

Abstract
Deficits in synaptic function, particularly through NMDA receptors (NMDARs), are linked to late-stage cognitive impairments in Alzheimer's disease (AD). At earlier disease stages, however, there is evidence for altered endoplasmic reticulum (ER) calcium signaling in human cases and in neurons from AD mouse models. Despite the fundamental importance of calcium to synaptic function, neither the extent of ER calcium dysregulation in dendrites nor its interaction with synaptic function in AD pathophysiology is known. Identifying the mechanisms underlying early synaptic calcium dysregulation in AD pathogenesis is likely a key component to understanding, and thereby preventing, the synapse loss and downstream cognitive impairments. Using two-photon calcium imaging, flash photolysis of caged glutamate, and patch-clamp electrophysiology in cortical brain slices, we examined interactions between synaptically and ER-evoked calcium release at glutamatergic synapses in young AD transgenic mice. We found increased ryanodine receptor-evoked calcium signals within dendritic spine heads, dendritic processes, and the soma of pyramidal neurons from 3xTg-AD and TAS/TPM AD mice relative to NonTg controls. In addition, synaptically evoked postsynaptic calcium responses were larger in the AD strains, as were calcium signals generated from NMDAR activation. However, calcium responses triggered by back-propagating action potentials were not different. Concurrent activation of ryanodine receptors (RyRs) with either synaptic or NMDAR stimulation generated a supra-additive calcium response in the AD strains, suggesting an aberrant calcium-induced calcium release (CICR) effect within spines and dendrites. We propose that presenilin-linked disruptions in RyR signaling and subsequent CICR via NMDAR-mediated calcium influx alters synaptic function and serves as an early pathogenic factor in AD.
Introduction
Neurons use calcium for a myriad of roles, with functional specificity often determined by cellular localization; for example, calcium release in the nucleus may drive gene transcription, whereas in dendrites it modulates synaptic transmission and plasticity (Collin et al., 2005; Berridge, 2006). The source of calcium is also an important factor: extracellular calcium entry occurs through plasma membrane channels such as NMDA receptors (NMDARs), and voltage-gated calcium channels (VGCCs), whereas intracellular reserves from the endoplasmic reticulum (ER) are liberated through inositol triphosphate (IP3R) or ryanodine receptors (RyRs) (Verkhratsky, 2005). Importantly, calcium exposure at these channels can facilitate subsequent release through the process of calcium-induced calcium release (CICR). Given the complexity of interactions among calcium sources and their regenerative potential, neurons tightly regulate calcium signaling to ensure proper function and viability.
Dysregulated calcium signaling is linked to Alzheimer's disease (AD) pathogenesis through a variety of mechanisms (LaFerla, 2002; Stutzmann, 2007; Bezprozvanny and Mattson, 2008). The most pronounced effects are associated with mutant presenilin (PS), which causes early-onset AD and increases ER calcium release. In recent studies, we proposed that RyR-evoked calcium release is preferentially enhanced by PS mutations through increased RyR2 expression (Stutzmann et al., 2006; Chakroborty et al., 2009), whereas channel studies have shown that mutant PS alters IP3R gating properties (Cheung et al., 2008, 2010) or ER leak channel function (Tu et al., 2006; Nelson et al., 2007). Regardless of mechanism, the functional implications of increased CICR in 3xTg-AD mouse models are pronounced, resulting in aberrant recruitment of RyR-calcium stores in basal synaptic transmission, SK channel activation, enhanced presynaptic vesicle release, and long-term plasticity in hippocampal neurons (Chakroborty et al., 2009). These signaling changes are present before β-amyloid formation, tau deposits, or memory deficits, and may represent early pathogenic processes that contribute to the histopathological and synaptic deficits that define the later disease stages.
What is not understood is how RyR-mediated calcium increases are triggered in spines and dendrites, and how this affects synaptic responses in AD mice. Therefore, in this study, we examine interactions among several calcium sources in pyramidal neurons from AD and NonTg mouse models, and compare the RyR-mediated contribution in each to gauge differences in calcium dynamics within neuronal microdomains. We found that in mutant PS1-expressing neurons, there is a profound RyR-mediated calcium increase within dendritic processes and spines that can be elicited through direct RyR activation, basal synaptic transmission, NMDAR activation, and synergistic interactions among these stimuli. Notably, calcium signals triggered by VGCC activity are not upregulated, suggesting that enhanced intracellular calcium signals are mediated largely through glutamatergic pathways. Particularly relevant for AD is the involvement of NMDAR-calcium influx and subsequent CICR via aberrant RyR recruitment. This is consistent with a larger body of literature targeting NMDAR-mediated toxicity in AD pathology (Harkany et al., 1999; Shankar et al., 2007; Bezprozvanny and Mattson, 2008; Demuro et al., 2010) and the hypothesis that breakdown of synapses is a causative factor in AD-linked cognitive decline (Terry et al., 1991; Masliah, 1995; Selkoe, 2008).
Materials and Methods
Transgenic AD mice.
Two AD mouse models were used in this study to validate and confirm the aberrant calcium dynamics in more than model: (1) 3xTg-AD mice generated from the PS1M146VKI mouse and also expressing APPSWE and TauP130L (Oddo et al., 2003); and (2) TAS/TPM double-transgenic mice coexpressing PS1M146V and APPSWE mutations (Howlett et al., 2004). Age-matched (4–6 weeks old) NonTg control mice, based on the same background strain (J29/C57BL6), also were used. Both male and female mice were used in these studies. Animals were cared for and used in accordance with protocols approved by Rosalind Franklin University of Medicine and Science Animal Care and Use Committee.
Electrophysiology.
Brain slices (300 μm) containing the prefrontal cortex were prepared as previously described (Stutzmann et al., 2004) and superfused at 2 ml/min with standard artificial CSF (ACSF) solution containing the following (in mm): 125 NaCl, 2.5 KCl, 2 CaCl2, 1.2 MgSO4, 1.25 NaH2PO4, 25.0 NaHCO3, 10 d-dextrose and 0.05 picrotoxin (Sigma-Aldrich) and equilibrated with 95% O2 and 5% CO2 (pH 7.3–7.4) at room temperature (27°C). Osmolarity was maintained at 310 mOsm. Patch pipettes (4–5 MΩ) were filled with intracellular solution containing the following substances (in mm): 135 K-gluconate, 10 HEPES, 10 Na-phosphocreatine, 2 MgCl2, 4 NaATP, and 0.4 NaGTP, pH adjusted to 7.3–7.4 with KOH (Sigma), and 50 μm fura-2 (Invitrogen). In experiments using photolysis of Montreal Neurological Institute (MNI)-caged-l-glutamate (500 μm; Tocris Bioscience) for NMDAR activation, K-gluconate was replaced with Cs-methylsulfonate in the intracellular solution, and the extracellular solution contained the following (in μm): CNQX 20, picrotoxin 20, and TTX 1 (all from Sigma-Aldrich).
Prefrontal cortical layer V pyramidal neurons were identified visually via infrared (IR) differential interference contrast optics, and electrophysiologically by their passive membrane properties and spike frequency accommodation. Membrane potentials were obtained in current-clamp mode acquired at 10 kHz with a Digidata 1322 A-D converter and Multiclamp 700B amplifier, and were recorded and analyzed using pClamp 10.2 (Molecular Devices). Series resistance was monitored throughout the experiment and was <10 MΩ. Synaptic stimulation of layer V cortical pyramidal neurons was accomplished by placing a monopolar stimulating electrode in cortical layer II ∼50–100 μm lateral to the apical dendrite of the layer V neuron. Current pulses were generated using an A360 stimulus isolator (WPI) with a pulse duration of 100 μs and intensity ranging from 25 to 75 μA. In all experiments, the stimulus intensity was adjusted to evoke a standard reference EPSP of ∼4 mV before treatment. Membrane potential was continuously monitored, and spontaneous EPSPs were recorded in current-clamp mode concurrent with calcium imaging. To verify glutamatergic transmission, EPSPs were blocked by addition of 20 μm CNQX (Sigma-Aldrich) at the conclusion of each experiment. Analysis of spontaneous EPSPs was accomplished with MiniAnalysis software (Synaptosoft).
Calcium imaging and flash photolysis.
Ca2+ imaging within individual neurons was performed in brain slice preparations using a custom-made video-rate multiphoton-imaging system based on an upright Olympus BX51 microscope frame (Stutzmann and Parker, 2005). Individual neurons were filled with the calcium indicator, fura-2 (50 μm) via the patch pipette as described previously (Stutzmann et al., 2004). In these studies, fura-2 is not used as a ratiometric dye (primarily because the Ti:sapphire laser cannot tune between two excitation wavelengths rapidly enough), but, rather, excitation at a single wavelength is used to measure relative changes in fluorescence. We excite fura-2 at the equivalent 380 nm wavelength, which generates a bright signal at low calcium levels, allowing us to clearly see small compartments such as spines at resting levels. Upon calcium increases, fluorescence decreases, but it is the relative change in fluorescence that is measured. Laser excitation was provided by trains (80 MHz) of ∼100 frames/s pulses at 780 nm from a Ti:sapphire laser (Mai Tai Broadband, Spectra-Physics). The laser beam was scanned by a resonant galvanometer (General Scanning Lumonics), allowing rapid (7.9 kHz) bidirectional scanning in the x-axis and by a conventional linear galvanometer in the y-axis, to provide a full-frame scan rate of 30 frames/s. The laser beam was focused onto the tissue through a 40× water-immersion objective (numerical aperture = 0.8). Emitted fluorescence light was detected by a wide-field photomultiplier (Electron Tubes) to derive a video signal that was captured and analyzed by Video Savant 5.0 software (IO Industries). Further analysis of background corrected images was performed using MetaMorph software. For clarity, pseudocolored images of fura-2 fluorescence are expressed as inverse pseudo-ratios so that increases in [Ca2+] correspond to increasing ratios: F0/ΔF (where F0 is the average resting fluorescence at baseline, and ΔF is the decrease of fluorescence upon Ca2+ release). Data indicating relative percentage changes in fluorescent intensity were calculated as the percentage over baseline: (F0/ΔF − 1)*100. UV flash photolysis was accomplished using an X-Cite 120 Fluorescence Illumination system (Photonic Solution) and UV filter cube (340–400 nm) in a light path separate from the IR laser input, with exposure time controlled through electronic shutters (Uniblitz) operated and synchronized through digital outputs (Digidata 1322) controlled by pClamp 10 software.
Statistics.
Unless otherwise specified, comparison of data across the three transgenic groups used a one-way ANOVA with a Tukey post hoc analysis to determine significance between groups. For analysis of cumulative event histograms, the Kolmogorov–Smirnov (KS) test was used. Statistical significance was set at p < 0.05 or z < 0.05.
Immunoblot analysis.
Cortical tissue was harvested from 1- to 3-month-old 3xTg and NonTg animals. Tissue was homogenized on ice in Tissue Protein Extraction Reagent (Invitrogen) containing protease inhibitors (Rouche). Total cortical protein was quantified and separated by SDS-PAGE on 4–12% Bis-Tris NuPAGE gradient gels (Invitrogen). Protein was transferred onto polyvinylidene difluoride membranes (Hybond-P; GE Healthcare) at 30 V for 2 h under reducing conditions. Membranes were blocked with 5% nonfat milk in TBS for 1 h at room temperature. Mouse anti-NMDAR1 and rabbit anti-β-actin primary antibodies (Millipore and Cell Signaling Technologies, respectively) were diluted 1:1000 in 2.5% nonfat milk and applied to membranes for 24 h at 4°C with shaking. HRP-conjugated goat secondary antibodies were applied for 1 h at room temperature. Protein expression was evaluated using Novex ECL chemoluminescent substrate (Invitrogen) and a Versa Doc imaging system (Bio-Rad). Densitometric analysis was conducted using Versa Doc software. NMDAR levels are represented as protein expression relative to β-actin.
Results
Membrane properties and basal synaptic transmission in AD transgenic neurons
In 3xTg-AD neurons, increased ER calcium release triggers downstream effects on membrane excitability likely via activation of calcium-activated SK channels (Brennan et al., 2008; Chakroborty et al., 2009). Here, we further investigated how these altered calcium dynamics affect passive and active membrane properties in neurons from the frontal cortex, a brain region highly vulnerable to AD pathology. In control ACSF solution, there were no significant differences in the resting membrane potential (F(2,82) = 1.2, p = 0.31) or membrane input resistance (F(2,43) = 0.69, p = 0.51), among the three mouse lines (Table 1) (number of cells/group: 32, 15, and 36 for the NonTg, TAS/TPM, and 3xTg-AD strains, respectively). However, stimulating RyRs with caffeine (10 mm) significantly increased the membrane hyperpolarization within each mouse model, and the steady-state hyperpolarization was greater in the AD-Tg strains (3xTg-AD, n = 12; and TAS/TPM, n = 15) compared with the NonTgs (n = 13) (F(2,43) = 21.4, p < 0.001). Caffeine did not significantly change membrane input resistance (p > 0.05).
Table 1.
Membrane properties of cortical pyramidal neurons from NonTg, 3xTg-AD, and TAS/TPM mice
| NonTg | 3xTg | TAS | |
|---|---|---|---|
| RMP (mV) | |||
| ACSF | −71.5 ± 0.65 | −71.8 ± 0.64 | −73.5 ± 1.54 |
| Caffeine | −74.9 ± 1.38 | −78.0 ± 0.88 | −79.0 ± 1.98 |
| Ri (MΩ) | |||
| ACSF | 180.9 ± 7.29 | 197.6 ± 14.74 | 179.2 ± 14.90 |
| Caffeine | 186.0 ± 8.58 | 187.2 ± 9.75 | 175.9 ± 17.36 |
RMP, Resting membrane potential; Ri, membrane input resistance.
To measure effects of altered ER calcium release on synaptic responses, a stimulating electrode was placed in the layer II cortical region adjacent to the apical dendrite of the layer V neuron to evoke subthreshold EPSPs. Synaptically evoked calcium responses were imaged from spiny apical dendrites ∼500 μm from the soma. For each neuron, the stimulus intensity was adjusted to evoke an EPSP of ∼4 mV from a membrane potential of −70 mV (Fig. 1). In control ACSF, the requisite stimulus intensity was not significantly different (F(2,82) = 0.08; p = 0.92) among the NonTg (65 ± 9.2 μA, n = 32), TAS/TPM (60 ± 12.7 μA, n = 15), and 3xTg-AD lines (62 ± 10.7 μA, n = 36). In NonTg mice, 10 mm caffeine did not significantly alter the requisite stimulus intensity (49 ± 4.7 μA; n = 13; p > 0.05). However, in the AD mice, RyR activation by 10 mm caffeine significantly reduced the stimulus intensity required to evoke a 4 mV EPSP (TAS/TPM, 33 ± 7.6 μA, n = 15; 3xTg-AD, 34 ± 2.32 μA, n = 12; p < 0.05), indicating that RyR activation can increase synaptic excitability in young AD transgenic strains but not in normal NonTg mice. Although synaptic excitability appears normal in AD transgenic mice under control conditions, stimulation with caffeine reveals increased sensitivity to RyR-mediated potentiation in AD transgenics. It is possible this is attributable to the increased RyR2 levels observed both the 3xTg-AD mice (Chakroborty et al., 2009), as well as in the TAS/TPM mice as demonstrated with qRT-PCR (p < 0.05; data not shown).
Figure 1.

Glutamatergic synaptic signaling is differentially affected by RyR activation in the AD Tg neurons. A, Representative traces demonstrate synaptically evoked glutamatergic EPSPs in a layer V pyramidal neuron. B, The stimulation intensities required to evoke a 4 mV EPSP in the NonTg, TAS/TPM, and 3xTg-AD mice are similar in control ACSF solution; however, upon concurrent RyR activation with 10 mm caffeine, significantly less stimulation is required in the AD transgenic mice relative to ACSF conditions and to the NonTg values in caffeine. *p < 0.05.
To determine an optimal stimulation protocol that generates a postsynaptic Ca2+ response yet does not interfere with long-term alterations in presynaptic vesicle release properties, we compared a paired-pulse facilitation (PPF) protocol (70 ms intervals) at low frequency (0.05 Hz) before and after application of stimulation trains of either 30 or 100 Hz for 1.5 s. PPF after 30 Hz stimulation show no differences from prestimulus conditions across transgenic animal group (data not shown; p > 0.05); however, PPF was significantly decreased after 100 Hz stimulation (p < 0.05), suggesting that high-frequency stimulation altered presynaptic release properties. Therefore, 30 Hz stimulation for 1.5 s was chosen for subsequent synaptic stimulation/calcium imaging experiments (data not shown).
Effect of enhanced RyR-evoked calcium release on spontaneous vesicle release properties
The ER extends into presynaptic regions, and RyRs are found in some synaptic terminals (Bouchard et al., 2003). Based on this, we next examined how synaptic signaling properties may be affected by enhanced RyR-evoked calcium release in the AD transgenic mice. Spontaneous EPSPs were measured both with and without TTX (1 μm) in the bath, and, regardless of caffeine treatment and transgenic strain, there were no differences in their frequency or amplitude with sodium channels blocked (p = 0.65). This likely reflects the lack of spontaneous action potentials within the circuits measured from these cortical brain slice preparations. Therefore, subsequent experiments omitted TTX. In control ACSF, spontaneous EPSP frequency and amplitude were similar across all animal groups (F(2,38) = 0.40; p > 0.05 and F(2,38) = 0.15; p > 0.05, respectively) (Fig. 2). As shown in Figure 2, B and C, there were no significant differences in the cumulative amplitude and frequency histograms (KS test, p > 0.05), indicating that spontaneous vesicle release probabilities appear similar between AD transgenic animals and NonTg controls under basal conditions. AMPA/kainite glutamatergic synaptic transmission was confirmed at the end of each recording with bath application of CNQX to block spontaneous events. The number of cells per group was 8, 10, and 21, respectively, for the NonTg, TAS/TPM, and 3xTg-AD strains.
Figure 2.
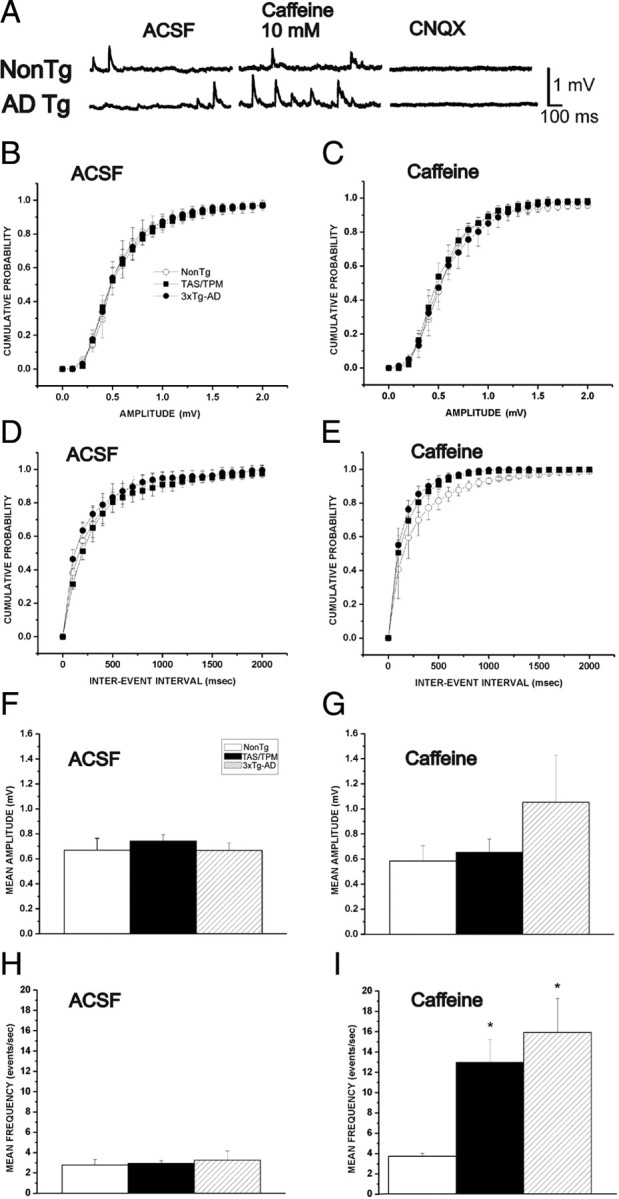
RyR stimulation increases the frequency but not amplitude of glutamatergic miniature potentials in AD transgenic mice. A, Representative individual traces of spontaneous miniature potentials demonstrate effects of 10 mm caffeine in NonTg and 3xTg-AD neurons. Confirmation of glutamatergic spontaneous events is determined by blocking with CNQX. B–D, Cumulative probability histograms show amplitude (B, C) and interevent intervals (D, E) of mEPSP events. Both frequency and amplitude of spontaneous release events are similar between NonTg and AD Tg neurons in control ACSF conditions (B, D), while caffeine increases the frequency but not amplitude of events in the AD Tg neurons only (C, E), as indicated by a leftward shift in the cumulative interevent histogram. F, Average amplitude in control ACSF. G, Average amplitude in 10 mm caffeine. H, Average frequency in control ACSF. I, Average amplitude in 10 mm caffeine. *Significantly different from NonTg, p < 0.05.
We next captured spontaneous events in the presence of 10 mm caffeine to determine whether RyR activation unmasks differences in glutamate release at the AD transgenic synapses. Under these conditions, we observe a pronounced increase in the average frequency (F(2,38) = 6.27, p < 0.05) of spontaneous events but not in their amplitude (F(2,38) = 1.29,p = 0.33) in both AD transgenic mouse lines compared with the NonTg controls. This is demonstrated by a significant leftward shift in the interevent cumulative probability histogram (Fig. 2E) [KS test, 3xTg (n = 21) vs NonTg (n = 8), z = 1.25 p < 0.05; TAS/TPM (n = 10) vs NonTg (n = 8), z = 1.8, p < 0.01]. These data indicate that both the passive membrane properties and synaptic transmission appear normal in both AD transgenic mouse lines under basal conditions at 4–6 weeks of age. Yet, synaptic excitability is significantly increased for the AD transgenic mice upon RyR activation, likely via increased presynaptic glutamate release.
Relative differences in RyR-mediated ER Ca2+ release within cellular compartments
Neurons are highly compartmentalized cells that have distinct calcium signaling requirements within subcellular regions. Therefore, we investigated whether the mutant PS1-mediated calcium alterations were of a uniform magnitude across neuronal compartments, or whether regional differences exist as demonstrated in hippocampal CA1 pyramidal neurons from 3xTg-AD mice (Chakroborty et al., 2009). We compared ER calcium release from the soma, proximal dendrites, and dendritic spine heads from distal apical branches using bath application of 10 or 20 mm caffeine in the NonTg, 3xTg-AD, and TAS/TPM mice. These caffeine concentrations were chosen to establish near-threshold calcium response levels in the NonTg neurons, while still capturing the full range of calcium responses in the AD transgenic mice. Representative pseudocolored images of relative calcium changes in response to RyR stimulation within the soma, dendrites, and spines from NonTg, TAS/TPM, and 3xTg-AD neurons are shown in Figure 3A,B. When measuring somatic signals, the region overlying the nucleus was excluded. The 10 mm caffeine produced threshold RyR-evoked calcium responses in the NonTg neurons; therefore, this concentration was used for the remaining experiments unless otherwise indicated. For the TAS/TPM and 3xTg-AD neurons, significantly greater calcium responses in all compartments were observed relative to the NonTg neurons, with the exception of the soma for the TAS/TPM neurons [ANOVA: soma, F(2,16) = 4.32; p < 0.05; NonTg, n = 5; TAS/TPM, n = 6; 3xTg, n = 6: dendrites (F(2,22) = 10.45; p < 0.05; NonTg, n = 8; TAS/TPM, n = 9; 3xTg, n = 6: spines (F(2,120) = 49.44; p < 0.05; NonTg, n = 31; TAS/TPM, n = 44; 3xTg, n = 47]. With 20 mm caffeine, the AD transgenic mice generate significantly greater calcium responses than NonTg mice in all compartments (ANOVA: soma (F(2,17) = 4.98; p < 0.05, NonTg, n = 5; TAS/TPM, n = 6; 3xTg, n = 6: dendrites, F(2,44) = 3.63; p < 0.05; NonTg, n = 14; TAS/TPM, n = 18; 3xTg, n = 13: spines, F(2,147) = 17.36; p < 0.05; NonTg, n = 53; TAS/TPM, n = 39; 3xTg, n = 56). Here, the TAS/TPM and 3xTg-AD mice had similar calcium responses in the soma and dendrites, but the 3xTg-AD mice still generated greater RyR-evoked calcium signals in the spine heads (p < 0.05). In previous studies, we had confirmed that the caffeine application was underlying the aberrant calcium responses and was not working through other signaling pathways (Chakroborty et al., 2009). Here we also used ryanodine in the pipette during caffeine application, which reduced the evoked calcium response and verifies that caffeine stimulation is triggering the calcium response (data not shown). Given the critical role of dendritic calcium release in modulating synaptic signaling, this scenario sets the stage for altered synaptic function in the AD mice as a consequence of localized aberrant calcium release.
Figure 3.
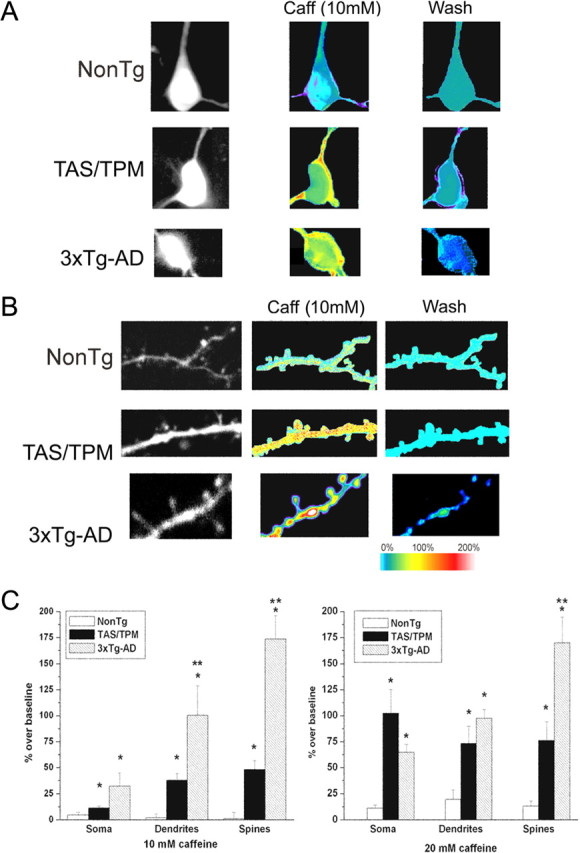
RyR-evoked increase in ER calcium release within neuronal compartments in AD Tg neurons. A, Representative fura-2 images of resting calcium levels (left column) within the soma of NonTg (top), TAS/TPM (middle), and 3xTg-AD (bottom) neurons. The middle column represents maximal relative calcium changes after 10 mm caffeine application; the right column shows relative fluorescence intensity after a 10 min washout. Each 2-photon image represents an average of 100 frames captured at a frame rate of 30 Hz. Resulting percentage over baseline values are shown as pseudocolored images corresponding to color scale bar. B, Same as in A, but detailing the dendrites and spines. C, Bar graphs show averaged data (mean ± SE) for each transgenic group for caffeine concentrations of 10 mm (left) and 20 mm (right) in the soma, dendrites, and spines. *Significantly different from NonTg within compartment; **significantly different from TAS/TPM (p < 0.05).
Localized feed-forward CICR through RyR predominates in dendrites
RyR sensitivity appears to be markedly increased in the AD mice, thus relatively small increases in postsynaptic calcium may be sufficient to activate CICR. Previous studies in 3xTg-AD neurons showed that increased RyR-mediated CICR is required for the increased IP3R-evoked ER calcium response, as exaggerated IP3R-evoked calcium response was normalized to NonTg levels by blocking the RyR during IP3R activation (Stutzmann et al., 2006). Here, we tested the converse—whether blocking the IP3R while stimulating the RyR could similarly “normalize” the aberrant RyR-mediated calcium release. RyR stimulation with caffeine was repeated in the presence of the IP3R antagonist heparin infused into the neuron through the patch pipette (Fig. 4). Isolating the RyR ER–calcium response in this manner resulted in markedly different calcium responses both within and between mouse models, and was based largely on cellular compartment. Heparin significantly lowered the caffeine-evoked calcium signal only in the soma of 3xTg-AD neurons (F(1,12) = 12.77; p < 0.01). However, unlike the complementary experiment in which dantrolene normalized the IP3-evoked calcium response to NonTg levels, heparin did not completely reverse the RyR–calcium response back to NonTg levels (Fig. 4), and a significantly greater RyR response remained in the 3xTg-AD soma (n = 6) relative to the NonTg soma (n = 7) (F(1,12) = 5.08, p = < 0.05).
Figure 4.

Recruitment of somatic IP3-sensitive stores with RyR stimulation in AD neurons. A (soma) and B (distal dendrites, spines) show pseudocolored images of relative calcium changes in a representative 3xTg-AD neuron evoked by caffeine (10 mm) in the presence of heparin. C, Bar graph shows averaged (mean ± SE) calcium responses for each compartment per transgenic/treatment group in the soma, dendrites, and spines. *Significantly different from Non Tg/caffeine, p < 0.05; **significantly different from 3xTg caffeine, p < 0.05.
In dendritic compartments, heparin had little effect on the caffeine-evoked RyR-calcium response in either the NonTg (dendrites, n = 16; spines, n = 83) or 3xTg-AD neurons (dendrites, n = 10; F(1,25) = 0.23; p = 0.97; and spines: n = 68; F(1,150) = 0.43; p = 0.52). Notably, heparin's reduction of the somatic ER calcium response in 3xTg neurons occurred in the absence of exogenously applied or stimulated IP3, suggesting that endogenous basal levels of IP3 may be sufficient to generate an IP3R response when combined with calcium release through RyR.
Synergistic calcium dynamics between synaptic stimulation and RyR activation in dendrites and spines of AD transgenic mice
In light of the pronounced RyR-evoked calcium responses in synaptic regions from mutant PS1-expressing mice, we next explored how this may affect synaptically generated signals from both a calcium signaling and electrical excitability perspective. Here, synaptically evoked calcium signals were elicited by stimulating afferent fibers impinging on layer V cortical pyramidal neurons using a 30 Hz stimulus frequency. Evoked calcium signals were captured in the following sequence: 30 s in control ACSF, followed by 1.5 s of 30 Hz stimulation, a 30 s delay, 1 min of bath-applied caffeine (10 mm), followed by 1.5 s of synaptic stimulation in caffeine, then several minutes (2–10 min) of washout. The averaged maximal calcium response and representative EPSPs during synaptic stimulation are shown for each condition in Figure 5. Interestingly, 30 Hz synaptic stimulation alone generates a greater postsynaptic calcium response in spine heads and distal processes from the AD mice compared with NonTg mice. Although this occurs in the absence of exogenous RyR activation, the increased calcium is mediated via CICR as ryanodine (30 μm) in the patch pipette blocks this effect. To further examine the interaction between RyR and synaptically evoked calcium responses, we then stimulated presynaptic fibers in 10 mm caffeine and measured the combined calcium response in postsynaptic dendrites and spines integrated over a 1.5 s time period. In the AD transgenic mice, there was a pronounced synergistic calcium response with coincident RyR and synaptic stimulation that exceeded either calcium response in isolation (ANOVA; F(2,319) = 50.6; p < 0.01; n = 132, 122, and 166, respectively, for NonTg, TAS/TPM, and 3xTg-AD strains). It should be noted that when data are shown for the synaptic stimulation plus caffeine conditions, the caffeine-evoked calcium response has been subtracted out, thereby isolating the calcium signal resulting from the coincident stimulation. In NonTg spines and dendrites, concurrent RyR and synaptic stimulation did not trigger additional calcium release, consistent with a lower threshold for CICR. We further confirmed the contribution of RyR-mediated calcium release in the AD mice by including ryanodine in the patch pipette and repeating the synaptic stimulation protocol. Blocking the RyR eliminates both the isolated and synergistic elevation of calcium in the AD neurons F(2,262) = 150.6 (p < 0.001; n = 78, 102, and 83, respectively, for NonTg, TAS/TPM, and 3xTg-AD strains).
Figure 5.

Synergistic calcium interactions between RyR and glutamatergic synaptic transmission in AD Tg neurons. A, Pseudocolored images of relative calcium changes in representative NonTg (top), TAS/TPM (middle), and 3xTg-AD (bottom) neurons in the following conditions (from left to right): baseline (ACSF), 30 Hz synaptic stimulation (1.5 s), caffeine alone (10 mm), 30 Hz synaptic stimulation plus caffeine, and washout. B, Representative Ca2+ response traces after 30 Hz synaptic stimulation (voltage trace shown in top) shown as percentage over baseline, in control ACSF (left panels) and in 10 mm caffeine (right panels) for NonTg (top), 3xTg-AD (middle), and TAS/TPM (bottom) neurons. C, Bar graphs show averaged (mean ± SE) Ca2+ responses integrated over a 1.5 s time period of 30 Hz synaptic stimulation in control ACSF (left grouping), synaptic stimulation plus caffeine (center), and synaptic stimulation with ryanodine in the pipette (right grouping) for the NonTg, TAS/TPM, and 3xTg-AD neurons. Statistically significant differences are indicated by asterisks (one-way ANOVA, p < 0.05). *Significantly different from NonTg within treatment group; **significantly different from synaptic stimulation in ACSF within transgenic strain.
NMDA-mediated activation of CICR
NMDAR-mediated calcium influx provides a significant source of calcium entry through plasma membrane channels, and upregulation of NMDAR activity is implicated as a mechanism in AD pathology. Indeed, several therapeutic approaches either currently in use or under investigation to preserve cognitive function in AD target NMDAR (Burns and Jacoby, 2008; Doody et al., 2008; Yamin, 2009). However, direct observation of NMDAR-mediated calcium signaling in AD models has yet to be examined. In this series of experiments, this is addressed by pharmacologically isolating and subsequently activating NMDAR through photolysis of caged glutamate in the presence of CNQX and picrotoxin, and comparing whole-cell currents and dendritic calcium responses in AD and NonTg neurons. The membrane potential was voltage clamped at −70 mV, and APV was included at the conclusion of all trials to confirm NMDAR activity. As shown in Figure 6, isolated NMDA currents were not different among the mouse strains; however, the evoked dendritic spine calcium responses were significantly greater in both AD strains relative to NonTg (ANOVA (F(2,360) = 26.53; p < 0.05: n = 110, 134, and 117, respectively, in the NonTg, TAS/TPM, and 3xTg-AD neurons). Blocking the RyR in AD neurons prevented the enhanced NMDA calcium responses and normalized them to NonTg levels. This indicates that the additional calcium came from RyR-mediated CICR in the AD mice, and, that this CICR facilitation via NMDAR activation does not occur in the NonTg mice (Fig. 6A,B) (ANOVA; F(2,207) = 8.817, p < 0.05: n = 136, 70, and 102, respectively, in the NonTg, TAS/TPM and 3xTg-AD neurons).
Figure 6.

NMDAR-mediated calcium influx triggers CICR in AD neurons. A, Representative pseudocolored images of maximal calcium responses evoked from NMDAR activation in NonTg (top), TAS/TPM (middle), and 3xTg-AD (bottom) neurons in control ACSF (left), 10 mm caffeine (middle), and caffeine + APV (right). B, Bar graphs showing the averaged maximal calcium response evoked from NMDAR activation for NonTg, TAS/TPM, and 3xTg-AD neurons in control ACSF (left), with concurrent RyR activation with 10 mm caffeine (middle), and NMDAR activation with ryanodine in the pipette (right). C, Left, individual representative traces of evoked calcium responses from a NonTg neuron (blue) and 3xTg-AD neuron (red) from NMDAR activation alone (left), NMDAR + 10 mm caffeine (middle), and NMDAR + caffeine + APV. Right, individual representative whole-cell current traces evoked from a NonTg neuron (blue) and 3xTg-AD neuron (red) from NMDAR activation alone (left), NMDAR + 10 mm caffeine (middle), and NMDAR + caffeine + APV. D, Bar graphs on left show averaged whole-cell currents in NonTg, TAS/TPM, and 3xTg-AD mice evoked from NMDAR activation. Bars on right (same order as on left) show averaged whole-cell currents evoked from concurrent NMDAR plus RyR activation. E, Bar graph shows no differences between steady-state NMDAR protein levels in the cortex of NonTg and 3xTg-AD mice. Inset above shows representative immunoblot images from a NonTg and 3xTg-AD sample. *Significantly different from NonTg (p < 0.05).
We next asked whether direct RyR activation concurrent with NMDAR stimulation can generate supra-additive calcium responses in the dendrites of AD neurons, as observed with synaptic stimulation. The MNI-glutamate photolysis protocol was repeated as above but now included bath application of 10 mm caffeine. Similar to the dynamics observed with synaptic stimulation, the coincident activation of RyR and NMDAR resulted in a further calcium increase in the AD Tg mice only; there were no additional effects in the NonTg mice under these conditions (Fig. 6A,B) (ANOVA; F(2,360) = 26.53; p < 0.001: n = 110, 134, and 117, respectively, in the NonTg, TAS/TPM and 3xTg-AD neurons). The evoked NMDA whole-cell currents were not significantly different among mouse lines, nor did caffeine exposure change the magnitude of the currents (Fig. 6C). This supports increased CICR via the RyR as the mechanism underlying the NMDAR-mediated enhanced calcium responses rather than increased calcium flux directly through NMDAR. Consistent with these findings, cortical NMDAR protein levels were not different between the NonTg mice (n = 6) and the 3xTg-AD mice (n = 7) (Fig. 6D,E).
VGCC signaling in distal dendrites and spines of AD transgenic neurons
Calcium entry through VGCC provides another large calcium source in dendrites and spines. We wished to extend the observations made with synaptically mediated CICR changes in the AD mice and determine whether VGCC entry is similarly upregulated and plays a role in the increased synaptically evoked calcium responses. VGCCs were activated by dendritic depolarization via back-propagating action potentials (bAPs) generated at the soma. A series of 7–10 action potentials was triggered by depolarizing current injections (500 ms, 40 pA over threshold current) with picrotoxin (10 μm) and APV (50 μm) included in the bath solution. Under these conditions, there were no significant differences in the integrated or peak calcium responses in dendritic spines among the mouse strains (Fig. 7) (ANOVA: F(2,79) = 2.13; p = 0.23: n = 25, 28, and 37, respectively, in the NonTg, TAS/TPM, and 3xTg-AD neurons), consistent with previous studies examining bAP responses in the soma of AD neurons (Stutzmann et al., 2004, 2006). To determine whether RyR stimulation in parallel with VGCC activation can generate exaggerated CICR, caffeine was bath applied concurrent with bAPs, and calcium responses measured in the same spines. Again, there were no significant differences among the mouse strains, nor were any supra-additive calcium responses generated (ANOVA: F(2,79) = 3.43; p = 0.12: n = 25, 28, and 37, respectively, in the NonTg, TAS/TPM and 3xTg-AD neurons). Last, the calcium channel blocker Cd2+ (100 μm) was washed in to confirm VGCC activity.
Figure 7.
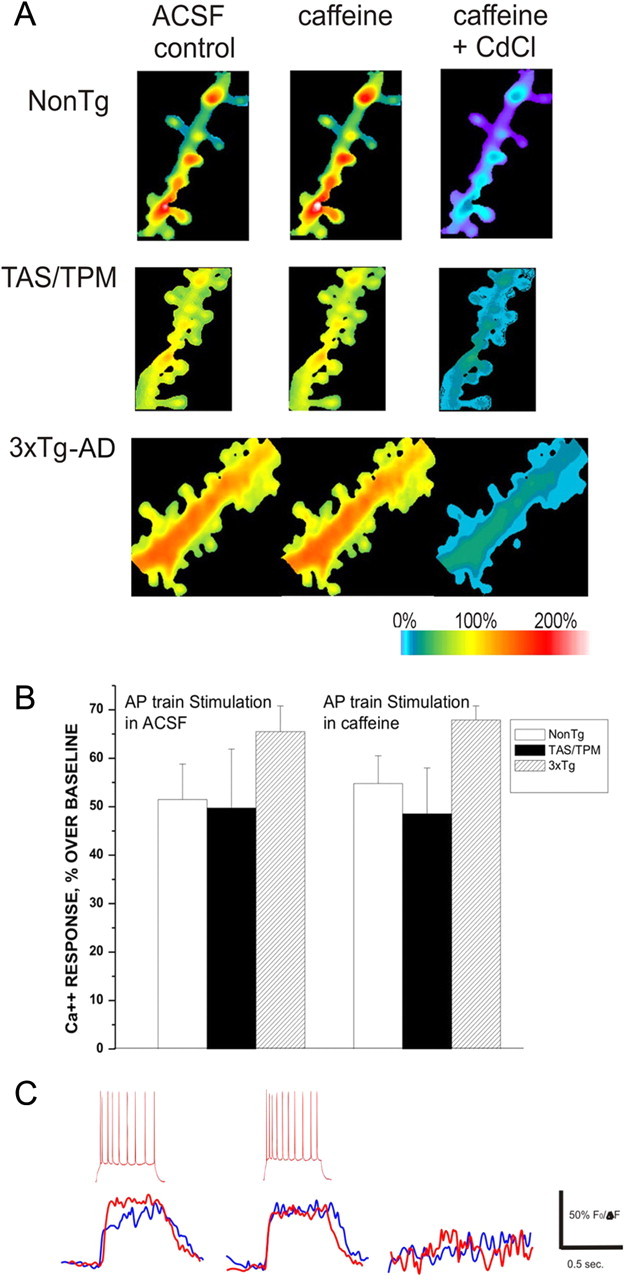
Calcium entry through VGCC does not trigger additional CICR in AD neurons. A, Representative pseudocolored images of maximal calcium responses evoked from a train of back-propagating action potentials in NonTg (top), TAS/TPM (middle), and 3xTg-AD (bottom) neurons in control ACSF (left), 10 mm caffeine (middle), and caffeine + CdCl(right). B, Bar graph shows averaged calcium response evoked from a train of action potentials measured from dendritic spines in control ACSF (left) and 10 mm caffeine (right). C, Individual representative calcium traces evoked from a train of action potentials (inset, above) in a NonTg neuron (blue) and 3xTg-AD neuron (red) in control ACSF (left), 10 mm caffeine (middle), and caffeine + CdCl (right).
Discussion
Increased ER calcium release in mutant PS-expressing neurons is well documented, and the contribution of calcium dyshomeostasis to AD pathogenesis is increasingly validated (LaFerla, 2002; Stutzmann, 2007; Bezprozvanny and Mattson, 2008). Previous studies have demonstrated increases in somatic RyR-evoked calcium release in mutant PS1-expressing neurons, which is consistent with increased RyR expression (Smith et al., 2005; Stutzmann et al., 2006) and increased RyR2 mRNA levels specifically (Chakroborty et al., 2009). Elevated RyRs have also been observed in human AD cases early in the disease process (Kelliher et al., 1999; O'Neill et al., 2001). With this, calcium dysregulation in sporadic AD is also evident, with Aβ peptides, hyperphosphorylated tau, and ApoE4 expression altering neuronal calcium signaling (Stutzmann, 2007).
Despite the evidence implicating dysregulated calcium signaling in AD pathogenesis, the corresponding downstream consequences on neurophysiology have yet to be explored in detail. Considering that loss of synapses is correlated with the degree of cognitive impairment in AD and that calcium signaling is fundamental to synaptic function and integrity, we explored the extent of calcium signaling dysregulation within spines and dendrites in AD neurons and tested the hypothesis that aberrant CICR via the RyR underlies synaptic signaling pathology before the onset of cognitive deficits. We found the following in cortical neurons from presymptomatic TAS/TPM and 3xTg-AD mice relative to NonTg mice: (1) that the patterns of RyR-evoked calcium elevation differ significantly within different neuronal compartments, with dendrites and spines displaying the greatest sensitivity to RyR activation in the AD mice; (2) that postsynaptic calcium responses are exaggerated in dendritic processes and spine heads in response to synaptic stimulation, as are calcium responses evoked by NMDAR activation; (3) that there is likely a RyR-dependent presynaptic effect on neurotransmitter vesicle release that is unique to the AD transgenic mice; and (4) that dendritic processes and spines show synergistic CICR upon coactivation of synaptic inputs and RyRs.
Increased calcium release in dendrites and spine heads corresponds with altered synaptic transmission
In pyramidal neurons, the RyR is found throughout the neuron including presynaptic terminals, distal dendritic processes, and dendritic spine heads—of which ∼50% contain smooth ER (Spacek and Harris, 1997; Hertle and Yeckel, 2007). In these subcellular domains, RyRs are optimally positioned to modulate synaptic signaling and plasticity through tuning of calcium release within both spatial and temporal domains. We observe here a significant increase in the RyR-evoked calcium response in several cellular compartments in the AD mice, with the greatest relative increases occurring in distal dendrites and spines, which are the key sites of synaptic contacts that support neurotransmission and plasticity. The mechanism underlying the increased calcium release may reflect an impaired ER leak channel ascribed to mutant presenilin, thereby resulting in a general increase in ER calcium store levels (Tu et al., 2006; Nelson et al., 2007). The broader implications of these findings are consistent with the RyR-mediated alterations in short and long forms of hippocampal plasticity and synaptic strength in young 3xTg-AD neurons (Chakroborty et al., 2009).
Reciprocal CICR between RyR and IP3R in AD transgenic neurons
Reciprocal CICR dynamics between IP3R- and RyR-mediated calcium release can exist in neurons, such that calcium released through IP3R can facilitate RyR-release and vice versa. This may be the mechanism by which photoreleased IP3 generates an enhanced RyR calcium response in mutant PS1-expressing neurons. In this scenario, blocking the RyR normalizes the IP3-evoked calcium response in mutant PS1-expressing neurons but has no effect in NonTg neurons (Stutzmann et al., 2006). Here, we examined whether RyR-evoked calcium release can facilitate an IP3R calcium response in the AD mice, thereby questioning whether there is an equivalent degree of CICR reciprocity between the channels. We found that blocking the IP3R reduced the RyR-calcium signal in the soma of 3xTg-AD neurons; yet, the total evoked calcium response was still greater than the NonTg response. This suggests that mutant PS can facilitate the sensitivity of both the IP3 and Ry channels to calcium, but the effect is most profound on the RyR and only observed in the soma. This is consistent with the IP3R distribution in pyramidal neurons, which is predominantly in the soma and proximal dendrites, whereas RyRs are also found in distal processes and spines (Hertle and Yeckel, 2007; Nicolay et al., 2007; Fitzpatrick et al., 2009). This experiment also allows us to indirectly examine whether mutant presenilin sensitizes IP3 channel-gating properties, as proposed by Cheung et al. (2008, 2010). In the absence of exogenously applied IP3, an IP3–calcium response is generated upon RyR-evoked calcium release in mutant PS1-expressing neurons, yet not in the NonTg neurons, consistent with enhanced IP3R modal gating (Cheung et al., 2008, 2010). As calcium is a coagonist of the IP3R, the increased RyR-evoked calcium release in concert with basal levels of endogenous IP3 exposure may now be sufficient to evoke an IP3R response in AD but not NonTg mice.
Evoked and spontaneous synaptic transmission in NonTg and AD transgenic neurons
In light of the profound RyR-mediated calcium increases in synapse-dense regions, we next asked how this might impact synaptic function. Baseline synaptic transmission and passive membrane properties in young AD transgenic mice appeared similar to NonTg mice under control conditions. Yet, the appearance of normalcy may reflect compensatory mechanisms normalizing an aberrant RyR-mediated calcium contribution (Chakroborty et al., 2009). With RyR stimulation, differences between NonTg and AD transgenics now emerge in synaptic transmission and membrane excitability properties. For example, RyR activation generates a greater hyperpolarized membrane potential in the AD transgenic mice than in NonTg controls. This may be related to an increased coupling efficiency between the RyR- and the calcium-activated SK channel in mutant PS1-expressing neurons (Stutzmann et al., 2006; Brennan et al., 2008: Chakroborty et al., 2009). Presynaptic differences in RyR-evoked calcium release are also inferred from a selective increase in the frequency, but not amplitude, of miniature EPSPs (mEPSPs). These presynaptic dynamics are consistent with increased transmitter release upon RyR activation, and therefore may also relate to the reduced current strength required to generate a similar postsynaptic EPSP. At CA3-CA1 synapses, presenilin has a selective presynaptic role in activity-dependent neurotransmitter release that is dependent upon RyR-sensitive calcium stores (Zhang et al., 2009). Purported “gain-of-function” PS mutations may therefore result in upregulated calcium release in presynaptic terminals and accelerated vesicle depletion, which is consistent with the increased contribution of RyR stores to synaptic transmission and presynaptic plasticity in hippocampal slices from mutant PS1-expressing mice (Chakroborty et al., 2009).
Glutamatergic synaptic transmission evokes enhanced postsynaptic calcium release in AD cortical neurons
Independent of exogenous RyR activation, physiological synaptic stimulation results in exaggerated dendritic calcium signals in the AD transgenic neurons. The underlying mechanism may reflect a reduced CICR threshold, such that Ca2+ entry through plasma membrane channels is sufficient to elicit a RyR-evoked calcium response and to amplify local calcium levels. In neocortical pyramidal cells, the main sources of calcium influx in spines are voltage-dependent calcium channels (Sabatini and Svoboda, 2000) and NMDARs (Keller et al., 2008). Although it is possible that these calcium sources are also being altered by mutant PS1 expression, previous studies have shown that voltage-dependent calcium dynamics and calcium decay kinetics are not different between NonTg and mutant PS1-expressing models (Stutzmann et al., 2004, 2006). Likewise, NMDA-mediated currents were not different in the AD mice, nor were NMDA protein levels.
Altered CICR dynamics in spines and dendrites from mutant PS-expressing neurons were also observed when RyRs were activated concurrently with synaptic stimulation. Here, a far greater and synergistic calcium response is generated in the dendrites and spines of AD transgenic neurons, with only marginal interactions observed in the NonTg mice. This aberrant CICR effect is observed with both synaptically evoked calcium transients, as well as isolated NMDA activation, implicating glutamatergic transmission while excluding involvement of voltage-sensitive calcium channels. Direct molecular interactions between the RyR2 isoform and NR2B subunits have been described in cardiac muscle (Seeber et al., 2004); if such a complex exists in central neurons, it is possible that this coupling or functional interaction is enhanced in the AD models, although this has yet to be examined. The downstream effects of this increased calcium response may exert more global physiological effects such as a net decrease in membrane excitability via increased activation of calcium-dependent medium afterhyperpolarization currents via SK channels (Stutzmann et al., 2006; Brennan et al., 2008). The intermittent yet lifelong exposure to grossly elevated calcium transients within spine heads and distal processes may result in structural defects and functional abnormalities in synaptic transmission and contribute to the devastating cognitive deficits in AD (Matsuzaki et al., 2004; Korkotian and Segal, 2007; Jones et al., 2008).
Footnotes
This work was supported by the Alzheimer's Association, The American Federation for Aging Research, and National Institutes of Health Grant AG030205. We would like to acknowledge Jill C. Richardson (GlaxoSmithKline) for the development of the TAS/TPM mice. We thank Clark A. Briggs and Shreaya Chakroborty for editorial assistance.
References
- Berridge MJ. Calcium microdomains: organization and function. Cell Calcium. 2006;40:405–412. doi: 10.1016/j.ceca.2006.09.002. [DOI] [PubMed] [Google Scholar]
- Bezprozvanny I, Mattson MP. Neuronal calcium mishandling and the pathogenesis of Alzheimer's disease. Trends Neurosci. 2008;31:454–463. doi: 10.1016/j.tins.2008.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouchard R, Pattarini R, Geiger JD. Presence and functional significance of presynaptic ryanodine receptors. Prog Neurobiol. 2003;69:391–418. doi: 10.1016/s0301-0082(03)00053-4. [DOI] [PubMed] [Google Scholar]
- Brennan AR, Dolinsky B, Vu MA, Stanley M, Yeckel MF, Arnsten AF. Blockade of IP3-mediated SK channel signaling in the rat medial prefrontal cortex improves spatial working memory. Learn Mem. 2008;15:93–96. doi: 10.1101/lm.767408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns A, Jacoby R. Dimebon in Alzheimer's disease: old drug for new indication. Lancet. 2008;372:179–180. doi: 10.1016/S0140-6736(08)61046-6. [DOI] [PubMed] [Google Scholar]
- Chakroborty S, Goussakov I, Miller MB, Stutzmann GE. Deviant ryanodine receptor-mediated calcium release resets synaptic homeostasis in presymptomatic 3xTg-AD mice. J Neurosci. 2009;29:9458–9470. doi: 10.1523/JNEUROSCI.2047-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung KH, Shineman D, Müller M, Cárdenas C, Mei L, Yang J, Tomita T, Iwatsubo T, Lee VM, Foskett JK. Mechanism of Ca2+ disruption in Alzheimer's disease by presenilin regulation of InsP3 receptor channel gating. Neuron. 2008;58:871–883. doi: 10.1016/j.neuron.2008.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung KH, Mei L, Mak DO, Hayashi I, Iwatsubo T, Kang DE, Foskett JK. Gain-of-function enhancement of IP3 receptor modal gating by familial Alzheimer's disease-linked presenilin mutants in human cells and mouse neurons. Sci Signal. 2010;3:ra22. doi: 10.1126/scisignal.2000818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collin T, Marty A, Llano I. Presynaptic calcium stores and synaptic transmission. Curr Opin Neurobiol. 2005;15:275–281. doi: 10.1016/j.conb.2005.05.003. [DOI] [PubMed] [Google Scholar]
- Demuro A, Parker I, Stutzmann GE. Calcium signaling and amyloid toxicity in Alzheimer's disease. J Biol Chem. 2010;285:12463–12468. doi: 10.1074/jbc.R109.080895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doody RS, Gavrilova SI, Sano M, Thomas RG, Aisen PS, Bachurin SO, Seely L, Hung D. Effect of dimebon on cognition, activities of daily living, behaviour, and global function in patients with mild-to-moderate Alzheimer's disease: a randomised, double-blind, placebo-controlled study. Lancet. 2008;372:207–215. doi: 10.1016/S0140-6736(08)61074-0. [DOI] [PubMed] [Google Scholar]
- Fitzpatrick JS, Hagenston AM, Hertle DN, Gipson KE, Bertetto-D'Angelo L, Yeckel MF. IP3 receptor-dependent Ca2+ waves in pyramidal neuron dendrites propagate through hot spots and cold spots. J Physiol. 2009;587:1439–1459. doi: 10.1113/jphysiol.2009.168930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harkany T, Hortobágyi T, Sasvári M, Kónya C, Penke B, Luiten PG, Nyakas C. Neuroprotective approaches in experimental models of beta-amyloid neurotoxicity: relevance to Alzheimer's disease. Prog Neuropsychopharmacol Biol Psychiatry. 1999;23:963–1008. doi: 10.1016/s0278-5846(99)00058-5. [DOI] [PubMed] [Google Scholar]
- Hertle DN, Yeckel MF. Distribution of inositol-1,4,5-trisphosphate receptor isotypes and ryanodine receptor isotypes during maturation of the rat hippocampus. Neuroscience. 2007;150:625–638. doi: 10.1016/j.neuroscience.2007.09.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howlett DR, Richardson JC, Austin A, Parsons AA, Bate ST, Davies DC, Gonzalez MI. Cognitive correlates of Abeta deposition in male and female mice bearing amyloid precursor protein and presenilin-1 mutant transgenes. Brain Res. 2004;1017:130–136. doi: 10.1016/j.brainres.2004.05.029. [DOI] [PubMed] [Google Scholar]
- Jones VC, McKeown L, Verkhratsky A, Jones OT. LV-pIN-KDEL: a novel lentiviral vector demonstrates the morphology, dynamics and continuity of the endoplasmic reticulum in live neurons. BMC Neurosci. 2008;9:10. doi: 10.1186/1471-2202-9-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller DX, Franks KM, Bartol TM, Jr, Sejnowski TJ. Calmodulin activation by calcium transients in the postsynaptic density of dendritic spines. PLoS One. 2008;3:e2045. doi: 10.1371/journal.pone.0002045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelliher M, Fastbom J, Cowburn RF, Bonkale W, Ohm TG, Ravid R, Sorrentino V, O'Neill C. Alterations in the ryanodine receptor calcium release channel correlate with Alzheimer's disease neurofibrillary and beta-amyloid pathologies. Neuroscience. 1999;92:499–513. doi: 10.1016/s0306-4522(99)00042-1. [DOI] [PubMed] [Google Scholar]
- Korkotian E, Segal M. Morphological constraints on calcium dependent glutamate receptor trafficking into individual dendritic spine. Cell Calcium. 2007;42:41–57. doi: 10.1016/j.ceca.2006.11.006. [DOI] [PubMed] [Google Scholar]
- LaFerla FM. Calcium dyshomeostasis and intracellular signalling in Alzheimer's disease. Nat Rev Neurosci. 2002;3:862–872. doi: 10.1038/nrn960. [DOI] [PubMed] [Google Scholar]
- Masliah E. Mechanisms of synaptic dysfunction in Alzheimer's disease. Histol Histopathol. 1995;10:509–519. [PubMed] [Google Scholar]
- Matsuzaki M, Honkura N, Ellis-Davies GC, Kasai H. Structural basis of long-term potentiation in single dendritic spines. Nature. 2004;429:761–766. doi: 10.1038/nature02617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson O, Tu H, Lei T, Bentahir M, de Strooper B, Bezprozvanny I. Familial Alzheimer disease-linked mutations specifically disrupt Ca2+ leak function of presenilin 1. J Clin Invest. 2007;117:1230–1239. doi: 10.1172/JCI30447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolay NH, Hertle D, Boehmerle W, Heidrich FM, Yeckel M, Ehrlich BE. Inositol 1,4,5 trisphosphate receptor and chromogranin B are concentrated in different regions of the hippocampus. J Neurosci Res. 2007;85:2026–2036. doi: 10.1002/jnr.21328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Neill C, Cowburn RF, Bonkale WL, Ohm TG, Fastbom J, Carmody M, Kelliher M. Dysfunctional intracellular calcium homoeostasis: a central cause of neurodegeneration in Alzheimer's disease. Biochem Soc Symp. 2001:177–194. doi: 10.1042/bss0670177. [DOI] [PubMed] [Google Scholar]
- Oddo S, Caccamo A, Shepherd JD, Murphy MP, Golde TE, Kayed R, Metherate R, Mattson MP, Akbari Y, LaFerla FM. Triple-transgenic model of Alzheimer's disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron. 2003;39:409–421. doi: 10.1016/s0896-6273(03)00434-3. [DOI] [PubMed] [Google Scholar]
- Sabatini BL, Svoboda K. Analysis of calcium channels in single spines using optical fluctuation analysis. Nature. 2000;408:589–593. doi: 10.1038/35046076. [DOI] [PubMed] [Google Scholar]
- Seeber S, Humeny A, Herkert M, Rau T, Eschenhagen T, Becker CM. Formation of molecular complexes by N-methyl-d-aspartate receptor subunit NR2B and ryanodine receptor 2 in neonatal rat myocard. J Biol Chem. 2004;279:21062–21068. doi: 10.1074/jbc.M313009200. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. Soluble oligomers of the amyloid beta-protein impair synaptic plasticity and behavior. Behav Brain Res. 2008;192:106–113. doi: 10.1016/j.bbr.2008.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shankar GM, Bloodgood BL, Townsend M, Walsh DM, Selkoe DJ, Sabatini BL. Natural oligomers of the Alzheimer amyloid-β protein induce reversible synapse loss by modulating an NMDA-type glutamate receptor-dependent signaling pathway. J Neurosci. 2007;27:2866–2875. doi: 10.1523/JNEUROSCI.4970-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith IF, Hitt B, Green KN, Oddo S, LaFerla FM. Enhanced caffeine-induced Ca2+ release in the 3xTg-AD mouse model of Alzheimer's disease. J Neurochem. 2005;94:1711–1718. doi: 10.1111/j.1471-4159.2005.03332.x. [DOI] [PubMed] [Google Scholar]
- Spacek J, Harris KM. Three-dimensional organization of smooth endoplasmic reticulum in hippocampal CA1 dendrites and dendritic spines of the immature and mature rat. J Neurosci. 1997;17:190–203. doi: 10.1523/JNEUROSCI.17-01-00190.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stutzmann GE. The pathogenesis of Alzheimers disease is it a lifelong “calciumopathy”? Neuroscientist. 2007;13:546–559. doi: 10.1177/1073858407299730. [DOI] [PubMed] [Google Scholar]
- Stutzmann GE, Parker I. Dynamic multiphoton imaging: a live view from cells to systems. Physiology. 2005;20:15–21. doi: 10.1152/physiol.00028.2004. [DOI] [PubMed] [Google Scholar]
- Stutzmann GE, Caccamo A, LaFerla FM, Parker I. Dysregulated IP3 signaling in cortical neurons of knock-in mice expressing an Alzheimer's-linked mutation in presenilin1 results in exaggerated Ca2+ signals and altered membrane excitability. J Neurosci. 2004;24:508–513. doi: 10.1523/JNEUROSCI.4386-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stutzmann GE, Smith I, Caccamo A, Oddo S, Laferla FM, Parker I. Enhanced ryanodine receptor recruitment contributes to Ca2+ disruptions in young, adult, and aged Alzheimer's disease mice. J Neurosci. 2006;26:5180–5189. doi: 10.1523/JNEUROSCI.0739-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terry RD, Masliah E, Salmon DP, Butters N, DeTeresa R, Hill R, Hansen LA, Katzman R. Physical basis of cognitive alterations in Alzheimer's disease: Synapse loss is the major correlate of cognitive impairment. Ann Neurol. 1991;30:572–580. doi: 10.1002/ana.410300410. [DOI] [PubMed] [Google Scholar]
- Tu H, Nelson O, Bezprozvanny A, Wang Z, Lee SF, Hao YH, Serneels L, De Strooper B, Yu G, Bezprozvanny I. Presenilins form ER Ca2+ leak channels, a function disrupted by familial Alzheimer's disease-linked mutations. Cell. 2006;126:981–993. doi: 10.1016/j.cell.2006.06.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verkhratsky A. Physiology and pathophysiology of the calcium store in the endoplasmic reticulum of neurons. Physiol Rev. 2005;85:201–279. doi: 10.1152/physrev.00004.2004. [DOI] [PubMed] [Google Scholar]
- Yamin G. NMDA receptor-dependent signaling pathways that underlie amyloid beta-protein disruption of LTP in the hippocampus. J Neurosci Res. 2009;87:1729–1736. doi: 10.1002/jnr.21998. [DOI] [PubMed] [Google Scholar]
- Zhang C, Wu B, Beglopoulos V, Wines-Samuelson M, Zhang D, Dragatsis I, Südhof TC, Shen J. Presenilins are essential for regulating neurotransmitter release. Nature. 2009;460:632–636. doi: 10.1038/nature08177. [DOI] [PMC free article] [PubMed] [Google Scholar]