

Abstract
The feasibility of preparing microparticles with high insulin loading suitable for needle-free ballistic drug delivery by spray-freeze-drying (SFD) was examined in this study. The aim was to manufacture dense, robust particles with a diameter of around 50 µm, a narrow size distribution and a high content of insulin. Atomization using ultrasound atomizers showed improved handling of small liquid quantities as well as narrower droplet size distributions over conventional two-fluid nozzle atomization. Insulin nanoparticles were produced by SFD from solutions with a low solid content (<10 mg ml−1) and subsequent ultra-turrax homogenization. To prepare particles for needle-free ballistic injection, the insulin nanoparticles were suspended in matrix formulations with a high excipient content (>300 mg ml−1) consisting of trehalose, mannitol, dextran (10 kDa) and dextran (150 kDa) (abbreviated to TMDD) in order to maximize particle robustness and density after SFD. With the increase in insulin content, the viscosity of the nanosuspensions increased. Liquid atomization was possible up to a maximum of 250 mg of nano-insulin suspended in a 1.0 g matrix. However, if a narrow size distribution with a good correlation between theoretical and measurable insulin content was desired, no more than 150 mg nano-insulin could be suspended per gram of matrix formulation. Particles were examined by laser light diffraction, scanning electron microscopy and tap density testing. Insulin stability was assessed using size exclusion chromatography (SEC), reverse phase chromatography and Fourier transform infrared (FTIR) spectroscopy. Densification of the particles could be achieved during primary drying if the product temperature (Tprod) exceeded the glass transition temperature of the freeze concentrate (Tg′) of −29.4°C for TMDD (3∶3∶3∶1) formulations. Particles showed a collapsed and wrinkled morphology owing to viscous flow of the freeze concentrate. With increasing insulin loading, the d (v, 0.5) of the SFD powders increased and particle size distributions got wider. Insulin showed a good stability during the particle formation process with a maximum decrease in insulin monomer of only 0.123 per cent after SFD. In accordance with the SEC data, FTIR analysis showed only a small increase in the intermolecular β-sheet of 0.4 per cent after SFD. The good physical stability of the polydisperse particles made them suitable for ballistic injection into tissue-mimicking agar hydrogels, showing a mean penetration depth of 251.3 ± 114.7 µm.
Keywords: spray-freeze-drying, needle-free injection, insulin delivery, nanosuspensions, protein formulation
1. Introduction
Needle-free ballistic particle injection is a relatively new drug and vaccine delivery technique that accelerates drug particles to a sufficient momentum using high-velocity gas jets to enable their penetration into epidermal and subepidermal layers of the skin (Burkoth et al. 1999). Its efficacy and safety, in particular in the area of vaccination, is well established and needle-free particle injection has shown many advantages over conventional needle and syringe injection: (i) it eliminates the possibility of needle and syringe re-use, (ii) needle-stick injuries are impossible, (iii) injection can be performed by everybody, not only trained personnel, (iv) a cold chain of refrigeration is not required for dry powders, and (v) for vaccines, much less antigen is required since it can be delivered directly to the epidermis (Dean & Chen 2004; Maa et al. 2004). Two different types of disposable handheld devices have been described in the literature and are referred to as (i) the bursting-membrane device and (ii) the Venturi device (Costigan et al. 2004). Actuation of both device types is achieved by rupturing a helium gas cylinder of 30–60 bar pressure which leads to an acceleration of particles to exit velocities of up to 1000 m s−1 (Liu et al. 2008). Skin penetration depth, one of the main criteria during particle formulation for needle-free ballistic injection, has been described as a function of particle density (ρ), particle velocity (v) and particle radius (r) (Kendall et al. 2004). According to Kendall et al. (2004), a threshold value of ρvr of approximately 7 kg m−1 s−1 is needed before particles begin to breach the stratum corneum. If the objective is the delivery of particles into the viable epidermis, a value of ρvr of 7–12 kg m−1 s−1 is required (Kendall et al. 2004).
Two different types of particles have been described for needle-free ballistic injection with protein-based drugs or antigens: (i) small, dense gold particles with a diameter of 1–3 µm and (ii) large sugar-polymer particles with diameters from 20 to 70 µm (Dean et al. 2003; Chen et al. 2004). Gold particles are limited in the amount of active ingredient they can deliver, as the therapeutic agent has to be precipitated onto their surface; therefore, they are preferred for small doses of antigen, as large drug doses cannot be delivered this way (Chen et al. 2001). However, owing to their high density of 19 300 kg m−3, the small diameter of 1–3 µm is sufficient to provide enough momentum density for penetration into the epidermis (Dean et al. 2003; Kendall et al. 2004). To achieve similar penetration depths, sugar or polymer-based particles have to be larger in size, usually 20–70 µm in diameter, owing to their relatively low density compared with gold (Kendall et al. 2000; Dean et al. 2003). Successful epidermal powder delivery has been reported using particles with a d(v, 0.5) = 39 ± 4 µm and a tap density of 720 ± 0.07 kg m−3 (Maa et al. 2004). A substantial advantage of the larger particles is that the protein is embedded into a sugar-polymer matrix allowing higher drug-loading factors, protein stabilization by excipients and, if desired, modification of release kinetics. In general, the requirements for particles manufactured from organic excipients, such as sugars and polymers, are: they should be between 40 and 100 µm with a density of at least 700 kg m−3, the mechanical robustness must be sufficient in order to avoid particle break-up during acceleration and impact on the skin and they have to contain excipients which are acceptable in epidermal and subepidermal drug administration. Particle diameters above 100 µm have been reported to cause pain sensation and bleeding (Burkoth et al. 1999).
Spray-freeze-drying (SFD) has been the method of choice to form large protein or vaccine-loaded sugar-polymer particles for needle-free ballistic injection. Maa et al. (2004) have successfully reformulated the flu vaccine Fluvirin into particles for powder injection using the excipients trehalose, mannitol and dextran (10 kDa) with SFD. Phase I clinical trials showed successful particle delivery to the epidermis and elicited humoral immune response in humans (Dean & Chen 2004). SFD is a modification of the lyophilization procedure with an additional unit operation, spray-freezing, to create discrete spherical particles of a defined size. Briefly, the process consists of the atomization of a liquid solution into a cryogenic gas or liquid with instant freezing of the generated droplets followed by sublimation of the ice at low temperature and pressure during freeze-drying. The benefits of SFD over its related particle manufacturing techniques, such as classical freeze-drying or spray-drying, are that the particle size of the final powder can be well controlled, as it mostly depends on the atomization conditions during spray-freezing (Schiffter 2007). In contrast, the final particle size during spray-drying will be influenced by a number of factors including atomization conditions, solution concentration and evaporation rate. Furthermore, particles with a diameter of 50 µm and narrow size distribution cannot be achieved easily in a laboratory spray-drying system, as the residence time of large droplets in the drying chamber is not sufficiently long enough (Masters 1991; Maury et al. 2005). Classical freeze-drying does not lead to distinct particles, but rather to the well-known cake structure that has to be disaggregated in a subsequent step, usually leading to a very wide particle size distribution with a substantial fine powder fraction that cannot be used for needle-free delivery. A summary of other methods for the manufacture of powders for needle-free injection can be found in the literature (Burkoth et al. 1999; Ziegler 2008).
However, SFD often can result in porous fragile particles that are not suitable for needle-free ballistic delivery because of low density and mechanical robustness (Costantino et al. 2000, 2002; Schiffter 2007). High concentrations of sugars and polymers in the initial liquid formulation are necessary (often around 350 mg ml−1) to obtain sufficient particle density and mechanical strength (Sonner et al. 2002; Maa et al. 2004). In particular, polymers such as dextran or hydroxyethyl starch in the formulation are essential ingredients for the maximization of particle robustness (Rochelle & Lee 2007). As current injection devices feature a maximum injectable powder amount of 1–2 mg, the administrable dose of the active pharmaceutical ingredient is limited to that amount, but often even more due to the fact that the addition of essential formulation excipients has to be taken into account. Further limitations may occur if the drug is poorly soluble in the sugar-polymer matrix formulation (Burkoth et al. 1999).
In this work, we present for the first time how mechanically robust and dense particles with a high insulin loading for needle-free ballistic injection can be manufactured by SFD of nanosuspensions and how changes in the process conditions affect the physical particle properties as well as the protein stability. SFD of suspensions was first described in a patent by Gombotz et al. (1991) and used in work published by Johnson et al. (1997) as well as Costantino et al. (2000) for the preparation of protein-loaded controlled release microspheres made of poly(lactic-co-glycolic) acid (PLGA). Here we present a modified version of this process for the preparation of immediate release microparticles with a high loading factor of a poorly soluble protein, in our case insulin. As the primary aim of the final microparticles is needle-free ballistic injection, the above-mentioned requirements in regard to mechanical robustness, particle size and powder density as well as the stability of the insulin during processing are the main focus of this work. In more detail, we first show the impact of formulation composition and process conditions—atomization, freezing and freeze-drying—on the size and density of sugar-polymer-based placebo spheres and the need to control these parameters tightly. We provide novel data about how atomization conditions using two-fluid nozzles or ultrasonic atomizers should be chosen in order to achieve large droplets, and therefore ultimately large particle diameters between 50 and 100 µm. Additionally, we introduce and investigate a new concept that can be applied to increase the density of sugar-polymer-based microparticles during freeze-drying by simple alteration of the lyophilization conditions in the primary drying stage. Second, we investigate the SFD of insulin nanosuspensions in a sugar-polymer solution and show new data about the influence of the suspended insulin on particle properties and particle morphology. Nanoparticulate and microparticulate insulin at different concentrations will be suspended in sugar-polymer-based formulations and the maximum suspendable insulin amount that still allows successful particle formation by SFD will be determined. We will also provide, for the first time in this area of research, new and extensive datasets about the density and mechanical robustness of the final placebo and insulin-loaded particles and use these data to calculate the ρvr value for each of the manufactured powders. Third, we examine the biological stability of insulin during each unit operation of the SFD process to provide a comprehensive understanding of the impact of processing conditions on the structural stability of the protein.
The results presented here show new information on how physically and biologically stable particles for needle-free ballistic powder delivery with high content of a poorly soluble protein drug can be manufactured. They will further advance understanding of the relationship of processing conditions and particle properties and how this dependence can be used to increase the size, density and mechanical robustness of particles for ballistic delivery. The work will provide guidance on how (i) atomization conditions should be chosen in order to achieve the desired particle size for needle-free injection, (ii) freeze-drying conditions can be used to increase the final density and mechanical robustness, and (iii) dosing problems due to payload limitations of current devices can be overcome.
2. Material and methods
2.1. Materials
Insulin from bovine pancreas with 28 USP units mg−1 (product no. I5500, lot no. 096K1527) and dextran from Leuconostoc mesenteroides with an average molecular weight of 10 kDa (product no. D9260) and with an average molecular weight of 150 kDa (product no. D4876) were purchased from Sigma-Aldrich Co. Ltd (Gillingham, UK). Trehalose dihydrate (Acros Organics, product no. 18255-1000) and mannitol (Acros Organics, product no. 42392-0010) were purchased from Fisher Scientific (Loughborough, UK). The density of all substances as determined by helium (He)-pycnometry is listed in table 1. All substances used for analytical procedures—glacial acetic acid, acetonitrile, l-arginine, phosphoric acid, sodium sulphate, ethanolamine and water—were purchased as HPLC grade from Fisher Scientific.
Table 1.
Density of formulation excipients determined by He-pycnometry.
| substance | measured density (kg m−3) | reference densitya (kg m−3) |
|---|---|---|
| trehalose dihydrate | 1526 | 1540 |
| mannitol | 1521 | 1510 |
| dextran 10 kDa | 1023 | 1040 |
| dextran 150 kDa | 1505 | — |
| bulk insulin | 1324 | — |
aAccording to the supplier.
2.2. Methods
2.2.1. Preparation of insulin nanoparticles by SFD
Insulin or an insulin–trehalose (1∶1) mixture was dissolved at concentrations of 5 or 10 mg ml−1, respectively, in double-distilled and filtered (0.2 µm) 0.01 M HCl. Ten millilitres of the solution was atomized into the vapour over a stainless steel bowl filled with liquid nitrogen (LN2) using a 60 kHz ultrasound nozzle (Sono-Tek, Milton, NY, USA) at an atomization power of 3.1 W and a flow rate of 0.5 ml min−1 provided by a peristaltic pump (Minipuls, Gilson Inc., Middleton, WI, USA). After atomization, the bowl was topped up with LN2 and subsequently transferred onto the pre-cooled shelves (−40°C) of an FTS Lyostar I freeze-drying system (SP Industries, Warminster, PA, USA). Primary drying was achieved at a shelf temperature of −20°C for 48 h followed by secondary drying at 20°C for 12 h. The chamber pressure was kept constant at 100 mtorr (=0.133 mbar) throughout the drying stages. At the end of the freeze-drying cycle, the vacuum was released with scientific air (Scientific Synthetic Air 5.5, H2O ≤ 2 ppm, BOC). The powders were transferred from the stainless steel bowls into 20 cm3 serum tubing vials in the controlled environment of a glove box with 0.5 per cent relative humidity at 20°C. The vials were stoppered, sealed, crimped and frozen to −80°C until further usage.
2.2.2. Preparation of insulin-loaded microparticles by SFD
Insulin-loaded microspheres with immediate release properties were manufactured by SFD of insulin nanosuspensions. A process originally described by Johnson et al. (1997) and Costantino et al. (2000) was modified in order to achieve the desired particle characteristics necessary for needle-free ballistic delivery. In detail, the formulation excipients, trehalose, mannitol and dextran, were dissolved in the desired concentrations in double-distilled and filtered (0.2 µm) water. The pH of the solution was set to either pH 2.0 or pH 7.0 using 0.1 M HCl or 0.1 M NaOH. Purchased bulk insulin or insulin nanoparticles were dissolved and suspended in 1.0 g of matrix formulation. Homogenization was performed by vortexing or by using an Ultra-Turrax homogenizer at 20 000 r.p.m. for 3 minutes. The final formulation was pumped at a liquid flow rate of 0.5 ml min−1 to either a 48 or a 25 kHz ultrasound nozzle (Sono-Tek). Atomization was performed at sufficient atomization power directly into the vapour over a small stainless steel bowl filled with LN2. At the end of the atomization operation, the bowls were once more topped up with LN2 and then kept under ambient conditions until the LN2 had completely boiled off to avoid boiling retardation occurring when transferred too early onto warmer surfaces. Immediately afterwards, the bowls were closed with a Gore Lyoguard cap (W. L. Gore & Associates, Newark, DE, USA) and transferred onto the pre-cooled shelves (−40°C) of an FTS Lyostar I freeze-drying system. Primary drying was performed at −10°C, −20°C or −30°C for 48 h and secondary drying was performed at 20°C for 24 h. A chamber pressure of 100 mtorr (=0.133 mbar) was maintained throughout the freeze-drying cycles. At the end of each cycle, the vacuum in the freeze-dryer chamber was released using scientific air (Scientific Synthetic Air 5.5, H2O ≤ 2 ppm, BOC). On opening of the freeze-dryer, the stainless steel bowls were immediately transferred into the 0.5 per cent relative humidity environment of a glove box at room temperature. The powders were transferred from the stainless steel bowls into 20 cm3 serum tubing vials and handled as outlined in the nanoparticle manufacturing procedure.
2.2.3. Karl–Fischer titration analysis
Residual moisture content of all powders after SFD was determined using coulometric Karl–Fischer titration in a DL39 titrator with a Stromboli oven and autosampler system (Mettler Toledo, Leicester, UK). A 50 mg sample was used for each analysis. Titration was performed for 30 min at an oven temperature of 120°C. All analyses were performed in triplicate.
2.2.4. Laser light diffraction
Droplet size distributions of the sprays generated with the ultrasound and two-fluid atomizers at different operation parameters as well as particle size distribution of the final freeze-dried powders were determined by laser light diffraction using a Malvern Mastersizer S (Malvern Instruments Ltd, Malvern, UK). A 300F lens was used for the droplet size distribution measurements. The nozzle was mounted directly over the pathway of the laser beam at a distance of 1.5 cm from the lens. Droplet size distribution was calculated as an average of six measurements with 6000 data points using the Mie theory. For particle size distribution measurements, the instrument was equipped with a 300RF lens with a backscatter detector and a small sample dispersion unit. The final dried product was suspended in cyclohexane at a concentration so as to achieve an obscuration between 10 and 15 per cent. The small volume dispersion unit was set to 2000 r.p.m. Particle size distribution was calculated as an average of six experiments using the Mie theory. Refractive indices 1.4262 for cyclohexane and 1.3903 for the sugar matrix were used. The median diameter, d(v, 0.5), and the span of the volume distribution were used to characterize the powders. The span is defined as
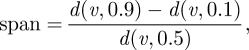 |
2.1 |
with d(v, 0.1) and d(v, 0.9) describing the particle diameter at 10 and 90 per cent screen underflow.
2.2.5. He-pycnometry and bulk density measurements
Powder density was analysed using a Pycnomatic ATR helium pycnometer (ThermoFisher, Loughborough, UK) with a small sample volume unit of 6.95255 cm3. Analyses were performed with 0.8–1.0 g of the freeze-dried sample. Density measurements were carried out subsequent to 20 initial cleaning cycles with analytical grade helium at 20.0°C. Particle density was calculated as an average of six measurements with a maximum relative standard deviation of 0.50 per cent. Powder bulk and tap density measurements were performed according to method 5.0/2.9.15 of the European Pharmacopoeia with the exception of using a 5 ml volumetric flask due to the small sample volumes available. The Hausner ratio was calculated from the tap density divided by the poured bulk density of the powder. The porosity of the powders was calculated using the density measured by He-pycnometry and the tap density according to the following equation:
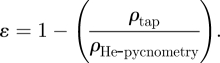 |
2.2 |
2.2.6. Wide angle X-ray diffraction and differential scanning calorimetry
Wide angle X-ray diffraction (WAXD) analysis was performed on a Philips model X'pert MPD with Cu Kα radiation at 40 kW and 40 mA from 100 mg of powder. Differential scanning calorimetry (DSC) was used to determine the glass transition temperature of the freeze concentrate, Tg′, of each formulation. Measurements were performed on a Mettler Toledo DSC 822. For each analysis, an approximate volume of 10 µl liquid formulation was filled in a DSC pan. The pan was crimped and frozen at a rate of 10 K min−1 in the sample DSC chamber of the instrument. Subsequently, temperature scans at a heating rate of 10 K min−1 were performed. Tg′ was determined as the temperature of the inflexion point of the endothermic shift in heat capacity on the thermogram.
2.2.7. UV/VIS spectrophotometry
Insulin content of the spray-freeze-dried powders was determined by UV absorbance at 280 nm and 20°C using a CaryBio 50 spectrophotometer with a Peltier element for measurements at constant temperature (Varian, Yarnton, UK). Calibration was performed by dissolving pure insulin at concentrations of 0.020, 0.052, 0.130, 0.260, 0.480, 1.140 and 1.700 mg ml−1 in 0.01 M HCl. The final regression line had a correlation coefficient of 0.9989, and 5–10 mg of each sample after SFD was dissolved in 0.01 M HCl directly before the analysis. The insulin content was calculated as a percentage of the weighted powder excluding the residual humidity determined by prior Karl–Fischer analysis.
2.2.8. Size exclusion chromatography
Size exclusion chromatography (SEC) was performed on a Varian HPLC system with two Prostar 210 solvent delivery systems, a Prostar 325 UV-detector, a Prostar 500 column valve oven and a model 410 autosampler (Varian, Yarnton, UK). To obtain sufficient selectivity and resolution, a 30 cm × 7.8 mm Protein Pak 125 column (Waters) was used for the SEC analysis. The procedure was performed according to method 5.0/0838 of the European Pharmacopoeia. Briefly, the mobile phase consisted of 15 per cent glacial acetic acid, 20 per cent acetonitrile and 65 per cent of an aqueous l-arginine solution (1.0 g l−1). The flow rate of the mobile phase was 0.5 ml min−1 and the sample volume injected 100 µl of 1.0 mg ml−1 insulin in 0.01 M HCl. The eluent was detected using UV absorbance at 276 nm.
2.2.9. Reverse-phase chromatography
Reverse-phase chromatography was performed on the Varian HPLC system described in §2.2.7. Reverse-phase chromatography was conducted using a 250 × 4.6 mm Jupiter 5 µm C18300 Å column (Phenomenex, Macclesfield, UK) according to monograph 5.0/0838 of the European Pharmacopoeia. Briefly, mobile phase A consisted of 28.4 g sodium sulphate made up to 1000 ml with HPLC grade water plus 2.7 ml phosphoric acid (85%). The pH was adjusted to 2.3 with ethanolamine. Mobile phase B consisted of 550 ml mobile phase A plus 450 ml acetonitrile. The flow rate was 1.0 ml min−1. The elution conditions were 42 per cent A and 58 per cent B for 30 min. Then, a linear gradient from 42 per cent A to 11 per cent A and 58 per cent B to 89 per cent B over 14 min was used. This was followed by 11 per cent A and 89 per cent B for an additional 6 min. The detection wavelength was 214 nm and the injected volume was 20 µl of 1.0 mg ml−1 insulin solution.
2.2.10. Determination of protein secondary structure by Fourier transform infrared spectroscopy
Protein secondary structure was determined by Fourier transform infrared (FTIR) spectroscopy. FTIR spectra of aqueous insulin solutions were recorded on an Omnic Nicolet Magna IR 550 spectrophotometer (Thermo Fisher, Dreieich, Germany) using a Proteus CaF2 transmission cell with optical pathlength of 6 µm. SFD particles were dissolved in 0.01 M HCl at pH 2.0, resulting in insulin concentrations of 1.0–1.5% (weight/volume). Pure matrix solutions without insulin for background subtraction were prepared accordingly. For each measurement, 42 scans were collected at the slowest mirror speeds possible. The measurements were repeated three times and averaged to reduce the baseline effects and spectral noise. Between measurements, the cell was purged with 0.01 M HCl until no signal was detectable. Subtraction of the background signal was performed manually with a subtraction factor between 0.95 and 1.05. During the whole process, the spectrometer was constantly purged with dry air to avoid effects due to water vapour. Insulin spectra were focused on the area-normalized, baseline-corrected amide I band (1700–1600 cm−1). Quantification was performed by peak fitting using the program PeakFit (Systat Software GmbH, Erkrath, Germany). Peak positions were determined by second derivative spectra and matched with the shape of the amide I band in an iteration process. The resulting fit was considered final when a correlation coefficient of ≥0.999 was accomplished.
2.2.11. Particle penetration testing and light microscopy
Particle penetration was tested by injecting 500 µg spray-freeze-dried particles into 3 per cent (w/v) agar hydrogels using the inline Venturi needle-free injection device with a 5 ml helium cartridge at 60 bar. The device is shown in figure 1 together with a photograph of the powder cassette components. The design of the device and the Venturi effect have already been described in the literature (Liu 2007; Liu et al. 2008). Briefly, a supersonic nozzle inside the device is used to create a high-velocity, low-pressure jet. This jet flows into a chamber with a slightly larger diameter to create a pressure difference across the powder cassette. At highest gas velocity, the pressure difference reaches a maximum and the drug particles are drawn from the cassette (which has been manually opened before device actuation) into the gas flow where they are accelerated. To achieve a uniform particle distribution on the target rather than a particle deposition in the centre of the injection plane, an element of swirl is introduced to the helium gas flow by a specially perforated swirl disc inside the device.
Figure 1.

Photograph of the inline Venturi injection device with an 8 mm exit diameter used for the needle-free ballistic particle injection.
The particles for injection were weighted into the powder cassettes using a microbalance (MT5, Mettler Toledo) and the cassette was subsequently inserted into the body of the needle-free device. The tissue-mimicking agar hydrogels were produced by dissolving dry agar powder in water under moderate heating and subsequent degassing at a constant pressure. The resulting liquid agar formulation was injected into specifically designed moulds using a 10 ml syringe and refrigerated for at least 3 h to ensure complete solidification prior to ballistic particle injection. The cylindrical hydrogel phantoms had a diameter of 22 mm and a height of 5 mm. Post-injection, transverse agar hydrogel slices of around 200 µm in thickness were prepared and examined with light microscopy using a Nikon Eclipse Ti-inverted microscope (Nikon Instruments Europe B.V., Kingston, UK). Particle penetration depth was determined as distance of the particle from the original agar surface.
2.2.12. Scanning electron microscopy
Particle morphology of all dried particles was examined using an Amray 1810T scanning electron microscope (Amray; Bedford, USA) at 20 kV. Samples were fixed on aluminium stubs (G301, Plano) with self-adhesive films and sputtered with gold at 20 mA/5 kV (Hummer JR Technics; Munich, Germany) for 1.5 min.
3. Results and discussion
3.1. Influence of atomization conditions on droplet size distribution
The d(v, 0.5) and span of the volume distribution as well as the d(n, 0.5) and span of the number distributions of the aqueous sprays from four different ultrasound atomizers and one two-fluid nozzle at different atomization conditions are listed in table 2. The droplet size distribution of a spray generated with a two-fluid nozzle depends on the nozzle geometry, surface tension and viscosity of the formulation, the liquid flow rate and the atomizing air flow rate (Reitz & Bracco 1986; Lefebvre 1989). The Reynolds number, describing the inertial forces to viscous forces, and the Weber number, describing a fluids inertia compared with its surface tension, govern the atomization regime. The calculated Reynolds numbers for the condition set in table 2 were between 4.53 × 105 and 0.91 × 105. Correspondingly, the calculated Weber numbers were between 4.90 × 103 and 1.95 × 103. The mean droplet size of all sprays generated with the two-fluid nozzle was between 11.92 and 131.93 µm and, therefore, substantially smaller than the nozzle exit diameter of 0.7 mm. This shows that all spraying conditions were within the atomization regime. To obtain a spray containing large droplets with a d(v,0.5) > 40 µm, high liquid flow rates of 10 ml min−1 in combination with low atomizing air flow rates (180–500 l h−1) were necessary. However, all two-fluid nozzle sprays showed a wide droplet size distribution with spans larger than 1.626. As high flow rates of 10 ml min−1 were needed to achieve the desired droplet size, handling of samples with a volume smaller than 2 ml proved very difficult. The use of the peristaltic pump with these flow rates showed a substantial pulsation of the liquid inside the tubing as well as during atomization at the nozzle tip explaining the wide droplet size distributions. A parallel decrease in liquid and atomizing flow rates from the above values with the aim to keep the d(v, 0.5) larger than 40 µm could not be achieved and led to incomplete atomization with randomly sized droplets which reduced the yield and feasibility of this spraying process.
Table 2.
Droplet size characteristics of water and TMDD (3 : 3 : 3 : 1) 35% (w/w) sprays generated with a Buchi two-fluid nozzle with an orifice diameter of 0.7 mm and four ultrasound nozzles with operational frequencies of 120, 60, 48 and 25 kHz. MBT, microbore tubing; SSA, specific surface area.
| nozzle type | operational parameters | SSA (m2 g−1) | volume distribution |
number distribution |
||
|---|---|---|---|---|---|---|
| d(v, 0.5) (µm) | span | d(n, 0.5) (µm) | span | |||
| two-fluid nozzle (water at a liquid flow rate of 10 ml min−1) | ||||||
| Buchi 0.7 mm | air flow 900 l h−1 | 0.675 | 11.92 | 1.813 | 2.78 | 2.213 |
| air flow 540 l h−1 | 0.192 | 39.41 | 1.758 | 9.06 | 2.163 | |
| air flow 300 l h−1 | 0.069 | 88.50 | 1.506 | 32.24 | 1.626 | |
| air flow 180 l h−1 | 0.056 | 131.93 | 1.884 | 37.38 | 1.822 | |
| ultrasound atomizer (water at a liquid flow rate of 0.5 ml min−1) | ||||||
| 120 kHz | power 4.7 W | 0.239 | 26.39 | 1.127 | 17.01 | 1.016 |
| 60 kHz | power 2.9 W | 0.178 | 35.34 | 0.843 | 26.25 | 0.849 |
| 48 kHz | power 3.1 W | 0.146 | 42.24 | 0.819 | 33.14 | 0.758 |
| 25 kHz | power 1.5 W | 0.094 | 67.29 | 0.851 | 48.45 | 0.908 |
| 25 kHz (MBT) | power 1.5 W | 0.112 | 55.74 | 0.689 | 46.54 | 0.660 |
| ultrasound atomizer (TMDD (3 : 3 : 3 : 1) 350 mg/g solution at a liquid flow rate of 0.5 ml min−1) | ||||||
| 25 kHz (MBT) | power 1.5 W | 0.096 | 63.50 | 0.543 | 56.38 | 0.548 |
In contrast to two-fluid nozzles, ultrasonic atomizers operate at a specific resonant frequency. Atomization is achieved by sustained vibration due to a standing, sinusoidal longitudinal wave. Droplet size is governed by the frequency of nozzle vibration with surface tension and viscosity of the atomized liquid solution being of secondary importance (Berger 2006). With decreasing nozzle operation frequency, droplet size increases. Our work showed that liquid flow rates of as low as 0.25 ml min−1 could be handled in combination with ultrasound atomizers still achieving droplet diameters of 30–60 µm. Size distributions of all measurements were substantially narrower than seen in any of the two-fluid nozzle atomization experiments. Atomization at 48 and 25 kHz generated droplets with a d(v, 0.5) and d(n, 0.5) > 40 µm. As these droplets were in the size range suitable for needle-free ballistic injection, these two nozzle frequencies were chosen for the actual particle manufacturing by SFD. Equipping a 25 kHz atomizer with internal microbore tubing (diameter = 0.5 µm) at a liquid flow rate of 0.5 ml min−1, a d(v, 0.5) of 63.50 µm could be achieved with 92 per cent of the droplets being larger than 40 µm. The narrow size distribution is reflected in the small difference between mean volume and number diameter and a span (volume) of 0.543 (table 2). In a monodisperse spray, the mean number and volume diameter would be equal. This could not be achieved for any of the used nozzle systems.
3.2. Influence of formulation and freeze-drying conditions on particle size and density
Composition and solid concentration of the liquid feed influence size and morphology of particles prepared by SFD (Costantino et al. 2000; Yu et al. 2002). SFD of pure insulin solutions with concentrations of 5 mg ml−1 using a 60 kHz ultrasound atomizer resulted in powders with a mean tap density of 3.23 ± 0.48 kg m−3 (table 3). A highly porous powder structure with a large, irregular and rough surface was clearly visible in the scanning electron microscopy (SEM) photographs (figure 2a). No discrete particles existed in the powder. Similar observations were published by Costantino et al. (2000) and Yu et al. (2004) for spray-freeze-dried bovine serum albumin, the specific surface area (SSA) of their final BSA powder being 19.2 and 97.7 m2 g−1. These large surface areas are characteristic for spray-freeze-dried products derived from liquid solutions with concentrations less than 50 mg ml−1 (Leuenberger 2002; Yu et al. 2002, 2004). The particle size measurements of insulin powders, obtained from SFD of pure insulin formulations with a 5 mg ml−1 solid content, showed that the highly porous structure seen in figure 2a could be broken down into nanoparticles with a d(v, 0.5) of 238 ± 39 nm by homogenization with an Ultra-Turrax at 20 000 r.p.m. in acetone or cyclohexane (figure 3). In comparison, the bulk insulin, as received from the supplier, had substantially larger particles with a mean d(v, 0.5) of 12.34 ± 0.51 µm before and 2.65 ± 0.35 µm after the same homogenization procedure (table 3). If trehalose was added as excipient to the 5 mg ml−1 insulin solution to achieve a final formulation with a solid content of 10 mg ml−1 (insulin–trehalose ratio = 1 : 1), the resulting nanoparticles were slightly larger in diameter with a mean d(v, 0.5) of 285 ± 58 nm after homogenization in cyclohexane. An increase in concentration of drug or addition of formulation excipients, such as sugars or polymers, increases the solution viscosity and volume fraction of the solute, thus leading to larger particles after SFD at constant operational parameters. However, neither the nanoparticulate SFD insulin powders nor the purchased bulk insulin was directly suitable for needle-free ballistic injection. Injection tests into agar hydrogel targets did not show any particle penetration. All powders could be recovered on the agar surface. The ρvr values calculated for these powders were clearly below the well-accepted threshold value of 7.0 (table 3) necessary for particle penetration after needle-free injection and confirmed these observations. Therefore, particles with larger diameter and/or density had to be prepared in order to achieve successful drug delivery by needle-free particle injection.
Table 3.
Size distribution and density values for bulk insulin and spray-freeze-dried insulin formulations with low insulin concentration (5 mg ml−1) and atomization at 60 kHz. Hausner ratio and porosity were calculated from the density values. ρvr values were calculated with a velocity of 750 m s−1.
| formulation | size distribution d(v, 0.5) (µm)/span (—) |
density from He-pycnometry (g cm−3) | bulk density (g cm−3) | tap density (g cm−3) | Hausner ratio | porosity (%) | ρvr value (calculated from tap density) | |
|---|---|---|---|---|---|---|---|---|
| before homogenization | after homogenization | |||||||
| bulk insulin (as supplied) | 12.34/1.546 | 2.65/1.839 | 1.324 | 0.265 | 0.400 | 1.509 | 69.8 | 1.85 |
| insulin after SFD, 5 mg ml−1 unformulated at pH 2.0 | 8.91/1.496 | 0.24/1.208 | 1.322 | 3.23 × 10−3 | 4.36 × 10−3 | 1.350 | 99.7 | 1.46 × 10−2 |
| insulin after SFD, 5 mg ml−1 + trehalose 5 mg ml−1 at pH 2.0 | 10.29/1.572 | 0.29/1.251 | 1.429 | 3.45 × 10−3 | 4.63 × 10−3 | 1.342 | 99.7 | 1.79 × 10−2 |
Figure 2.

Representative SEM pictures from spray-freezing into vapour over LN2 using (a) 60 kHz and (b–d) 48 kHz ultrasound nozzles with subsequent freeze-drying. (a) Pure insulin powder from a 5 mg ml−1 insulin solution at pH 2.0 (magnification 2500×), (b) SFD microparticles from a pure TMDD (3 : 3 : 3 : 1) 35% (w/w) solution freeze-dried at −10°C and 100 mtorr (magnification 1000×) and (c) at –30°C and 100 mtorr (magnification 2500×). (d) SFD microspheres from a TMDD (3 : 3 : 3 : 1) 35% (w/w) solution loaded with 25% insulin (magnification 500×).
Figure 3.

Volume size distributions of insulin and TMDD (3 : 3 : 3 : 1) particles after SFD and homogenization. Insulin nanoparticles after SFD (filled triangle) of a 5 mg ml−1 insulin solution at pH 2.0, purchased bulk insulin before SFD (open triangle), TMDD (3 : 3 : 3 : 1) after SFD with atomization at 48 kHz and primary drying at −10°C (filled circle) and TMDD (3 : 3 : 3 : 1) after SFD with atomization at 25 kHz and primary drying at −10°C (open square).
SFD particles containing a mixture of trehalose, mannitol and dextran (10 kDa) prepared from a liquid solution with a concentration of 350 mg ml−1 have been described by Maa et al. (2004) for use with needle-free particle injectors. Maa's particles with a d(v, 0.5) of 39 µm and a tap density of 720 kg m−3 were successfully used in phase I clinical trials for epidermal powder immunization of the reformulated influenza vaccine Fluvirin (Dean & Chen 2004). Applying the concept of ρvr with an estimated exit velocity of 750 m s−1 reported for the device, the ρvr value was 10.52 kg m−1 s−1 (Kendall et al. 2004). This value underestimated the real ρvr value, as the tap density of the powder is used for the calculation rather than the apparent particle density. As the target tissue for these flu vaccine-loaded particles was the epidermis with its substantial amount of Langerhans cells, particles for systemic insulin delivery need to have a larger ρvr value, achievable by an increase either in size or in density or both, to penetrate into subepidermal regions. However, the maximum particle size is limited to approximately 100 µm, as otherwise the occurrence of a strong pain sensation has been reported upon injection (Burkoth et al. 1999). To investigate the density of the SFD excipients prior to processing, He-pycnometry was used and showed that the bulk density for dextran with a molecular weight of 150 kDa is higher than for dextran with 10 kDa (table 1). These findings were used to develop a new particle formulation with increased density in comparison with previously published data (Maa et al. 2004). To test in particular the influence of the different dextrans on the particle density, the ratio of dextran 10 kDa to dextran 150 kDa was altered from 100 to 0 per cent in steps of 25 per cent. Consequently, the tap density of the TMDD (trehalose, mannitol, dextran 10 kDa, dextran 100 kDa) formulations after SFD using a 48 kHz nozzle increased from 0.752 ± 0.021 to 0.829 ± 0.013 µm with increasing amounts of large molecular weight dextran. However, in the same way, an increase in viscosity was visible with increasing amounts of high molecular weight dextran complicating the atomization procedure. Therefore, a ratio of dextran 10 kDa to dextran 150 kDa of 3 : 1 or 2 : 2 was chosen for all the following experiments. All final powders were completely amorphous in X-ray examinations (WAXD diagrams not shown) even though mannitol was present. Dextran and other polymers such as polyvinylpyrrolidone (PVP) have been reported to prevent mannitol crystallization during freeze-drying, in particular if fast freezing with a cryogenic liquid, such as LN2, is performed (Cavatur et al. 2002; Rochelle & Lee 2007).
The final SFD particles of pure TMDD (3 : 3 : 3 : 1) 350 mg/g formulations dried at −10°C or at −20°C primary drying temperature showed an irregularly shaped, contracted and wrinkled surface morphology (figure 2b). This has been explained by Sonner et al. (2002) as the result of collapse of the amorphous TMD10 kDa phase during primary drying. No change in particle appearance could be seen in the SEM pictures with increasing dextran (150 kDa) content. This type of collapsed morphology has been described in the literature for formulations containing dextran or hydroxyethyl starch and depends on the viscosity of the amorphous structure above its glass transition temperature (Sonner et al. 2002; Maa et al. 2004; Rochelle & Lee 2007). Primary drying temperatures above the glass transition temperature (Tg′) of the amorphous freeze concentrate of frozen SFD formulations can lead to particle morphologies deviating from the original spherical shape of the frozen droplets, in particular, if polymers such as dextran or hydroxyethyl starch are present. Measurement of the Tg′ of the TMDD (3 : 3 : 3 : 1) 350 mg/g formulation using DSC gave a value of −29.4°C. Choosing a temperature of −30°C as the shelf temperature during primary drying the spherical shape of the particles could be preserved, as the product temperature stayed well below Tg′ during drying (figure 2c). However, it could clearly be seen that an increase in primary drying temperature, and as a result the collapse and shrinkage of the particles, led to a beneficial increase in the tap density of the final powders. Values for the tap density increased from 662 ± 19 kg m−3 at −30°C primary drying to 791 ± 15 kg m−3 at −10°C primary drying due to shrinkage of the particles at the higher drying temperatures. Measurements of the product temperature Tprod using a thermocouple arrangement in a 20 ml standard glass freeze-drying vial with 3 g spray-frozen droplets showed that Tprod exceeded Tg′ for the last 2590 min of the 2880 min primary drying phase (figure 4). Measurement of vial weight during lyophilization using a freeze-drying microbalance inside the drying chamber showed the completion of ice sublimation after approximately 2200 min of primary drying. This confirmed the thermocouple data as the temperature measured inside the vial reached a constant value above the shelf temperature at the same time point. As the collapse temperature (TC) of an amorphous freeze concentrate is usually 2°C above Tg′, plastic flow of the TMDD phase may occur when Tprod exceeds Tg′ and even TC during primary drying (Tang & Pikal 2004). At temperatures higher than Tg′, the viscosity of an amorphous matrix decreases drastically and can be described by the Williams–Landel–Ferry equation as a function of (T − Tg), where in our case T is Tprod and Tg is Tg′ of the freeze concentrate (Hancock & Zografi 1997). As viscosity decreases to a level that facilitates plastic flow, matrix deformation and collapse of pores can occur (Levi & Karel 1995; Rochelle & Lee 2007). Therefore, it seems that the collapsed and shrunken appearance of the TMDD particles is due to the plastic flow of the formulation during primary drying when Tprod exceeds Tg′ and even TC. Usually, during regular freeze-drying cycles, the collapse of the product during primary drying has to be avoided to obtain a high-quality, porous cake structure. However, if particles are prepared by SFD and intended to be used for needle-free ballistic powder injection, plastic flow during lyophilization seems to be beneficial in regard to increasing particle density and mechanical robustness. Therefore, a primary drying shelf temperature of −10°C was chosen for all microparticle manufacturing experiments in this work.
Figure 4.

Process data recorded during lyophilization of TMDD (3 : 3 : 3 : 1) 350 mg ml−1 after spray-freezing of 3 g solution with atomization at 48 kHz and quench freezing of 3 g solution in LN2. Frozen droplets and solutions were freeze-dried in 20 ml serum tubing vials. (a) Thermocouple data showing Tprod after freezing (solid line, shelf; line with open circle, spray-frozen; line with inverted triangle, bulk-frozen), (b) weight of 20 ml serum tubing vials with 3 g of spray-frozen solution and (c) calculated sublimation rate of spray-frozen TMDD solution during lyophilization at −20°C and 100 mtorr primary drying.
3.3. Insulin loading of TMDD formulations
The bulk insulin used in this work contained 28 IU (international units) of insulin in 1 mg of powder supplied by Sigma-Aldrich. Therefore, 1 IU insulin was equivalent to 35.7 µg of the bulk insulin powder. For needle-free ballistic particle injection, a maximum area loading factor, defined as the area covered by particles divided by the total skin area, has been determined to avoid skin damage. The area loading factor and target area fix the powder payload for a given particle size and density. The Venturi device used in these experiments had a nozzle exit diameter of 8 mm and a maximum payload of 1 mg calculated for monodisperse particles with a diameter of 48 µm. As the dry bulk insulin available on the market exhibits inferior particle characteristics for needle-free ballistic delivery in terms of particle size, density and physical robustness (figure 3 and table 3), it had to be reformulated using the developed matrix formulation with trehalose, mannitol, dextran 10 kDa and dextran 150 kDa as already described in §3.2. To obtain a single dose for needle-free insulin injection with, for example, 5 IU, the reformulated particles need an insulin loading of 17.9 per cent (as the maximum device payload equals 1 mg). This is equivalent to 76.3 mg insulin mixed with 1000 mg of the 350 mg/g TMDD (3 : 3 : 3 : 1) solution. The solubility of insulin is lowest around its isoelectric point at pH 5.3–5.4. Solubility can be increased to 20 mg ml−1 at pH values lower than 4.0 and higher than 7.0 (Joshi & Huh 2004). However, this solubility is still much lower than that needed to dissolve 76.3 mg in less than 1 ml of TMDD solution. Furthermore, acid formulation conditions can cause insulin to aggregate and form fibrils (Bouchard et al. 2000). To obtain a high drug loading in the reformulated particles for needle-free ballistic injection, suspensions of the pre-prepared insulin nanoparticles in the TMDD matrix solution were spray-freeze-dried. The concept of SFD of suspensions has been reported before by Johnson et al. (1997) and Costantino et al. (2000), however, applied to the manufacture of protein-loaded PLGA microspheres for controlled release delivery. In their work, the nano-size protein particles were suspended in a PLGA solution in methylene chloride and the resulting suspension was sprayed into a vessel containing frozen ethanol overlaid with LN2. The vessel was afterwards transferred into a −80°C freezer to allow the microspheres to harden for 3 days before they were collected by filtration and dried under vacuum. However, as the present work was focused on the preparation of immediate release microparticles with high density and mechanical robustness, the preformed insulin nanoparticles (obtained from SFD of 0.5% insulin solutions) were suspended in the newly developed TMDD (3 : 3 : 3 : 1) 350 mg/g matrix solution (§3.2) and were homogenized using the Ultra-Turrax homogenizer at 20 000 r.p.m. for 3 minutes. Particle formation was then performed by directly atomizing the suspension into the vapour over LN2 filling a stainless steel bowl (as explained in §2.2.2) and subsequently subjecting the frozen droplets to a freeze-drying cycle designed to cause particle densification by controlled plastic flow at −10°C primary drying temperature, as discussed in §3.2. All new placebo and insulin formulations are shown in table 4. Suspensions with up to 150 mg insulin could still be atomized well with the ultrasound nozzles even though high-power inputs of 8.0–12.0 W were necessary for liquid break-up. For comparison, the power needed for the pure TMDD formulations was 1.5 W for the 25 kHz nozzle and 3.1 W for the 48 kHz nozzle. Also, the internal microbore tubing of the 25 kHz atomizer could only be used up to an insulin content of 20 per cent before clogging. A comparative study using the purchased bulk insulin in the suspensions showed that on average 2.3 W or more atomization power was needed to achieve droplet formation with the ultrasound nozzles in contrast to nano-insulin. Furthermore, atomization of formulations 6 and 7, where 233 mg and 250 mg insulin, respectively, was suspended in 1.0 g TMDD, could not be achieved with purchased untreated insulin, but instead with preformed insulin nanoparticles. Therefore, only insulin nanoparticles were used in the formulations listed in table 4. The measured insulin content after rehydration corresponded well to the theoretical values for all suspensions with an insulin content of 30 per cent or less. Above this value, the formulation consistency resembled a paste rather than a suspension. Liquid processing and continuous atomization were increasingly difficult even though preformed insulin nanoparticles were used. The 48 kHz ultrasound atomizers needed a maximum power input of 15 W to achieve liquid break-up. However, during atomization larger droplets in the millimetre size range were seen detaching from the nozzle tip and could later be detected as large particles in the final powder. The measured insulin content of particles with a theoretical insulin loading of 40 per cent (formulation 6) deviated by an average of 3.3 per cent. A theoretical insulin loading of 50 per cent was accomplished by decreasing the solid content of the TMDD matrix solution to 250 mg/g. However, the final insulin content of the particles showed a high deviation of −5.2 per cent from its theoretical value. Higher insulin content on bases of a 250 or 350 mg/g TMDD formulation could not be achieved as atomization was no longer possible. The 25 kHz atomizer did not manage to form any droplets in its usual size range from formulations 6 and 7 and could therefore not be used.
Table 4.
Insulin loading and physical properties of particles after SFD and needle-free ballistic injection at 60 bar helium.
| formulation | preparation | theoretical insulin content (%) | no. | nozzle | measured insulin content (%) | size distribution d (v, 0.5) (µm)/span (—) | physical stability d (v, 0.5) (µm)/span (—) |
|---|---|---|---|---|---|---|---|
| 1.000 g TMDD (3331) 35% (w/w) | pH 7.0—solution | 0.0 | 1a | 25 kHz | — | 53.12/0.646 | 49.65/0.956 |
| 0.000 g insulin | 1b | 48 kHz | — | 46.81/0.921 | 43.71/1.215 | ||
| 1.000 g TMDD (3331) 35% (w/w) | pH 7.0—suspension vortexing | 10.0 | 2a | 25 kHz | — | — | — |
| 0.039 g insulin | 2b | 48 kHz | 9.9 ± 0.3 | 47.43/0.987 | 43.32/1.223 | ||
| 1.000 g TMDD (3331) 35% (w/w) | pH 7.0—suspension vortexing | 20.0 | 3a | 25 kHz | — | — | — |
| 0.088 g insulin | 3b | 48 kHz | 19.7 ± 0.3 | 48.98/1.023 | 48.42/1.212 | ||
| 1.000 g TMDD (3331) 35% (w/w) | pH 7.0—suspension homogenizer | 25.0 | 4a | 25 kHz | 25.3 ± 1.2 | 61.67/0.801 | 53.42/1.076 |
| 0.117 g insulin | 4b | 48 kHz | 25.1 ± 0.9 | 49.57/1.026 | 42.89/1.231 | ||
| 1.000 g TMDD (3331) 35% (w/w) | pH 7.0—suspension homogenizer | 30.0 | 5a | 25 kHz | 29.7 ± 0.5 | 60.43/0.792 | 51.32/1.103 |
| 0.150 g insulin | 5b | 48 kHz | 30.4 ± 0.9 | 50.01/1.061 | 44.23/1.273 | ||
| 1.000 g TMDD (3331) 35% (w/w) | pH 7.0—suspension homogenizer | 40.0 | 6a | 25 kHz | — | — | — |
| 0.233 g insulin | 6b | 48 kHz | 36.7 ± 1.5 | 50.21/1.031 | 44.01/1.301 | ||
| 1.000 g TMDD (3331) 25% (w/w) | pH 2.0—suspension homogenizer | 50.0 | 7a | 25 kHz | — | — | — |
| 0.250 g insulin | 7b | 48 kHz | 44.8 ± 2.2 | 76.28/1.379 | 41.09/1.643 | ||
| 1.000 g TMDD (3322) 35% (w/w) | pH 7.0—suspension homogenizer | 30.0 | 8a | 25 kHz | 30.3 ± 0.8 | 59.02/0.753 | 54.19/0.938 |
| 0.150 g insulin | 8b | 48 kHz | 30.1 ± 0.6 | 51.75/1.005 | 46.98/1.198 |
Particle size distributions after SFD showed an increase in d(v, 0.5) with an increase in insulin content. The span of the distribution increased slightly by 0.14 from 0 to 30 per cent insulin content for the 48 kHz nozzle and by 0.146 for the 25 kHz nozzle. Particle morphology of the suspension particles was similar to the insulin-free variant. The particles also showed a collapsed and wrinkled surface morphology. However, it could clearly be seen that the shrinkage of the insulin particles was not as strong as of the pure TMDD particles (figure 2d). The main reason for this is the higher overall solid content of the formulations with solid suspended insulin nanoparticles within their structure in comparison with those without. The total solid content increased from 35 per cent for formulations 1a and 1b to 47.3 per cent for formulation 6b (based on the theoretical values). The higher solid content also led to less void volume and pores after ice sublimation and therefore less influence of the pore collapse during primary drying if Tprod exceeded Tg′/TC. Furthermore, the bulk density and tap density increased with increasing content of suspended insulin and showed the highest value for formulation 8b with a value of 827 kg m−3, which resulted in a calculated ρvr value of 16.05 kg m−1 s−1 (table 5). In the same manner, the density of the particles measured with He-pycnometry decreased slightly owing to displacement of the denser TMDD matrix formulation by the less dense insulin nanoparticles. As a consequence of all of the above-described results, the porosity of the powders decreased with increasing insulin content of the particles to reach 41.1 per cent for formulation 8b. Visually, none of the suspended insulin nanoparticles could be seen on or sticking out of the surface of the reformulated particles (figure 2d).
Table 5.
Bulk density, tap density and real density of insulin formulations after SFD. Hausner ratio and porosity were calculated with the measured density values. ρvr values were calculated with a velocity of 750 m s−1. For formulation composition, refer to table 4.
| formulation | density from He-pycnometry (g cm−3) | bulk density (g cm−3) | tap density (g cm−3) | Hausner ratio (—) | porosity (%) | ρvr value (calculated from tap density) |
|---|---|---|---|---|---|---|
| 1a | 1.439 | 0.586 | 0.736 | 1.256 | 48.9 | 14.66 |
| 1b | 1.434 | 0.632 | 0.791 | 1.251 | 44.8 | 13.89 |
| 2a | — | — | — | — | — | — |
| 2b | 1.432 | 0.629 | 0.790 | 1.256 | 44.8 | 14.05 |
| 3a | — | — | — | — | — | — |
| 3b | 1.419 | 0.634 | 0.790 | 1.246 | 44.3 | 14.51 |
| 4a | 1.415 | 0.591 | 0.739 | 1.250 | 47.8 | 17.09 |
| 4b | 1.410 | 0.641 | 0.791 | 1.234 | 43.9 | 14.70 |
| 5a | 1.401 | 0.601 | 0.746 | 1.241 | 46.8 | 16.91 |
| 5b | 1.394 | 0.648 | 0.789 | 1.218 | 43.4 | 14.80 |
| 6a | — | — | — | — | — | — |
| 6b | 1.388 | 0.652 | 0.802 | 1.230 | 42.2 | 15.10 |
| 7a | — | — | — | — | — | — |
| 7b | 1.382 | 0.452 | 0.636 | 1.407 | 54.0 | 18.19 |
| 8a | 1.402 | 0.645 | 0.793 | 1.229 | 43.4 | 17.55 |
| 8b | 1.405 | 0.680 | 0.827 | 1.216 | 41.1 | 16.05 |
3.4. Physical stability of the suspension particles during needle-free ballistic injection
Particle injection into an attrition chamber with subsequent collection and size measurement using laser light diffraction showed a good physical stability for all batches from formulations 1–6 and formulation 8 (table 4). Figure 5 compares the changes in number and volume size distribution of the particles from formulation 4b before and after device actuation. It can clearly be seen that the small shift of the number distribution indicates the good physical robustness without substantial break-up of the large particles into small fragments. This is in contrast to the findings published by Ziegler (2007) showing a substantial size reduction of the d(n, 0.5) from approximately 20 to 12.5 µm after injection into an attrition chamber at 60 bar and microscopic particle size determination. In our experiments, the mechanical stability of the particles decreased slightly with increasing insulin content seen in the increasing difference between the d(v, 0.5) before and after injection as well as increasing span values. The widening of the particle size distribution was regarded as an indication of particle breakdown. Formulation 7 with the decreased TMDD solid content showed substantial break-up of the particles with a decrease in mean volume diameter from 76 to 41 µm. From these results, it can clearly be seen that the high solid content in the novel matrix formulation of 350 mg/g prior to freeze-drying is essential for providing sufficient mechanical stability and robustness of the particles (table 4). The application of the concept of shrinkage during primary drying is also beneficial towards increased physical stability of the newly developed particles as it reduces porosity (table 5).
Figure 5.

Particle size distribution of formulation 4b (TMDD (3 : 3 : 3 : 1) with 25% insulin, 48 kHz) before and after injection into an attrition chamber using the Venturi device with a 60 bar helium cartridge. Number distribution before injection (filled circle) and number distribution after injection (open circle). Volume distribution before injection (filled triangle) and volume distribution after injection (open triangle).
To test particle penetration properties, injections into tissue mimicking 3 per cent agar hydrogel phantoms were carried out with the 8 mm inline Venturi device loaded with a 60 bar helium cylinder. Agar is a polysaccharide complex extracted from the agarocytes of red algae and is composed of about 70 per cent agarose and 30 per cent agaropectin. Agarose is the neutral gelling fraction in agar consisting of a linear polymer of alternating d-galactose and 3,6-anhydrogalactose units, whereas agaropectin is the non-gelling fraction made of 1,3-glycosidically linked d-galactose units, some of which are sulphated at position 6 (Scott & Eagleson 1988). Agar provides mechanical material properties that are relatively close to human skin. Human skin typically has a bulk density between 1000 and 1100 kg m−3 (Duck 1990), and its Young's modulus has been reported to vary between 5 and 150 kPa (Sanders 1973; Diridollou et al. 2000; Hendriks et al. 2003; Boyer et al. 2007; Zahouani et al. 2009). The values strongly depend on the anatomic site, skin colour and age as well as on the method of measurement. In comparison, the density of a 3 per cent agar hydrogel, at 1020 kg m−3, is very similar to that of human skin and its Young's modulus, which has been published to be between 30 and 100 kPa, also matches well the requirements of a skin tissue-mimicking phantom (Chen et al. 2003; Nitta et al. 2003; Samani et al. 2003). Additionally, the use of agar hydrogels for testing particle penetration properties provides a number of benefits over using excised human skin: (i) the agar phantoms are easily reproducible in a known quality without changes in mechanical properties providing a system for objective comparison of penetration depth studies, (ii) the properties of the hydrogels can be varied by change of agar concentration by addition of other components such as gelatin, and (iii) the agar gels can be sliced with a regular scalpel into thin slices to enable a rapid microscopic determination of penetration depth without the necessity for staining methods. For these reasons, agar hydrogels have been the material most commonly used in research on needle-free ballistic powder injection (Condliffe et al. 2010).
After injection of the pure TMDD particles (formulation 1) into 3 per cent agar hydrogels, neither particles nor particle fragments were seen in the microscopic examination of the sliced agar targets. The particles dissolved immediately owing to the high aqueous content of the agar hydrogel (97%). However, injection channels were still visible in the agar slices under the microscope (picture not shown). Figure 6 shows the microscopic pictures of monodisperse polystyrene spheres (d = 48 µm) and insulin particles (formulation 4) injected into agar hydrogel test beds. It can be seen that the morphologies of the insulin-loaded particles differ from those seen in the SEM pictures (figure 2d). The injected particles had an elongated, disrupted and slightly porous appearance. Higher microscopic magnification showed that only particulate clusters of very small particles were still remaining in the agar. It seemed that the highly soluble matrix of TMDD was dissolved rapidly, leaving clusters of the originally suspended small and poorly soluble insulin nanoparticles behind. The mean penetration depth of the polystyrene spheres was calculated to be 312.8 ± 73.6 µm with some particles penetrating as deep as 450 µm. The mean penetration depth of particles from formulation 4 was 251.3 ± 114.7 µm. All the SFD powders used in this study were polydisperse. It can clearly be seen from figure 6b that, owing to the polydispersity of the SFD particles following a lognormal distribution, the high amount of small particles only penetrated to depths less than 250 µm. However, it was found in the microscopic studies that larger insulin-loaded particles can penetrate to depths of up to 450 µm. For injection sites such as the inside of the forearm, the capillary region of the dermis is reported to start more than 150–250 µm below the skin surface (Aulton 2007).
Figure 6.

Light microscopy pictures of (a) monodisperse polystyrene spheres (48 µm) and (b) polydisperse particles of formulation 4b (TMDD (3 : 3 : 3 : 1) with 25% insulin/48 kHz) into 3% agar hydrogel test beds. Injection was performed with a 0.250 mg powder payload in an 8 mm Venturi device with a helium gas cylinder at 60 bar. 10× magnification; scale bars, (a,b) 300 µm.
3.5. Insulin stability during SFD and after particle injection
The particle formation process in this work consisted of four main unit operations: (i) preparation of the insulin suspension, in particular homogenization of nano-sized insulin in TMDD, (ii) atomization of the resulting suspension through a 48 or 25 kHz nozzle, (iii) fast freezing of the generated spray in LN2 at –196°C, and (iv) freeze-drying of the frozen microdroplets at −10°C and 100 mtorr. The presence of insulin aggregates was examined using SEC. A-21-Desamido insulin content was monitored by reverse-phase chromatography. Table 6 presents the loss in insulin monomer and the amount of 21-desamido insulin for the insulin nanoparticle preparations as well as for the TMDD insulin formulations. Insulin showed very little aggregation and degradation during the nanoparticle formation process. The isolated spraying process into air caused more aggregation than spray-freezing with subsequent thawing or freeze-drying plus rehydration. This phenomenon has frequently been reported for other proteins during SFD (Sonner et al. 2002; Yu et al. 2006). Yu et al. (2006) showed a 21.7 per cent decrease in lysozyme activity and a 13.7 per cent increase in optical density after atomization using a two-fluid nozzle. X-ray spectroscopy (XPS) of surface composition showed an increased nitrogen content of 3.34 per cent in a lysozyme–trehalose formulation (1 : 20) in contrast to the theoretically expected 0.94 per cent for a homogeneous distribution which showed substantial surface excess of protein molecules prior to freezing and drying. Similar findings were published by Webb et al. (2002) for rhINF-γ. A loss of 12 per cent of soluble protein was consistent with the amount of protein measured by XPS on the surface of the final powder. These observations have been explained by the different time frames which surface active proteins have to access the air–water interface which is longer for the isolated spraying process than for the spray-freezing process. As peptides and proteins are often surface active, interfaces may contribute to protein aggregation (Niven et al. 1996; Webb et al. 2002). It has been reported that protein molecules absorb at the hugely increased air–water surface during atomization processes resulting in structural unfolding with subsequent aggregate formation (Adler & Lee 1999). When sprayed into the vapour over LN2, droplets start freezing as they pass through the cryogenic gas and freeze completely upon contact with the LN2 surface. This rapid cooling and freezing process first decreases and then inhibits molecular mobility, and, therefore, surface access and potential aggregation of protein molecules (Webb et al. 2002). Another explanation for the higher amount of native protein after spray-freezing given by Sonner et al. (2002) was potential recovery of protein during freezing that aggregated during atomization.
Table 6.
Influence of processing, atomization, spray-freezing and SFD on insulin aggregation and degradation determined by size exclusion and reverse-phase chromatography.
| insulin formulation | preparation | loss of insulin monomer (%) | A-21-desamido insulin (%) |
|---|---|---|---|
| bulk insulin (unprocessed) | — | — | 1.680 |
| insulin after atomization | 5 mg ml−1, unformulated at pH 2.0 | 0.101 | 1.786 |
| 5 mg ml−1, plus trehalose 5 mg ml−1 at pH 2.0 | 0.066 | 1.699 | |
| insulin after atomization, freezing and thawing | 5 mg ml−1, unformulated at pH 2.0 | 0.044 | 1.721 |
| 5 mg ml−1, plus trehalose 5 mg ml−1 at pH 2.0 | 0.031 | 1.722 | |
| insulin after SFD | 5 mg ml−1, unformulated at pH 2.0 | 0.078 | 1.795 |
| 5 mg ml−1, plus trehalose 5 mg ml−1 at pH 2.0 | 0.049 | 1.779 | |
| insulin directly after homogenization in TMDD | pure insulin nanoparticles | 0.092 | 1.799 |
| insulin–trehalose (1 : 1) nanoparticles | 0.075 | 1.781 | |
| formulation 2b | 0.104 | 1.962 | |
| formulation 3b | 0.095 | 1.917 | |
| formulation 4a | 0.114 | 1.901 | |
| formulation 4b | 0.095 | 1.768 | |
| formulation 5a | 0.098 | 1.894 | |
| formulation 5b | 0.120 | 1.823 | |
| formulation 6b | 0.123 | 1.856 | |
| formulation 7b | 0.110 | 2.080 | |
| formulation 8a | 0.094 | 1.871 | |
| formulation 8b | 0.089 | 1.798 |
The complete SFD of pure 5 mg ml−1 insulin formulations for nanoparticle manufacture showed an additional loss of insulin monomer and an increase in A-21-desamido insulin in comparison with the values obtained after isolated spray freezing (table 6). This further decrease in insulin stability resulted from the dehydration stress during primary and secondary drying. Addition of 5 mg ml−1 trehalose to the pure insulin solutions could reduce the formation of protein aggregates and degradation products during all unit operations of the SFD process. Trehalose is one of the preferred cryoprotectants and lyoprotectants in modern freeze-drying. It can stabilize the native form of proteins by preferential hydration, thus increasing their thermodynamic stability. Additionally, the dehydrated form of proteins and other biological structures are protected via water replacement and their functionality can be restored as soon as the hydration and temperature conditions return to normal (Gil et al. 1996; Arakawa et al. 2001; Lins et al. 2004). Among the various osmolytes in nature used to overcome deleterious environmental effects, one of the main benefits of trehalose is that it does not have internal hydrogen bonds, therefore allowing a more flexible formation of hydrogen bonds with the biomolecules.
The insulin-loaded TMDD formulations showed an additional decrease in insulin monomer between 0.011 and 0.045 per cent which was due to the homogenization conditions as well as the SFD for microparticle formation (table 6). Formulation 6 had the highest final value of 0.123 per cent monomer loss. The amount of A-21-desamido insulin increased slightly from 1.680 per cent of the unprocessed insulin to a maximum of 2.080 per cent during the entire particle manufacture process. Formulation 7 showed the highest value of A-21-desamido insulin. This was most likely due to the formulation of the particles at pH 2.0, which promotes protein deamidation (Aulton 2007). It can be seen from all experiments that the formulation excipients trehalose, mannitol and dextran improved stability and prevented an extensive insulin aggregation or degradation. However, most of the insulin in the final SFD formulations for the manufacture of the ballistic particles is not in solution, but suspended in the TMDD matrix, in particular in formulations with a pH of 7.0. Denaturation energies are higher for protein molecules in the solid state than in liquid solution (Lee 2002). Therefore, SFD stability of the insulin suspension is expected to be improved in comparison with insulin solutions.
FTIR spectroscopy used to examine the protein secondary structure during processing did not show any substantial changes of the amide I band after SFD and subsequent rehydration (figure 7). A slight decrease in α-helix content by 1.52 per cent with an increase in the intermolecular β-sheet by 0.40 per cent could be detected in formulation 6b, which showed the highest amount of damage in the HPLC studies. The intramolecular β-sheet content stayed constant. Using spray-freezing into LN2 (SFL) instead of a conventional SFD process, Yu et al. (2002) could only determine an increase in insulin dimer in excipient-free insulin powders. Surprisingly, their formulated SFL insulin preparations showed equal or even lower dimer contents than the unprocessed bulk substance. Their amount of A-21-desamido insulin in their final SFL powders stayed constant at 3.04 per cent ± 0.01 with an increase to 3.72 per cent for an insulin/tyloxapol/lactose formulation.
Figure 7.

FTIR spectrum (amide I band) of a 5 mg ml−1 insulin solution at pH 2.0. (a) Second derivative spectrum. (b) Amide I band with Gaussian peak fitting. Peak positions were determined by second derivatives (upper spectrum) and assigned to secondary structures: α-helix 1656 cm−1, intramolecular β-sheet 1636 cm−1, intermolecular β-sheet 1614 cm−1, other 1678 and 1689 cm−1. Percentile structural contents were then quantified by peak fitting (lower spectrum) assuming a Gaussian peak shape.
4. Conclusions
A new SFD technique for the manufacture of particles suitable for needle-free ballistic delivery with a high load of poorly soluble protein drugs has been described. The manufactured microparticles consist of a water-soluble sugar-polymer matrix with suspended nanoparticulate drug inside. The unit operations involved in the manufacturing process have been examined in detail in regard to their impact on physical particle characteristics, such as size, density and particle break-up during injection, as well as biological protein stability.
The ultrasound atomizers with an operational frequency of 25 kHz and 48 kHz showed a superior behaviour over two-fluid nozzles in regard to (i) the manufacture of droplet sprays with a d(v, 0.5) larger than 40 µm at low liquid flow rates between 0.25 and 0.50 ml min−1, (ii) the handling of small liquid volumes below 2.0 ml, and (iii) their suitability for laboratory-scale SFD. The ultrasound nozzles produced a ‘soft spray’ with low droplet velocities that did not cause violent boiling of the LN2 unlike that observed with two-fluid nozzles. Insulin showed a good overall stability during ultrasound atomization with a loss of only 0.101 per cent of the insulin monomer and an increase in A-21-desamido insulin by 0.106 per cent. Addition of trehalose as a formulation and stabilization excipient reduced these values to 0.066 and 0.019 per cent, respectively.
SFD of 5 mg ml−1 insulin and 10 mg ml−1 insulin–trehalose solutions resulted in highly porous powders that were broken up into nanoparticles by subsequent homogenization with an Ultra-Turrax tool. The received nanoparticles had a mean d(v, 0.5) of 238 and 285 nm with low tapped powder densities of 4.36 and 4.63 kg m−3, respectively. Both powders were not suitable for needle-free injection and showed no penetration into agar hydrogel targets. However, particles prepared by SFD of a concentrated 350 mg/g solution of trehalose, mannitol and two dextrans with 10 and 150 kDa molecular weight (ratio 3 : 3 : 3 : 1) resulted in distinct microparticles. Application of a primary drying temperature of −10°C induced a viscous flow of the TMDD matrix during drying with pore collapse, particle shrinkage and densification of the particles in comparison with primary drying at a shelf temperature of −30°C. Measurement of Tg′ of the formulation and product temperature during freeze-drying showed that Tprod exceeded Tg′ for almost all of the primary drying phase. No particle fusion due to the viscous flow could be observed for any of the formulations. As a result, tap density increased by 129 to 791 kg m−3 with a decrease in powder porosity by 8.9 per cent (calculated from the tap density values).
Microparticles with high insulin loading of up to 36.7 per cent insulin (for a 350 mg/g TMDD matrix formulation) and of up to 44.8 per cent (for a 250 mg/g TMDD matrix formulation) could be achieved by SFD of the matrix formulation with suspended insulin nanoparticles. However, changes in the consistency and viscosity of the suspension formulation with increasing insulin content led to deviation between the theoretical insulin content and the actual value determined after SFD. Up to an insulin loading of 30 per cent, a close correlation between theoretical and actual amount could be achieved. Above this value, the suspension had a paste-like consistency. The SEM pictures of the final product showed little difference from particles prepared from completely dissolved placebo formulations without insulin. Insulin-loaded particles showed a larger particle size and density due to increases in formulation viscosity and overall solid content. The largest ρvr values were observed for formulation 7b with 44.8 per cent insulin content due to its large particle size. However, formulation 7b showed a wide size distribution with a span of 1.379 and substantial particle break-up after particle injection of the device into the attrition chamber (table 3). Therefore, formulation 8b with 30 per cent insulin content proved to be the best formulation prepared with a 48 kHz nozzle from a 350 mg/g TMDD (3 : 3 : 2 : 2) solution. The values were 51.75 µm for the d(v, 0.5) and 827 kg m−3 for the tap density, resulting in a ρvr of 16.05 kg m−1 s−1. In needle-free injection and penetration tests, the particles showed little particle break-up with a decrease of the d(v, 0.5) by only 4.83 µm and penetration into agar hydrogels to mean depths of 251.3 µm, and therefore sufficiently deep enough to overcome the stratum corneum and enter the viable dermis.
The complete SFD of the insulin nanoparticle suspension formulations resulted in only very little protein damage. The largest value for the loss in insulin monomer that was observed was measured by SEC to 0.123 per cent. The largest detected amount of 21-desamido insulin was 2.080 per cent, determined by reverse-phase chromatography. However, the unprocessed bulk insulin (as supplied) already showed 1.680 per cent.
The particle manufacturing technique described here provides the possibility of manufacturing microparticles with a stable, poorly soluble protein drug in a high concentration. As protein molecules in the solid state are thermodynamically more stable than dissolved ones in liquid solution, SFD from protein nanosuspensions could be a promising particle formulation procedure for other poorly soluble and labile active pharmaceutical ingredients in the future. This will be of particular interest if the overall dose that can be administered with a given medical device is limited to low milligram or even microgram quantities. In this work, the application was focused on needle-free ballistic particle injection, where the device used was limited to a powder dose of 1–2 mg, but other areas such as nasal delivery or pulmonary delivery (if smaller microparticles are manufactured) could be of interest, too. Future drug systems could include, for example, glucagon, also a poorly soluble protein injected into the viable dermis or subcutaneous tissue at a dose of 1 mg, for the treatment of severe hypoglycaemia.
Acknowledgements
The authors would like to thank Particle Therapeutics Ltd for supporting this research with their device expertise and for providing the needle-free injection devices used in this research.
Footnotes
One contribution to a Theme Supplement ‘Scaling the heights—challenges in medical materials: an issue in honour of William Bonfield, Part I. Particles and drug delivery’.
References
- Adler M., Lee G. 1999. Stability and surface activity of lactate dehydrogenase in spray-dried trehalose. J. Pharm. Sci. 88, 188–208. [DOI] [PubMed] [Google Scholar]
- Arakawa T., Prestrelskia S. J., Kenneya W. C., Carpenter J. F. 2001. Factors affecting short-term and long-term stabilities of proteins. Adv. Drug Deliv. Rev. 46, 307–326. ( 10.1016/S0169-409X(00)00144-7) [DOI] [PubMed] [Google Scholar]
- Aulton M. E. 2007. Pharmaceutics. The design and manufacture of medicines, pp. 565–597, 3rd edn London, UK: Churchill Livingston. [Google Scholar]
- Berger H. L. 2006. Ultrasonic liquid atomization: theory and application, pp. 1–177, 2nd edn Hyde Park, NY: Partrige Hill. [Google Scholar]
- Bouchard M., Zurdo J., Nettleton E. J., Dobson C. M., Robinson C. V. 2000. Formation of insulin amyloid fibrils followed by FTIR simultaneously with CD and electron microscopy. Protein Sci. 9, 1960–1967. ( 10.1110/ps.9.10.1960) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyer G., Zahouani H., Le Bot A., Laquieze L. 2007. In vivo characterization of viscoelastic properties of human skin using dynamic micro-indentation. In Proc. Engineering in Medicine and Biology Society, EMBS 2007, 29th Annual Int. Conf. of the IEEE, pp. 4584–4587. [DOI] [PubMed] [Google Scholar]
- Burkoth T. L., Bellhouse B. J., Hewson G., Muddle A. G., Sarphie D. F. 1999. Transdermal and transmucosal powdered drug delivery. Crit. Rev. Ther. Drug Deliv. Syst. 16, 331–384. [DOI] [PubMed] [Google Scholar]
- Cavatur R., Vemuri N., Pyne A., Chrzan Z., Toledo-Velasquez D., Suryanarayanan R. 2002. Crystallization behavior of mannitol in frozen aqueous solution. Pharm. Res. 19, 894–900. ( 10.1023/A:1016177404647) [DOI] [PubMed] [Google Scholar]
- Chen D., Weis K. F., Chu Q., Erickson C., Endres R., Lively C. R., Osorio J., Payne L. G. 2001. Epidermal powder immunization induces both cytotoxic T-lymphocyte and antibody responses to protein antigens of influenza and hepatitis B viruses. J. Virol. 75, 11 630–11 640. ( 10.1128/JVI.75.23.11630-11640.2001) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Q., Suki B., An N. 2003. Dynamic mechanical properties of agarose gel by a fractional derivative model. In Proc. Summer Bioengineering Conf., Sonesta Beach Resort, Key Biscayne, Florida, USA, 25–29 June. [Google Scholar]
- Chen D., Burger M., Chu Q., Endres R., Zuleger C., Dean H., Payne L. G. 2004. Epidermal powder immunization: cellular and molecular mechanisms for enhancing vaccine immunogenicity. Virus Res. 1103, 147–153. [DOI] [PubMed] [Google Scholar]
- Condliffe J., Schiffter H. A., Cleveland R. O., Coussios C. C. 2010. An acoustic microscopy technique to assess particle size and distribution following needle-free injection. J. Acoust. Soc. Am. 127, 2252–2261. ( 10.1121/1.3314252) [DOI] [PubMed] [Google Scholar]
- Costantino H. R., et al. 2000. Protein spray-freeze drying. Effect of atomization conditions on particle size and stability. Pharm. Res. 17, 1374–1383. ( 10.1023/A:1007570030368) [DOI] [PubMed] [Google Scholar]
- Costantino H. R., Firouzabadian L., Hogeland K., Wu C., Carrasquillo K. G., Griebenow K., Zale S. E., Tracy M. A. 2002. Protein spray freeze drying. 2. Effect of formulation variables on particle size and stability. J. Pharm. Sci. 91, 388–395. ( 10.1002/jps.10059) [DOI] [PubMed] [Google Scholar]
- Costigan G., Liu Y., Brown G. L., Carter F. V., Bellhouse B. J. 2004. Evolution of the design of Venturi devices for the delivery of dry particles to skin or mucosal tissue. In Proc. 24th Int. Symp. Shock Waves, Beijing, PR China. [Google Scholar]
- Dean H. J., Chen D. 2004. Epidermal powder immunization against influenza. Vaccine 23, 681–686. ( 10.1016/j.vaccine.2004.06.041) [DOI] [PubMed] [Google Scholar]
- Dean H. J., Fuller D., Osorio J. E. 2003. Powder and particle-mediated approaches for delivery of DNA and protein vaccines into the epidermis. Comp. Immunol. Microbiol. Infect. Dis. 26, 337–388. [DOI] [PubMed] [Google Scholar]
- Diridollou S., Patat F., Gens F., Vaillant L., Black D., Lagarde J. M., Gall Y., Berson M. 2000. In vivo model of the mechanical properties of the human skin under suction. Skin Res. Technol. 6, 214–221. ( 10.1034/j.1600-0846.2000.006004214.x) [DOI] [PubMed] [Google Scholar]
- Duck F. A. 1990. Physical properties of tissue. A comprehensive reference book. New York, NY: Academic Press. [Google Scholar]
- Gil A. M., Belton P. S., Felix V. 1996. Spectroscopic studies of solid alpha–alpha trehalose. Spectrochim. Acta Part A 52, 1649–1659. ( 10.1016/0584-8539(96)01728-X) [DOI] [Google Scholar]
- Gombotz W., Healy M., Brown L. 1991. Very low temperature casting of controlled release microspheres. US Patent 5019400.
- Hancock B., Zografi G. 1997. Characteristics and significance of the amorphous state in pharmaceutical systems. J. Pharm. Sci. 86, 1–12. ( 10.1021/js9601896) [DOI] [PubMed] [Google Scholar]
- Hendriks F. M., Brokken D., Van Eemeren J., Oomens C. W. J., Baaijens F. P. T., Horsten J. B. A. M. 2003. A numerical–experimental method to characterize the non linear mechanical behaviour of human skin. Skin Res. Technol. 9, 274–283. ( 10.1034/j.1600-0846.2003.00019.x) [DOI] [PubMed] [Google Scholar]
- Johnson O. L., et al. 1997. The stabilization of human growth hormone into biodegradable microspheres. Pharm. Res. 14, 730–735. ( 10.1023/A:1012142204132) [DOI] [PubMed] [Google Scholar]
- Joshi A., Huh H. 2004. Evaluation of small-scale titration techniques for protein solubility characterization. In Proc. AAPS Annual Meeting, Baltimore, MD, USA [Google Scholar]
- Kendall M., Wrighton-Smith P., Bellhouse B. 2000. Transdermal ballistic delivery of microparticles: investigation into skin penetration. In Proc. World Congress of Medical Physics and Biomedical Engineering, Chicago. Piscataway, NJ: IEEE. [Google Scholar]
- Kendall M., Mitchell T., Wrighton-Smith P. 2004. Intradermal ballistic delivery of microparticles into excised human skin for pharmaceutical applications. J. Biomech. 37, 1733–1741. ( 10.1016/j.jbiomech.2004.01.032) [DOI] [PubMed] [Google Scholar]
- Lee G. 2002. Spray-drying of proteins. Pharm. Biotechnol. 13, 135–158. [DOI] [PubMed] [Google Scholar]
- Lefebvre A. H. 1989. Atomization and sprays, pp. 27–78. New York, NY: Hemisphere Publishing Corporation. [Google Scholar]
- Leuenberger H. 2002. Spray freeze-drying—the process of choice for low water soluble drugs. J. Nanoparticle Res. 4, 111–119. ( 10.1023/A:1020135603052) [DOI] [Google Scholar]
- Levi G., Karel M. 1995. Volumetric shrinkage (collapse) in freeze-dried carbohydrates above their glass transition temperature. Food Res. Int. 28, 145–151. ( 10.1016/0963-9969(95)90798-F) [DOI] [Google Scholar]
- Lins R. D., Pereira S. C., Hunenberger P. H. 2004. Trehalose protein interaction in aqueous solution. Proteins Struct. Funct. Bioinformatics 55, 177–186. ( 10.1002/prot.10632) [DOI] [PubMed] [Google Scholar]
- Liu Y. 2007. Utilization of the venturi effect to introduce micro-particles for epidermal vaccination. Med. Eng. Phys. 29, 390–397. ( 10.1016/j.medengphy.2006.05.015) [DOI] [PubMed] [Google Scholar]
- Liu Y., Costigan G., Bellhouse B. J. 2008. Swirling effects on the performance of the microparticle acceleration and penetration: parametric studies. Powder Technol. 183, 189–195. ( 10.1016/j.powtec.2007.07.025) [DOI] [Google Scholar]
- Maa Y. F., Ameri M., Shu C., Payne L. G., Chen D. 2004. Influenza vaccine powder formulation development: spray-freeze-drying and stability evaluation. J. Pharm. Sci. 93, 1913–1922. [DOI] [PubMed] [Google Scholar]
- Masters K. 1991. Spray drying handbook. Harlow, UK: Longman Scientific & Technical. [Google Scholar]
- Maury M., Murphy K., Kumar S., Shi L., Lee G. 2005. Effects of process variables on the powder yield of spray-dried trehalose on a laboratory spray-dryer. Eur. J. Pharm. Biopharm. 59, 565–573. ( 10.1016/j.ejpb.2004.10.002) [DOI] [PubMed] [Google Scholar]
- Nitta T., Endo Y., Haga H., Kawabata K. 2003. Microdomain structure of agar gels observed by mechanical-scanning probe microscopy. J. Electron Microsc. 52, 277–281. ( 10.1093/jmicro/52.3.277) [DOI] [PubMed] [Google Scholar]
- Niven R. W., Prestrelski S. J., Treuheit M. J., Ip A. Y., Arakawa T. 1996. Protein nebulization. Part 2. Stabilization of G-CSF to air-jet nebulization and the role of protectants. Int. J. Pharm. 127, 191–201. ( 10.1016/0378-5173(95)04209-1) [DOI] [Google Scholar]
- Reitz D. R., Bracco F. V. 1986. Mechanisms of breakup of round liquid jets. In Encyclopedia of fluid mechanics (ed. Cheremisinoff N. P.), pp. 233–249. Houston, TX: Gulf Publishing Co. [Google Scholar]
- Rochelle C., Lee G. 2007. Dextran or hydroxyethyl starch in spray-freeze-dried trehalose/mannitol microparticles intended as ballistic particulate carriers for proteins. J. Pharm. Sci. 96, 2296–2309. ( 10.1002/jps.20861) [DOI] [PubMed] [Google Scholar]
- Samani A., Bishop J., Luginbuhl C., Plewes D. B. 2003. Measuring the elastic modulus of ex vivo small tissue samples. Phys. Med. Biol. 48, 2183–2198. ( 10.1088/0031-9155/48/14/310) [DOI] [PubMed] [Google Scholar]
- Sanders R. 1973. Torsional eleasticity of human skin in vivo. Pflügers Arch. 342, 255–260. ( 10.1007/BF00591373) [DOI] [PubMed] [Google Scholar]
- Schiffter H. 2007. Spray-freeze-drying in the manufacture of pharmaceuticals. Eur. Pharm. Rev. 12, 67–71. [Google Scholar]
- Scott T., Eagleson M. 1988. Concise encyclopedia: biochemistry, p. 18, 2nd edn New York, NY: Walter de Gruyter. [Google Scholar]
- Sonner C., Maa Y. F., Lee G. 2002. Spray freeze drying for protein powder preparation: particle characterization and a case study with trypsinogen stability. J. Pharm. Sci. 91, 2122–2139. ( 10.1002/jps.10204) [DOI] [PubMed] [Google Scholar]
- Tang X., Pikal M. 2004. Design of freeze drying processes for pharmaceuticals: practical advice. Pharm. Res. 21, 191–200. ( 10.1023/B:PHAM.0000016234.73023.75) [DOI] [PubMed] [Google Scholar]
- Webb S. D., Golledge S. T., Cleland J. L., Carpenter J. F., Randolph T. W. 2002. Surface adsorption of recombinant human interferon-γ in lyophilized and spray-lyophilized formulations. J. Pharm. Sci. 91, 1474–1487. ( 10.1002/jps.10135) [DOI] [PubMed] [Google Scholar]
- Yu Z., Rogers T. L., Hu J., Johnston K. P., Williams R. O., III 2002. Preparation and characterization of microparticles containing peptide produced by a novel process: spray freezing into liquid. Eur. J. Pharm. Biopharm. 54, 221–228. ( 10.1016/S0939-6411(02)00050-4) [DOI] [PubMed] [Google Scholar]
- Yu Z., Garcia A. S., Johnston K. P., Williams R. O., III 2004. Spray freezing into liquid nitrogen for highly stable protein nanostructured microparticles. Eur. J. Pharm. Biopharm. 58, 529–537. ( 10.1016/j.ejpb.2004.04.018) [DOI] [PubMed] [Google Scholar]
- Yu Z., Johnston K. P., Williams R. O. 2006. Spray freezing into liquid versus spray-freeze drying: influence of atomization on protein aggregation and biological activity. Eur. J. Pharm. Sci. 27, 9–18. ( 10.1016/j.ejps.2005.08.010) [DOI] [PubMed] [Google Scholar]
- Zahouani H., Pailler-Mattei C., Sohm B., Vargiolu R., Cenizo V., Debret R. 2009. Characterization of the mechanical properties of a dermal equivalent compared with human skin in vivo by indentation and static friction tests. Skin Res. Technol. 15, 68–76. ( 10.1111/j.1600-0846.2008.00329.x) [DOI] [PubMed] [Google Scholar]
- Ziegler A. 2007. Needle-free injection: attrition forces acting on spray-dried and spray-freeze-dried protein-loaded particles with needle-free injectors. In Monographs in pharmaceutics (ed. Lee G.), pp. 147–151. Aachen, Germany: Shaker Publishing. [Google Scholar]
- Ziegler A. 2008. Needle-free delivery of powdered protein vaccines: a new and rapidly developing technique. J. Pharm. Innov. 3, 204–213. ( 10.1007/s12247-008-9039-x) [DOI] [Google Scholar]