

Abstract
Congenital heart block (CHB) is a conduction abnormality that affects hearts of fetuses and/or newborn to mothers with autoantibodies reactive with the intracellular soluble ribonucleoproteins 48kD La, 52kD Ro, and 60kD Ro. CHB carries substantial mortality and morbidity, with more than 60% of affected children requiring lifelong pacemakers. Several hypotheses have been proposed to explain the pathogenesis of CHB. These can be grouped under three main hypotheses: Apoptosis, Serotoninergic and Ca channel hypothesis. Here we discuss these hypotheses and provide recent scientific thinking that will most likely dominate the future of this field of research.
Introduction
CHB is a conduction abnormality which affects the sino-atrial (SA) node, the atrioventricular (AV) node and the ventricles of the fetal heart with no structural abnormalities. Maternal autoantibodies’ (anti-Ro/La antibodies also referred to as positive IgG) effects on the SA node are manifested as sinus bradycardia (sometimes transient), on the AV node as different degrees of AV block and on the ventricles as heart failure.
Various degrees of AV block as well as bradycardia represent the clinical findings in CHB children. Third degree AV block is irreversible with mortality approaching 30%. Some of the reported CHB deaths are due to heart failure [1, 2]. Recently, a previously under-appreciated sinus bradycardia unrelated to AV block was reported in CHB animal models [3, 4]. This was subsequently confirmed by Brucato et al., [5] and Hamilton et al. [6], who reported sinus bradycardia in infants born to mothers seropositive to anti-Ro positive maternal antibodies. The high incidence of sinus bradycardia in mouse CHB models and in affected infants indicates that the spectrum of conduction abnormalities in CHB extends beyond the AV node to also affect the SA node. Further support for this hypothesis comes from autopsies showing calcification of the SA node in CHB human fetal heart [7]. Because AV block has been the hallmark of CHB, the AV node, rather than the SA node, was then the main focus of previous publications [3, 4, 8] and perhaps even during clinical diagnosis of CHB [5]. The electrophysiological basis of sinus bradycardia has been recently established by the demonstration that maternal antibodies inhibition of L-type Ca current, ICa-L [9, 10], and the T-type Ca current, ICa-T in the sinus node myocyte [9]. Interestingly, the potassium current, IK, and pacemaker current, If which are both involved in the sinus node automaticity were not affected [9]. Maternal antibodies’ inhibition of both ICa-L and ICa-T in the sinus node leads to a slower slope of phase 4 depolarization thereby resulting in a slower heart rate [9].
AV block is the most serious and irreversible manifestation of CHB. The non cardiac manifestations of CHB are skin rash, cytopenias and hepatitis [11]. All manifestations except CHB are transient and resolve at about 6 months with the disappearance of maternal autoantibodies from the neonatal circulation [12, 13]. The transient features reflect the effect of the autoantibodies in organs that have the capacity of continual regeneration. Interestingly, despite being exposed to the same autoantibodies, no complete AV block has been reported in the mother’s heart [14–16].
Histology
The pathogenic autoantibodies causing CHB are associated with progressive destruction of the AV node [17, 18] and calcification of the SA node in human fetal heart [7]. Autopsy revealed fibrosis and calcification of the AV node [7, 19]. Histological studies demonstrated antibodies in cardiac tissue [7, 20]. Deposition of complement, lymphocytic infiltrates, calcification and fibrosis has also been found in fetuses dying from CHB [7, 19–21]. CHB fetal hearts eluates were found to contain autoantibodies to SSA/Ro 52 and 60 kDa [22]. The observation of antibody deposition, fibrosis and calcification has been found in the entire myocardium and not restricted to the AV node [7, 20, 23, 24]. The overall effects these autoantibodies include myocarditis and dialated cardiomyopathy [17, 25–28]; a feature found in few cases of CHB. Prolongation of the QT interval has also been described [29, 30].
Incidence
Because of the rarity and complex etiology of CHB, the incidence is not well established. A generally accepted mean incidence is 1:17,000 in the 1970s [31] and 1:11,000 in the latter decade [32, 33]. However, this incidence dramatically increases to about 5% in Lupus patients and to 18% in subsequent pregnancies [34]. This indicates that the incidence of CHB in the latter decades [32, 35] was higher than previously reported likely due to more effective detection of CHB during pregnancy using fetal ultrasound and to the improved diagnostics. The recurrence rate is 18%, [32, 36, 37] supporting the need for close echocardiographic monitoring in all subsequent pregnancies with heightened surveillance between 18 and 24 weeks of gestation. Despite the persisting antibodies, such a recurrence level indicates that the antibodies are necessary but other fetal factors are involved in the susceptibility to CHB.
Mortality and Morbidity
The mortality rate of 20% in CHB children of mothers seropositive for anti-SSA/Ro and anti-SSB/La has been reported [34]. The survival is proportional to the gestational age at birth; as such, the children born at a later gestational age had a lower mortality. Infants who survive the neonatal period have an excellent prognosis. In fact, more than 60% of affected children require lifelong pacemakers before entering adulthood [32, 38]. Most deaths reported thereafter seem to be related to heart failure [39]. Therefore, most deaths in CHB occur in utero or in the first 3 months of life leading to a mortality of 20%.
Proposed hypothesis of CHB
Based on the facts that there is no convincing evidence that maternal antibodies can cross the sarcolemma of a normal cardiac myocyte, two categories of mechanisms for CHB have been put forward. The first is abnormal surface expression of intracellular Ro/La antigens and the second is the cross-reactivity of maternal antibodies with targets other than Ro/La antigens. To explain the pathogenesis of CHB based on these two categories of mechanisms, three major hypotheses have been proposed. The apoptosis hypothesis [cell surface expression of the Ro/La antigen] [40], the serotoninergic hypothesis [41] and the Ca channel hypothesis [cross-reactivity of maternal antibodies with cell surface receptors and Ca channel proteins respectively] [42].
Apoptosis Hypothesis
The apoptosis hypothesis proposes abnormal (opsonization) surface expression of intracellular Ro/La antigens which become accessible to circulating maternal antibodies on the surface of the cardiac myocytes. It is suggested that opsonization converts the physiologic process of apoptosis to circumstances in which an inflammatory component is evoked [43]. Miranda et al., [40, 43] demonstrated that induction of apoptosis results in surface translocation of Ro/La antigens in human fetal cardiac myocytes.
To examine whether antibodies reactive to Ro/La antigen system indeed bind to the surface of human fetal myocytes and to assess the consequences of this surface bindings, the authors used biotinylation of cell surface proteins, scanning electron microscopy of immunogold labeled cells and examined the consequences of the release of the inflammatory cytokine [43]. They confirmed that induction of apoptosis results in surface accessibility of all Ro-La antigens for recognition by tumor necrosis factor (TNF) from macrophages co-cultured with apoptotic fetal cardiac myocytes. It was concluded that opsonized apoptotic fetal cardiac myocytes promote an inflammatory response by macrophages causing damage to surrounding conducting tissue. Clancy et al. demonstrated that healthy fetal cardiocytes are involved in physiologic clearance of apoptotic cells and that surface binding by anti-SSA/Ro and -SSB/La antibodies inhibits uptake of apoptotic cells by the healthy cardiocytes. The result is accumulation of apoptotic cells promoting further inflammation and subsequent scarring [44].
It is however difficult to correlate the above findings to AV conduction abnormalities seen in CHB because the experiments were performed in ventricular myocytes, not in conduction system myocytes such as AV node myocytes. However, evidence for the subcellular translocation of La autoantigen during physiologic apoptosis in the fetal mouse conducting system has been provided [45]. Other experimental evidence has been proposed to account for the translocation of Ro/La to the cell surface, including viral infection [46], UV light and interferon (IFN) treatment [47].
Serotoninergic Hypothesis
Accessibility of intracellular target antigens to circulating maternal antibodies remains a challenge. This lead to the consideration of alternative hypotheses such as cross-reaction between one or any of the Ro-La components and a cell surface cardiac receptor and/or channel. Indeed, support for this second hypothesis is the report by Eftekhari et al. that antibodies reactive with the serotoninergic 5-hydroxytryptamine (5-HT4A receptor, cloned from human adult atrium, also bind 52kD Ro [41]. Moreover, affinity-purified 5-HT4 antibodies antagonized the serotonin induced L-type Ca channel activation in human atrial cells [41]. The finding that functional beta-adrenoceptors in experimental animals appear late in the gestation stage, makes this hypothesis more intriguing, since 5-HT4 receptors could functionally replace the beta-adrenergic receptor during development. Two peptides in the C terminus of 52kD Ro, aa365-382 and aa380-396, were identified that shared some similarity with the 5-HT4 receptor. The former was recognized by sera from mothers of children with neonatal lupus and it was this 52kD Ro peptide that was reported to be cross reactive with antibodies to peptide aa165-185, derived from the second extracellular loop of the 5-HT4 receptor [41]. These findings are of particular importance, since over 75% of serum from mothers whose children have CHB contain antibodies to 52kD Ro. Given the intriguing possibility that antibodies to the 5-HT4 receptor might represent the hitherto elusive reactivity which could directly contribute to AV block, we initiated a study to determine the prevalence of anti-5-HT4 antibodies in mothers whose children have CHB [48]. RT-PCR was employed to examine the mRNA expression of the 5-HT4 receptor in the human fetal heart. One hundred sixteen sera were evaluated for anti-5 HT4 reactivity. The biochemical results demonstrated mRNA expression of the 5-HT4 receptor in the human fetal atrium. The electrophysiologic studies established that human fetal atrial cells express functional 5-HT4 receptors [48]. Moreover, sera from 116 mothers, whose children have CHB, were evaluated. Ninety-nine (85%) of these maternal sera contained antibodies to Ro, 84% of which were reactive with the 52kD Ro component by immunoblot. However, none of the 116 sera were reactive with the peptide spanning aa165-185 of the serotoninergic receptor. Due to discrepancies between studies involving reactivity of maternal antibodies and the 5-HT4 receptor, a reassessment was carried to assess the prevalence of anti-5-HT4 receptor autoantibodies in mothers of affected children [49]. This study showed that only 16% of the sera from mothers of children with congenital heart block were positive for anti-5-HT4 receptor autoantibodies. The discrepancy was attributed to differences in pH and epitope exposure in the ELISA plates. Nonetheless, it was concluded that additional risk factors are needed to contribute to the development of CHB. Although 5-HT4 receptors are present and functional in the human fetal heart, maternal antibodies to the 5-HT4 receptor were only rarely present in sera from affected children and do not seem to be strongly associated with the development of CHB [48].
Ca channel Hypothesis
The formulation of Ca channel hypothesis was driven by the fact that AV node electrogenesis is under the control of L-type Ca Channel which is responsible for conduction between the atria and the ventricle. Inhibition or blockade of this channel ultimately leads to AV block reminiscent of conduction abnormalities seen in CHB. The Ca channel hypothesis states that circulating maternal antibodies recognize L-type Ca channel pore forming protein α1-subunit to which they bind and prevent Ca ions from entering the myocyte [42]. Consequently, conduction of the SA impulse through the AV node to the ventricle will be hampered leading to delay in conduction or worst to complete AV block.
Voltage Gated Calcium Channels
Two L-type Ca channels are expressed in the heart are: Cav1.2 (α1C) and Cav1.3 (α1D). Cav1.2 or α1C is ubiquitously expressed in the heart and essentially mediates cardiac excitation-contraction coupling. Cav1.3 or α1D on the other hand, is restricted to the supraventricular tissue in the adult with the highest expression in the SA node and AV node. Cav1.2 ICa-L activates at more positive (−40 and −30 mV) potentials, and accounts for the conduction electrogenesis at the AV node, whereas Cav1.3 ICa-L activation occurs between −60 and −40 mV at a range in which diastolic depolarization at the SAN operates [50]. Thus, Cav1.3 ICa-L is essential for normal cardiac pacemaker activity. In fact, genetic deletion of Cav1.3 exhibits sinus bradycardia and AV blocks similar to what is reported in CHB [51]. It is therefore logical that Ca channel blockade at the SA and AV nodes by maternal anti-SSA/Ro –SSB/La antibodies will be expected to interrupt the sinus rhythm and conduction of the impulse to the ventricles.
Experimental models of congenital heart block
There are a number of in vivo and ex vivo models developed to understand the mechanism and the pathogenesis in CHB. The in vivo models are divided into passive and active immunization of female mice, rats or rabbits. The ex vivo studies reproduce CHB by adding maternal autoantibodies to Langendorff perfused hearts. This section will describe each model used with an overview of the data and their success in inducing conduction abnormalities
Passive model of CHB
A passive model of CHB was developed in BALB/c mice [3]. Timed pregnancies were injected with anti-SSA/Ro –SSB/La autoantibodies purified from human sera from mothers with CHB children. ECG screening of the pups from the injected mothers showed sinus bradycardia and AV block as compared to controls. IgG from mothers with CHB children induced I degree AV block in 88% (14/16), 90% (9/10) and 47% (14/30) in pups injected at 8, 11 or 16 days gestation, respectively [3]. Sinus bradycardia was also present in 44% (7/16), 70% (7/10) and 33% (10/30) of pups injected at 8, 11, or 16 days gestation [3]. Interestingly, no complete AV block was observed.
Active model of CHB
An active model of CHB was created by immunizing female mice with human or murine SSA/Ro 52, SSA/Ro 60 or SSB/La 48 antigens [8, 52]. A high incidence of advanced degrees of AV block (II and III) was observed with the immunization using SSA/Ro 52. The average of the incidence of I degree AV block in these models was around 21% which is similar to the incidence reported in humans. However the incidence of III degree AV block was 2.5%. Specifically, in the study by Miranda-Carus et al., the authors used female BALB/c mice that were immunized with human recombinant 48-kDa SSB/La, 60-kDa SSA/Ro, 52-kDa SSA/Ro (52α), and 52 β(amino acids 169–245 deleted) as well as with murine recombinant 52-kDa SSA/Ro. They showed that I degree AV block was detected in 7% of 27 pups born to mothers immunized with 48La, 20% of 54 pups born to 60Ro-immunized mothers, 6% of 56 pups born to 52α immunized mothers, 7% of 86 pups born to 52 β-immunized mothers, and 9% of 22 pups born to mothers immunized with murine 52Ro. Advanced conduction abnormalities were only identified in offspring of 52α- or 52 β-immunized mice. In the 52β group, five pups had complete block [8].
Using a peptide from the SSA/Ro 52 spanning amino acids 200-238 referred to as p200, I degree AV block was induced in 19% (10/52) rat pups [53]. Eftekhari et al. immunized female mice with four peptide from the putative cross reactive sites in the 5-HT4 serotonin receptor. They demonstrated that pups from mothers immunized with 5-HT4 peptides showed bradycardia and AV block (13% of pups) [54]. Immunization with SSA/Ro 52β in Balb/c mice induced AV block in 12% (10/86) in which 5/86 pups had complete III AV block [8]. SSA/Ro 60 and SSB/La 48 induce I degree AV block in 17% (16/97) for SSA/Ro 60 and 7% (4/55) for SSB/La (averaged total from 2 studies Miranda-Crus 1998 and Suzuki 2005 [55]). As suggested from human studies, SSA/Ro 60 and SSB/La 48 seem to be less associated with CHB induction [56, 57]. Xiao et al immunized female rabbits with human 52 kDa SSA/Ro and identified that 1 pup (0.7%) had second degree AV block with 2:1 patterns, 7 pups (4.6%) showed sinus bradycardia, 8 pups (5.3%) showed first degree AV block, and 5 pups (3.3%) had both sinus bradycardia and AV block. They also noticed that 31 out of 152 pups (20.4%) were born dead [52].
The results provide strong evidence for the pathogenic role of maternal autoantibodies, particularly anti-SSA/Ro 52 autoantibodies in the development of CHB.
Maternal antibodies induce sinus bradycardia and AV block in Langendorff perfused whole hearts
A question was raised whether direct perfusion of an isolated beating rat heart with maternal IgG would also result in electrocardiographic conduction abnormalities similar to those seen in affected infants. The effects of maternal IgG containing anti-SSA/Ro-SSB/La antibodies on the ECG recording of an isolated rat heart perfused by the Langendorff technique were assessed. Recordings were done using a conventional ECG machine in lead I. After 5 min of perfusion with maternal IgG (800 mg/ml), there was bradycardia associated with 2:1 second-degree AV block that degenerated into complete AV block at about 15 min of perfusion. The QRS complex is absent but the P waves were clearly seen. After 25 min of reperfusion with Tyrode’s solution, only partial recovery was seen. In contrast, perfusion of the heart with normal IgG from healthy mothers with healthy children did not alter ECG parameters. The sinus bradycardia and AV block was similarly demonstrated in Langendorff perfused human hearts [4] and by other groups [58–61].
Maternal antibodies specifically inhibit L-type Ca current (ICa-L ) in single cardiac myocytes
The effect of maternal IgG on the L-type Ca current was tested in ventricular cardiomyocytes, SA and AV nodal cells. Maternal antibodies inhibited the ICa-L by 62%, 46.2% and 51% in ventricular myocytes, SA and AV nodal cells respectively. The maternal antibodies had no effect on the transient outward K current (Ito) [62], the delayed rectifier K current (IKs) [62] and the fast Na current (INa) [62] indicating specificity for Ca channels.
Maternal antibodies autoantibodies inhibited Cav1.3 ICa-L in native cardiomyocytes
The challenge of investigating the electrophysiological effects of the autoantibodies on individual L-type Ca channels has been difficult because 1) both Cav1.2 and Cav1.3 Ca channels contribute to the total ICa-L [63] in the native cardiomyocytes, 2) both are sensitive to the Ca channel blockers and 3) there is no biophysical method to functionally separate the Cav1.2 from Cav1.3 Ca channels. The use of transgenic mouse models has been limited due to embryonic lethality at E14.5 of the Cav1.2 knockout mouse [63]. Therefore, the study and characterization of postnatal Cav1.3 in isolated cardiac cells by Cav1.2 gene deletion is not possible.
As a result, the study of Cav1.3 ICa-L has been limited to expression systems such as tsA201 cells which do not express endogenous Ca channels thus allowing for the individual expression of either Cav1.3 ICa-L or Cav1.2 ICa-L. In this regard, we recently reported a novel model of effective lentiviral silencing of Cav1.2 that allowed for the investigation and characterization of the Cav1.3 Ca channels in native cardiomyocytes [64]. The addition of maternal autoantibodies following the silencing of the Cav1.2 gene inhibited the Cav1.3 ICa-L by 35%. This observation provided the first evidence that the Cav1.3 ICa-L is inhibited in native cardiomyocytes.
Maternal antibodies directly recognize the L-type Ca channel proteins (Cav1.2 and Cva1.3)
To demonstrate that IgG from mothers with CHB children binds directly to the L-type Ca channels α1C and α1D protein, Western blot following immunoprecipitation was used. Immunoprecipitated α1C from human fetal hearts was probed using commercial anti-α1C antibody and IgG. Both recognized the same molecular weight band. This effect was not observed with IgG lacking the anti-SSA/Ro –SSB/La antibodies [52]. Similarly, cross-reactivity of maternal antibodies with L-type Ca channel α1D protein was detected. Western blot experiments were performed on proteins extract with IgG and anti- α1D antibody. Anti- α1D antibody recognized the 190-kDa band corresponding to the α1D Ca channel protein. IgG from mothers with CHB children but not healthy mothers recognized the same 190-kDa α1D Ca channel protein band.
To further dissect the site of action of maternal antibodies on the α1D subunit, we generated GST fusion proteins corresponding to the extracellular loop S5–S6 of each of the four domains that form the pore of the Ca channel α1D subunit and tested their reactivity with sera from mothers who have children with CHB. The results demonstrated that a fraction (14.4%) of maternal sera whose children have CHB reacted specifically with the extracellular loop of S5–S6 of the first, but not the second, third or fourth domain of the α1D subunit as demonstrated by both ELISA and Western blots [65]. Furthermore, the ELISA positive sera inhibited the expressed α1D Ca current. We concluded that the mere presence of anti-α1D Ca channel antibodies in the sera of mothers with CHB children suggest that additional risk factors may contribute to the pathogenesis of CHB.
Possible reversal of electrocardiographic abnormalities by upregulation of Ca channels
Recently, we hypothesize that if inhibition of the Ca current is critical in cardiac conduction disorders found in CHB patients, then upregulation of the Ca current should rescue or reverse the electrocardiographic abnormalities seen in CHB. The cardiac specific overexpression of the Cav1.2 α1C subunit of the L-type Ca channel was achieved. Detailed molecular, hemodynamic and electrophysiological characteristics of these transgenic (TG) mice are reported elsewhere [66]. Briefly, the Cav1.2 α1C transcript was increased by 2.8 fold. Similarly, the density of ICa-L increased by 44% to 52% in cardiac myocytes from the TG mice. This percent increase is ideal since maternal autoantibodies inhibit ICa-L within the same range, 40–60% [10, 62]. Therefore, we postulated that immunization of TG mice overexpressing Cav1.2 should give birth to pups with no or fewer electrocardiographic abnormalities. Indeed, a lesser degree of sinus bradycardia and fewer AV conduction abnormalities were observed in TG pups from immunized mothers overexpressing Cav1.2 (unpublished data). The finding that TG pups from Cav1.2 overexpression mothers had reduced conduction abnormalities following immunization with SSA/Ro and SSB/La, points to the importance of finding a suitable Ca channel agonist that could help infants diagnosed with CHB and ameliorate the severity of conduction abnormalities. The therapeutic implications can be directed towards the restoration of Ca channel function by the use of L-type Ca channel agonists which will only enhance the fetal, not the mother’s Ca channels. In this regard, we have recently shown that Bay K8644, an L-type Ca channel agonist, was not only able to reverse the inhibitory effect of maternal antibodies of Ca current but restored it above the basal level [10]. Efforts are being directed now towards the development of L-type Ca channel agonists which will only enhance the fetal Ca channels.
All together, the Ca channel hypothesis provides evidence supporting an etiologic role of maternal antibodies involvement in the pathogenesis of CHB and point to an essential role of Ca channels in the functional development of CHB.
Heart failure
It is interesting that around 10% of children with CHB not only exhibit conduction abnormalities but also mechanical failure leading to heart failure likely due to inhibition of the ventricular Ca channels. Cav1.2 is the major channel involved in the EC coupling. However, Cav1.3, which is absent in the adult ventricle, is expressed in the ventricles during fetal stage. It has been suggested that the presence of the Cav1.3 Ca channel in the ventricles during fetal life is needed to aid in the EC coupling since fetal cardiomyocytes lacks the normal Ca induced Ca release (CICR) found in the adult heart. CICR plays a critical role in cardiac contractile function as it is a major source of Ca. However, there are striking morphological and functional differences between the mature and developing heart sarcoplasmic reticulum (SR). In contrast to the adult heart, the SR is sparse and less functional during fetal life. It has been proposed that as a consequence of the underdeveloped SR, the sarcolemmal Ca channels play a major role for Ca delivery to the contractile proteins in the fetal heart. Therefore, the presence of both Cav1.2 and Cav1.3 during fetal stages is necessary to compensate for the lack of a developed SR in order to trigger the contractile function of the myocardium. Conversely, blockade of the Ca channels in the fetal heart by maternal antibodies will diminish the contractile force and worsen cardiac function eventually leading to heart failure even in the presence of a pacemaker.
Proposed Hypothesis for the Pathogenesis of CHB
Two distinct consequences of Ca channel blockade by maternal antibodies can be identified and are summarized in Figure 1: Acute (minutes) and chronic (weeks) effects. We proposed that the in utero consequences of this chronic inhibition of ICa-L could lead to the following scenario: Fetal heart Ca channels are chronically exposed to maternal antibodies during pregnancy (starting at about the 12th week of gestation when significant amounts of IgGs are detected). Our hypothesis is that this chronic exposure of Ca channels to maternal antibodies could lead to internalization, degradation and eventually to cell death since Ca channels, as pointed earlier, have been reported to play a vital role in fetal EC coupling. This is consistent with the finding that cross-linking of adjacent ion channels by the two Fab arms of IgG increases the rate of normal internalization of the target protein/antibody complex and thereby decreases the channel density on the cell surface [67]. Cells from fetuses whose Ca channel density is too low (step 4) will eventually die and those whose Ca channel density, although reduced (step 3 and 3a), but still provides the necessary Ca entry for cell function will survive (3a) and will allow AV conduction to occur. This scenario could account for the discordance in twins. Ca dysregulation/apoptosis (5) could favor the translocation of Ro/La to the membrane where they are accessible to maternal antibodies (6) leading to inflammation, cell death, fibrosis and calcification. All together, autoantibodies and Ca channels are causally related to the development of CHB but the range and frequency of conduction defects suggest that additional factors may be necessary to explain the full spectrum of CHB.
Figure 1.
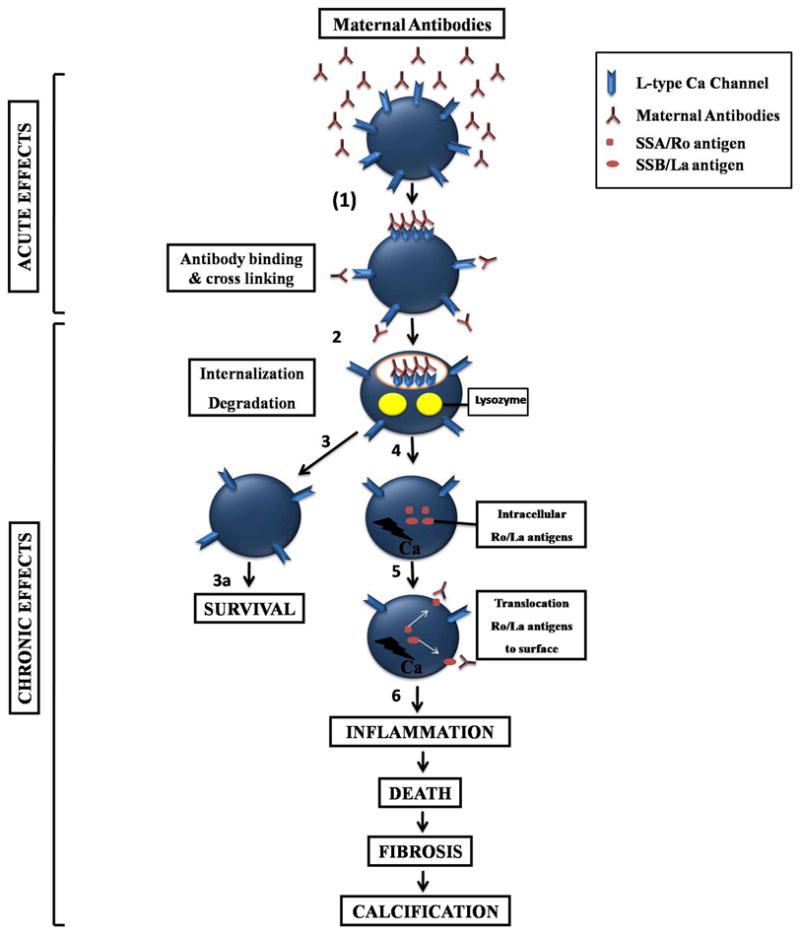
Schematic representation of maternal antibodies interaction with cardiac Ca channels. The acute events are initiated by binding of the circulating maternal antibodies to surface L-type Ca channels. This leads to cross-linking the antibody-antigen complexes (step 1). As a result of the interaction, the surface complex is internalized and degraded by lysozymes (step 2). In step 3 and 3a, two scenarios are proposed: fetal cells with enough remaining Ca channels will survive (fetal discordance of twins) and secure atrioventricular conduction; those with severe depletion of Ca channels (step 4) will result in Ca dysregulation and apoptosis (step 5). This may lead to the translocation of the intracellular SSA/Ro and SSB/La to the cell surface to bind to their cognate antibodies (step 5). Altogether, this process will eventually trigger inflammation (step 6), cell death, fibrosis and calcification (see text for more details).
Future directions.
During the recent few years, significant advances have been made in the understanding of the pathogenesis of CHB. The limitations associated with each of the above hypotheses to explain the full spectrum of CHB will likely guide the future efforts in the search for a unifying hypothesis for CHB pathophysiology. Indeed none of the above mentioned mechanisms can yet account for the low incidence of CHB in infants from mothers with Ro/La antibodies and for the vulnerability of the fetal, but not the mother’s heart to CHB. Identification of the extracellular loop of domain I S5-S6 of L-type Ca channel Cav1.3 subunit as a target for autoantibodies from mothers with CHB children will provide novel insights into the development of therapeutic peptides that could bind to the pathogenic antibodies, thereby, preventing the initiation and progression of CHB. The finding that Cav1.2 overexpression mice reduced the conduction abnormalities following immunization with SSA/Ro and SSB/LA, points to the importance of finding a suitable Ca channel agonist that could help infants diagnosed with CHB and reverse the severity of conduction abnormalities.
Acknowledgments
SOURCES OF FUNDING
This work was supported by the National Institutes of Health (R01-HL-077494) and the Veterans Affairs MERIT grants to Dr. Boutjdir.
ACKNOWLEDGMENTS
None
Footnotes
CONFLICT
No Conflict to disclose
References
- 1.Waltuck J, Buyon JP. Autoantibody-associated congenital heart block: outcome in mothers and children. Ann Intern Med. 1994 Apr 1;120:544–51. doi: 10.7326/0003-4819-120-7-199404010-00003. [DOI] [PubMed] [Google Scholar]
- 2.Anandakumar C, Biswas A, Chew SS, Chia D, Wong YC, Ratnam SS. Direct fetal therapy for hydrops secondary to congenital atrioventricular heart block. Obstet Gynecol. 1996 May;87:835–7. [PubMed] [Google Scholar]
- 3.Mazel JA, El-Sherif N, Buyon J, Boutjdir M. Electrocardiographic abnormalities in a murine model injected with IgG from mothers of children with congenital heart block. Circulation. 1999 Apr 13;99:1914–8. doi: 10.1161/01.cir.99.14.1914. [DOI] [PubMed] [Google Scholar]
- 4.Boutjdir M, Chen L, Zhang ZH, et al. Arrhythmogenicity of IgG and anti-52-kD SSA/Ro affinity-purified antibodies from mothers of children with congenital heart block. Circ Res. 1997 Mar;80:354–62. doi: 10.1161/01.res.80.3.354. [DOI] [PubMed] [Google Scholar]
- 5.Brucato A, Cimaz R, Catelli L, Meroni P. Anti-Ro-associated sinus bradycardia in newborns. Circulation. 2000 Sep 12;102:E88–9. doi: 10.1161/01.cir.102.11.e88. [DOI] [PubMed] [Google Scholar]
- 6.Menon A, Silverman ED, Gow RM, Hamilton RM. Chronotropic competence of the sinus node in congenital complete heart block. Am J Cardiol. 1998 Nov 1;82:1119–21. A9. doi: 10.1016/s0002-9149(98)00569-4. [DOI] [PubMed] [Google Scholar]
- 7.Litsey SE, Noonan JA, O’Connor WN, Cottrill CM, Mitchell B. Maternal connective tissue disease and congenital heart block. Demonstration of immunoglobulin in cardiac tissue. N Engl J Med. 1985 Jan 10;312:98–100. doi: 10.1056/NEJM198501103120206. [DOI] [PubMed] [Google Scholar]
- 8.Miranda-Carus ME, Boutjdir M, Tseng CE, DiDonato F, Chan EK, Buyon JP. Induction of antibodies reactive with SSA/Ro-SSB/La and development of congenital heart block in a murine model. J Immunol. 1998 Dec 1;161:5886–92. [PubMed] [Google Scholar]
- 9.Hu K, Qu Y, Yue Y, Boutjdir M. Functional basis of sinus bradycardia in congenital heart block. Circ Res. 2004 Mar 5;94:e32–8. doi: 10.1161/01.RES.0000121566.01778.06. [DOI] [PubMed] [Google Scholar]
- 10.Qu Y, Baroudi G, Yue Y, Boutjdir M. Novel molecular mechanism involving alpha1D (Cav1.3) L-type calcium channel in autoimmune-associated sinus bradycardia. Circulation. 2005 Jun 14;111:3034–41. doi: 10.1161/CIRCULATIONAHA.104.517326. [DOI] [PubMed] [Google Scholar]
- 11.Buyon JP. Neonatal lupus and autoantibodies reactive with SSA/Ro-SSB/La. Scand J Rheumatol Suppl. 1998;107:23–30. doi: 10.1080/03009742.1998.11720702. [DOI] [PubMed] [Google Scholar]
- 12.Watson RM, Lane AT, Barnett NK, Bias WB, Arnett FC, Provost TT. Neonatal lupus erythematosus. A clinical, serological and immunogenetic study with review of the literature. Medicine (Baltimore) 1984 Nov;63:362–78. [PubMed] [Google Scholar]
- 13.Laxer RM, Roberts EA, Gross KR, et al. Liver disease in neonatal lupus erythematosus. J Pediatr. 1990 Feb;116:238–42. doi: 10.1016/s0022-3476(05)82880-x. [DOI] [PubMed] [Google Scholar]
- 14.O’Neill TW, Mahmoud A, Tooke A, Thomas RD, Maddison PJ. Is there a relationship between subclinical myocardial abnormalities, conduction defects and Ro/La antibodies in adults with systemic lupus erythematosus? Clin Exp Rheumatol. 1993 Jul–Aug;11:409–12. [PubMed] [Google Scholar]
- 15.Behan WM, Behan PO, Reid JM, Doig W, Gairns J. Family studies of congenital heart block associated with Ro antibody. Br Heart J. 1989 Oct;62:320–4. doi: 10.1136/hrt.62.4.320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dorner T, Hiepe F, Goldner B, Apostoloff E. Investigations into Ro-specific antibody-associated congenital cardiac conduction defects. Clin Investig. 1992 Jun;70:492–6. doi: 10.1007/BF00210230. [DOI] [PubMed] [Google Scholar]
- 17.Jaeggi ET, Hornberger LK, Smallhorn JF, Fouron JC. Prenatal diagnosis of complete atrioventricular block associated with structural heart disease: combined experience of two tertiary care centers and review of the literature. Ultrasound Obstet Gynecol. 2005 Jul;26:16–21. doi: 10.1002/uog.1919. [DOI] [PubMed] [Google Scholar]
- 18.Jaeggi ET, Hamilton RM, Silverman ED, Zamora SA, Hornberger LK. Outcome of children with fetal, neonatal or childhood diagnosis of isolated congenital atrioventricular block. A single institution’s experience of 30 years. J Am Coll Cardiol. 2002 Jan 2;39:130–7. doi: 10.1016/s0735-1097(01)01697-7. [DOI] [PubMed] [Google Scholar]
- 19.Ho SY, Esscher E, Anderson RH, Michaelsson M. Anatomy of congenital complete heart block and relation to maternal anti-Ro antibodies. Am J Cardiol. 1986 Aug 1;58:291–4. doi: 10.1016/0002-9149(86)90064-0. [DOI] [PubMed] [Google Scholar]
- 20.Lee LA, Coulter S, Erner S, Chu H. Cardiac immunoglobulin deposition in congenital heart block associated with maternal anti-Ro autoantibodies. Am J Med. 1987 Oct;83:793–6. doi: 10.1016/0002-9343(87)90918-1. [DOI] [PubMed] [Google Scholar]
- 21.Clancy RM, Kapur RP, Molad Y, Askanase AD, Buyon JP. Immunohistologic evidence supports apoptosis, IgG deposition, and novel macrophage/fibroblast crosstalk in the pathologic cascade leading to congenital heart block. Arthritis Rheum. 2004 Jan;50:173–82. doi: 10.1002/art.11430. [DOI] [PubMed] [Google Scholar]
- 22.Reichlin M, Brucato A, Frank MB, et al. Concentration of autoantibodies to native 60-kd Ro/SS-A and denatured 52-kd Ro/SS-A in eluates from the heart of a child who died with congenital complete heart block. Arthritis Rheum. 1994 Nov;37:1698–703. doi: 10.1002/art.1780371120. [DOI] [PubMed] [Google Scholar]
- 23.Meckler KA, Kapur RP. Congenital heart block and associated cardiac pathology in neonatal lupus syndrome. Pediatr Dev Pathol. 1998 Mar-Apr;1:136–42. doi: 10.1007/s100249900017. [DOI] [PubMed] [Google Scholar]
- 24.Piercecchi-Marti MD, Mohamed H, Chau C, Liprandi A, Fredouille C. Congenital atrioventricular block: histological aspects. Forensic Sci Int. 2003 Sep 9;136:12–5. doi: 10.1016/s0379-0738(03)00224-x. [DOI] [PubMed] [Google Scholar]
- 25.Eronen M, Heikkila P, Teramo K. Congenital complete heart block in the fetus: hemodynamic features, antenatal treatment, and outcome in six cases. Pediatr Cardiol. 2001 Sep-Oct;22:385–92. doi: 10.1007/s002460010256. [DOI] [PubMed] [Google Scholar]
- 26.Taylor-Albert E, Reichlin M, Toews WH, Overholt ED, Lee LA. Delayed dilated cardiomyopathy as a manifestation of neonatal lupus: case reports, autoantibody analysis, and management. Pediatrics. 1997 May;99:733–5. doi: 10.1542/peds.99.5.733. [DOI] [PubMed] [Google Scholar]
- 27.Nield LE, Silverman ED, Smallhorn JF, et al. Endocardial fibroelastosis associated with maternal anti-Ro and anti-La antibodies in the absence of atrioventricular block. J Am Coll Cardiol. 2002 Aug 21;40:796–802. doi: 10.1016/s0735-1097(02)02004-1. [DOI] [PubMed] [Google Scholar]
- 28.Nield LE, Silverman ED, Taylor GP, et al. Maternal anti-Ro and anti-La antibody-associated endocardial fibroelastosis. Circulation. 2002 Feb 19;105:843–8. doi: 10.1161/hc0702.104182. [DOI] [PubMed] [Google Scholar]
- 29.Gordon PA, Khamashta MA, Hughes GR, Rosenthal E. Increase in the heart rate-corrected QT interval in children of anti-Ro-positive mothers, with a further increase in those with siblings with congenital heart block: comment on the article by Cimaz et al. Arthritis Rheum. 2001 Jan;44:242–3. doi: 10.1002/1529-0131(200101)44:1<242::AID-ANR34>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 30.Cimaz R, Stramba-Badiale M, Brucato A, Catelli L, Panzeri P, Meroni PL. QT interval prolongation in asymptomatic anti-SSA/Ro-positive infants without congenital heart block. Arthritis Rheum. 2000 May;43:1049–53. doi: 10.1002/1529-0131(200005)43:5<1049::AID-ANR13>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 31.Michaelsson M, Engle MA. Congenital complete heart block: an international study of the natural history. Cardiovasc Clin. 1972;4:85–101. [PubMed] [Google Scholar]
- 32.Buyon JP, Hiebert R, Copel J, et al. Autoimmune-associated congenital heart block: demographics, mortality, morbidity and recurrence rates obtained from a national neonatal lupus registry. J Am Coll Cardiol. 1998 Jun;31:1658–66. doi: 10.1016/s0735-1097(98)00161-2. [DOI] [PubMed] [Google Scholar]
- 33.Hamilton RMGR. Disorders of heart rate and rhythm. London: Springer-Verlag Ltd., Neonatal Heart Disease; [Google Scholar]
- 34.Buyon JPCR. Neonatal Lupus. 7. Philadelphia: Lippincott Williams & Wilkins; 2006. [Google Scholar]
- 35.Siren MK, Julkunen H, Kaaja R. The increasing incidence of isolated congenital heart block in Finland. J Rheumatol. 1998 Sep;25:1862–4. [PubMed] [Google Scholar]
- 36.Eronen M, Siren MK, Ekblad H, Tikanoja T, Julkunen H, Paavilainen T. Short-and long-term outcome of children with congenital complete heart block diagnosed in utero or as a newborn. Pediatrics. 2000 Jul;106:86–91. doi: 10.1542/peds.106.1.86. [DOI] [PubMed] [Google Scholar]
- 37.Brucato A, Gasparini M, Vignati G, et al. Isolated congenital complete heart block: longterm outcome of children and immunogenetic study. J Rheumatol. 1995 Mar;22:541–3. [PubMed] [Google Scholar]
- 38.Michaelsson M, Riesenfeld T, Jonzon A. Natural history of congenital complete atrioventricular block. Pacing Clin Electrophysiol. 1997 Aug;20:2098–101. doi: 10.1111/j.1540-8159.1997.tb03636.x. [DOI] [PubMed] [Google Scholar]
- 39.Guereta LG, Burgueros M, Moreno F. Outcome of isolated congenital heart block diagnosed in utero. Heart. 1997 Jul;78:95–6. doi: 10.1136/hrt.78.1.95-a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Miranda ME, Tseng CE, Rashbaum W, et al. Accessibility of SSA/Ro and SSB/La antigens to maternal autoantibodies in apoptotic human fetal cardiac myocytes. J Immunol. 1998 Nov 1;161:5061–9. [PubMed] [Google Scholar]
- 41.Eftekhari P, Salle L, Lezoualc’h F, et al. Anti-SSA/Ro52 autoantibodies blocking the cardiac 5-HT4 serotoninergic receptor could explain neonatal lupus congenital heart block. Eur J Immunol. 2000 Oct;30:2782–90. doi: 10.1002/1521-4141(200010)30:10<2782::AID-IMMU2782>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 42.Boutjdir M. Molecular and ionic basis of congenital complete heart block. Trends Cardiovasc Med. 2000 Apr;10:114–22. doi: 10.1016/s1050-1738(00)00059-1. [DOI] [PubMed] [Google Scholar]
- 43.Miranda-Carus ME, Askanase AD, Clancy RM, et al. Anti-SSA/Ro and anti-SSB/La autoantibodies bind the surface of apoptotic fetal cardiocytes and promote secretion of TNF-alpha by macrophages. J Immunol. 2000 Nov 1;165:5345–51. doi: 10.4049/jimmunol.165.9.5345. [DOI] [PubMed] [Google Scholar]
- 44.Clancy RM, Neufing PJ, Zheng P, et al. Impaired clearance of apoptotic cardiocytes is linked to anti-SSA/Ro and -SSB/La antibodies in the pathogenesis of congenital heart block. J Clin Invest. 2006 Sep;116:2413–22. doi: 10.1172/JCI27803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tran HB, Ohlsson M, Beroukas D, et al. Subcellular redistribution of la/SSB autoantigen during physiologic apoptosis in the fetal mouse heart and conduction system: a clue to the pathogenesis of congenital heart block. Arthritis Rheum. 2002 Jan;46:202–8. doi: 10.1002/1529-0131(200201)46:1<202::AID-ART10062>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 46.Baboonian C, Venables PJ, Booth J, Williams DG, Roffe LM, Maini RN. Virus infection induces redistribution and membrane localization of the nuclear antigen La (SS-B): a possible mechanism for autoimmunity. Clin Exp Immunol. 1989 Dec;78:454–9. [PMC free article] [PubMed] [Google Scholar]
- 47.Furukawa F, Kashihara-Sawami M, Lyons MB, Norris DA. Binding of antibodies to the extractable nuclear antigens SS-A/Ro and SS-B/La is induced on the surface of human keratinocytes by ultraviolet light (UVL): implications for the pathogenesis of photosensitive cutaneous lupus. J Invest Dermatol. 1990 Jan;94:77–85. doi: 10.1111/1523-1747.ep12873930. [DOI] [PubMed] [Google Scholar]
- 48.Buyon JP, Clancy R, Di Donato F, et al. Cardiac 5-HT(4) serotoninergic receptors, 52kD SSA/Ro and autoimmune-associated congenital heart block. J Autoimmun. 2002 Aug-Sep;19:79–86. doi: 10.1006/jaut.2002.0594. [DOI] [PubMed] [Google Scholar]
- 49.Kamel R, Eftekhari P, Clancy R, Buyon JP, Hoebeke J. Autoantibodies against the serotoninergic 5-HT4 receptor and congenital heart block: a reassessment. J Autoimmun. 2005 Aug;25:72–6. doi: 10.1016/j.jaut.2005.04.005. [DOI] [PubMed] [Google Scholar]
- 50.Koschak A, Reimer D, Huber I, et al. alpha 1D (Cav1.3) subunits can form l-type Ca2+ channels activating at negative voltages. J Biol Chem. 2001 Jun 22;276:22100–6. doi: 10.1074/jbc.M101469200. [DOI] [PubMed] [Google Scholar]
- 51.Mangoni ME, Couette B, Bourinet E, et al. Functional role of L-type Cav1.3 Ca2+ channels in cardiac pacemaker activity. Proc Natl Acad Sci U S A. 2003 Apr 29;100:5543–8. doi: 10.1073/pnas.0935295100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Xiao GQ, Qu Y, Hu K, Boutjdir M. Down-regulation of L-type calcium channel in pups born to 52 kDa SSA/Ro immunized rabbits. FASEB J. 2001 Jul;15:1539–45. doi: 10.1096/fj.01-0052com. [DOI] [PubMed] [Google Scholar]
- 53.Salomonsson S, Sonesson SE, Ottosson L, et al. Ro/SSA autoantibodies directly bind cardiomyocytes, disturb calcium homeostasis, and mediate congenital heart block. J Exp Med. 2005 Jan 3;201:11–7. doi: 10.1084/jem.20041859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Eftekhari P, Roegel JC, Lezoualc’h F, Fischmeister R, Imbs JL, Hoebeke J. Induction of neonatal lupus in pups of mice immunized with synthetic peptides derived from amino acid sequences of the serotoninergic 5-HT4 receptor. Eur J Immunol. 2001 Feb;31:573–9. doi: 10.1002/1521-4141(200102)31:2<573::aid-immu573>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 55.Suzuki H, Silverman ED, Wu X, et al. Effect of maternal autoantibodies on fetal cardiac conduction: an experimental murine model. Pediatr Res. 2005 Apr;57:557–62. doi: 10.1203/01.PDR.0000155947.82365.E4. [DOI] [PubMed] [Google Scholar]
- 56.Julkunen H, Miettinen A, Walle TK, Chan EK, Eronen M. Autoimmune response in mothers of children with congenital and postnatally diagnosed isolated heart block: a population based study. J Rheumatol. 2004 Jan;31:183–9. [PubMed] [Google Scholar]
- 57.Salomonsson S, Dorner T, Theander E, Bremme K, Larsson P, Wahren-Herlenius M. A serologic marker for fetal risk of congenital heart block. Arthritis Rheum. 2002 May;46:1233–41. doi: 10.1002/art.10232. [DOI] [PubMed] [Google Scholar]
- 58.Restivo M, Kozhevnikov DO, Boutjdir M. Optical mapping of activation patterns in an animal model of congenital heart block. Am J Physiol Heart Circ Physiol. 2001 Apr;280:H1889–95. doi: 10.1152/ajpheart.2001.280.4.H1889. [DOI] [PubMed] [Google Scholar]
- 59.Garcia S, Nascimento JH, Bonfa E, et al. Cellular mechanism of the conduction abnormalities induced by serum from anti-Ro/SSA-positive patients in rabbit hearts. J Clin Invest. 1994 Feb;93:718–24. doi: 10.1172/JCI117025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hamilton RM, Lee-Poy M, Kruger K, Silverman ED. Investigative methods of congenital complete heart block. J Electrocardiol. 1998;30( Suppl):69–74. doi: 10.1016/s0022-0736(98)80035-6. [DOI] [PubMed] [Google Scholar]
- 61.Viana VS, Garcia S, Nascimento JH, et al. Induction of in vitro heart block is not restricted to affinity purified anti-52 kDa Ro/SSA antibody from mothers of children with neonatal lupus. Lupus. 1998;7:141–7. doi: 10.1191/096120398678919895. [DOI] [PubMed] [Google Scholar]
- 62.Boutjdir M, Chen L, Zhang ZH, Tseng CE, El-Sherif N, Buyon JP. Serum and immunoglobulin G from the mother of a child with congenital heart block induce conduction abnormalities and inhibit L-type calcium channels in a rat heart model. Pediatr Res. 1998 Jul;44:11–9. doi: 10.1203/00006450-199807000-00002. [DOI] [PubMed] [Google Scholar]
- 63.Seisenberger C, Specht V, Welling A, et al. Functional embryonic cardiomyocytes after disruption of the L-type alpha1C (Cav1.2) calcium channel gene in the mouse. J Biol Chem. 2000 Dec 15;275:39193–9. doi: 10.1074/jbc.M006467200. [DOI] [PubMed] [Google Scholar]
- 64.Karnabi E, Qu Y, Mancarella S, Yue Y, Wadgaonkar R, Boutjdir M. Silencing of Cav1.2 gene in neonatal cardiomyocytes by lentiviral delivered shRNA. Biochem Biophys Res Commun. 2009 Jul 10;384:409–14. doi: 10.1016/j.bbrc.2009.04.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Karnabi E, Qu Y, Wadgaonkar R, et al. Congenital Heart Block: Identification of Autoantibody Binding Site on the Extracellular Loop (Domain I, S5-S6) of α1D L-type Ca Channel. Journal of Autoimmunity. 2009 doi: 10.1016/j.jaut.2009.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Muth JN, Yamaguchi H, Mikala G, et al. Cardiac-specific overexpression of the alpha(1) subunit of the L-type voltage-dependent Ca(2+) channel in transgenic mice. Loss of isoproterenol-induced contraction. J Biol Chem. 1999 Jul 30;274:21503–6. doi: 10.1074/jbc.274.31.21503. [DOI] [PubMed] [Google Scholar]
- 67.Lennon VA, Kryzer TJ, Griesmann GE, et al. Calcium-channel antibodies in the Lambert-Eaton syndrome and other paraneoplastic syndromes. N Engl J Med. 1995 Jun 1;332:1467–74. doi: 10.1056/NEJM199506013322203. [DOI] [PubMed] [Google Scholar]