

Abstract
The TOR (target of rapamycin) signal transduction network monitors intra- and extracellular conditions that favor cell growth. Research during the last decade has revealed a modular structure of the TOR signaling network. Each signaling module senses a particular set of signals from the cellular milieu and exerts regulatory control towards TOR activity. The TOR pathway responds to growth factor signals, nutrient availability, and cellular stresses like hypoxia and energy stress. The signaling modules and their molecular components constituting the TOR network are remarkably conserved in both sequence and function across species. In yeast, roundworms, flies, and mice, the TOR pathway has been shown to regulate lifespan. Correspondingly, genetic, dietary or pharmacological manipulation of individual signaling modules as well as TOR activity itself extends lifespan in these model organisms. We discuss the potential impact of manipulating TOR activity for human health and lifespan.
Keywords: Longevity, aging, ageing, lifespan, TOR, TORC1
Introduction
The achievements of medicine and public health over the past century and a half have resulted in an increased life expectancy, most notably in developed countries. Since 1840, lifespan expectancy has increased nearly linearly without showing signs of slowing down (Christensen et al., 2009). Much of this increase in the early part of the 20th century was due to improvements in infant and childhood survival. However, since the midpoint of the last century, lifespan improvements have largely been due to a reduction of mortality among the elderly. The probability of dying within a year for 80 and 90 year-olds has steadily declined since 1950. This increase in lifespan, accompanied by a decrease in fecundity, has lead to an aging population in many developed countries (Christensen et al., 2009).
Along with an aging population comes an increase in age-related diseases, such as cancer, cardiovascular disease, type 2 diabetes, osteoporosis, and Alzheimer’s, among many others (Christensen et al., 2009; Drachman, 2006; Holroyd et al., 2008). As a result, identifying effective methods to treat or prevent these diseases has become a public health priority. The observation that lifespan and many age-related diseases have a substantial heritable component in humans has encouraged the search for the genetic underpinnings of these processes (Fallin and Matteini, 2009). Data from model organisms have also indicated that the genetic pathways regulating lifespan exhibit remarkable functional conservation across multiple species, possibly even extending to humans (Bell et al., 2009; Partridge, 2009). The identification of genes regulating lifespan can reveal the biological basis of age-related diseases, potentially leading to better prognosis and therapies.
In this review, we summarize evidence from model organisms that lifespan can be influenced by the TOR pathway, an ancient genetic pathway functionally conserved across many species. The TOR pathway plays a well established role in growth control and lifespan. In yeast, roundworms, flies, and mice, the TOR pathway has been shown to regulate lifespan (Kenyon, 2005). Deregulation of TOR signaling has also been consistently observed in age-related diseases such as type-2 diabetes and a wide variety of cancers (Brugge et al., 2007; Guertin and Sabatini, 2007). In this review, we first describe the regulatory structure of the TOR pathway and emphasize its modularity. Second, we turn to the genetic evidence from model organisms associating each signaling module with lifespan control. We will conclude by discussing the promises and complications for future studies of the TOR pathway’s role in human longevity.
The Structure of the TOR Signaling Network
The ability to integrate and balance a multitude of signals from the intra- and extracellular environment is reflected in the modular structure of the TOR pathway (Figure 1). A central signaling core receives stimulatory and inhibitory inputs from an array of signaling branches. In turn, signals are relayed through the signaling core towards the regulation of translation, autophagocytosis (see accompanying review from Koga et al. in this issue), apoptotic regulators, mitochondrial oxidative phosphorylation, and stress resistance. In general, each signaling branch is specialized to sense one particular signal that is relayed to the TOR signaling core. However, some components of the signaling core are activated by multiple signaling branches, and some signaling branches have targets other than the TOR signaling core. Akt, for example, becomes activated by classic growth factor signaling as well as by oxidative stress, and AMPK has targets other than Tsc2 and Raptor. Below, we describe the biochemical composition of each of the individual modules and their links to the TOR signaling pathway (Figure 1).
Figure 1. The Tor signaling network.

The signaling core of the Tor pathway integrates the activity of growth factor signaling like insulin and wnt, hypoxia, energy stress and amino acid sufficiency to regulate translation initiation. The individual signaling modules survey the intra- and extracellular environment for their particular stimuli, and relay their signal to key components of the Tor signaling core, Tsc2 and the Tor complex 1 (TORC1). Among other processes, Tor regulates the biosynthetic capacity of the cell by controlling translational initiation. For species-dependent differences, please see main text.
The TOR Signaling Core
This molecular signal transduction pathway serves as the integrative backbone that all other branches of the signaling network connect to (Inoki et al., 2005; Manning et al., 2002). The signaling pathway ultimately controls the activity of TORC1, which is composed of the Serine/Threonine kinase TOR, along with its associated proteins Raptor and mLst8 (Guertin and Sabatini, 2005). Akt activates TORC1 by directly phosphorylating two separate targets that regulate TORC1 activity: the GTPase activating protein (GAP) Tsc2 (tuberous sclerosis complex protein 2) and the inhibitor of TORC1, PRAS40 (Sancak et al., 2007). In its unphosphorylated state, Tsc2, in complex with Tsc1 and Melted, inhibits the activity of the small GTPase Rheb by stimulating the turnover of Rheb-associated GTP to GDP (Manning, 2004; Pan et al., 2004; Teleman et al., 2005; Zhang et al., 2003). To reverse this process, Rheb potentially requires a guanine nucleotide exchange factor (GEF), possibly TCTP, but recent results raise questions as to the identity of Rheb’s GEF (Hsu et al., 2007; Rehmann et al., 2008; Wang et al., 2008). GTP bound Rheb activates TORC1 (Harris and Lawrence, 2003). Thus, Akt phosphorylation and inhibition of Tsc2 results in Rheb-GTP activating TORC1. The second Akt target, PRAS40, binds the mTOR kinase domain and inhibits mTOR activity (Vander Haar et al., 2007). Upon phosphorylation by Akt, PRAS40 dissociates from mTOR, thereby alleviating its inhibition (Vander Haar et al., 2007). TORC1 controls downstream targets by activating S6 Kinase (S6K) and repressing the translational regulator 4E-BP, both by a direct phosphorylation mechanism.
While the Tor signaling core is generally considered to be highly conserved across species, some prominent differences, apart from the increased number of paralogs in higher organisms, stand out. In yeast, S. cerevisiae does not contain functional orthologs of tsc1, tsc2, or rheb, while these genes are present in S. pombe. The tsc1 and tsc2 genes are also absent in C. elegans (see below). Finally, two tor genes are present inthe genomes of S. cerevisiae (encoding TOR1 and TOR2) and S. pombe (encoding Tor1p and Tor2p), while all other model organisms discussed here only have one. The differential incorporation of TOR1 and TOR2 into distinct complexes gave rise to the discovery of TORC1 and TORC2 (Loewith et al., 2002).
Five Signaling Inputs
1. Growth factor signaling
Stimulation of receptor tyrosine kinases (RTKs) by growth factors, especially IGF or insulin, results in a PI3K-dependent activation of Akt, and subsequently, TORC1. The molecular structure and lifespan effects of the insulin signaling pathway are extensively reviewed in the accompanying review by Ziv et al. in this issue.
2. The energy stress signaling branch
Low cellular energy levels result in a repression of TORC1 activity. Signals indicating a drop in the cell’s energy content, such as the inhibition of either glycolysis or mitochondrial oxidative phosphorylation, lead to the accumulation of AMP and result in the activation of the AMP-dependent kinase AMPK. AMP directly binds to the gamma-subunit of AMPK, thereby allowing LKB1 or CaMKK1 to place an activating phosphate on Threonine 172 of the catalytic subunit, AMPKα (Hardie, 2007; Shaw, 2009). Activated AMPK reduces the activity of TORC1 through at least two mechanisms. First, AMPK directly phosphorylates Tsc2, resulting in the stimulation of Tsc2’s GAP activity and the inactivation of TORC1 (Corradetti et al., 2004; Inoki et al., 2003; Shaw et al., 2004). Second, AMPK directly phosphorylates Raptor, resulting in a repression of TORC1 activity (Gwinn et al., 2008).
Phosphorylation of Tsc2 at different sites by distinct kinases can either activate or repress Tsc2’s GAP activity. AMPK-mediated phosphorylation of Tsc2 results in activation, whereas Akt-mediated phosphorylation of Tsc2 results in inhibition. The differential effect of phosphorylation results from the specific sites that are phosphorylated by the two kinases. Akt phosphorylates multiple sites on Tsc2, including Ser939, Ser1130, and Thr1462, whereas AMPKα phosphorylates Thr1227 and Ser1345 (Inoki et al., 2002; Inoki et al., 2003; Manning et al., 2002; Potter et al., 2002). In this manner, differential phosphorylation of Tsc2 through various signaling branches can activate or inhibit TORC1 activity.
3. The hypoxia signaling branch
The onset of hypoxia results in a strong inhibition of TORC1. Under normoxic conditions, prolyl-hydroxylase domain containing proteins (PHD) hydroxylate HIF1α on specific prolyl residues, marking HIF1α for recognition by the von Hippel-Lindau (VHL) protein (Epstein et al., 2001). VHL acts as a recognition subunit for an ubiquitin ligase and targets HIF1α for ubiquitin-dependent degradation (Cockman et al., 2000; Kamura et al., 2000; Maxwell et al., 1999). Conversely, hypoxic conditions created by low intracellular oxygen concentration result in the stabilization of the transcription factor HIF1α, and the activation of the hypoxic response (Kaelin, 2008; Kaelin and Ratcliffe, 2008). Stabilized HIF1α forms a heterodimer with the aryl hydrocarbon receptor nuclear translocator (ARNT) and activates the transcription of the hypoxic response genes, most notably REDD1 (Brugarolas et al., 2004; Reiling and Hafen, 2004). REDD1 releases Tsc2 from the growth-factor induced association with 14-3-3 proteins, thereby activating the Tsc1/Tsc2 complex and leading to the inhibition of TORC1 activity (DeYoung et al., 2008). In turn, the transcription and translation of HIF1α is controlled by TORC1 activity (Bernardi et al., 2006; Hui et al., 2006). This is supported by results of cell culture studies indicating that rapamycin treatment results in a failure to elicit the hypoxic response (Thomas et al., 2006).
4. The nutrient signaling branch
Withdrawal of amino acids acts as a powerful inhibitor of TORC1 activity, even in the presence of acute growth factor stimulation (Hara et al., 1998). The mammalian Akt-TOR signaling network primarily senses the levels of L-Glutamine, which acts synergistically with essential amino acids to activate TORC1 (Nicklin et al., 2009). Recently, two small heterodimeric GTPases, RagA/RagC and RagB/RagD, have been shown to relay the signal of amino acid sufficiency to TORC1 (Kim et al., 2008; Sancak et al., 2008). In response to abundant amino acids, the heterodimeric RagA-C or RagB-D complexes become GTP-loaded and bind directly to Raptor, resulting in the relocalization of TORC1 to a subcellular endomembrane where the activator Rheb resides (Sancak and Sabatini, 2009).
5. The Wnt signaling branch
Wnt signaling is involved in various aspects of embryonic development, cell fate decisions, and cell movement, but its role in cell proliferation and stem cell maintenance raises the possibility that Wnt might regulate cellular and organismal lifespan as well (van Amerongen and Nusse, 2009). Supporting this possibility, it has been shown that Wnt signaling results in the activation of TORC1. Stimulation of the Wnt receptors in the Frizzled and LRP protein family culminates in the inhibition of Glycogen synthase kinase 3 (Gsk3) (van Amerongen and Nusse, 2009). Active Gsk3 has been shown to directly phosphorylate and activate Tsc2 when primed by an AMPK-dependent phosphorylation (Inoki et al., 2006). Therefore, Wnt-mediated inactivation of Gsk3 activates Tsc2’s GAP activity, resulting in TORC1 activation.
Tor signaling output
Translational Initiation Output
TORC1 activity stimulates cap-dependent mRNA translation by regulating translation initiation and elongation (Proud, 2007; Scheper et al., 2007; Wang and Proud, 2006). Translation is initiated by the 40S ribosomal subunit binding to the mRNA 5′ end, scanning for the initiation codon, and joining with the 60S ribosomal subunit to form the catalytically competent 80S ribosome. Prior to mRNA binding, the 40S ribosomal subunit complexes with eukaryotic initiation factors (eIFs) and the initiator tRNA bound to eIF2 to form a 43S pre-initiation complex. The eIF4F complex is composed of several proteins bound to the mRNA 5′ end, including eIF4A, eIF4B, eIF4E, and eIF4G, and recruits the 43S pre-initiation complex to mRNA. eIF4G acts as a scaffold protein that assembles the eIF4F complex and bridges the poly(A) binding proteins (PABPs) on the mRNA 3′ end with eIF4E on the 5′ end. This leads to the circularization of mRNAs, which has a synergistic effect on the rate of translation (Gebauer and Hentze, 2004; Sonenberg, 2000).
The TOR pathway regulates the circularization of mRNAs through the direct phosphorylation of eukaryotic initiation factor 4E binding protein (4E-BP) (Brunn et al., 1997). Reduced TORC1 activity results in hypo-phosphorylated 4E-BP that binds to eIF4E (Brunn et al., 1997). 4E-BP’s binding of eIF4E blocks the association of eIF4E with eIF4G, thus preventing eIF4F complex formation and mRNA circularization (Haghighat et al., 1995). Active TORC1 directly phosphorylates 4E-BP, which prevents 4E-BP from binding eIF4E and alleviates the repressive effects of 4E-BP on translation (Brunn et al., 1997; Harris and Lawrence, 2003; Shamji et al., 2003).
The TOR pathway also regulates translation initiation and elongation through the activation of S6K (Ma and Blenis, 2009). To control translation initiation, S6K regulates PDCD4 and eIF4B. S6K phosphorylates PDCD4, an inhibitor of the initiation factor eIF4A, marking it for Ubiquitin-mediated destruction (Dorrello et al., 2006). Upon phosphorylation by S6K, eIF4B is recruited to the translation preinitiation complex, where it is thought to partner with eIF4A to form a fully functional mRNA helicase (Holz et al., 2005; Rogers et al., 2001; Shahbazian et al., 2006). To control translational elongation, S6K regulates SKAR (S6K1 Aly/REF-like Target) and eEF2K (elongation Factor 2 Kinase). S6K activates SKAR, a component of the exon-junction complex (EJC), which serves to enhance translation of spliced mRNAs (Holz et al., 2005). Furthermore, S6K directly phosphorylates and inhibits eEF2K (Wang et al., 2001). When active, eEF2K phosphorylates and inhibits the translation elongation factor eEF2 (Redpath et al., 1993). Thus, active TORC1 results in the activation of S6K and stimulates translation initiation as well as translational elongation.
Genetic Regulation of Longevity by TOR Pathway Components
Genetic experiments exploring the effects of longevity genes in a diverse group of model organisms are bound to exploit distinct readouts. In contrast to the seemingly apparent measurement of lifespan in multicellular organisms, lifespan in unicellular organisms is measured as replicative and/or chronological lifespan. Replicative lifespan measures the number of times a mother cell can produce a daughter cell (Mortimer and Johnston, 1959). Chronological lifespan is measured by the length of time cells can survive in a quiescent state while retaining the ability to reenter the cell cycle upon stimulation by appropriate cues (Fabrizio and Longo, 2003).
In this section, we review evidence that link the reduction of TORC1 activity to the extension of lifespan. Furthermore, we will discuss whether the lifespan effects caused by genetic manipulation of single or multiple signaling branches of the TOR pathway are consistent with predictions made from the biochemical structure of the pathway.
The longevity phenotype of the TOR signaling core
Across all model organisms tested so far, reduced TORC1 signaling extends lifespan. This impressive track record comes with the caveat that null mutants in the nutrient-sensing TOR pathway display pleiotropic phenotypes. Drosophila larvae lacking TOR show phenotypes similar to animals subjected to amino acid starvation: reduced nuclear size, developmental arrest, and lipid vesicle aggregation in the larval fat body (Oldham et al., 2000; Zhang et al., 2000). Similarly, C. elegans lacking CeTOR (let363) and or daf-15 (the worm ortholog of the TORC1 protein Raptor) are developmentally arrested, display intestinal hypotrophy and accumulate fat (Jia et al., 2004; Long et al., 2002; Vellai et al., 2003). CeTOR activity reduced by RNAi (Hansen et al., 2007; Pan et al., 2007; Syntichaki et al., 2007; Vellai et al., 2003) extends lifespan in C. elegans. Interestingly, even the developmentally arrested L3 larvae genetically mutant for CeTOR show an extended lifespan when compared to either wild type or larvae arrested in L3 by starvation (Vellai et al., 2003). Similarly, heterozygosity for daf-15/raptor or repression of rheb-1 expression using RNAi also extends lifespan in worms (Honjoh et al., 2009; Jia et al., 2004). In essence, reduction of TORC1 activity shows a longevity phenotype.
Of note, the longevity phenotype of daf-15/raptor is suppressed by daf-16/foxO (Henderson et al., 2006; Jia et al., 2004). In contrast, neither the developmental arrest nor the fat accumulation phenotype of homozygous daf-15/raptor mutants is suppressible by the FoxO ortholog daf-16, indicating that these phenotypes are parallel or downstream of the insulin signaling pathway in worms (Jia et al., 2004). These conflicting results raise the question of how daf-16/foxO and daf15/raptor are connected. Mechanistically, DAF-16/FoxO was shown to negatively regulate the transcription of daf-15/raptor in vitro and in vivo (Jia et al., 2004). This surprising finding shows the molecular connection between insulin and TOR signaling in C. elegans, depite the lack of functional Tsc1/Tsc2 orthologs in this organism. The genetic and molecular interactions of daf16/foxO with daf15/raptor demonstrate a complex relationship between insulin signaling and the TOR pathway.
In S. cerevisiae, deletion of tor1 (Bonawitz et al., 2007; Kaeberlein et al., 2005; Powers et al., 2006), or inhibition of TORC1 by rapamycin treatment (Medvedik et al., 2007; Powers et al., 2006) extends the chronological and replicative lifespan. In Drosophila, modulation of various genes that encode components of the TOR signaling core, including dTsc1, dTsc2, dTOR, and dS6K, extends lifespan (Honjoh et al., 2008; Kapahi et al., 2004; Luong et al., 2006). Similarly, rapamycin treatment of male and female flies causes extended median lifespan (Bjedov et al., 2010). Consistently, lifespan extension by inhibition of S6 Kinase is also observed in C. elegans, mice and yeast (Fabrizio et al., 2001; Hansen et al., 2007; Pan et al., 2007; Selman et al., 2009) (Table 1).
Table 1. TOR signaling network genes shown to affect lifespan in model organisms.
See text for detailed discussion.
| Human gene name | Model organism tested (gene name or drug treatment) | LOF lifespan phenotype (extend/shorten) | References |
|---|---|---|---|
| TOR signaling core | |||
| TOR | S. cerevisiae (TOR1) | extend | (Bonawitz et al., 2007; Kaeberlein et al., 2005; Powers et al., 2006; Smith et al., 2008) |
| C. elegans (let-363) | extend | (Hansen et al., 2007; Jia et al., 2004; Vellai et al., 2003) | |
| Drosophila (dTOR) | extend | (Kapahi et al., 2004; Luong et al., 2006) | |
| M. musculus (rapamycin) | extend | (Harrison et al., 2009; Chen et al., 2009) | |
| Raptor | C. elegans (daf-15) | extend | (Jia et al., 2004) |
| Rheb | C. elegans (CeRheb) | extend | (Honjoh et al., 2008) |
| Tsc1/2 | Drosophila (dTSC1/2) | extend ‡ | (Kapahi et al., 2004) |
| S6K | S. cerevisiae (SCH9) | extend | (Fabrizio et al., 2001; Kaeberlein et al., 2005) |
| C. elegans (rsks-1) | extend | (Hansen et al., 2007; Pan et al., 2007; Selman et al., 2009) | |
| Drosophila (dS6K) | extend ♫ | (Kapahi et al., 2004) | |
| M. musculus (S6K1) | extend | (Selman et al., 2009) | |
| TOR signaling output | |||
| 4E-BP | Drosophila (d4E-BP) | shorten | (Zid et al., 2009) |
| Mnk1/2 | Drosophila (Lk6) | extend * | (Reiling et al., 2005) |
| eIF2 β | C. elegans eIF2β (iftb-1) | extend | (Hansen et al., 2007) |
| eIF4A | S. cerevisiae (TIF1/2) | extend | (Smith et al., 2008) |
| C. elegans (inf-1) | extend | (Curran and Ruvkun, 2007) | |
| eIF4E | C. elegans (ife-2) | extend | (Hansen et al., 2007; Pan et al., 2007; Syntichaki et al., 2007) |
| eIF4G | S. cerevisiae (TIF4631) | extend | (Smith et al., 2008) |
| C. elegans (ifg-1) | extend | (Curran and Ruvkun, 2007; Hansen et al., 2007; Pan et al., 2007) | |
| Ribosomal proteins | S. cerevisiae (rp genes) | extend | (Kaeberlein et al., 2005; Smith et al., 2008; Steffen et al., 2008) |
| C. elegans (rps and rpl genes) | extend | (Curran and Ruvkun, 2007; Hansen et al., 2007) | |
| rRNA processing factors | S. cerevisiae (nop12, loc1, ssf1, rei1) | extend | (Kaeberlein et al., 2005; Steffen et al., 2008) |
| TOR inputs | |||
| HIF1-α | C. elegans (hif-1) | unclear | (Chen et al., 2009; Mehta et al., 2009; Zhang et al., 2009) |
| VHL1 | C. elegans (vhl-1) | extend | (Chen et al., 2009; Mehta et al., 2009; Zhang et al., 2009) |
| PHD proline hydroxylase | C. elegans (egl-9) | extend | (Chen et al., 2009; Mehta et al., 2009; Zhang et al., 2009) |
| AMPK | S. cerevisiae (snf1) | no effect | (Ashrafi et al., 2000) |
| C. elegans (aak-2) | shorten | (Apfeld et al., 2004; Greer et al., 2007; Narbonne and Roy, 2006, 2009) | |
| M. musculus (metformin treatment) | extend ‡ | (Anisimov et al., 2008) | |
| Lkb1 | C. elegans (par-4) | shorten | (Narbonne and Roy, 2009) |
| STRAD-alpha | C. elegans (strd-1) | shorten | (Narbonne and Roy, 2009) |
LOF: loss of function. See text for detailed discussion.
LOF lifespan phenotype observed under starvation conditions.
Gain of function experiment.
Expression of dominant negative allele.
The lifespan extension by inhibition of the TOR pathway is nutrient dependent in Drosophila (Kapahi et al., 2004), yeast (Kaeberlein et al., 2005) and C. elegans (Honjoh et al., 2008). While reduction of TOR activity extends lifespan under nutrient-rich conditions, these manipulations show a limited effect under conditions of dietary restriction (DR). This is most likely due to the inhibition of TORC1 activity by DR. Hence, genetic or pharmacological inactivation of TOR on top of DR is not expected to have much of an effect, although the combination of rapamycin and DR treatments can cause some additional effect when compared to each single treatment (Bjedov et al., 2010). This might be due to the incomplete inhibition on TORC1 activity by rapamycin towards 4E-BP (see below). Taken together, the data establishes at least a partial role of TORC1 signaling in mediating the effects of DR, and suggests that reduction of TORC1 activity mimics, at least to some extent, the effects of dietary restriction (see below).
The longevity phenotype of the hypoxia signaling branch
The hypoxic response centers on the stabilization of the HIF1α protein. Three recent publications analyzed the role of HIF1α in the regulation of lifespan under normoxic conditions in C. elegans (Chen et al., 2009b; Mehta et al., 2009; Zhang et al., 2009). As the aging experiments have been performed under different temperatures, genetic backgrounds, and food conditions, no consistent picture has emerged, but some trends have come into sight (Kaeberlein and Kapahi, 2009). Most noticeably, all three groups agree that Hif1α contributes to the genetic determination of lifespan, but disagree over whether the mechanism involves HIF1α stabilization, ablation or both.
Stabilization of HIF1α, either by overexpression of a degradation-resistant HIF1α mutant or by genetic deletion of the negative regulators egl-9 (the PHD ortholog) or VHL, results in an extended lifespan at 20°C (Mehta et al., 2009; Zhang et al., 2009). This effect has not been observed at 25° Celsius (Chen et al., 2009b). In contrast, lifespan extension in hif1α mutants or in hif1α RNAi treated animals is consistently observed by the groups of Kapahi and Powell-Coffmann, but not in the Kaeberlein laboratory (Chen et al., 2009; Mehta et al., 2009; Zhang et al., 2009). Furthermore, the mechanism of lifespan extension by mutants for hif1α has been shown to be either dependent or independent on daf-16/foxO by Zhang et al and Chen et al., respectively (Chen et al., 2009b; Zhang et al., 2009).
Taken together, the deletion or the overexpression of HIF1α extends lifespan under somewhat different conditions. It is likely that the loss and gain of function mutations elicit their effects on lifespan by distinct mechanisms (Zhang et al., 2009). Why temperature, food sources, and xenobiotic additives routinely used in C. elegans experiments make lifespan differentially sensitive to the activity of hif1α and daf-16/foxO remains an open issue. Alternatively, the possible lack of statistical power in the individual experiments presented by all three groups might have contributed towards the complex picture (Box 1). Notably, however, the group reporting no lifespan effect of hif1a loss of function reached a cumulative sample size of 558 (ia04) mutants and 563 wt controls in 11 individual experiments. In a combined analysis, this sample size would achieve sufficient statistical power to support their conclusions. However, a summary estimate of the 11 experiments was not provided, thus limiting the interpretation of the negative finding (Mehta et al., 2009). In summary, a potentially sufficiently powered study appears to find no effect, while two potentially underpowered studies report significant extensions of lifespan. This represents a conundrum warranting further investigation.
Box 1. Statistical power, cohort size and reproducibility of measured lifespan differences.
Insufficient statistical power is a commonly encountered issue in original and/or replication studies, including studies employing survival analysis. Power represents the probability to detect a significant difference between two groups when the true underlying difference is not zero. Typically, studies aim to achieve power of at least 0.8, meaning that 8 out of 10 independent experiments will detect a difference, if a true difference really exists. It is possible, albeit unlikely, for an underpowered study to detect a significant difference. However, an underpowered study that fails to detect a significant difference produces two possible interpretations: it either missed to detect a true underlying difference, or the true underlying difference was in fact zero.
Power is largely a function of effect size (the difference in lifespan between experimental and control groups) and sample size (the number of individual animals in the experimental and control groups, see Graph). The measure of an effect in survival analysis is the hazard ratio, which is equivalent to the ratio of the mean survival times of the experimental and control groups. If the hazard ratio equals 1.0, there is no lifespan difference between experimental and control groups. If the hazard ratio is 1.30, the average life span of the experimental group is 30% longer. Previous work performed power analysis using the Weibull distribution to model the distribution of survival probabilities (Heo et al., 1998). Here, we present a power analysis for the commonly-used, non-parametric log-rank test.
The bigger the effect size, the smaller the sufficiently powered experiment becomes. Conversely, smaller effect sizes require much larger sample sizes in order to achieve sufficient power. The experimentalist has to strike a balance to design trials with sufficient power without letting the cohort size (and workload) growing out of sight. If the mutant lives – on average – twice as long as the wild type (the hazard ratio is 2.0), 33 mutants and 33 controls provides power at the 0.8 level. If the difference in average lifespan between mutant and wild type strains is 30% (a hazard ratio of 1.3), a 0.8 powered study requires 229 mutants and 229 controls (see graph). The power curves in the graph show the power achieved using the non-parametric log-rank test, a commonly used method for survival analysis in model organism studies.
Box 1. Power for survival analysis.
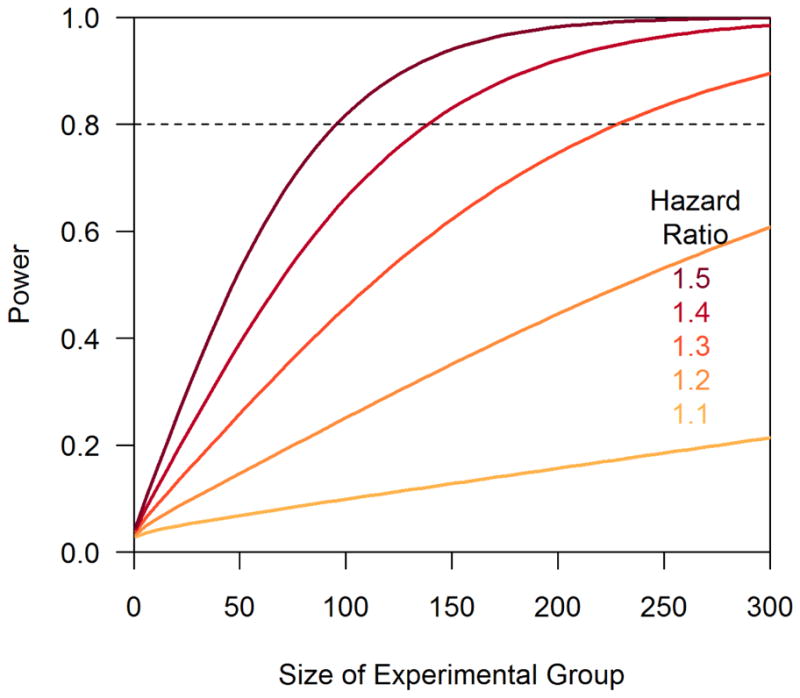
Power is calculated for the log-rank two-sided test under different hazard ratios assuming equal numbers in the experimental and control groups and no censoring, alpha=0.05, and no accrual period (Schoenfeld, 1981). The size of the experimental group is shown along the X-axis. The dashed line indicates power of 0.80.
In addition, the data by Chen et al. proposes that hif1α acts downstream of the TOR pathway to regulate lifespan in C. elegans. RNAi against hif1α does not further extend the lifespan of daf-15/raptor heterozygotes or rsks-1 (the C. elegans S6K ortholog) homozygotes. In a complementary experiment, HIF1α protein stabilization caused by egl-9/PHD mutations abolishes the lifespan extension of rsks-1 mutants (Chen et al., 2009b). This suggests that hif1α functions downstream of S6K to regulate lifespan, consistent with TORC1-dependent translational control of HIF1α mRNA in kidney cancer cells (Thomas et al., 2006).
The longevity phenotype of energy stress signaling
AMPK, an AMP-dependent kinase, is the ancient and central sensor of cellular energy stress. Once activated by low energy levels, it inhibits TORC1 activity and triggers a cellular response towards catabolic metabolism. The activation of AMPK represents a conserved and protective switch shielding the organism from the consequences of metabolic overspending in dire times.
Mutations in one of the two genes encoding the catalytic subunit of AMPK in C. elegans (aak-2), but not the paralogous aak-1, reduce lifespan (Apfeld et al., 2004; Greer et al., 2007; Narbonne and Roy, 2006). Consistent with the requirement of Lkb1-dependent phosphorylation to activate AMPK (Hardie, 2007), loss of function mutations in par-4 (the Lkb1 ortholog) or one of its cofactors, strd-1 (the STRAD-α ortholog), phenocopy the loss of aak-2 (Narbonne and Roy, 2009). Conversely, expression of a constitutively active AMPK catalytic subunit has been shown to extend lifespan (Apfeld et al., 2004). The genetic results in C. elegans are consistent with the biochemical structure of the TOR pathway. However, whether the AMPK-dependent effects on lifespan are mediated by the modulation of TORC1 activity remains to be addressed (see below).
Mutational analysis of the genes encoding the orthologs of the AMPK subunits in S. cerevisiae project a differentiated picture of AMPK’s role in lifespan regulation (Ashrafi et al., 2000). In yeast, snf1 encodes an ortholog of the catalytic subunit AMPKα. In addition, the three genes sip1, sip2 and gal83 are orthologs of the scaffold/glycogen-binding subunit AMPKβ, and snf4 encodes the ortholog of the ATP/AMP binding subunit AMPKγ (Table 1). To distinguish between short lifespan phenotypes due to accelerated aging versus a reduction in cellular fitness, Ashrafi et al. measured markers of cellular aging such as sterility, changes in nucleolar morphology, and the redistribution of the Sir transcriptional silencing complex. Consistent with expectations, the sip2 deletion mutants have a shorter lifespan accompanied by all of the markers of accelerated aging (Ashrafi et al., 2000). Contrary to expectations, however, the deletion of snf4 displays a moderate increase in replicative lifespan, sip1 mutants have no effect, and snf1 and gal83 mutants show a shortened replicative lifespan without the accelerated aging phenotype of sip2 mutants (Ashrafi et al., 2000). Even more surprisingly, the overexpression of SNF1, the yeast ortholog of the catalytic subunit AMPKα, results in a shortened lifespan with an accelerated aging phenotype (Ashrafi et al., 2000), and not the extended lifespan that was observed when AMPKα was overexpressed in C. elegans (Apfeld et al., 2004).
The metabolic master-regulator AMPK is far from a one-trick pony, and TORC1 is only one of its many targets (Hardie, 2007). This is exemplified by the requirement of AMPK for the lifespan extension of daf-2 mutants (the InR ortholog in C. elegans) (Narbonne and Roy, 2006). The daf-2, aak-2 double mutant display a short lifespan due to a failure to downregulate the adipose triglyceride lipase ATGL-1 and the lack of appropriate osmoregulation (Narbonne and Roy, 2006). The multitude of direct AMPK targets and their associated effects in vivo, which have been called surprising (Cunningham and Ashrafi, 2009), constitute a formidable challenge to dissect out the relationship of TORC1 and AMPK for lifespan regulation in the future (see below).
The longevity phenotype of nutrient signaling
To our knowledge, no experimental lifespan data connecting genetic manipulation of the components of the amino acid sensing pathway to TORC1 signaling is available as of yet. Nevertheless, reducing amino acid availability by modifying the feeding regiment is a commonly applied form of dietary restriction (DR) and probably represents the most robust mechanism to extend lifespan across all species tested. In the following paragraph, we will briefly examine the evidence that amino acid restriction mediates lifespan extension via the TOR pathway, and refer to a more extensive review on the subject for further reading (Kapahi et al., Cell Metabolism 2010).
In Drosophila and C. elegans, amino acid deprivation phenocopies the loss of TORC1 function (Long et al., 2002; Oldham et al., 2000; Zhang et al., 2000), and DR has been shown to inhibit the biochemical activity of TORC1 in mice (Estep et al., 2009). In conjunction with cell culture experiments that show a strong inhibition of TORC1 upon amino acid removal (Hara et al., 1998), the data strongly supports the hypothesis that the TOR network acts as a primary nutrient sensor.
If TOR mediates the effects of DR on longevity, the model predicts that, in the ideal case, the DR-dependent lifespan extension cannot be further extended by genetically reducing TOR function. This concept has at least two complications: 1) the looming lethality of full TORC1 inhibition, limiting the experimental approach to either non-lethal (redundant) genes or hypomorphic alleles, and 2) the hope that combining two weak TORC1 inhibiting treatments canproduce an additive effect on lifespan. Nevertheless, in Drosophila, C. elegans and S. cerevisiae, experimental data show that a further reduction of TOR activity often fails to extend the lifespan of DR (Kaeberlein et al., 2005; Kapahi et al., 2004; Powers et al., 2006), although some combinatorial effects of rapamycin treatment and DR have been observed (Bjedov et al., 2010). In summary, even with the caveat that DR is executed differently among laboratory protocols and model organisms, there is strong evidence that the lifespan-extending effect of nutrient restriction is mediated by the reduction of TOR signaling (Kapahi et al., 2010).
The longevity phenotype of Wnt signaling
Biochemical evidence shows that the TOR pathway receives stimulating input from the Frizzled family of Wnt receptors, leading to the prediction that active Wnt signaling could result in the shortening of lifespan (Figure 1). Much of the functional genetic evidence implicating the Wnt pathway in the regulation of lifespan comes from the Klotho mouse model of accelerated aging. Klotho was originally discovered as a mutated gene in a mouse strain that exhibited accelerated aging and premature death (Kuro-o et al., 1997). While loss of function mutations in Klotho result in accelerated aging (Kuro-o et al., 1997), overexpression of Klotho extends lifespan (Kurosu et al., 2005). The Klotho gene encodes a transmembrane protein that appears to act as a humoral factor that regulates multiple pathways, including the insulin, Wnt, and fibroblast growth factor (FGF) signaling pathways, among others (Wang and Sun, 2009). Evidence initially suggested that Klotho exerted its effects on aging through the insulin signaling pathway (Kurosu et al., 2005). Subsequent studies found that Klotho also acts as a Wnt antagonist, and Klotho mutants displayed enhanced Wnt signaling along with stem cell depletion (Liu et al., 2007). It might appear counter-intuitive that Wnt signaling, a process known to be involved in stem cell self-renewal (Klaus and Birchmeier, 2008; Nusse, 2008), can result in stem cell depletion and accelerated aging. However, the continuous exposure to Wnt has been shown to induce cellular senescence of both stem cells and differentiated cells (Castilho et al., 2009; Liu et al., 2007). It has been postulated that the continuous activation of cellular growth by Wnt signaling can activate a protective mechanism that leads to cellular senescence (Castilho et al., 2009). Linking Wnt-mediated aging to the TOR pathway, Wnt-induced senescence of epidermal stem cells in mice was shown to be dependent on mTor (Castilho et al., 2009). Perhaps, prolonged Wnt exposure exhausts the stem cell niche or causes cell senescence of differentiated cells, leading to premature aging of the entire organism.
Despite the published evidence that the activation of TOR by Wnt can shorten cellular lifespan, there is currently no published data showing that Wnt regulates organismal lifespan in a TOR-dependent manner. It has been shown that the transcriptional effects of Wnt signaling are required for daf-16/foxO-dependent lifespan regulation and dauer formation in worms (Curran and Ruvkun, 2007; Essers et al., 2005). These results, in conjunction with the interplay between TOR and Insulin signaling and the control of cellular survival through the Wnt-induced regulation of TOR, raise the prospect that Wnt might regulate TOR-dependent lifespan regulation.
The longevity phenotype of translational Initiation factors
Regulation of translation is the best characterized output of TOR signaling that regulates lifespan (Table 1). TORC1 regulates translation by activating S6 Kinase (S6K) and repressing the translational inhibitor 4E-BP, both by a direct phosphorylation mechanism (Harris and Lawrence, 2003). The binding of 4E-BP with eIF4E represses translation initiation by preventing eIF4F complex formation and mRNA circularization (see above). The inhibition of TORC1, either through genetic manipulation or through dietary restriction (DR), allows 4E-BP to repress translation initiation (see above). Consistent with biochemical experiments, genetic studies in Drosophila demonstrate that a null mutation in d4E-BP abrogates the lifespan-extending effects of DR, and overexpression of activated alleles of d4E-BP lengthens lifespan under nutrient rich conditions (Zid et al., 2009).
When 4E-BP is active under conditions of low TORC1 activity, overall translational output is reduced as assessed by 35S-methionine incorporation (Zid et al., 2009). However, regulation of translational output by the TOR pathway is not as simple as reducing translation from all mRNAs. Genome-wide translational profiles in Drosophila under normal and DR conditions revealed that mRNAs with complex secondary structure in their 5′ UTRs were sensitive to the repressive effects of 4E-BP, while mRNAs with simple 5′ UTRs escaped the translational repression of 4E-BP (Zid et al., 2009). Among the mRNAs not repressed by 4E-BP under DR conditions were nuclear-encoded mitochondrial mRNAs encoding components of the electron transport chain (ETC). This suggests that oxidative phosphorylation is favored over glycolysis when nutrient levels are low, and DR-induced elevated mitochondrial ETC activity has been reported across many species (Guarente, 2008). Oxidative phosphorylation produces up to 36 moles of ATP per mole of glucose while metabolizing glucose to lactate via the glycolytic chain generates only 2 moles of ATP per mole of glucose. Hence, the ETC can be rightfully viewed as a more efficient way to produce ATP. Alternatively, the catabolic macromolecular precursor molecules NADPH, Acetyl-CoA and the glycolytic intermediates produced by glycolysis might not be required or might even be detrimental during times of restricted resources during DR (Vander Heiden et al., 2009). This shift in metabolism appears to be required for the lifespan extending effects of DR (Zid et al., 2009). Future investigation will be required to determine how a shift towards oxidative phosphorylation results in lifespan extension.
Studies using C. elegans and S. cerevisae have demonstrated that the inhibition of several components of the translation initiation complex, including eIF2, eIF4A, eIF4E, and eIF4G can extend lifespan (Curran and Ruvkun, 2007; Hansen et al., 2007; Henderson et al., 2006; Kaeberlein et al., 2005; Pan et al., 2007; Smith et al., 2008; Steffen et al., 2008; Syntichaki et al., 2007). From 55 C. elegans genes known to cause developmental arrest phenotypes similar to ifg-1 (human eIF4G) (Kamath et al., 2003), 24 genes were found to extend lifespan upon RNAi-based inhibition just during adulthood (Chen et al., 2007). Many genes identified were involved in translation initiation and ribosomal biogenesis. Ribosomal proteins and rRNA processing factors have also been shown to regulate lifespan both in worms and yeast (Table 1). Genes involved in translation and nutrient sensing display significant degrees of functional conservation even between yeast and worms, organisms separated by approximately 1.5 billion years of evolution. Yeast orthologs of worm aging genes are significantly enriched for genes that affect replicative lifespan and protein translation in yeast (Smith et al., 2008).
Among the four known S6K targets (SKAR, PDCD4, eIF4B, and eEF2K), only SKAR (Richardson et al., 2004) and eIF4B (Hernandez et al., 2004) have been shown to control cell growth. There is no published data showing that SKAR or eIF4B regulate organismal lifespan, or that PDCD4 or eEF2K control cellular or organismal lifespan.
The activity of eIF4E is not only regulated by 4E-BP, but also by mitogen-activated protein kinases (MAPK)-interacting kinases 1 and 2 (Mnk1 and Mnk2). In mammals, Mnk1/2 phosphorylates eIF4E at Serine209 (Scheper et al., 2001). The exact physiological role for this phosphorylation in mammals is not completely understood. Mice deficient for Mnk1 and Mnk2 are viable and develop in the absence of any detectable eIF4E phosphorylation (Ueda et al., 2004). In contrast, Mnk1/2 dependent phosphorylation of eIF4E becomes essential for cell survival and tumor progression in a cancer context (Wendel et al., 2007). Taken together, it seems that eIF4E phosphorylated by Mnk1/2 becomes limiting in certain pathological settings with high requirements on translation (e.g. cancer). In these stress contexts, the Ser209 phosphorylated eIF4E can be viewed as an activated form of this translational initiation factor, and Mnk1/2 as an eIF4E activating regulator. Although the precise mechanism remains unknown, it appears that TOR might promote eIF4E phosphorylation through Mnk1/2 (Wendel et al., 2007).
Interestingly, eIF4E phosphorylation has been shown to be important for growth in Drosophila (Lachance et al., 2002). Consistently, Lk6, the single homolog of Mnk1/2 in Drosophila, regulates growth in a dose and nutrient-dependent manner (Reiling et al., 2005). Lk6 mutants show an extension of lifespan only under starvation conditions where translational repression is especially important in order to survive such harsh conditions (Reiling et al., 2005). In summary, Lk6 deficient flies phenocopy the reduced activity of TORC1 under nutrient stress circumstances. Although the TORC1-LK6 link is not established biochemically, this phenotype is consistent with MNK’s role of regulating translation downstream of TOR.
The longevity phenotype of autophagy
In addition to translational regulation, the TOR pathway controls lifespan by regulating autophagy. Macro-autophagy is the degradation of organelles, allowing cellular macromolecules to be catabolized and recycled. The lifespan extending effects of DR and TORC1 inhibition require autophagy (Hansen et al., 2008; Jia and Levine, 2007). The role of autophagy in lifespan regulation is extensively reviewed in the accompanying article by Koga and colleagues.
Of Drugs, Mice and Men
Studying lifespan in relatively long-lived mammals is challenging due to the high costs of long-term experiments that place large demands on laboratory space. These challenges have been taken on in a few recent studies using pharmacological and genetic interventions inhibiting the TOR pathway (Harrison et al., 2009; Selman et al., 2009). The results are supporting a role for TORC1 activity on lifespan determination in mice. In this section, we discuss the effects of metformin and rapamycin, two small molecules affecting TORC1 activity. We also review the effects of genetic ablation of S6K1 on mammalian lifespan, and briefly summarize the sex-specific effects of the resulting alteration of the TOR signaling network.
Lifespan extension by metformin treatment, an AMPK activator
Metformin, a relatively simple and inexpensive biguanide commonly prescribed to treat type 2 diabetes, activates AMP-dependent Kinase by an unknown mechanism. Physiologically, metformin acts in three ways: by reducing the resorption of carbohydrates in the intestine, by inhibiting gluconeogenesis in the liver, and by sensitizing peripheral tissue to insulin. Due to its relatively mild side effects and positive effect on cardiovascular disease in diabetic patients, 40 million prescriptions of metformin were filled in the US in 2008, making metformin the #1 ranked line of treatment among type 2 diabetes patients, and #10 among all prescribed generics (SDI/Verispan and VONA, 2008). On a cellular level, the activation of AMPK causes a general switch from anabolic to catabolic metabolism (Hardie, 2007). In addition to other targets, AMPK phosphorylates Tsc2 and Raptor (Figure 1), resulting in the downregulation of TORC1 activity (Gwinn et al., 2008).
The life extending properties of biguanides on rodents have been known for some time (Dilman and Anisimov, 1980). Consistent with its ability to activate AMPK, metformin has been reported to extend median lifespan in C. elegans dependent on par-4/lkb1 and aak-2/ampk (Onken and Driscoll). Furthermore, metformin treatment extends the median and maximal lifespan of female wild-type mice (Anisimov et al., 2008). Interestingly, a growing body of evidence also suggests that a series of mouse tumor models treated with metformin display delayed or reduced tumor load (Buzzai et al., 2007; Huang et al., 2008). Consistently, retrospective studies of type 2 diabetics demonstrated that taking metformin is associated with a reduced risk for pancreatic and breast cancer in humans (Evans et al., 2005; Li et al., 2009; Libby et al., 2009). The ability of metformin to reduce tumor load may suggest that the lifespan-extending effects might be due to a reduced cancer burden. However, at least in wild-type mice, this seems not to be the case (Anisimov et al., 2008). The incidence of tumors was similar in metformin treated and non-treated controls, while metformin-treated mice had a significant lifespan extension. However, tumor sizes of untreated controls and metformin treated mice were not examined in this study. Metformin’s physiological mechanism to extend lifespan remains enigmatic, but its reduction of TORC1 activity might provide a plausible explanation. As of yet, there are no reports on the effect of metformin on human lifespan.
Polyphenols, including Resveratol, act as powerful activators of AMPK (Zang et al., 2006). They also have been reported to activate Sirtuins, especially SIRT2 (see accompanying review by G. Tranah in this issue). Several studies have reported increased healthspan (defined as the reduction of age-related diseases) in mice treated with Resveratol, but to date it remains unclear if these effects are mediated by the activation of AMPK, of Sirtuins, or both (Finkel et al., 2009). Newly developed compounds activating Sirtuins without affecting AMPK activity should help to resolve this matter (Baur et al., 2006; Milne et al., 2007).
Lifespan extension by rapamycin treatment, a TORC1 inhibitor
Rapamycin and its direct chemical derivatives (“rapalogs”) (Guertin and Sabatini, 2009) have long been used as allosteric TORC1 inhibitors in the lab and as immunosuppressants in the clinic. Some rapalogs have recently been FDA approved for the treatment of a few selected cancers (Guertin and Sabatini, 2007). In a recent study, Harrison and colleagues showed that the administration of rapamycin late in life (600 days) significantly extends the mean life span of male and female mice cohorts by 9% and 13%, respectively (Harrison et al., 2009). Like the metformin study, Harrington et al. also report no significant changes in cancer onset and frequency between treated and control cohorts. A separate study started at a later time and which is therefore still underway, investigates the effects of rapamycin fed mice started at 270 days of age, and already notes a significant decrease in mortality risk of the treated mice at a time point when 51% of females and 68% of the males had died (Harrison et al., 2009). Examination of ribosomal protein S6 (rpS6) phosphorylation, a frequently used surrogate marker for TORC1 activity, in visceral fat tissue showed a 75–80% reduction of TORC1 activity in rapamycin treated mice. However, rapamycin has been reported to accumulate in fat, so this might represent an overestimate of rapamycin’s effect on TORC1 activity in other tissues. In an independent study, old mice (22–24 months) were fed with rapamycin for six weeks only. A comparison to vehicle-fed controls revealed a significant increase in lifespan, restored hematopoietic stem cell function and increased immunity function (Chen et al., 2009a). These results support the hypothesis that inhibition of TORC1 activity might contribute to lifespan extension in mammals.
The precise biochemical consequences of long-term rapamycin treatment are complex. For one, extended rapamycin treatment has been shown to inhibit TORC2 (a Tor-containing protein complex with functions distinct from TORC1) in addition to TORC1 in some mammalian cell lines (Sarbassov et al., 2006). Secondly, chronic rapamycin treatment inhibits TORC1 activity towards S6K permanently, while TORC1-dependent phosphorylation of 4E-BP1 rebounds after a transient phase of inhibition (Choo et al., 2008; Feldman et al., 2009; Thoreen et al., 2009). This contrasts with the previous view that rapamycin inhibits TORC1 kinase activity towards all substrates. Taken into consideration, this suggests that rapamycin treatment might mimic the specific inhibition of S6K (see below). Lastly, inhibition of TORC1 has been shown to release the negative feedback inhibition towards InR/IGFR/IRS activity, resulting in an increase of PI3K, Akt and serum glucocorticoid-induced kinase (SGK) activity (Alessi et al., 2009; Manning, 2004). Dissecting out the relevant biochemical events and target tissues required for the rapamycin-dependent lifespan extension will be an important challenge. The newly developed ATP competitive inhibitors of mTOR and dual-specificity inhibitors of mTOR and PI3K might prove to be valuable tools to address these questions, in addition to mice with genetically altered TORC1 signaling (Feldman et al., 2009; Thoreen et al., 2009).
Lifespan extension by genetic deletion of S6K1
S6K1 is a direct TORC1 substrate, and the phosphorylation of S6K1 by TORC1 is strictly rapamycin sensitive (Choo and Blenis, 2009; Thoreen and Sabatini, 2009). Mice carrying a genetic deletion of S6K1 are viable, and this finding gave rise to the discovery of S6K2 (Lee-Fruman et al., 1999; Shima et al., 1998). The putative S6K1 - S6K2 redundancy might contribute to the compensation for the loss of S6K1. In support of this hypothesis, S6K2 activity was found to be upregulated in S6K1 deficient cells (Lee-Fruman et al., 1999; Pende et al., 2004; Shima et al., 1998).
A recently published study utilizing a S6K1 deficient cohort of mice showed a 19% increase of median lifespan in females when compared to isogenic wild-type controls, without any effects on tumor incidence rates. Of note, there was no significant improvement for male mice (Selman et al., 2009). The result of Selman et al. establishes S6K1 as a major mediator of TORC1-dependent lifespan in mice. However, no measurement of S6K2 activity in S6K1 mutant mice is provided (Selman et al., 2009). Nevertheless, the data suggest that it is S6K1 that plays a relevant role in mammalian lifespan regulation downstream of TORC1.
The AMPK-TORC1-S6K subcircuit loop
Overall, the data from mice show that AMPK activation, TORC1 inhibition and S6K1 deletion share the common phenotype of extended lifespan. The signal transduction from AMPK to TORC1 and, subsequently, to S6K is solidly established. Importantly, a recent report sheds light on a previously underappreciated connection of S6K back to AMPK. In skeletal muscle isolated from S6K1−/− mice, high AMPK activity has been observed (Aguilar et al., 2007). Data in C. elegans by Selman et al. also suggest elevated AMPK activity in S6K mutants. Consistently, the prolonged lifespan of S6K mutant worms depends on AMPK (aak2 in C. elegans). The biochemical circuitry of the TOR signaling network predicts that loss of S6K1 triggers elevated AMPK activity and results in suppression of TORC1 by AMPK-dependent phosphorylation of Tsc2 and Raptor (Gwinn et al., 2008). Therefore, inhibition of S6K1, either by genetic deficiency or by pharmacological means, triggers a negative feedback loop that acts to reduce TORC1 activity. How this translates back into mice remains to be explored, as a true epistatic relationship placing S6K1 and AMPK into a negative feedback loop culminating in TORC1 inhibition and thereby controlling lifespan in mice remains to be proven, but is tantalizing nonetheless.
Female vs. male lifespan differences
The model of the AMPK-TORC1-S6K circuit would explain the power of the S6K1 deletion to extend lifespan in female mice, but leaves very few clues as to why this effect does not apply to male mice. Moreover, the sex-specific differences in the rapamycin study are less pronounced (Harrison et al., 2009). The metformin study does not illuminate the issue any further, as it presents data exclusively from female mice. The effect of AMPK activation on lifespan in male mice remains unknown (Anisimov et al., 2008). Of note, IGFR deficient mice also display the tendency towards longer lived females (Holzenberger et al., 2003).
Are similar sex-specific patterns also observed in other model organisms? Lifespan experiments in C. elegans utilize hermaphrodites, and the effects of mating type differences in yeasts on lifespan are unexplored. However, in Drosophila, sex-specific effects in lifespan are observed, and the manipulation of the Insulin signaling pathway (Clancy et al., 2001; Tatar et al., 2001) or the TOR pathway (Bjedov et al., 2010; Zid et al., 2009) leads to sex-specific effects. Sex-specific phenotypes of elevated TORC1 signaling have also been noted in human disease. Loss-of-function of the tsc1 or tsc2 genes induces lyphangioleiomyomatosis (LAM), only diagnosed in women for unknown reasons (Henske, 2003). The relation to the observed differences in lifespan extension between male and female mice by rapamycin treatment or S6K1 deletion and the female-specific pathology of high TORC1 activity remains to be explored.
Mechanisms of TOR-dependent lifespan extension
Although a careful and comprehensive exploration of the possible and probable mechanisms of TOR-dependent lifespan extension is beyond the scope of this review, several key observations point in the direction of at least four consistent effects of reduced TORC1 signaling: 1) upregulated mitochondrial oxidative phosphorylation (Bonawitz et al., 2007; Guarente, 2008; Zid et al., 2009) 2) increased resistance to oxidative stress (Fabrizio et al., 2003), 3) a cell-protective metabolic response (Wei et al., 2009), and 4) the initiation of autophagy (Salminen and Kaarniranta, 2009). The multitude of responses poses the pressing questions of how much each individual response contributes towards lifespan extension upon the reduction of TORC1 activity, and if the individual responses act synergistically or additively. For example, while increasing the protection against superoxide seems to contribute towards extended lifespan in yeast and flies (Parkes et al., 1998), this mechanism seems to be only partially responsible for the lifespan extension mediated by the sch9 (S6K ortholog) mutant in yeast (Fabrizio et al., 2003). Quantitative studies employing pharmacological and genetic tools in model organisms will again be the method of choice to answer these questions.
Outlook: The TOR signaling network and human lifespan
The data summarized in this review, covering all individual signaling modules of the TOR pathway in multiple model organisms, including rodents, strongly suggest that reduced TORC1 signaling carries the benefit of an extended lifespan. Considering the growing amount of these consistent results in experimental settings, the time has become mature to evaluate the relevance of TORC1 signaling in humans. As aging is the single largest risk factor for human disease in developed countries, interventions to postpone its progression will have tremendous impact on individual and public health. It is intuitive to investigate the effects of mTOR antagonists or agonists (e.g. rapamycin and metformin) on human health. However, such studies encounter many significant limitations. Using data from patients prescribed metformin and rapamycin face confounding from selection bias, as these agents are prescribed to treat serious life-shortening diseases. Furthermore, rapamycin and its derivatives carry a long and gruesome list of side effects, excluding or at least seriously limiting the initiation of prospective longitudinal studies in healthy individuals with this class of drugs. In contrast, the relatively mild side effects of metformin might represent less of a hurdle to initiate a long-term study in healthy individuals, yet it may still take a very long time to observe measurable effects.
Alternatively, identifying specific genetic variants affecting variation in human lifespan or longevity may present a feasible approach to address this question. The data from model organisms can be crafted into the hypothesis that long-lived humans might carry genetic variants in genes of the TOR signaling network that cause reduced TORC1 activity. For instance, comparing the genetic profiles (e.g. DNA sequence variations or methylation patterns) of genes constituting the human TOR network in a sufficiently large cohort of centenarians to appropriate controls may reveal if specific variants of certain genes are significantly associated with the long-lived phenotype. Subsequently, the identified variants could be tested for their effects on TORC1 activity by biochemical means and transplanted into model organisms for genetic effects on lifespan. With the ever dropping cost and increasing power of modern sequencing technology, this approach, which was out of reach just a couple of years ago, is firmly within the reach of today’s science. Perhaps, we might live to see if TOR signaling never gets old.
Acknowledgments
This study was supported in part the National Institute on Aging K01 AG022782, R01 AG023692, U19 AG023122 and REAC Award 38107-521859-430000. DSE is supported by a National Institutes of Health training grant T32 DK007418.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aguilar V, Alliouachene S, Sotiropoulos A, Sobering A, Athea Y, Djouadi F, Miraux S, Thiaudiere E, Foretz M, Viollet B, et al. S6 kinase deletion suppresses muscle growth adaptations to nutrient availability by activating AMP kinase. Cell Metab. 2007;5:476–87. doi: 10.1016/j.cmet.2007.05.006. [DOI] [PubMed] [Google Scholar]
- Alessi DR, Pearce LR, Garcia-Martinez JM. New insights into mTOR signaling: mTORC2 and beyond. Sci Signal. 2009;2:pe27. doi: 10.1126/scisignal.267pe27. [DOI] [PubMed] [Google Scholar]
- Anisimov VN, Berstein LM, Egormin PA, Piskunova TS, Popovich IG, Zabezhinski MA, Tyndyk ML, Yurova MV, Kovalenko IG, Poroshina TE, et al. Metformin slows down aging and extends life span of female SHR mice. Cell Cycle. 2008;7:2769–73. doi: 10.4161/cc.7.17.6625. [DOI] [PubMed] [Google Scholar]
- Apfeld J, O’Conner G, McDonagh T, DiStefano P, Curtis R. The AMP-activated protein kinase AAK-2 links energy levels and insulin-like signals to lifespan in C. elegans. Genes & Development. 2004;18:3004–3009. doi: 10.1101/gad.1255404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashrafi K, Lin SS, Manchester JK, Gordon JI. Sip2p and its partner snf1p kinase affect aging in S. cerevisiae. Genes Dev. 2000;14:1872–85. [PMC free article] [PubMed] [Google Scholar]
- Baur JA, Pearson KJ, Price NL, Jamieson HA, Lerin C, Kalra A, Prabhu VV, Allard JS, Lopez-Lluch G, Lewis K, et al. Resveratrol improves health and survival of mice on a high-calorie diet. Nature. 2006;444:337–42. doi: 10.1038/nature05354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell R, Hubbard A, Chettier R, Chen D, Miller JP, Kapahi P, Tarnopolsky M, Sahasrabuhde S, Melov S, Hughes RE. A human protein interaction network shows conservation of aging processes between human and invertebrate species. PLoS Genet. 2009;5:e1000414. doi: 10.1371/journal.pgen.1000414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernardi R, Guernah I, Jin D, Grisendi S, Alimonti A, Teruya-Feldstein J, Cordon-Cardo C, Simon MC, Rafii S, Pandolfi PP. PML inhibits HIF-1alpha translation and neoangiogenesis through repression of mTOR. Nature. 2006;442:779–85. doi: 10.1038/nature05029. [DOI] [PubMed] [Google Scholar]
- Bjedov I, Toivonen J, Kerr F, Slack C, Jacobson J, Foley A, Partridge L. Mechanisms of Life Span Extension by Rapamycin in the Fruit Fly Drosophila melanogaster. Cell Metabolism. 2010;11:35–46. doi: 10.1016/j.cmet.2009.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonawitz ND, Chatenay-Lapointe M, Pan Y, Shadel GS. Reduced TOR signaling extends chronological life span via increased respiration and upregulation of mitochondrial gene expression. Cell Metab. 2007;5:265–77. doi: 10.1016/j.cmet.2007.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brugarolas J, Lei K, Hurley RL, Manning BD, Reiling JH, Hafen E, Witters LA, Ellisen LW, Kaelin WG., Jr Regulation of mTOR function in response to hypoxia by REDD1 and the TSC1/TSC2 tumor suppressor complex. Genes Dev. 2004;18:2893–904. doi: 10.1101/gad.1256804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brugge J, Hung MC, Mills GB. A new mutational AKTivation in the PI3K pathway. Cancer Cell. 2007;12:104–7. doi: 10.1016/j.ccr.2007.07.014. [DOI] [PubMed] [Google Scholar]
- Brunn GJ, Hudson CC, Sekulic A, Williams JM, Hosoi H, Houghton PJ, Lawrence JC, Jr, Abraham RT. Phosphorylation of the translational repressor PHAS-I by the mammalian target of rapamycin. Science. 1997;277:99–101. doi: 10.1126/science.277.5322.99. [DOI] [PubMed] [Google Scholar]
- Buzzai M, Jones RG, Amaravadi RK, Lum JJ, DeBerardinis RJ, Zhao F, Viollet B, Thompson CB. Systemic treatment with the antidiabetic drug metformin selectively impairs p53-deficient tumor cell growth. Cancer Res. 2007;67:6745–52. doi: 10.1158/0008-5472.CAN-06-4447. [DOI] [PubMed] [Google Scholar]
- Castilho RM, Squarize CH, Chodosh LA, Williams BO, Gutkind JS. mTOR mediates Wnt-induced epidermal stem cell exhaustion and aging. Cell Stem Cell. 2009;5:279–89. doi: 10.1016/j.stem.2009.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Liu Y, Zheng P. mTOR regulation and therapeutic rejuvenation of aging hematopoietic stem cells. Sci Signal. 2009a;2:ra75. doi: 10.1126/scisignal.2000559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen D, Pan KZ, Palter JE, Kapahi P. Longevity determined by developmental arrest genes in Caenorhabditis elegans. Aging Cell. 2007;6:525–33. doi: 10.1111/j.1474-9726.2007.00305.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen D, Thomas EL, Kapahi P. HIF-1 modulates dietary restriction-mediated lifespan extension via IRE-1 in Caenorhabditis elegans. PLoS Genet. 2009b;5:e1000486. doi: 10.1371/journal.pgen.1000486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choo AY, Blenis J. Not all substrates are treated equally: implications for mTOR, rapamycin-resistance and cancer therapy. Cell Cycle. 2009;8:567–72. doi: 10.4161/cc.8.4.7659. [DOI] [PubMed] [Google Scholar]
- Choo AY, Yoon SO, Kim SG, Roux PP, Blenis J. Rapamycin differentially inhibits S6Ks and 4E-BP1 to mediate cell-type-specific repression of mRNA translation. Proc Natl Acad Sci U S A. 2008;105:17414–9. doi: 10.1073/pnas.0809136105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen K, Doblhammer G, Rau R, Vaupel JW. Ageing populations: the challenges ahead. Lancet. 2009;374:1196–208. doi: 10.1016/S0140-6736(09)61460-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clancy DJ, Gems D, Harshman LG, Oldham S, Stocker H, Hafen E, Leevers SJ, Partridge L. Extension of life-span by loss of CHICO, a Drosophila insulin receptor substrate protein. Science. 2001;292:104–6. doi: 10.1126/science.1057991. [DOI] [PubMed] [Google Scholar]
- Cockman ME, Masson N, Mole DR, Jaakkola P, Chang GW, Clifford SC, Maher ER, Pugh CW, Ratcliffe PJ, Maxwell PH. Hypoxia inducible factor-alpha binding and ubiquitylation by the von Hippel-Lindau tumor suppressor protein. J Biol Chem. 2000;275:25733–41. doi: 10.1074/jbc.M002740200. [DOI] [PubMed] [Google Scholar]
- Corradetti MN, Inoki K, Bardeesy N, DePinho RA, Guan KL. Regulation of the TSC pathway by LKB1: evidence of a molecular link between tuberous sclerosis complex and Peutz-Jeghers syndrome. Genes Dev. 2004;18:1533–8. doi: 10.1101/gad.1199104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham KA, Ashrafi K. Fat rationing in dauer times. Cell Metab. 2009;9:113–4. doi: 10.1016/j.cmet.2009.01.008. [DOI] [PubMed] [Google Scholar]
- Curran SP, Ruvkun G. Lifespan regulation by evolutionarily conserved genes essential for viability. PLoS Genet. 2007;3:e56. doi: 10.1371/journal.pgen.0030056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeYoung MP, Horak P, Sofer A, Sgroi D, Ellisen LW. Hypoxia regulates TSC1/2-mTOR signaling and tumor suppression through REDD1-mediated 14-3-3 shuttling. Genes Dev. 2008;22:239–51. doi: 10.1101/gad.1617608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dilman VM, Anisimov VN. Effect of treatment with phenformin, diphenylhydantoin or L-dopa on life span and tumour incidence in C3H/Sn mice. Gerontology. 1980;26:241–6. doi: 10.1159/000212423. [DOI] [PubMed] [Google Scholar]
- Dorrello NV, Peschiaroli A, Guardavaccaro D, Colburn NH, Sherman NE, Pagano M. S6K1- and betaTRCP-mediated degradation of PDCD4 promotes protein translation and cell growth. Science. 2006;314:467–71. doi: 10.1126/science.1130276. [DOI] [PubMed] [Google Scholar]
- Drachman DA. Aging of the brain, entropy, and Alzheimer disease. Neurology. 2006;67:1340–52. doi: 10.1212/01.wnl.0000240127.89601.83. [DOI] [PubMed] [Google Scholar]
- Epstein AC, Gleadle JM, McNeill LA, Hewitson KS, O’Rourke J, Mole DR, Mukherji M, Metzen E, Wilson MI, Dhanda A, et al. C. elegans EGL-9 and mammalian homologs define a family of dioxygenases that regulate HIF by prolyl hydroxylation. Cell. 2001;107:43–54. doi: 10.1016/s0092-8674(01)00507-4. [DOI] [PubMed] [Google Scholar]
- Essers MA, de Vries-Smits LM, Barker N, Polderman PE, Burgering BM, Korswagen HC. Functional interaction between beta-catenin and FOXO in oxidative stress signaling. Science. 2005;308:1181–4. doi: 10.1126/science.1109083. [DOI] [PubMed] [Google Scholar]
- Estep PW, 3rd, Warner JB, Bulyk ML. Short-term calorie restriction in male mice feminizes gene expression and alters key regulators of conserved aging regulatory pathways. PLoS One. 2009;4:e5242. doi: 10.1371/journal.pone.0005242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans JM, Donnelly LA, Emslie-Smith AM, Alessi DR, Morris AD. Metformin and reduced risk of cancer in diabetic patients. BMJ. 2005;330:1304–5. doi: 10.1136/bmj.38415.708634.F7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabrizio P, Liou LL, Moy VN, Diaspro A, Valentine JS, Gralla EB, Longo VD. SOD2 functions downstream of Sch9 to extend longevity in yeast. Genetics. 2003;163:35–46. doi: 10.1093/genetics/163.1.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabrizio P, Longo VD. The chronological life span of Saccharomyces cerevisiae. Aging Cell. 2003;2:73–81. doi: 10.1046/j.1474-9728.2003.00033.x. [DOI] [PubMed] [Google Scholar]
- Fabrizio P, Pozza F, Pletcher SD, Gendron CM, Longo VD. Regulation of longevity and stress resistance by Sch9 in yeast. Science. 2001;292:288–90. doi: 10.1126/science.1059497. [DOI] [PubMed] [Google Scholar]
- Fallin MD, Matteini A. Genetic epidemiology in aging research. J Gerontol A Biol Sci Med Sci. 2009;64:47–60. doi: 10.1093/gerona/gln021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldman ME, Apsel B, Uotila A, Loewith R, Knight ZA, Ruggero D, Shokat KM. Active-site inhibitors of mTOR target rapamycin-resistant outputs of mTORC1 and mTORC2. PLoS Biol. 2009;7:e38. doi: 10.1371/journal.pbio.1000038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finkel T, Deng CX, Mostoslavsky R. Recent progress in the biology and physiology of sirtuins. Nature. 2009;460:587–91. doi: 10.1038/nature08197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gebauer F, Hentze MW. Molecular mechanisms of translational control. Nat Rev Mol Cell Biol. 2004;5:827–35. doi: 10.1038/nrm1488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greer EL, Dowlatshahi D, Banko MR, Villen J, Hoang K, Blanchard D, Gygi SP, Brunet A. An AMPK-FOXO pathway mediates longevity induced by a novel method of dietary restriction in C. elegans. Curr Biol. 2007;17:1646–56. doi: 10.1016/j.cub.2007.08.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guarente L. Mitochondria--a nexus for aging, calorie restriction, and sirtuins? Cell. 2008;132:171–6. doi: 10.1016/j.cell.2008.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guertin DA, Sabatini DM. An expanding role for mTOR in cancer. Trends Mol Med. 2005;11:353–61. doi: 10.1016/j.molmed.2005.06.007. [DOI] [PubMed] [Google Scholar]
- Guertin DA, Sabatini DM. Defining the role of mTOR in cancer. Cancer Cell. 2007;12:9–22. doi: 10.1016/j.ccr.2007.05.008. [DOI] [PubMed] [Google Scholar]
- Guertin DA, Sabatini DM. The pharmacology of mTOR inhibition. Sci Signal. 2009;2:pe24. doi: 10.1126/scisignal.267pe24. [DOI] [PubMed] [Google Scholar]
- Gwinn DM, Shackelford DB, Egan DF, Mihaylova MM, Mery A, Vasquez DS, Turk BE, Shaw RJ. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell. 2008;30:214–26. doi: 10.1016/j.molcel.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haghighat A, Mader S, Pause A, Sonenberg N. Repression of cap-dependent translation by 4E-binding protein 1: competition with p220 for binding to eukaryotic initiation factor-4E. EMBO J. 1995;14:5701–9. doi: 10.1002/j.1460-2075.1995.tb00257.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen M, Chandra A, Mitic LL, Onken B, Driscoll M, Kenyon C. A role for autophagy in the extension of lifespan by dietary restriction in C. elegans. PLoS Genet. 2008;4:e24. doi: 10.1371/journal.pgen.0040024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen M, Taubert S, Crawford D, Libina N, Lee SJ, Kenyon C. Lifespan extension by conditions that inhibit translation in Caenorhabditis elegans. Aging Cell. 2007;6:95–110. doi: 10.1111/j.1474-9726.2006.00267.x. [DOI] [PubMed] [Google Scholar]
- Hara K, Yonezawa K, Weng QP, Kozlowski MT, Belham C, Avruch J. Amino acid sufficiency and mTOR regulate p70 S6 kinase and eIF-4E BP1 through a common effector mechanism. J Biol Chem. 1998;273:14484–94. doi: 10.1074/jbc.273.23.14484. [DOI] [PubMed] [Google Scholar]
- Hardie DG. AMP-activated/SNF1 protein kinases: conserved guardians of cellular energy. Nat Rev Mol Cell Biol. 2007;8:774–85. doi: 10.1038/nrm2249. [DOI] [PubMed] [Google Scholar]
- Harris TE, Lawrence JC., Jr TOR signaling. Sci STKE 2003. 2003:re15. doi: 10.1126/stke.2122003re15. [DOI] [PubMed] [Google Scholar]
- Harrison DE, Strong R, Sharp ZD, Nelson JF, Astle CM, Flurkey K, Nadon NL, Wilkinson JE, Frenkel K, Carter CS, et al. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature. 2009;460:392–5. doi: 10.1038/nature08221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson ST, Bonafe M, Johnson TE. daf-16 protects the nematode Caenorhabditis elegans during food deprivation. J Gerontol A Biol Sci Med Sci. 2006;61:444–60. doi: 10.1093/gerona/61.5.444. [DOI] [PubMed] [Google Scholar]
- Henske EP. Metastasis of benign tumor cells in tuberous sclerosis complex. Genes Chromosomes Cancer. 2003;38:376–81. doi: 10.1002/gcc.10252. [DOI] [PubMed] [Google Scholar]
- Heo M, Faith MS, Allison DB. Power and sample size for survival analysis under the Weibull distribution when the whole lifespan is of interest. Mech Ageing Dev. 1998;102:45–53. doi: 10.1016/s0047-6374(98)00010-4. [DOI] [PubMed] [Google Scholar]
- Hernandez G, Vazquez-Pianzola P, Zurbriggen A, Altmann M, Sierra JM, Rivera-Pomar R. Two functionally redundant isoforms of Drosophila melanogaster eukaryotic initiation factor 4B are involved in cap-dependent translation, cell survival, and proliferation. Eur J Biochem. 2004;271:2923–36. doi: 10.1111/j.1432-1033.2004.04217.x. [DOI] [PubMed] [Google Scholar]
- Holroyd C, Cooper C, Dennison E. Epidemiology of osteoporosis. Best Pract Res Clin Endocrinol Metab. 2008;22:671–85. doi: 10.1016/j.beem.2008.06.001. [DOI] [PubMed] [Google Scholar]
- Holz MK, Ballif BA, Gygi SP, Blenis J. mTOR and S6K1 mediate assembly of the translation preinitiation complex through dynamic protein interchange and ordered phosphorylation events. Cell. 2005;123:569–80. doi: 10.1016/j.cell.2005.10.024. [DOI] [PubMed] [Google Scholar]
- Holzenberger M, Dupont J, Ducos B, Leneuve P, Geloen A, Even PC, Cervera P, Le Bouc Y. IGF-1 receptor regulates lifespan and resistance to oxidative stress in mice. Nature. 2003;421:182–7. doi: 10.1038/nature01298. [DOI] [PubMed] [Google Scholar]
- Honjoh S, Yamamoto T, Uno M, Nishida E. Signalling through RHEB-1 mediates intermittent fasting-induced longevity in C. elegans. Nature. 2008 doi: 10.1038/nature07583. [DOI] [PubMed] [Google Scholar]
- Honjoh S, Yamamoto T, Uno M, Nishida E. Signalling through RHEB-1 mediates intermittent fasting-induced longevity in C. elegans. Nature. 2009;457:726–30. doi: 10.1038/nature07583. [DOI] [PubMed] [Google Scholar]
- Hsu YC, Chern JJ, Cai Y, Liu M, Choi KW. Drosophila TCTP is essential for growth and proliferation through regulation of dRheb GTPase. Nature. 2007;445:785–8. doi: 10.1038/nature05528. [DOI] [PubMed] [Google Scholar]
- Huang X, Wullschleger S, Shpiro N, McGuire VA, Sakamoto K, Woods YL, McBurnie W, Fleming S, Alessi DR. Important role of the LKB1-AMPK pathway in suppressing tumorigenesis in PTEN-deficient mice. Biochem J. 2008;412:211–21. doi: 10.1042/BJ20080557. [DOI] [PubMed] [Google Scholar]
- Hui AS, Bauer AL, Striet JB, Schnell PO, Czyzyk-Krzeska MF. Calcium signaling stimulates translation of HIF-alpha during hypoxia. FASEB J. 2006;20:466–75. doi: 10.1096/fj.05-5086com. [DOI] [PubMed] [Google Scholar]
- Inoki K, Corradetti MN, Guan KL. Dysregulation of the TSC-mTOR pathway in human disease. Nat Genet. 2005;37:19–24. doi: 10.1038/ng1494. [DOI] [PubMed] [Google Scholar]
- Inoki K, Li Y, Zhu T, Wu J, Guan KL. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat Cell Biol. 2002;4:648–57. doi: 10.1038/ncb839. [DOI] [PubMed] [Google Scholar]
- Inoki K, Ouyang H, Zhu T, Lindvall C, Wang Y, Zhang X, Yang Q, Bennett C, Harada Y, Stankunas K, et al. TSC2 integrates Wnt and energy signals via a coordinated phosphorylation by AMPK and GSK3 to regulate cell growth. Cell. 2006;126:955–68. doi: 10.1016/j.cell.2006.06.055. [DOI] [PubMed] [Google Scholar]
- Inoki K, Zhu T, Guan KL. TSC2 mediates cellular energy response to control cell growth and survival. Cell. 2003;115:577–90. doi: 10.1016/s0092-8674(03)00929-2. [DOI] [PubMed] [Google Scholar]
- Jia K, Chen D, Riddle DL. The TOR pathway interacts with the insulin signaling pathway to regulate C. elegans larval development, metabolism and life span. Development. 2004;131:3897–906. doi: 10.1242/dev.01255. [DOI] [PubMed] [Google Scholar]
- Jia K, Levine B. Autophagy is required for dietary restriction-mediated life span extension in C. elegans. Autophagy. 2007;3:597–9. doi: 10.4161/auto.4989. [DOI] [PubMed] [Google Scholar]
- Kaeberlein M, Kapahi P. The hypoxic response and aging. Cell Cycle. 2009;8:2324. doi: 10.4161/cc.8.15.9126. [DOI] [PubMed] [Google Scholar]
- Kaeberlein M, Powers RW, 3rd, Steffen KK, Westman EA, Hu D, Dang N, Kerr EO, Kirkland KT, Fields S, Kennedy BK. Regulation of yeast replicative life span by TOR and Sch9 in response to nutrients. Science. 2005;310:1193–6. doi: 10.1126/science.1115535. [DOI] [PubMed] [Google Scholar]
- Kaelin WG., Jr The von Hippel-Lindau tumour suppressor protein: O2 sensing and cancer. Nat Rev Cancer. 2008;8:865–73. doi: 10.1038/nrc2502. [DOI] [PubMed] [Google Scholar]
- Kaelin WG, Jr, Ratcliffe PJ. Oxygen sensing by metazoans: the central role of the HIF hydroxylase pathway. Mol Cell. 2008;30:393–402. doi: 10.1016/j.molcel.2008.04.009. [DOI] [PubMed] [Google Scholar]
- Kamath RS, Fraser AG, Dong Y, Poulin G, Durbin R, Gotta M, Kanapin A, Le Bot N, Moreno S, Sohrmann M, et al. Systematic functional analysis of the Caenorhabditis elegans genome using RNAi. Nature. 2003;421:231–7. doi: 10.1038/nature01278. [DOI] [PubMed] [Google Scholar]
- Kamura T, Sato S, Iwai K, Czyzyk-Krzeska M, Conaway RC, Conaway JW. Activation of HIF1alpha ubiquitination by a reconstituted von Hippel-Lindau (VHL) tumor suppressor complex. Proc Natl Acad Sci U S A. 2000;97:10430–5. doi: 10.1073/pnas.190332597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapahi P, Chen D, Rogers AN, Katewa S, Wai-Lun Li P, Kockel L. Less is more with TOR. The emerging role of the conserved nutrient sensing TOR pathway in aging. Cell Metab. 2010 doi: 10.1016/j.cmet.2010.05.001. submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapahi P, Zid BM, Harper T, Koslover D, Sapin V, Benzer S. Regulation of lifespan in Drosophila by modulation of genes in the TOR signaling pathway. Curr Biol. 2004;14:885–90. doi: 10.1016/j.cub.2004.03.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenyon C. The plasticity of aging: insights from long-lived mutants. Cell. 2005;120:449–60. doi: 10.1016/j.cell.2005.02.002. [DOI] [PubMed] [Google Scholar]
- Kim E, Goraksha-Hicks P, Li L, Neufeld TP, Guan KL. Regulation of TORC1 by Rag GTPases in nutrient response. Nat Cell Biol. 2008;10:935–45. doi: 10.1038/ncb1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klaus A, Birchmeier W. Wnt signalling and its impact on development and cancer. Nat Rev Cancer. 2008;8:387–98. doi: 10.1038/nrc2389. [DOI] [PubMed] [Google Scholar]
- Kuro-o M, Matsumura Y, Aizawa H, Kawaguchi H, Suga T, Utsugi T, Ohyama Y, Kurabayashi M, Kaname T, Kume E, et al. Mutation of the mouse klotho gene leads to a syndrome resembling ageing. Nature. 1997;390:45–51. doi: 10.1038/36285. [DOI] [PubMed] [Google Scholar]
- Kurosu H, Yamamoto M, Clark JD, Pastor JV, Nandi A, Gurnani P, McGuinness OP, Chikuda H, Yamaguchi M, Kawaguchi H, et al. Suppression of aging in mice by the hormone Klotho. Science. 2005;309:1829–33. doi: 10.1126/science.1112766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lachance PE, Miron M, Raught B, Sonenberg N, Lasko P. Phosphorylation of eukaryotic translation initiation factor 4E is critical for growth. Mol Cell Biol. 2002;22:1656–63. doi: 10.1128/MCB.22.6.1656-1663.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee-Fruman KK, Kuo CJ, Lippincott J, Terada N, Blenis J. Characterization of S6K2, a novel kinase homologous to S6K1. Oncogene. 1999;18:5108–14. doi: 10.1038/sj.onc.1202894. [DOI] [PubMed] [Google Scholar]
- Li D, Yeung SC, Hassan MM, Konopleva M, Abbruzzese JL. Antidiabetic therapies affect risk of pancreatic cancer. Gastroenterology. 2009;137:482–8. doi: 10.1053/j.gastro.2009.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Libby G, Donnelly LA, Donnan PT, Alessi DR, Morris AD, Evans JM. New users of metformin are at low risk of incident cancer: a cohort study among people with type 2 diabetes. Diabetes Care. 2009;32:1620–5. doi: 10.2337/dc08-2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Fergusson MM, Castilho RM, Liu J, Cao L, Chen J, Malide D, Rovira II, Schimel D, Kuo CJ, et al. Augmented Wnt signaling in a mammalian model of accelerated aging. Science. 2007;317:803–6. doi: 10.1126/science.1143578. [DOI] [PubMed] [Google Scholar]
- Loewith R, Jacinto E, Wullschleger S, Lorberg A, Crespo JL, Bonenfant D, Oppliger W, Jenoe P, Hall MN. Two TOR complexes, only one of which is rapamycin sensitive, have distinct roles in cell growth control. Mol Cell. 2002;10:457–68. doi: 10.1016/s1097-2765(02)00636-6. [DOI] [PubMed] [Google Scholar]
- Long X, Spycher C, Han ZS, Rose AM, Muller F, Avruch J. TOR deficiency in C. elegans causes developmental arrest and intestinal atrophy by inhibition of mRNA translation. Curr Biol. 2002;12:1448–61. doi: 10.1016/s0960-9822(02)01091-6. [DOI] [PubMed] [Google Scholar]
- Luong N, Davies CR, Wessells RJ, Graham SM, King MT, Veech R, Bodmer R, Oldham SM. Activated FOXO-mediated insulin resistance is blocked by reduction of TOR activity. Cell Metab. 2006;4:133–42. doi: 10.1016/j.cmet.2006.05.013. [DOI] [PubMed] [Google Scholar]
- Ma XM, Blenis J. Molecular mechanisms of mTOR-mediated translational control. Nat Rev Mol Cell Biol. 2009;10:307–18. doi: 10.1038/nrm2672. [DOI] [PubMed] [Google Scholar]
- Manning BD. Balancing Akt with S6K: implications for both metabolic diseases and tumorigenesis. J Cell Biol. 2004;167:399–403. doi: 10.1083/jcb.200408161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manning BD, Tee AR, Logsdon MN, Blenis J, Cantley LC. Identification of the tuberous sclerosis complex-2 tumor suppressor gene product tuberin as a target of the phosphoinositide 3-kinase/akt pathway. Mol Cell. 2002;10:151–62. doi: 10.1016/s1097-2765(02)00568-3. [DOI] [PubMed] [Google Scholar]
- Maxwell PH, Wiesener MS, Chang GW, Clifford SC, Vaux EC, Cockman ME, Wykoff CC, Pugh CW, Maher ER, Ratcliffe PJ. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature. 1999;399:271–5. doi: 10.1038/20459. [DOI] [PubMed] [Google Scholar]
- Medvedik O, Lamming DW, Kim KD, Sinclair DA. MSN2 and MSN4 link calorie restriction and TOR to sirtuin-mediated lifespan extension in Saccharomyces cerevisiae. PLoS Biol. 2007;5:e261. doi: 10.1371/journal.pbio.0050261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta R, Steinkraus KA, Sutphin GL, Ramos FJ, Shamieh LS, Huh A, Davis C, Chandler-Brown D, Kaeberlein M. Proteasomal regulation of the hypoxic response modulates aging in C. elegans. Science. 2009;324:1196–8. doi: 10.1126/science.1173507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milne JC, Lambert PD, Schenk S, Carney DP, Smith JJ, Gagne DJ, Jin L, Boss O, Perni RB, Vu CB, et al. Small molecule activators of SIRT1 as therapeutics for the treatment of type 2 diabetes. Nature. 2007;450:712–6. doi: 10.1038/nature06261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortimer RK, Johnston JR. Life span of individual yeast cells. Nature. 1959;183:1751–2. doi: 10.1038/1831751a0. [DOI] [PubMed] [Google Scholar]
- Narbonne P, Roy R. Inhibition of germline proliferation during C. elegans dauer development requires PTEN, LKB1 and AMPK signalling. Development. 2006;133:611–9. doi: 10.1242/dev.02232. [DOI] [PubMed] [Google Scholar]
- Narbonne P, Roy R. Caenorhabditis elegans dauers need LKB1/AMPK to ration lipid reserves and ensure long-term survival. Nature. 2009;457:210–4. doi: 10.1038/nature07536. [DOI] [PubMed] [Google Scholar]
- Nicklin P, Bergman P, Zhang B, Triantafellow E, Wang H, Nyfeler B, Yang H, Hild M, Kung C, Wilson C, et al. Bidirectional transport of amino acids regulates mTOR and autophagy. Cell. 2009;136:521–34. doi: 10.1016/j.cell.2008.11.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nusse R. Wnt signaling and stem cell control. Cell Res. 2008;18:523–7. doi: 10.1038/cr.2008.47. [DOI] [PubMed] [Google Scholar]
- Oldham S, Montagne J, Radimerski T, Thomas G, Hafen E. Genetic and biochemical characterization of dTOR, the Drosophila homolog of the target of rapamycin. Genes Dev. 2000;14:2689–94. doi: 10.1101/gad.845700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onken B, Driscoll M. Metformin induces a dietary restriction-like state and the oxidative stress response to extend C. elegans Healthspan via AMPK, LKB1, and SKN-1. PLoS One. 5:e8758. doi: 10.1371/journal.pone.0008758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan D, Dong J, Zhang Y, Gao X. Tuberous sclerosis complex: from Drosophila to human disease. Trends Cell Biol. 2004;14:78–85. doi: 10.1016/j.tcb.2003.12.006. [DOI] [PubMed] [Google Scholar]
- Pan KZ, Palter JE, Rogers AN, Olsen A, Chen D, Lithgow GJ, Kapahi P. Inhibition of mRNA translation extends lifespan in Caenorhabditis elegans. Aging Cell. 2007;6:111–9. doi: 10.1111/j.1474-9726.2006.00266.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkes TL, Elia AJ, Dickinson D, Hilliker AJ, Phillips JP, Boulianne GL. Extension of Drosophila lifespan by overexpression of human SOD1 in motorneurons. Nat Genet. 1998;19:171–4. doi: 10.1038/534. [DOI] [PubMed] [Google Scholar]
- Partridge L. Some highlights of research on aging with invertebrates, 2009. Aging Cell. 2009;8:509–13. doi: 10.1111/j.1474-9726.2009.00498.x. [DOI] [PubMed] [Google Scholar]
- Pende M, Um SH, Mieulet V, Sticker M, Goss VL, Mestan J, Mueller M, Fumagalli S, Kozma SC, Thomas G. S6K1(−/−)/S6K2(−/−) mice exhibit perinatal lethality and rapamycin-sensitive 5′-terminal oligopyrimidine mRNA translation and reveal a mitogen-activated protein kinase-dependent S6 kinase pathway. Mol Cell Biol. 2004;24:3112–24. doi: 10.1128/MCB.24.8.3112-3124.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potter CJ, Pedraza LG, Xu T. Akt regulates growth by directly phosphorylating Tsc2. Nat Cell Biol. 2002;4:658–65. doi: 10.1038/ncb840. [DOI] [PubMed] [Google Scholar]
- Powers RW, 3rd, Kaeberlein M, Caldwell SD, Kennedy BK, Fields S. Extension of chronological life span in yeast by decreased TOR pathway signaling. Genes Dev. 2006;20:174–84. doi: 10.1101/gad.1381406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proud CG. Amino acids and mTOR signalling in anabolic function. Biochem Soc Trans. 2007;35:1187–90. doi: 10.1042/BST0351187. [DOI] [PubMed] [Google Scholar]
- Redpath NT, Price NT, Severinov KV, Proud CG. Regulation of elongation factor-2 by multisite phosphorylation. Eur J Biochem. 1993;213:689–99. doi: 10.1111/j.1432-1033.1993.tb17809.x. [DOI] [PubMed] [Google Scholar]
- Rehmann H, Bruning M, Berghaus C, Schwarten M, Kohler K, Stocker H, Stoll R, Zwartkruis FJ, Wittinghofer A. Biochemical characterisation of TCTP questions its function as a guanine nucleotide exchange factor for Rheb. FEBS Lett. 2008;582:3005–10. doi: 10.1016/j.febslet.2008.07.057. [DOI] [PubMed] [Google Scholar]
- Reiling JH, Doepfner KT, Hafen E, Stocker H. Diet-dependent effects of the Drosophila Mnk1/Mnk2 homolog Lk6 on growth via eIF4E. Curr Biol. 2005;15:24–30. doi: 10.1016/j.cub.2004.12.034. [DOI] [PubMed] [Google Scholar]
- Reiling JH, Hafen E. The hypoxia-induced paralogs Scylla and Charybdis inhibit growth by down-regulating S6K activity upstream of TSC in Drosophila. Genes Dev. 2004;18:2879–92. doi: 10.1101/gad.322704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson CJ, Broenstrup M, Fingar DC, Julich K, Ballif BA, Gygi S, Blenis J. SKAR is a specific target of S6 kinase 1 in cell growth control. Curr Biol. 2004;14:1540–9. doi: 10.1016/j.cub.2004.08.061. [DOI] [PubMed] [Google Scholar]
- Rogers GW, Jr, Richter NJ, Lima WF, Merrick WC. Modulation of the helicase activity of eIF4A by eIF4B, eIF4H, and eIF4F. J Biol Chem. 2001;276:30914–22. doi: 10.1074/jbc.M100157200. [DOI] [PubMed] [Google Scholar]
- Salminen A, Kaarniranta K. Regulation of the aging process by autophagy. Trends Mol Med. 2009;15:217–24. doi: 10.1016/j.molmed.2009.03.004. [DOI] [PubMed] [Google Scholar]
- Sancak Y, Peterson TR, Shaul YD, Lindquist RA, Thoreen CC, Bar-Peled L, Sabatini DM. The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science. 2008;320:1496–501. doi: 10.1126/science.1157535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sancak Y, Sabatini DM. Rag proteins regulate amino-acid-induced mTORC1 signalling. Biochem Soc Trans. 2009;37:289–90. doi: 10.1042/BST0370289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sancak Y, Thoreen CC, Peterson TR, Lindquist RA, Kang SA, Spooner E, Carr SA, Sabatini DM. PRAS40 is an insulin-regulated inhibitor of the mTORC1 protein kinase. Mol Cell. 2007;25:903–15. doi: 10.1016/j.molcel.2007.03.003. [DOI] [PubMed] [Google Scholar]
- Sarbassov DD, Ali SM, Sengupta S, Sheen JH, Hsu PP, Bagley AF, Markhard AL, Sabatini DM. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol Cell. 2006;22:159–68. doi: 10.1016/j.molcel.2006.03.029. [DOI] [PubMed] [Google Scholar]
- Scheper GC, Morrice NA, Kleijn M, Proud CG. The mitogen-activated protein kinase signal-integrating kinase Mnk2 is a eukaryotic initiation factor 4E kinase with high levels of basal activity in mammalian cells. Mol Cell Biol. 2001;21:743–54. doi: 10.1128/MCB.21.3.743-754.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheper GC, van der Knaap MS, Proud CG. Translation matters: protein synthesis defects in inherited disease. Nat Rev Genet. 2007;8:711–23. doi: 10.1038/nrg2142. [DOI] [PubMed] [Google Scholar]
- SDI/Verispan and VONA. 2008 Top 200 generic drugs by total prescriptions. Drug Topics. 2008 http://drugtopics.modernmedicine.com/drugtopics/data/articlestandard//drugtopics/222009/599844/article.pdf.
- Selman C, Tullet JM, Wieser D, Irvine E, Lingard SJ, Choudhury AI, Claret M, Al-Qassab H, Carmignac D, Ramadani F, et al. Ribosomal protein S6 kinase 1 signaling regulates mammalian life span. Science. 2009;326:140–4. doi: 10.1126/science.1177221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shahbazian D, Roux PP, Mieulet V, Cohen MS, Raught B, Taunton J, Hershey JW, Blenis J, Pende M, Sonenberg N. The mTOR/PI3K and MAPK pathways converge on eIF4B to control its phosphorylation and activity. EMBO J. 2006;25:2781–91. doi: 10.1038/sj.emboj.7601166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shamji AF, Nghiem P, Schreiber SL. Integration of growth factor and nutrient signaling: implications for cancer biology. Mol Cell. 2003;12:271–80. doi: 10.1016/j.molcel.2003.08.016. [DOI] [PubMed] [Google Scholar]
- Shaw RJ. LKB1 and AMP-activated protein kinase control of mTOR signalling and growth. Acta Physiol (Oxf) 2009;196:65–80. doi: 10.1111/j.1748-1716.2009.01972.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw RJ, Bardeesy N, Manning BD, Lopez L, Kosmatka M, DePinho RA, Cantley LC. The LKB1 tumor suppressor negatively regulates mTOR signaling. Cancer Cell. 2004;6:91–9. doi: 10.1016/j.ccr.2004.06.007. [DOI] [PubMed] [Google Scholar]
- Shima H, Pende M, Chen Y, Fumagalli S, Thomas G, Kozma SC. Disruption of the p70(s6k)/p85(s6k) gene reveals a small mouse phenotype and a new functional S6 kinase. EMBO J. 1998;17:6649–59. doi: 10.1093/emboj/17.22.6649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith ED, Tsuchiya M, Fox LA, Dang N, Hu D, Kerr EO, Johnston ED, Tchao BN, Pak DN, Welton KL, et al. Quantitative evidence for conserved longevity pathways between divergent eukaryotic species. Genome Res. 2008;18:564–70. doi: 10.1101/gr.074724.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonenberg N, Hershey JWB, Mathews BM. In Translational control of gene expression. Cold Spring Harbor, New York: Cold Spring Harbor Laboratory Press; 2000. [Google Scholar]
- Steffen KK, MacKay VL, Kerr EO, Tsuchiya M, Hu D, Fox LA, Dang N, Johnston ED, Oakes JA, Tchao BN, et al. Yeast life span extension by depletion of 60s ribosomal subunits is mediated by Gcn4. Cell. 2008;133:292–302. doi: 10.1016/j.cell.2008.02.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Syntichaki P, Troulinaki K, Tavernarakis N. eIF4E function in somatic cells modulates ageing in Caenorhabditis elegans. Nature. 2007;445:922–6. doi: 10.1038/nature05603. [DOI] [PubMed] [Google Scholar]
- Tatar M, Kopelman A, Epstein D, Tu MP, Yin CM, Garofalo RS. A mutant Drosophila insulin receptor homolog that extends life-span and impairs neuroendocrine function. Science. 2001;292:107–10. doi: 10.1126/science.1057987. [DOI] [PubMed] [Google Scholar]
- Teleman AA, Chen YW, Cohen SM. Drosophila melted modulates FOXO and TOR activity. Dev Cell. 2005;9:271–81. doi: 10.1016/j.devcel.2005.07.004. [DOI] [PubMed] [Google Scholar]
- Thomas GV, Tran C, Mellinghoff IK, Welsbie DS, Chan E, Fueger B, Czernin J, Sawyers CL. Hypoxia-inducible factor determines sensitivity to inhibitors of mTOR in kidney cancer. Nat Med. 2006;12:122–7. doi: 10.1038/nm1337. [DOI] [PubMed] [Google Scholar]
- Thoreen CC, Kang SA, Chang JW, Liu Q, Zhang J, Gao Y, Reichling LJ, Sim T, Sabatini DM, Gray NS. An ATP-competitive mammalian target of rapamycin inhibitor reveals rapamycin-resistant functions of mTORC1. J Biol Chem. 2009;284:8023–32. doi: 10.1074/jbc.M900301200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thoreen CC, Sabatini DM. Rapamycin inhibits mTORC1, but not completely. Autophagy. 2009;5:725–6. doi: 10.4161/auto.5.5.8504. [DOI] [PubMed] [Google Scholar]
- Ueda T, Watanabe-Fukunaga R, Fukuyama H, Nagata S, Fukunaga R. Mnk2 and Mnk1 are essential for constitutive and inducible phosphorylation of eukaryotic initiation factor 4E but not for cell growth or development. Mol Cell Biol. 2004;24:6539–49. doi: 10.1128/MCB.24.15.6539-6549.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Amerongen R, Nusse R. Towards an integrated view of Wnt signaling in development. Development. 2009;136:3205–14. doi: 10.1242/dev.033910. [DOI] [PubMed] [Google Scholar]
- Vander Haar E, Lee SI, Bandhakavi S, Griffin TJ, Kim DH. Insulin signalling to mTOR mediated by the Akt/PKB substrate PRAS40. Nat Cell Biol. 2007;9:316–23. doi: 10.1038/ncb1547. [DOI] [PubMed] [Google Scholar]
- Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324:1029–33. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vellai T, Takacs-Vellai K, Zhang Y, Kovacs AL, Orosz L, Muller F. Genetics: influence of TOR kinase on lifespan in C. elegans. Nature. 2003;426:620. doi: 10.1038/426620a. [DOI] [PubMed] [Google Scholar]
- Wang X, Fonseca BD, Tang H, Liu R, Elia A, Clemens MJ, Bommer UA, Proud CG. Re-evaluating the roles of proposed modulators of mammalian target of rapamycin complex 1 (mTORC1) signaling. J Biol Chem. 2008;283:30482–92. doi: 10.1074/jbc.M803348200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Li W, Williams M, Terada N, Alessi DR, Proud CG. Regulation of elongation factor 2 kinase by p90(RSK1) and p70 S6 kinase. EMBO J. 2001;20:4370–9. doi: 10.1093/emboj/20.16.4370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Proud CG. The mTOR pathway in the control of protein synthesis. Physiology (Bethesda) 2006;21:362–9. doi: 10.1152/physiol.00024.2006. [DOI] [PubMed] [Google Scholar]
- Wang Y, Sun Z. Current understanding of klotho. Ageing Res Rev. 2009;8:43–51. doi: 10.1016/j.arr.2008.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei M, Fabrizio P, Madia F, Hu J, Ge H, Li LM, Longo VD. Tor1/Sch9-regulated carbon source substitution is as effective as calorie restriction in life span extension. PLoS Genet. 2009;5:e1000467. doi: 10.1371/journal.pgen.1000467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wendel HG, Silva RL, Malina A, Mills JR, Zhu H, Ueda T, Watanabe-Fukunaga R, Fukunaga R, Teruya-Feldstein J, Pelletier J, et al. Dissecting eIF4E action in tumorigenesis. Genes Dev. 2007;21:3232–7. doi: 10.1101/gad.1604407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zang M, Xu S, Maitland-Toolan KA, Zuccollo A, Hou X, Jiang B, Wierzbicki M, Verbeuren TJ, Cohen RA. Polyphenols stimulate AMP-activated protein kinase, lower lipids, and inhibit accelerated atherosclerosis in diabetic LDL receptor-deficient mice. Diabetes. 2006;55:2180–91. doi: 10.2337/db05-1188. [DOI] [PubMed] [Google Scholar]
- Zhang H, Stallock JP, Ng JC, Reinhard C, Neufeld TP. Regulation of cellular growth by the Drosophila target of rapamycin dTOR. Genes Dev. 2000;14:2712–24. doi: 10.1101/gad.835000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Gao X, Saucedo LJ, Ru B, Edgar BA, Pan D. Rheb is a direct target of the tuberous sclerosis tumour suppressor proteins. Nat Cell Biol. 2003;5:578–81. doi: 10.1038/ncb999. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Shao Z, Zhai Z, Shen C, Powell-Coffman JA. The HIF-1 hypoxia-inducible factor modulates lifespan in C. elegans. PLoS One. 2009;4:e6348. doi: 10.1371/journal.pone.0006348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zid BM, Rogers AN, Katewa SD, Vargas MA, Kolipinski MC, Lu TA, Benzer S, Kapahi P. 4E-BP extends lifespan upon dietary restriction by enhancing mitochondrial activity in Drosophila. Cell. 2009;139:149–60. doi: 10.1016/j.cell.2009.07.034. [DOI] [PMC free article] [PubMed] [Google Scholar]