067. doi: 10.1039/b416699j. [DOI] [PubMed] [Google Scholar]
- 11.Gaspar AR, Gamelas JAF, Evtuguin DV, Neto CP. Green Chem. 2007;9:717. [Google Scholar]
- 12.Litwinienko G, Ingold KU. Acc Chem Res. 2007;40:222. doi: 10.1021/ar0682029. [DOI] [PubMed] [Google Scholar]
- 13.For C-O activation mechanisms, see:; a) Choi J, Choliy Y, Zhang X, Emge TJ, Krogh-Jespersen K, Goldman AS. J Am Chem Soc. 2009;131:15627. doi: 10.1021/ja906930u. [DOI] [PubMed] [Google Scholar]; b) Ueno S, Mizushima E, Chatani N, Kakiuchi F. J Am Chem Soc. 2006;128:16516. doi: 10.1021/ja067612p. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.