

Abstract
Purpose
Current evidence indicates that an osteoblast lesion in prostate cancer is preceded by osteolysis. Thus, prevention of osteolysis would reduce complications of bone metastasis. Bone marrow-derived mesenchymal stem cells (MSC) have the ability to differentiate into osteoblast, and produce osteoprotegerin (OPG), a decoy receptor for the receptor activator for nuclear factor κ B ligand (RANKL), naturally. The present study examined the potential of unmodified MSC to prevent osteolytic bone lesions in a preclinical mouse model of prostate cancer.
Experimental design
The human prostate cancer cell line PC3 was implanted in tibiae of SCID mice. After establishment of the tumor, either unmodified or genetically-engineered MSC overexpressing OPG was injected at the site of tumor growth. The effects of therapy were monitored by bioluminescence imaging, micro-CT, immunohistochemistry and histomorphometry.
Results
Data indicated significant (P<0.001) inhibition of tumor growth and restoration of bone in mice treated with both unmodified and modified MSC. Detailed analysis suggested that the donor MSC inhibited tumor progression by producing woven bone around the growing tumor cells in the tibiae and by preventing osteoclastogenesis.
Conclusions
Overcoming the limitation of the number of MSC available in the bone can provide significant amelioration for osteolytic damage without further modification.
Statement of Translational Relevance.
Osteolytic bone damage is major cause of morbidity in several cancers. Due to the refractory nature of metastatic tumors in the bone, conventional chemotherapy combinations and radiation therapy fail to provide long-term cure. Further, even the modes effects offered by these therapies fail to restore bone destruction. Thus, new therapies are needed for the management of osteolytic bone damage in cancer patients. Results of the present study demonstrate that without any genetic modification, adult mesenchymal stem cells (MSC) are capable of inducing bone formation in response to cancer-associated bone loss. Since MSC are immune privileged, and their use in allogenic context has entered human clinical trials, results of this preclinical study is greatly poised as a potential alternative for cancer-induced bone damage not only for prostate cancer bone metastasis, which initiates with osteolytic events but also other cancers such carcinomas of the breast, thyroid, lung, kidney and myeloma.
INTRODUCTION
Prostate cancer is the second leading cause of cancer deaths in men behind lung cancer in the United States and metastasizes to bone in more than 70% of the cases during advance stages (1). Bone metastasis causes severe bone pain, pathological fractures and shortens life span by significant amount. Majority of the bone metastatic cancers (breast, lung, thyroid and kidney) generates osteolytic lesions whereas prostate cancer generates osteoblastic phenotype with an overall increase in bone volume (2–4). However, the appearance of osteoblastic lesions is preceded and/or accompanied by an osteolytic event, which is required for the establishment and growth of prostate cancer cells in the bone microenvironment (4, 5). The binding of receptor activator of nuclear factor-κB ligand (RANKL) to RANK on preosteoclasts or osteoclasts is essential for their maturation and activity (6, 7). Increased expression of RANKL has been observed in osteolytic malignancies and inhibition of osteoclastogenesis or metastasis has been considered as an intervention strategy. Osteoprotegerin (OPG) is a soluble decoy receptor for RANKL and prevents binding of RANKL to RANK, leading to inhibition of osteoclast activity and bone metastasis (8–10). OPG therefore promises tremendous hope for potential clinical use in the management of osteolytic bone metastasis. Systemic delivery of OPG has shown promise as a potential therapy in animal models, limiting hypercalcemia and osteolysis induced by myeloma, breast, lung or prostate cancer and reducing tumor establishment in bone (11–17).
Homing of adult bone marrow derived mesenchymal stem cells (MSC) to the sites of tumor growth is well known besides their ability to self-renew and differentiate into bone, cartilage, fat and of other tissue types (18). Systemic administration of MSC in mice has been shown to engraft within the tumor microenvironment in many cancers and thus represent an attractive cellular vehicle for cell therapy and gene therapy (19, 20). Since OPG is constitutively produced by MSC, we speculated that lack of MSC in sufficient quantities in the bone microenvironment is the reason for the inability to inhibit excess osteoclastogenesis and compensate for bone loss, and overcoming this limitation would provide therapy for osteolytic bone damage. The present study determined the potential of MSC that were unmodified as compared to that genetically engineered to over-express OPG in bone remodeling following osteolytic damage. Results indicated that naïve MSC inhibited tumor growth, comparable to MSC over-expressing OPG by formation of new bone around the tumor cells and by inhibiting osteoclast activation.
MATERIALS AND METHODS
Cell lines and reagents
Osteolytic prostate cancer cell line PC3 expressing firefly luciferase was a generous gift from Dr. Kenneth J. Pienta (University of Michigan, Ann Arbor, Michigan) and maintained in RPMI-1640 medium (Mediatech Inc. Hendron, VA) supplemented with 10% fetal bovine serum (Mediatech Inc.) and penicillin/streptomycin (Mediatech Inc). Osteoblastic prostate cancer cell line C4-2B was a generous gift from Dr. Marco G. Cecchini, Department of Urology and Department of Clinical Research, University of Bern, Switzerland and maintained in T-medium (Invitrogen, Carlsbad, CA) supplemented with 10% FBS and antibiotics. RAW 264.7 cells were obtained from the ATCC (Manassas, VA) and maintained in α-MEM supplemented with 10% FBS, 4 mM L-glutamine and antibiotics. MSC were maintained in Stem Line medium (Sigma-Aldrich Corp. St. Louis, MO), supplemented with 10% FBS, 4mM L-Glutamine and penicillin/streptomycin. HEK293 cells were purchased from ATCC and maintained in DMEM supplemented with 10% new born calf serum and penicillin/streptomycin. Isolation, purification and differentiation of mouse MSC from C57BL/6 mice were carried out as described recently (21). Antibodies for cytokeratin-18 and GFP were purchased from Abcam Ltd. (Cambridge, MA). Secondary immunodetection was performed using anti-rat and/or anti-rabbit ABC kits purchased from Vector Laboratories (Burlingame, CA). Total RNA was isolated using Trizol (Invitrogen, Carlsbad, CA) and purified using a Qiagen mini kit (Valencia, CA). iScript cDNA synthesis kit was purchased from Bio-Rad (Hercules, CA). Primers for RT-PCR analysis were designed using the Primer 3 (version 4.0) software and oligonucleotides were purchased from Integrated DNA Technologies, Inc. (Coralville, IA). cDNA samples were analyzed in Bio-Rad iCycler (Hercules, CA). Three-dimensional PC3 beads were prepared and supplied by Vivo Biosciences (Birmingham, AL). Proliferation of PC3 cells were determined by Vybrant ® MTT Cell Proliferation Assay Kit (Molecular Probes, Inc. Eugene, OR) as recommended by the manufacturer. Lentivirus encoding shRNA constructs for silencing OPG were designed and supplied by Sigma-Aldrich Corp (St. Louis, MO). Alkaline phosphatase enzyme activity was measured using a commercial kit (Sigma-Aldrich Corp. St. Louis, MO) following manufacturer’s instructions. Von Kossa staining was performed to detect calcium deposition following standard protocols (22).
Construction of recombinant plasmid and expression analysis
The recombinant OPG used in this study comprised the ligand-binding domain of human OPG (1–201 amino acids) fused to the Fc domain of human IgG. The OPG.Fc fusion gene was isolated from an adenoviral construct (kindly provided by Dr. Joanne Douglas, University of Alabama at Birmingham, Birmingham, AL) and subcloned into AAV plasmid under the control of CMV/chicken β-actin promoter. Expression of OPG.Fc as a secreted protein from the AAV plasmid was confirmed by transient transfection into HEK293 cells using lipofectamine-2000 reagent (Invitrogen Inc. Carlsbad, CA) and testing the supernatants on SDS-PAGE using a mouse monoclonal antibody against human OPG (Chemicon Inc., Temecula, CA).
Osteoclast assay
MSC were cultured for 2 days at confluency when conditioned media was collected, centrifuged at 4000 rpm to pellet any floating cells, and supernatant stored at −80°C. RAW 264.7 cells were cultured in 6-well culture dishes with 25 ng/ml of RANKL (SIGMA-Aldrich, St. Louis, MO) either in regular medium or MSC conditioned medium for 8 days. The culture medium was replaced every alternate day. After 8 days, cells were stained for tartrate resistant acid phosphatase (TRAP) to determine the effect of MSC conditioned media on osteoclast formation utilizing a leukocyte acid phosphatase kit (SIGMA-Aldrich, St Louis, MO).
Development of osteolytic bone metastasis model in the mouse
Six-week-old male SCID mice were purchased from the National Cancer Institute-Frederick Animal Production Facility (Frederick, MD). Maintenance of the animals was carried out following guidelines of the Institutional Animal Care and Use Committee (IACUC) and all experimental procedures were approved by the IACUC and the Occupational Health and Safety Department of the University of Alabama at Birmingham. The mice were acclimatized for a week following which 105 osteolytic PC3 prostate cancer cells, constitutively expressing luciferase, were implanted in the tibia of right leg (n=30) in 20μl PBS. The left tibia served as control and was injected with only PBS. A 3/10 cc (28 gauge) insulin syringe (BD Biosciences, Franklinlake, NJ) was used for the intra-tibial injection of the prostate cancer cells under isoflurane anesthesia.
Bioluminescence imaging
In vivo bioluminescence imaging was conducted in a cryogenically cooled IVIS-100 system (Xenogen Corp., Alameda, CA, USA) to detect luciferase expression using living imaging acquisition and analysis software (Xenogen Corp.) as described (11). For each animal, bioluminescence imaging were performed before, and 4 weeks after the initiation of MSC treatment. The intensity of light emission was represented with a pseudo color scaling of bioluminescent images. The bioluminescent images were overlaid on black and white photographs of the mice collected at the same time. Bioluminescence units were converted as counts/second for each animal and final counts were divided by the initial counts and plotted graphically as a measure of tumor growth.
Micro-CT analysis
Superficial CT scanning of whole skeleton was performed on live animals using MicroCAT II (Imtek, Inc. Knoxville, TN). For determination of the 3-D architecture of the trabecular and cortical bones, mice were sacrificed and tibia were harvested and analyzed in an advanced μCT instrument (μCT 40, Scanco Medical AG, Bassersdorf, Switzerland). Two scans were performed on each tibia, one for whole tibial bone with 16 μm resolution and one for trabecular analysis with a 6 μm resolution. For the whole tibia the scan was composed of 1129 slices with a threshold value of 265. A 3-dimensional reconstruction of the images was performed with the region of interest (ROI) consisting of both trabecular and cortical areas. The scan of the trabecular bone was performed below the growth plate. Each scan consisted of 209 slices of which 100 were used for the analysis. ROIs were drawn on each of the 100 slices just inside the cortical bone, to include only the trabecular bone and the marrow. Trabecular bone was set to a threshold at 327 to distinguish it from the marrow. The 3-D reconstruction was performed on the ROI which only contained trabecular bone; no cortical bone was present in these ROIs.
Bone histomorphometry
Soft tissues were fixed in 10% neutral buffered-formalin solution for 48 hours before embedding in paraffin for histological analysis. Bone tissues were decalcified in 0.5 mol/L EDTA in Ca2+- and Mg2+-free Dulbecco’s PBS (Cellgro) prior to embedding in paraffin. Six μm longitudinal serial sections were cut from the femur and tibia and stained with hematoxylin and eosin (H&E) or Goldner’s trichrome stain to determine the characteristics of tumor growth in the bone and the extent of osteolysis in response to different treatments. TRAP staining was performed on bone sections to determine osteoclast activity. Quantitative osteomeasurements of bones were performed using an Olympus BX51 microscope and Bioqaunt Image analysis Software (R&M Biometrics, Nashville, TN).
Biomechanical testing
Mice were sacrificed 4 weeks after the MSC treatments, and tibiae were collected and fresh-frozen. Specimens were tested to failure by three-point bending on 858 MiniBionix Materials Testing System (MTS Systems). Stiffness, peak load were calculated from the force displacement data.
Immunohistochemistry
Briefly, 6 μM paraffin sections of tibiae were deparaffinized in xylene, and hydrated through graded-alcohol. Antigen retrieval was performed in citrate buffer, pH 6.0, under steam for 20 min. Sections were cooled to room temperature and endogenous peroxidase was removed using 0.3% H2O2 in methanol for 30 min and blocked with 3% goat serum for 30 min. Tissue sections were then incubated with primary antibodies overnight at 4°C. Sections were washed in PBST and again incubated at room temperature with biotin-conjugated goat anti-rabbit/anti-rat secondary antibody for 2 hrs. After washing, sections were incubated with streptavidin-conjugated horseradish peroxidase for 1 hr at room temperature. After another wash with PBST, immunodetection was performed using DAB-H2O2 (Vector Labs, Burlingame, CA) and counterstained with hematoxylin.
OPG ELISA
For determination of OPG levels, secreted by the PC3 cells, MSC and MSC-OPG, cells were cultured separately for 72 hours and cell numbers were counted and 100 μl of culture media was subjected to ELISA (ALPCO Diagnostics, Wien, Germany) following manufacturer’s instructions. Each sample was analyzed in triplicate and the absorbance was measured in a micro-plate reader (BioTek Instruments Inc., Winooski, Vermont).
Statistical Analysis
Data were analyzed by one-way analysis of variance (ANOVA). A Tukey test was also applied for multiple comparisons wherever applicable. Values provided are the Mean ± SEM and the differences were considered significant if p<0.05.
RESULTS
Production of OPG by unmodified MSC and inhibition of osteoclastogenesis
Total RNA was isolated from mouse MSC and converted to cDNA and subjected to real-time PCR analysis. The result indicated expression of OPG mRNA by the MSC (Figure 1A). OPG immunostaining was also performed on cultured MSC, which clearly indicated production of OPG by the MSC (Figure 1B). Ability of MSC to inhibit osteoclastogenesis was tested in vitro by culturing pre-osteoclast RAW 264.7 cells in regular medium or MSC-conditioned medium in the presence of RANKL for seven days. TRAP staining indicated significant number of osteoclasts in RAW cells cultured in regular medium, comparable to RAW cells grown in MSC conditioned medium (Figure 1C). Levels of OPG produced in culture as secreted protein by PC3 cells, MSC and MSC genetically engineered to over-express OPG are given in Supplementary Figure 4.
Figure 1. Expression of OPG by MSC and its effects on osteoclast formation in vitro.
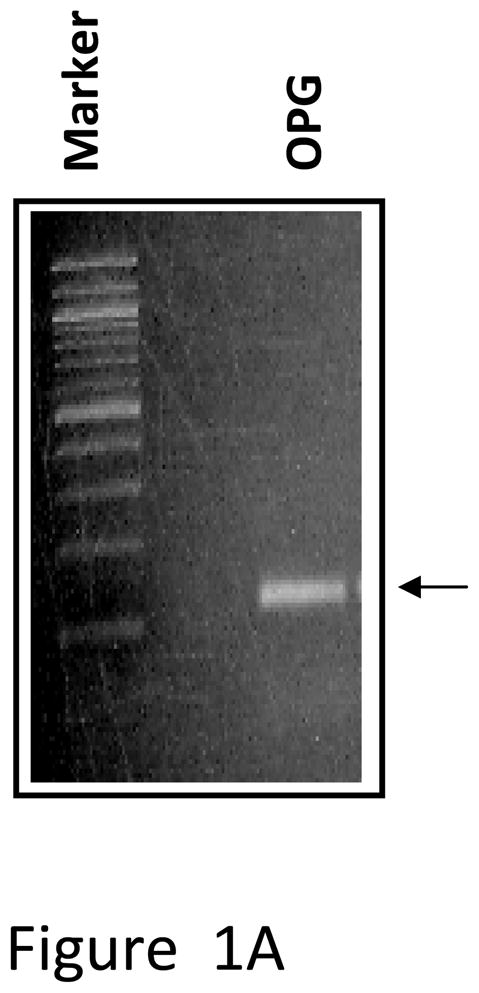
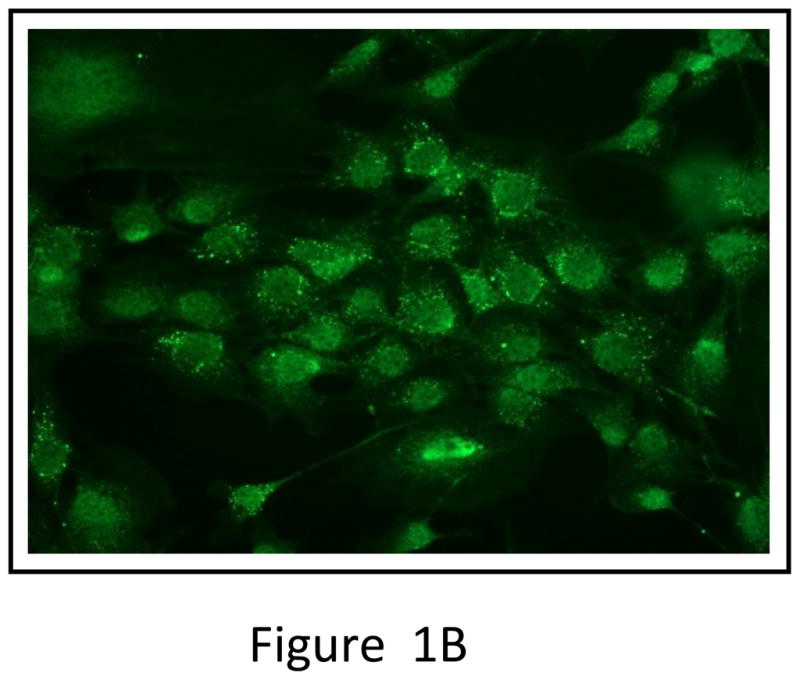


(A) RT-PCR analysis showing OPG mRNA expression in unmodified mouse MSC. (B) Immunocytochemical localization of OPG in cultured mouse MSC. (C) Pre-osteoclast RAW cells were cultured in MSC conditioned medium in the presence of RANKL for 7 days. TRAP staining indicated inhibition of osteoclastogenesis in RAW cells grown in MSC conditioned medium compared to RAW cells grown in regular medium in the presence of RANKL. Conditioned medium from OPG-silenced MSC (OPG-KO-MSC) failed to prevent osteoclast formation. TRAP, Tartrate resistant acid phosphatase; RM, regular media; CM, conditioned media. (D) Control MSC and OPG-KO-MSC were tested for differentiation into osteoblast lineage using osteoblast medium for 2 weeks. Von Kossa staining was performed to detect calcium deposits (black) to confirm that osteoblast lineage differentiation is compromised in OPG-KO-MSC (right panel) as compared to unmodified MSC (middle panel). There was no positive staining in MSC culture without the osteoblast medium (left panel).
Inhibition of tumor growth by MSC
To determine the effect of MSC in preventing the growth of prostate tumor in the bone, 6-week-old male SCID mice were injected intra-tibially with 105 osteolytic human prostate cancer cells, PC3, expressing firefly luciferase. The next day 5×105 bone marrow-derived mouse MSC, which were unmodified or over-expressing OPG (MSC-OPG) were injected in the tibia in proximity to the tumor cells. Growth of prostate tumor in the bone was evaluated by bioluminescence imaging four weeks after the administration of the MSC, which indicated almost 90% inhibition of tumor growth in both the treatment groups (Figure 2A&B). MSC-OPG did not show any advantage over unmodified MSC in preventing tumor progression.
Figure 2. Tumor growth following MSC therapy.



Non-invasive total body imaging was performed on the day of intra-tibial injection of PC3 cells (day 0) and 4 weeks after the intra-tibial administration of the MSC. (A) Mice represented in the left panel are the same mice that are represented in the right panel and they maintain the same order of alignment. (B) Quantitative analysis of luciferase expression as a measure of tumor growth, 4 weeks after the treatment with MSC or MSC modified to over-express OPG (**P<0.001). (C) When tumor cells were allowed to grow for 2 weeks followed by administration of MSC, therapeutic benefits are apparent but not statistically significant (P>0.05).
MSC therapy is ineffective for end-stage disease
To test if therapy with MSC is effective after the tumor has established in the bone microenvironment with high degree of oseteolytic damage, PC3 cells were injected in the tibia and allowed to grow for 2 weeks. Then, 5×105 MSC (unmodified or over-expressing OPG) were injected in the same location. Tumor progression was evaluated 4 weeks after the treatment and bioluminescence imaging indicated inhibition of tumor growth in some of the treated mice compared to the untreated ones, although data was not significantly different (P>0.05) between treated and the untreated animals when all the mice were taken into consideration for comparison (Figure 2C). Similar observations were made when histological architecture of the tibia was studied for bone loss due to MSC therapy (data not shown). These data suggest requirement of optimal number of MSC to prevent osteolysis in prostate cancer bone metastasis at an earlier time.
Effect of MSC therapy in bone remodeling
Micro computed tomography (μCT) of the skeleton showed significant loss of bone in the region of implantation of the PC3 cells, whereas complete restoration was observed in the tibiae treated with MSC and MSC-OPG (Figure 3A). Tibiae were harvested from the mice and studies were performed to understand the ultra-structure of the tibia. Three-dimensional μCT data indicated a significant decline in relative bone volume and trabecular connectivity density in untreated mice compared to age-matched normal mice. Both trabecular and cortical bone structures were completely restored in mice with PC3 cells in the tibia by the MSC therapy (Figure 3B). It is interesting to note that the relative bone volume and trabecular connectivity density in the treated mice significantly exceeded that observed in the tibiae of normal mice, indicating the effectiveness of the therapy. Bone restoration was highest in mice treated with MSC-OPG, which may be due to significant inhibition of osteoclastogenesis due to overproduction of OPG as compared to unmodified MSC treated mice (Figure 3B; Supplementary Figure S1 A&B). In MSC-OPG treated mice restoration of tibial bone resulted in limitation of marrow space and might compromise important event(s) like hematopoiesis, hence was excluded in further experiments. Histomorphometry supported the results obtained from bioluminescence imaging and μCT analysis. TRAP staining revealed highest number of osteoclasts in the untreated tibia, mainly at the tumor-bone interface, whereas the number and size of osteoclasts were significantly decreased in the tibia of the treated mice. Both the MSC and MSC-OPG treated mice indicated similar pattern of osteoclast staining (Figure 3C). Three-point mechanical testing of the tibial bone was performed to determine the mechanical strength after treatment. Data indicated similar bone strength in MSC treated mice compared to normal mice tibia (Supplementary Figure S2 A&B).
Figure 3. Histomorphometric analysis of bone.



(A) 3-dimensional scanning μCT of the mouse skeleton showing restoration of tibia following MSC therapy compared to untreated mice. (B) 3-dimensional transmission μCT of the bone showing significant osteolysis in the tibia due to the growth of PC3 cells, whereas MSC and MSC over-expressing OPG therapy prevented osteolysis and reduced tumor burden significantly. When compared to normal tibia, both the treated groups demonstrated higher relative bone volume and trabecular bone density. MSC over-expressing OPG treated mice showed the highest bone volume and trabecular density. This is likely due to higher inhibition of osteoclastogenesis. Sections of tibia stained with Goldner’s trichrome stain, where mineralized bone stains blue-green as shown in the bottom panel (Original magnification ×25). (C) Reduction of osteoclast activity following treatment as determined by TRAP staining. Both MSC and MSC over-expressing OPG demonstrated significantly less osteoclast activity at the tumor-bone interface (arrowheads) as compared to untreated mice. (Original magnification ×200)
Interaction between PC3 and MSC in vivo
Outcome of the previous experiment suggests that the therapeutic effects of MSC are not highly apparent when administered at the advanced stages of tumor-induced osteolytic bone lesion. It is likely that at a later stage the number of tumor cells in the tibia outnumbered the input MSC. MSC therapy did not influence the prostate tumor growth in the bone in a negative manner, even at later stages. This prompted us to study the interaction between the MSC and the PC3 cells in the bone in vivo. 105 PC3 cells were injected in the tibia and allowed to grow for 1 week for detectable tumors when 5×105 MSC were injected in the same site. MSC used here were derived from a GFP transgenic mouse. Mice were sacrificed 1 week after the injection of MSC and tibiae were harvested and subjected to histomorphometry. Results showed formation of new bone surrounding the tumor nests (Figure 4A). When analyzed under polarized light this newly formed bone comprised of randomly oriented collagen fibers, called woven bone and characteristic to fracture healing and prostate cancer bone metastasis in human (Figure 4A&B). Human epithelial cell marker cytokeratin-18 immunostaining was performed to identify the prostate tumors in the bone (Figure 4Cc), which also exposed a multiple layers of fibroblast-like cells arranged in concentric circle separating the tumor cells from the newly formed bone, which also negatively stained for cytokeratin-18. The outermost layer of these fibroblast-like cells were often seen to be embedded or being transformed into the newly formed bone (Figure 4Ca&c). These cells stained positively for GFP, which indicated new bone formation in the treated mice formed predominantly from exogenously administered MSC (Figure 4Cd). Formation of new bone around the tumor cells resulted in restricting the growth of prostate tumor cells in the tibia. No such woven bone formation was noticed in tibia injected with MSC in the absence of PC3 cells, suggesting bone formation in the PC3 injected tibia is triggered by the prostate tumor cells (Figure 4A). RT-PCR analysis of mRNA isolated from MSC obtained from a co-culture experiment with PC3 cells for 10 days showed no significant up-regulation of osteogenic genes in the MSC indicating differentiation of MSC towards an osteoblastic lineage may have been driven by osteolysis due to enhanced osteoclastogenesis (Figure 5A&B). In fact, TRAP staining indicated intense osteoclast activity at the tumor-bone interface (Figure 4Cb).
Figure 4. Mechanism of tumor inhibition following implantation of MSC in tibiae with PC3 tumors.



(A) Histomorphology of tibia showing presence or absence of new bone formation surrounding tumor nests in mice tibiae following implantation of unmodified MSC only, or following PC3 tumor cell implantation. Polarized light microscopy showing the newly formed bone, composed of randomly-oriented, mineralized collagen fibers (woven bone) (Original magnification ×100). When MSC were implanted into a normal tibia without the tumor cells, no such bone formation was observed (far left panel). When PC3 cells were injected in the tibia followed by implantation of MSC (OPG silenced) similarly no significant bone formation (far right panel) was observed suggesting the requirement of OPG for in vivo bone formation. (B) Graph showing the amount of woven bone formed in the tibia after treatment with MSC, OPG-KO-MSC or MSC-OPG. OPG-KO-MSC resulted in least amount of woven bone (*P<0.001) indicating a requirement for simultaneous inhibition of osteoclastogenesis while MSC differentiate into bone. (C) Hematoxylin and eosin (H&E) staining of the tibia showing spindle-like cells of mesenchymal origin bordering the tumor and the new bone (a). Significant osteoclast activity was noticed by TRAP staining at the tumor-bone interface most likely serving as the initiating factor for the MSC differentiation into osteoblasts (b). Immunostaining with the human epithelial marker cytokeratin 18 indicated tumor nests surrounded by the MSC (c). Staining with GFP antibody confirmed that differentiating MSC are of donor origin (d) (Original magnification ×20).
Figure 5. Expression of osteogenic genes.



(A) MSC were cultured for 10 days in either regular medium or PC3 conditioned medium. Total RNA was isolated, converted to cDNA and analyzed for up-regulation of osteogenic genes. Data showing no significant change in osteoblastic lineage differentiation after MSC were cultured in conditioned media obtained from PC3 cells. BSP, bone sialoprotein; ALP, alkaline phosphatase; Run×2, runt-related transcription factor; OC, osteocalcin; OP, osteopontin (B) MSC cultured in regular medium or PC3 conditioned medium showing equivalent alkaline phosphatase activity, indicating that PC3 cells did not initiate osteoblastic differentiation in the MSC directly. (C) PC3-MSC in vitro co-culture assay. PC3 cells were grown on hu-biogel matrix as 3D spheroids and cultured in a 0.8μm pore size transwell plate along with MSC in the lower chamber. After 72 hours the PC3 beads were collected and analyzed by MTT assay for cell proliferation. Data presented here are Mean±SEM (n=12 for each experimental conditions).
Characterization of osteoblastic phenotype in prostate cancer bone metastasis
We compared the growth kinetics of highly osteolytic PC3 cells and the osteoblastic C4-2B cells in the tibia of SCID mice. The rate of tumor progression was significantly lower in the tibia injected with C4-2B cells as compared to PC3 cells (Figure 6A&B). Although PC3 cells induced severe osteolysis by 1 month after inoculation, C4-2B cells produced osteoblastic events by 2 months after administration. There was a gradual switch from osteoblastic to osteolytic phenotype when C4-2B cells were allowed to grow for 6 months in the tibia (Figure 6C). These observations indicate that the growth kinetics of the cancer cells in the bone might be the determining factor favoring osteoblastic or osteolytic outcome.
Figure 6.



(A) Growth kinetics of osteoblastic C4-2B cells in the tibia of SCID mice. (B) Growth kinetics of PC3 cells in the tibia of SCID mice. (C) C4-2B injected tibia showing osteolytic lesions when allowed to grow for 6 months.
Role of MSC-produced OPG in bone formation
To test the significance of OPG in this process, an MSC line was generated where OPG expression was silenced using a lentivirus producing shRNA targeting (Supplementary Figure S3). OPG silenced MSC failed to differentiate into osteoblast in vitro as determined by von Kossa staining (Figure 1D) and no significant new bone formation was observed in the tibia when these MSC were tested against PC3 cells in a similar experiment mentioned in the previous section (Figure 4A). Moreover, conditioned medium from OPG silenced MSC failed to inhibit osteoclastogenesis when RAW cells were cultured for 8 days in presence of RANKL (Figure 1C), suggesting requirement of OPG for osteoblast differentiation and inhibition of osteoclastogenesis.
Effects of bone marrow microenvironment on the growth of PC3 cells
To test the effects of MSC on the prostate cancer cells, PC3 cells were co-cultured in a 3-dimension matrix with MSC in presence or absence of bone marrow conditioned medium for 3 days. MTT assay was performed to determine cell proliferation. Addition of naïve MSC to the co-culture did not alter PC3 cell proliferation compared to PC3 cultured only in bone marrow conditioned medium. An increase in cell proliferation was noted only when PC3 cells were cultured in bone marrow conditioned media along with MSC over-expressing OPG (*P<0.001; Figure 5C), supporting that OPG is also a survival factor for the prostate cancer cells (23). This suggests that the inhibitory effect of MSC on the growth of prostate cancer in vivo is an indirect effect and mediated by inhibition of osteoclastogenesis and differentiation into osteoblasts.
DISCUSSION
Results of the present study indicate the therapeutic potential of unmodified MSC in inhibiting the growth of prostate tumor in the bone and prevention of bone loss. MSC did not induce direct apoptosis of tumor cells, instead inhibition of tumor growth in the bone was mediated by new bone formation around them. Although significant therapeutic advantage can be obtained in osteolytic bone metastasis utilizing this approach, absence of a direct killing mechanism may help the metastasis re-establish itself in course of time. The beneficial effects of MSC can thus be further amplified by modifying them ex vivo to express tumoricidal genes besides retaining their ability to differentiate into bone. A recent study utilized MSC expressing urokinase-type plasminogen antagonist amino-terminal fragment (hATF) and showed inhibition of tumor growth by inhibiting angiogenesis prevented bone loss (24). Majority of the studies indicated that MSC promote tumor growth by participating in tumor stroma formation and establishment of pre-metastatic niche. Our in vitro studies showed an increase in proliferation rate of prostate cancer cells only when co-cultured with the MSC over-expressing OPG. However, the same MSC, over-expressing OPG tested provided a therapeutic effect in vivo. Hence, the observed in vitro effects could be attributed only to secretory proteins in the co-culture system and may not include other events in the tumor microenvironment. MSC inhibited the growth of prostate tumor in the bone in vivo by laying down new bone around the cancer cells, which slowed their rapid growth. Other factors such as cell contact in vivo may have also played important roles in addition to the effects of OPG in increasing osteogenesis.
Although the difference in tumor volume between unmodified MSC (MSC-GFP) and MSC over-expressing OPG was not statistically significant, there was an observable difference in the MSC-GFP group, which showed less tumor growth compared to MSC-OPG group. Since OPG may serve as a survival factor for tumor cells, this observation also indicates the importance of regulated expression of OPG for restoration of bone damage following osteolytic bone metastasis. Further, identification of TRAIL binding and RANK binding domains on OPG would allow genetic modifications in OPG to abolish TRAIL binding.
The present study also highlights the formation of woven bone in osteoblastic metastases (25–27). In our study, the therapeutic effect of the MSC is initially imparted by the woven bone formation surrounding the tumor nests. Formation of woven bone also characterizes osteoblastic metastases in prostate cancer and normal to fracture healing. Roudier et al (2008) performed a detailed histomorphic analysis of bone samples obtained from prostate cancer patients with bone metastasis and observed equal representation of osteoblastic and osteolytic components (2). They also showed that the woven bone formation in the osteoblastic metastases originated from the skeletal mesenchymal stem cells. The pattern of woven bone formation in our experiment truly matches the histological pattern observed in patient with prostate cancer bone metastasis as reported by Roudier et al, 2008. We speculate that the presentation of woven bone in prostate cancer bone metastasis is an endogenous therapeutic response by the resident MSC rather than a pathological outcome. Continuous presence of tumor cells in the bone induces osteolysis which in turn signals the MSC to initiate osteoblastogenesis and manifest the formation of woven bone as part of an early therapeutic response. As we compared growth kinetics of highly osteolytic PC3 cells vs. osteoblastic C4-2B cells in the tibia of SCID mice, the rate of tumor growth is significantly lower in tibia injected with C4-2B cells compared to PC3 cells. Therefore, it is likely that low turnover of C4-2B cells provides a therapeutic window for the endogenous MSC to induce bone formation to make up for the lost bone due to initial osteolytic event required by the cancer cells to colonize in the bone microenvironment. At this point we are unable to comment on other factors responsible for the slow in vivo turnover of the C4-2B cells. When mice were sacrificed 6-months after the implantation of the C4-2B cells, all the lesions in the tibiae were of osteolytic nature. Determination of osteoblastic and/or osteolytic phenotype may be dependent on factors such as growth kinetics of the cancer cells, phenotypic changes of the cancer cells, and events like hypoxia in the tumor microenvironment. Hypoxia is known to promote osteolytic bone metastasis and suppresses osteoblast differentiation (28), therefore slower turnover of C4-2B cells may have delayed the onset of hypoxia compared to PC3 cells, which may have delayed the formation of osteolytic lesions. Nyambo et al reported OPG produced by the bone marrow stromal cells inhibits TRAIL induced apoptosis of the tumor cells and favor growth of prostate cancer in vitro (29). Our in vitro co-culture assay showed bone marrow microenvironment and OPG produced by the MSC favored PC3 cell proliferation. Interestingly, MSC or MSC-OPG imparted therapeutic benefits when applied in vivo. Based on these findings we conclude that primary MSC has the potential to provide therapeutic advantage in limiting the establishment of prostate cancer in the bone at early stage by the virtue of its ability to differentiate into bone and inhibit osteoclastogenesis. These data signify that relatively abundant amounts of MSC in the tumor microenvironment can provide therapeutic effects by activating OPG and/or other factor(s) through interactions with prostate cancer cells. Since the amount of MSC in the bone microenvironment is extremely low (1 in 108 cells), strategies to endogenously mobilize and proliferate MSC upon bone metastasis of osteolytic cancers may provide significant therapy and reduce morbidity and mortality encountered in late stage prostate cancer patients. The aim of the present study was to determine potential of adult, bone marrow-derived MSC for the prevention of cancer osteolysis. Based on the limitations of the model used, it is imperative that additional studies should be performed with other osteolytic bone metastasis models having the bone defect in entire skeleton, in imuunocompetent animals, before translating the findings for humans.
Supplementary Material
(A) Graphical representation of relative bone volume after MSC and MSC-OPG therapy. The data shows significant restoration of bone (*P<0.001) in the treated animals as compared to untreated mice. (B) Quantitative data showing a significantly higher trabecular connectivity density in mice after MSC therapy (*P<0.001) when compared to untreated animals. It is interesting to note that both relative bone volume and trabecular connectivity density exceed values obtained from normal, age-matched mice. Data represented here are of the Mean ± SEM.
The peak load (S3A) and stiffness (S3B) of tibia following treatment with MSC in PC3 cell implanted groups were found to be statistically similar compared to tibia of age-matched control mice.
Immunofluorescence staining showing OPG expression in MSC and MSC with OPG silenced (magnification ×200).
Quantitative analysis of OPG production from cultures of PC cells, unmodified MSC and MSC over-expressing OPG. The assay was performed in triplicate (*p<0.05)
Acknowledgments
Financial support from the National Institutes of Health grants R01CA98817, R01AR50251, R01CA108585 and P30AR046031, and the U.S. Army Department of Defense grants BC044440 and PC050949 is gratefully acknowledged. We thank Dr. Maria Johnson and Xingsheng Li for excellent technical assistance in micro-CT measurements.
References
- 1.Jemal A, Siegel R, Ward E, Murray T, Xu H, Thun MJ. Cancer statistics. CA Cancer J Clin. 2007;57:43–66. doi: 10.3322/canjclin.57.1.43. [DOI] [PubMed] [Google Scholar]
- 2.Roudier MP, Morrissey C, True LD, Higano CS, Vessella RL, Ott SM. Histopathological assessment of prostate cancer bone osteoblastic metastases. J Urol. 2008;180:1154–60. doi: 10.1016/j.juro.2008.04.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ye L, Kynaston HG, Jiang WG. Bone metastasis in prostate cancer: molecular and cellular mechanisms. Int J Mol Med. 2007;20:103–11. [PubMed] [Google Scholar]
- 4.Guise TA, Yin JJ, Mohammad KS. Role of endothelin-1 in osteoblastic bone metastases. Cancer. 2003;97:779–84. doi: 10.1002/cncr.11129. [DOI] [PubMed] [Google Scholar]
- 5.Choueiri MB, Tu SM, Yu-Lee LY, Lin SH. The central role of osteoblasts in the metastasis of prostate cancer. Cancer Metastasis Rev. 2006;25:601–9. doi: 10.1007/s10555-006-9034-y. [DOI] [PubMed] [Google Scholar]
- 6.Blair JM, Zhou H, Seibel MJ, Dunstan CR. Mechanisms of disease: roles of OPG, RANKL and RANK in the pathophysiology of skeletal metastasis. Nat Clin Pract Oncol. 2006;3:41–9. doi: 10.1038/ncponc0381. [DOI] [PubMed] [Google Scholar]
- 7.Wittrant Y, Theoleyre S, Chipoy M, et al. RANKL/RANK/OPG: new therapeutic targets in bone tumours and associated osteolysis. Biochim Biophys Acta. 2004;1704:49–57. doi: 10.1016/j.bbcan.2004.05.002. [DOI] [PubMed] [Google Scholar]
- 8.Dougall WC, Chaisson M. The RANK/RANKL/OPG triad in cancer-induced bone diseases. Cancer Metastasis Rev. 2006;25:541–9. doi: 10.1007/s10555-006-9021-3. [DOI] [PubMed] [Google Scholar]
- 9.Whang PG, Schwarz EM, Gamradt SC, Dougall WC, Lieberman JR. The effects of RANK blockade and osteoclast depletion in a model of pure osteoblastic prostate cancer metastasis in bone. J Orthop Res. 2005;23:1475–83. doi: 10.1016/j.orthres.2005.05.004.1100230634. [DOI] [PubMed] [Google Scholar]
- 10.Simonet WS, Lacey DL, Dunstan CR, et al. Osteoprotegerin: A novel secreted protein involved in the regulation of bone density. Cell. 1997;89:309–19. doi: 10.1016/s0092-8674(00)80209-3. [DOI] [PubMed] [Google Scholar]
- 11.Chanda D, Isayeva T, Kumar S, et al. Systemic osteoprotegerin gene therapy restores tumor-induced bone loss in a therapeutic model of breast cancer bone metastasis. Mol Ther. 2008;16:871–78. doi: 10.1038/mt.2008.48. [DOI] [PubMed] [Google Scholar]
- 12.Holen I, Shipman CM. Role of osteoprotegerin (OPG) in cancer. Clin Sci (Lond) 2006;110:279–91. doi: 10.1042/CS20050175. [DOI] [PubMed] [Google Scholar]
- 13.Onyia JE, Galvin RJ, Ma YL, et al. Novel and selective small molecule stimulators of osteoprotegerin expression inhibit bone resorption. J Pharmacol Exp Ther. 2004;309:369–79. doi: 10.1124/jpet.103.057893. [DOI] [PubMed] [Google Scholar]
- 14.Vanderkerken K, De Leenheer E, Shipman C, et al. Recombinant osteoprotegerin decreases tumor burden and increases survival in a murine model of multiple myeloma. Cancer Res. 2003;63:287–9. [PubMed] [Google Scholar]
- 15.Zhang J, Dai J, Qi Y, et al. Osteoprotegerin inhibits prostate cancer-induced osteoclastogenesis and prevents prostate tumor growth in the bone. J Clin Invest. 2001;107:1235–44. doi: 10.1172/JCI11685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Morony S, Capparelli C, Sarosi I, Lacey DL, Dunstan CR, Kostenuik PJ. Osteoprotegerin inhibits osteolysis and decreases skeletal tumor burden in syngeneic and nude mouse models of experimental bone metastasis. Cancer Res. 2001;61:4432–6. [PubMed] [Google Scholar]
- 17.Capparelli C, Kostenuik PJ, Morony S, et al. Osteoprotegerin prevents and reverses hypercalcemia in a murine model of humoral hypercalcemia of malignancy. Cancer Res. 2000;60:783–7. [PubMed] [Google Scholar]
- 18.Kumar S, Chanda D, Ponnazhagan S. Therapeutic potential of genetically modified mesenchymal stem cells. Gene Ther. 2008;15:711–15. doi: 10.1038/gt.2008.35. [DOI] [PubMed] [Google Scholar]
- 19.Studeny M, Marini FC, Dembinski JL, et al. Mesenchymal stem cells: Potential precursors for tumor stroma and targeted delivery vehicles for anticancer agents. J Natl Can Inst. 2004;96:1593–603. doi: 10.1093/jnci/djh299. [DOI] [PubMed] [Google Scholar]
- 20.Nakamura K, Ito Y, Kawano Y, et al. Antitumor effect of genetically engineered mesenchymal stem cells in a rat glioma model. Gene Ther. 2004;11:1155–64. doi: 10.1038/sj.gt.3302276. [DOI] [PubMed] [Google Scholar]
- 21.Tropel P, Noël D, Platet N, Legrand P, Benabid AL, Berger F. Isolation and characterisation of mesenchymal stem cells from adult mouse bone marrow. Exp Cell Res. 2004;295:395–406. doi: 10.1016/j.yexcr.2003.12.030. [DOI] [PubMed] [Google Scholar]
- 22.Kumar S, Ponnazhagan S. Bone homing of mesenchymal stem cells by ectopic alpha 4 integrin expression. FASEB J. 2007;21:3917–27. doi: 10.1096/fj.07-8275com. [DOI] [PubMed] [Google Scholar]
- 23.Holen I, Croucher PI, Hamdy FC, Eaton CL. Osteoprotegerin (OPG) is a survival factor for human prostate cancer cells. Cancer Res. 2002;62:1619–23. [PubMed] [Google Scholar]
- 24.Fritz V, Noël D, Bouquet C, et al. Antitumoral activity and osteogenic potential of mesenchymal stem cells expressing the urokinase-type plasminogen antagonist amino-terminal fragment in a murine model of osteolytic tumor. Stem Cells. 2008;26:2981–2990. doi: 10.1634/stemcells.2008-0139. [DOI] [PubMed] [Google Scholar]
- 25.Dotan ZA. Bone imaging in prostate cancer. Nat Clin Pract Urol. 2008;5:434–44. doi: 10.1038/ncpuro1190. [DOI] [PubMed] [Google Scholar]
- 26.Kingsley LA, Fournier PG, Chirgwin JM, Guise TA. Molecular biology of bone metastasis. Mol Cancer Ther. 2007;6:2609–17. doi: 10.1158/1535-7163.MCT-07-0234. [DOI] [PubMed] [Google Scholar]
- 27.Guise TA, Mohammad KS, Clines G, et al. Basic mechanisms responsible for osteolytic and osteoblastic bone metastases. Clin Cancer Res. 2006;12:6213–16. doi: 10.1158/1078-0432.CCR-06-1007. [DOI] [PubMed] [Google Scholar]
- 28.Hiraga T, Kizaka-Kondoh S, Hirota K, Hiraoka M, Yoneda T. Hypoxia and hypoxia-inducible factor-1 expression enhance osteolytic bone metastases of breast cancer. Cancer Res. 2007;67:4157–63. doi: 10.1158/0008-5472.CAN-06-2355. [DOI] [PubMed] [Google Scholar]
- 29.Nyambo R, Cross N, Lippitt J, et al. Human bone marrow stromal cells protect prostate cancer cells from TRAIL-induced apoptosis. J Bone Miner Res. 2004;19:1712–21. doi: 10.1359/JBMR.040703. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(A) Graphical representation of relative bone volume after MSC and MSC-OPG therapy. The data shows significant restoration of bone (*P<0.001) in the treated animals as compared to untreated mice. (B) Quantitative data showing a significantly higher trabecular connectivity density in mice after MSC therapy (*P<0.001) when compared to untreated animals. It is interesting to note that both relative bone volume and trabecular connectivity density exceed values obtained from normal, age-matched mice. Data represented here are of the Mean ± SEM.
The peak load (S3A) and stiffness (S3B) of tibia following treatment with MSC in PC3 cell implanted groups were found to be statistically similar compared to tibia of age-matched control mice.
Immunofluorescence staining showing OPG expression in MSC and MSC with OPG silenced (magnification ×200).
Quantitative analysis of OPG production from cultures of PC cells, unmodified MSC and MSC over-expressing OPG. The assay was performed in triplicate (*p<0.05)