

Abstract
The mechanisms that underlie the potent Th1-adjuvant capacity of poly(methyl vinyl ether-co-maleic anhydride) nanoparticles (NPs) were investigated. Traditionally, polymer NPs have been considered delivery systems that promote a closer interaction between antigen and antigen-presenting cells (APCs). Our results revealed that poly(anhydride) NPs also act as agonists of various Toll-like receptors (TLRs) (TLR2, -4, and -5), triggering a Th1-profile cytokine release (gamma interferon [IFN-γ], 478 pg/ml versus 39.6 pg/ml from negative control; interleukin-12 [IL-12], 40 pg/ml versus 7.2 pg/ml from negative control) and, after incubation with dendritic cells, inducing a 2.5- to 3.5-fold increase of CD54 and CD86 costimulatory molecule expression. Furthermore, in vivo studies suggest that NPs actively elicit a CD8+ T-cell response. Immunization with empty NPs resulted in a significant delay in the mean survival date (from day 7 until day 23 postchallenge) and a protection level of 30% after challenge against a lethal dose of Salmonella enterica serovar Enteritidis. Taken together, our results provide a better understanding of how NPs act as active Th1 adjuvants in immunoprophylaxis and immunotherapy through TLR exploitation.
Vaccination and immunotherapy represent two of the major advances in modern medicine. However, the quest for better vaccines against pathogens, allergies, or cancer still remains a challenge. Antigen variability and suboptimal immunogenicity represent some of the crucial points that explain incomplete protection, and new vaccines will have to overcome these points. Currently numerous studies of the topic of new vaccination strategies are being carried out (9, 12).
Subunit vaccines represent one of these approaches since they have the potential to develop specific responses and entail the added benefits of allowing the incorporation of different antigens in a single vaccine (4, 5, 10, 51, 54). In addition, subunit vaccines avoid the infection risk of live attenuated and recombinant vaccines. However, they are poorly immunogenic and require the use of adjuvants (40). Within them, the alum salts, which are known to polarize the response toward Th2, have been the only approved adjuvants for human use for almost 1 century (7, 28). Currently new adjuvants are being approved, such as ASO4, included in Cervarix, a human papillomavirus vaccine (GlaxoSmithKline), containing Al(OH)3, and the immunomodulator 3-O-desacyl-4′monophosphoryl lipid A (MPL). Additionally, another adjuvant, MF59, has been approved in Europe for human use and is found in several vaccines, such as an influenza vaccine (37). Although it was initially referred to as a Th2-bias adjuvant (57), it was later defined as a Th0 adjuvant, which enhances whichever response is present, without biasing the profile (38).
The main challenges, such as immunotherapy against allergies and tumors or immunoprophylaxis against intracellular pathogens, require Th1 responses in order to be effective. For this reason a number of adjuvants that display the ability to bias the immune response toward the right Th profile are being investigated (8, 22, 50, 59).
Particulate delivery systems belong to the category of adjuvants that facilitate antigen uptake by antigen-presenting cells (APCs) or by increasing the influx of professional APCs into the injection site (21). Among the different types of particulated delivery systems, polymer nanoparticles (NPs) are a group of delivery systems with interesting abilities as adjuvants for both conventional and mucosal vaccination (29). Poly(anhydride) NPs made by the copolymer of methyl vinyl ether and maleic anhydride have demonstrated their efficacy as adjuvants in inducing Th1 immune responses (16, 17, 47-49). Poly(anhydride) NPs are actually licensed in the United Kingdom for oral drug delivery (46) and have proven adjuvant capacities in mentioned and other routes. Nanoparticles can enhance the delivery of the loaded antigen to the gut lymphoid cells due to their ability to be captured and internalized by cells of the gut-associated lymphoid tissue (GALT). Furthermore, these delivery systems offer a number of extra advantages, including controlled release properties (15). However, the ability of NPs to actively take part in immune potentiation through direct modulation of immune system cells had never been studied.
In the past few years, several immunostimulatory adjuvants have proved to be able to stimulate innate responses through the engagement of pathogen-associated molecular pattern (PAMP) receptors (PRR) initiating, amplifying, and directing the specific response (18, 19, 33, 39, 41, 56). PRR receptor families include the Toll-like receptors (TLRs), whose relationship to innate/adaptive immunity has been demonstrated widely (11, 18, 22, 26, 33, 34). TLR engagement has been shown to regulate the function of APCs, leading to the upregulation of costimulatory molecules, such as CD80 and CD86, and adhesion molecules that improve antigen presentation. TLRs are linked via adapter molecules to intracellular signaling pathways that generally lead to transcription of NF-κB target genes, such as cytokine genes that will modulate the immune response.
TLRs recognize conserved molecular motifs that are shared by infectious agents but which are absent in the host. Thus, TLR4 is the cell-based PRR that recognizes lipopolysaccharides (LPS) from Gram-negative bacteria (with LPS binding protein and cell surface CD14 as coreceptors). Bacterial peptidoglycan and lipopeptides are recognized by TLR2 in combination with TLR1 or TLR6. Flagellin is recognized by TLR5, CpG-containing DNA is recognized by TLR9, the TLR3 signal is in response to double-stranded RNA, and the TLR7 and TLR8 signals are in response to certain single-stranded RNA. Therefore, not only exogenous but also endogenous ligands have been shown to act as TLR activators (45). In addition to natural ligands, nonmicrobial, xenobiotic, or artificial ligands for these TLRs have been described (31). Therefore, our hypothesis was that poly(anhydride) NPs might act as an agonist of some TLRs in order to explain the mechanisms that underlay their potent Th1-adjuvant capacity (17, 35, 47-49).
In this study, the TLR ligation and the expression of costimulatory molecules and cytokine secretion by dendritic cells (DCs) after incubation with poly(anhydride) NPs were studied. Further, we studied the T-cell activation abilities of NP and ovalbumin (OVA) coadministration, evaluating T-CD4 and T-CD8 responses. Results revealed the NP-mediated enhancement of some TLRs that may be involved in the immune adjuvant properties of these particular delivery systems.
MATERIALS AND METHODS
Preparation of PVM/MA nanoparticles.
Poly(anhydride) NPs were prepared by a modification of the solvent displacement method (3). Briefly, 100 mg poly(methyl vinyl ether-co-maleic anhydride) (PVM/MA) copolymer were dissolved in 6 ml acetone. The acetone-copolymer mixture was poured into a solution containing 0.2 g mannitol in 5.8 ml of double-deionized water (H2Odd). All solvents were evaporated under reduced pressure by spray drying (mini-spray dryer; Büchi, Switzerland).
Characterization of nanoparticles.
The size and zeta potential of nanoparticles were determined by photon correlation spectroscopy and electrophoretic laser Doppler anemometry, respectively, employing a Zetamaster analyzer system (Malver Instruments Ltd., Worcestershire, United Kingdom). The diameter of the nanoparticles was determined after dispersion in ultrapure water (1:10) and measured at 25°C with a dynamic light scattering angle of 90°C. The zeta potential was determined as follows: 200 μl of the samples were diluted in 2 ml of a 0.1 mM KCl solution adjusted to pH 7.4 (25). The average particle size is expressed as the volume mean diameter (Vmd) in nanometers (nm), and the average surface charge as mV.
Differentiation of mouse dendritic cells from mouse femur bone marrow precursors.
Bone marrow dendritic cells (BMDC) were generated as follows: bone marrow from BALB/c mouse femur and tibia was extracted, and erythrocytes were lysated in ammonium chloride potassium (ACK) lysing buffer for 1 min (NH4Cl, 0.15 M; KHCO3, 10 mM; Na2 EDTA, 0.1 mM). Obtained cells were then washed by centrifugation in RPMI 1640 medium. The lymphocyte and granulocyte populations were then depleted by coincubation with 50 μg/ml of rabbit complement (Sigma Chem. Co.) and an antibody mixture against CD4 (GK1-5 hybridoma, 100 μg/ml), CD8 (H35.17.2 [142] hybridoma, 100 μg/ml), Ly-6G/Gr1 (10 μg/ml), and CD45R/B220 (50 μg/ml). (All reagents were from BD Pharmingen.) The mixture was incubated at 37°C for 50 min. After depletion, cells were washed, and all remaining cells were harvested in 24-well plates at 106 cells/ml. The medium (RPMI 1640 with 10% fetal calf serum [FCS], 2 mM glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin, and 5 × 10−5 mM 2-mercapthoethanol) was supplemented with 20 ng/ml of mouse granulocyte/macrophage colony-stimulating factor and interleukin 4 (IL-4) (Prepotech, London, United Kingdom) at 37°C and in 5% CO2. At days 2 and 5, two-thirds of the medium was replaced with fresh cytokine-supplemented medium. On the 7th day, in vitro assays for BMDC maturation markers were carried out by adding 500, 100, 20, and 4 μg/ml of poly(anhydride) NPs along with negative controls. Forty-eight hours later, supernatants were collected, and IL-12 (p70) and tumor necrosis factor alpha (TNF-α) measured by enzyme-linked immunosorbent assay (ELISA) (BD Pharmingen), according to the manufacturer's instructions. Finally, all cells were harvested and analyzed by flow cytometry by using anti-CD54 (clone 3E2), anti-CD86 (clone GL1), anti-CD11c (clone HL-3), and for the unspecific bind measurement, mouse fluorescein isothiocyanate (FITC)-labeled IgG1 (clone MOPC-21), at 4°C for 15 min. All reagents originated from BD Pharmingen. Then cells were washed, acquired on a FACScan cytometer (BD Biosciences), and analyzed using Cell Quest software (BD Biosciences). Finally, the gating was selected by comparison with positive and negative controls. The mean fluorescence intensity (MFI) of the control isotype (IgG1) was subtracted from the fluorescence data of its own group and then referred to the fluorescence of the negative control for the analyzed maturation marker, as follows: increase level = (sample MFImarker − sample MFIIgG1)/(untreated MFImarker − untreated MFIIgG1).
In vivo killing.
The system used to detect cytotoxic T lymphocyte (CTL) responses elicited after OVA-NP immunization is based on antigen presentation of OVA peptides on major histocompatibility (MHC) class I molecules. In vitro restimulation of splenic cells is performed with a specific OVA peptide characterized by its specific recognition MHC class I molecule. Thus, C57BL/6 mice were intraperitoneally (i.p.) immunized with 4 mg of empty poly(anhydride) NPs, 10 μg of OVA, or both 4 mg of NP and 10 μg OVA in different injection sites to reduce adsorptions. Due to the high ability of poly(anhydride) NPs to bind the amino-terminal part of proteins, NPs and OVA were administered in different sites of the abdomen, reducing possible OVA coating of poly(anhydride) NPs. At day 7, spleens from naïve mice were extracted and smashed, and erythrocytes lysed in ACK buffer. Resulting splenocytes were treated in the presence or in the absence of 10 μg/ml of the SIINFEKL peptide (encompassing the immunodominant H-2b-restricted CTL epitope from ovalbumin [amino acids 257 to 264], produced by Neomps, Strasbourg, France) for 30 min at 37°C. Splenic cells were then differentially labeled with 2.5 μM (SIINFEKL-pulsed targets) or 0.25 μM (nonpulsed splenocytes) of 5-(6)-carboxyfluorescein diacetate N-succinimidyl ester (CFSE) for 15 min at room temperature and used as target cells to detect in vivo cytotoxic activity. A dose of 107 cells per mouse (50% CFSE high and 50% CFSE low) was intravenously (i.v.) injected into the naïve and immunization groups. After 24 h spleens were removed, and the high-versus-low-CFSE lymphocyte population ratio was analyzed by flow cytometry. All lysis values correspond to triplicate samples. Specific lysis was calculated as follows: % specific lysis = 100 − {[% splenocytes CFSE high (from immunized mice)/% splenocytes CFSE low (from immunized mice)]/[% splenocytes CFSE high (from nonimmunized mice)/% splenocytes CFSE low (from nonimmunized mice)]} × 100.
Specific IFN-γ production.
To measure the production of gamma interferon (IFN-γ) in response to SIINFEKL, splenocytes from mice collected at day 8 after immunization were plated on 96-well plates at 8 × 105 cells/well with culture medium (CM) alone or with 30 μM SIINFEKL in a final volume of 0.25 ml of CM. Supernatants (50 μl) were removed 48 h later, and IFN-γ was measured by enzyme-linked immunospot (ELISPOT) assay (BD Pharmingen) according to the manufacturer's instructions.
TLR screening.
LPS-free empty NPs were shipped to Invivogen (France) to assay their capacity to stimulate TLR signaling. Briefly, samples and controls were tested on recombinant HEK293 cell lines that functionally overexpress a given TLR protein as well as a reporter gene which is a secreted alkaline phosphatase. The production of this reporter gene is driven by a NF-κβ-inducible promoter. Nanoparticles were incubated at 500 μg/ml, the highest nontoxic concentration, as recommended by Invivogen. Known TLR agonists used as positive controls include PAM2 (100 ng/ml) for TLR2; poly(I:C) (100 ng/ml) for TLR3; Escherichia coli K12 LPS (1 μg/ml) for TLR4; flagellin (1 μg/ml) for TLR5; R848 (10 μg/ml) for TLR7 and TLR8, and ODN 2006 (10 μg) for TLR9. All results are given as optical density (OD) values.
Protection studies of mice.
Phosphate-buffered saline (PBS) or empty poly(anhydride) NPs were administered to groups of 10 BALB/c mice. Ten days after administration, mice were i.p. infected with a lethal dose of Salmonella enterica serovar Enteritidis in stationary phase (1.6 × 102 CFU, in 100 μl phosphate-buffered saline). The number of dead mice after challenge was recorded daily. All mice were treated in accordance with institutional guidelines for treatment of animals (Ethical Committee for the Animal Experimentation, CEEA, of the University of Navarra).
IFN-γ and IL-4 release from splenic cells.
In order to determine the degree of activation of the two subset Th1/Th2 populations in immunized mice, IFN-γ and IL-4 release from splenic cells was determined. Three animals immunized with PBS or poly(anhydride) NPs were sacrificed at day 10 by cervical dislocation, and spleens were removed and placed in RPMI 1640 media (Gibco-BRL, Paisley, United Kingdom) under sterile conditions. Spleens were smashed in tissue culture dishes and centrifuged (400 × g, 10 min), the supernatant was discarded, and the sediment was washed twice in phosphate-buffered saline. The cellular pellet was resuspended in 2 ml RPMI 1640-HEPES medium (Gibco-BRL) supplemented with 500 μl of antibiotic-antimycotic solution (Sigma) and 10% (vol/vol) fetal bovine serum. In a 24-well microtiter plate (Costar, NY), cell suspensions were added at 800,000 cells/well, along with Salmonella serovar Enteritidis heat extracts (HE) at 100 μg/well (36). Negative controls without HE were also included. All wells were set up in duplicate. Cells were incubated (37°C, 48 h, 5% CO2), supernatants were collected, and the levels of IFN-γ and IL-4 released determined with a commercial sandwich ELISA kit (Biosource, Camrillo, CA).
Statistical analyses.
When required, the nonparametric Kruskal-Wallis test, followed by the Mann-Whitney U test, was used. Statistically significant differences were considered when P was <0.05. All data processing was performed using the GraphPadPrism 5 software.
RESULTS
Dendritic cell costimulatory molecule increase triggered by nanoparticles.
Nanoparticle formulations displayed a homogeneous volume mean diameter of 230 ± 5 nm, with a low polydispersity index. In addition, poly(anhydride) NPs displayed an electronegative surface charge with a Zeta potential value of −36 ± 2 mV.
After the differentiation of BMDC, an analysis of CD54 and CD86 markers was performed in order to confirm the induction of positive maturation triggered by NPs. Flow cytometry studies showed that incubation of 4 μg/ml of poly(anhydride) NPs induced 2.3- and 2.4-fold increases of expression of both CD54 and CD86, respectively, compared with BMDC incubated with medium alone (Fig. 1). When the concentration of empty NP was raised to 20 μg/ml, the expression of the costimulatory molecule CD54 reached a 3.6-fold increase relative to the negative control.
FIG. 1.

Flow cytometric analysis of BMDC maturation markers. Bars show the fold-increase of mean relative fluorescence over negative control of CD86 and CD54 after coincubation of BMDC with 500 μg of NP, 100 μg of NP, 20 μg of NP, and 4 μg of NP.
Induction of cytokine production of BMDC by NP stimulation.
We then examined whether the NP stimulation was able to stimulate bone marrow-derived DCs to produce the proinflammatory cytokine IL-12 or TNF-α. It was found that NP stimulation induced TNF-α release in a dose-dependent manner (Fig. 2A), varying from 27.8 pg/ml at 4 μg/ml of NP to 478.7 pg/ml at 500 μg/ml and being in all cases significantly higher (P < 0.001) than those found in supernatant from nonstimulated cells. The analysis of IL-12 production revealed also a dose-response pattern, being significant from 20 to 100 μg/ml. A higher concentration of NP (500 μg/ml) reduced the IL-12 release to basal levels (Fig. 2B).
FIG. 2.
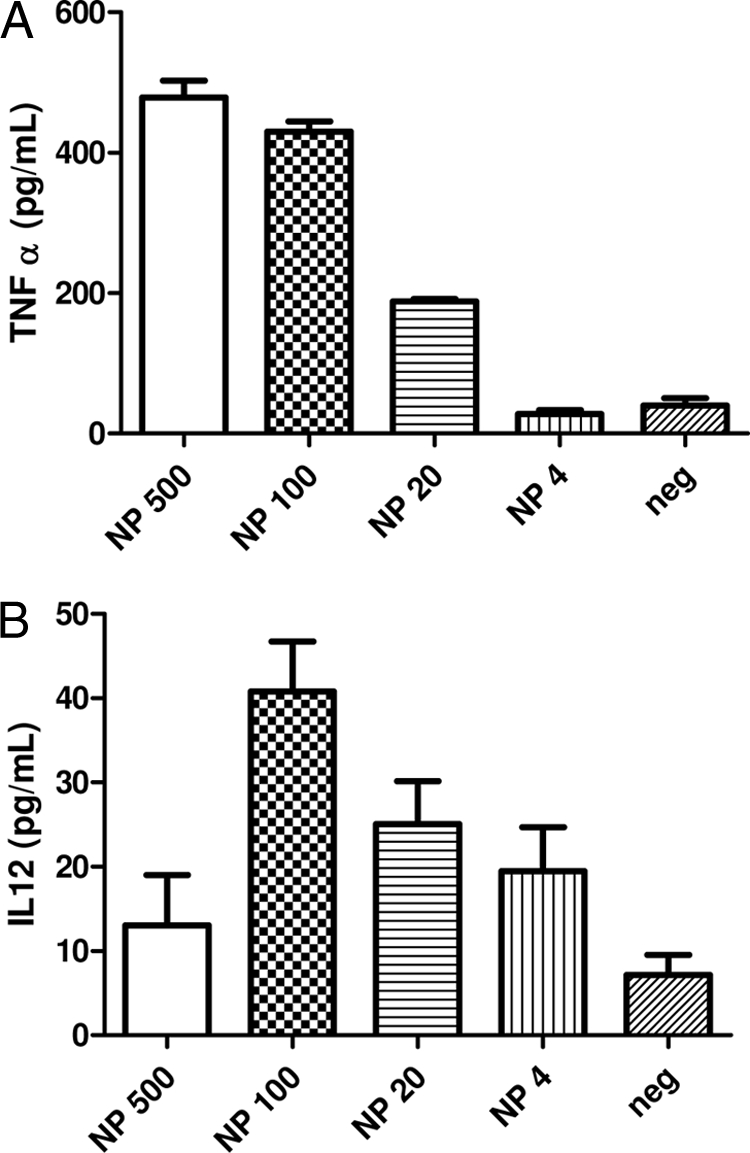
Proinflammatory cytokine detection in supernatants of BMDC. Levels of TNF-α (A) and IL-12 (B) released to the BMDC culture supernatant after 24 h of coincubation with 500 μg of NP, 100 μg of NP, 20 μg of NP, and 4 μg of NP. Nonstimulated controls are also included. All values are shown as arithmetic means ± standard errors of the means (SEM) (n = 3).
Cytotoxicity induced by NP uptake: in vivo killing.
An in vivo CTL assay was conducted to measure the capacity of NPs to activate OVA-specific CD8+ T cells using the specific SIINFEKL OVA peptide. C57BL/6 mice were immunized i.p. with OVA (10 μg), NP (4 mg), or a mixture of OVA and NP. At day 7 after immunization, mice were injected i.v. with a mixed population of CFSE-labeled splenocytes. Twenty-four hours after injection of the CFSE-labeled cells, splenocytes were analyzed by flow cytometry for the presence of the two populations of CFSE-labeled cells. Results indicate that the specific T-cell lysis response to SIINFEKL treatment was negligible in the absence of the NP adjuvant. However, when lymphocytes from mice immunized with NPs plus OVA were tested, the in vivo specific CTL response against SIINFEKL was 41.2% ± 16.5% (Fig. 3).
FIG. 3.
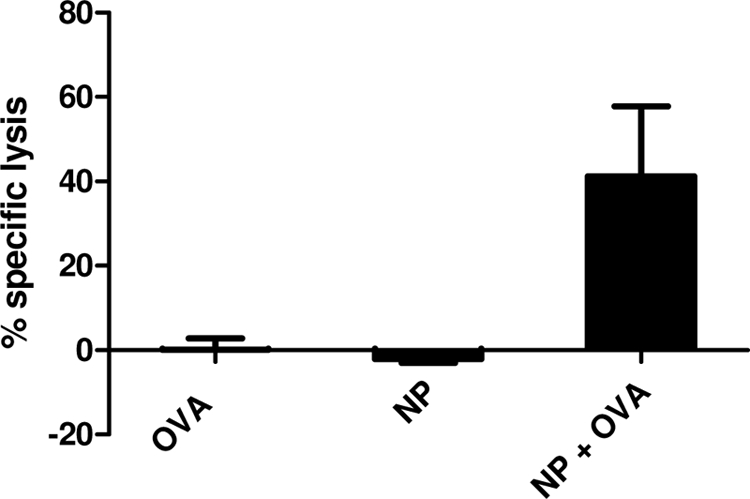
In vivo induction of CTL activity specific for SIINFEKL peptide. C57BL/6 mice were immunized with OVA, poly(anhydride) NPs, or OVA plus NP; 7 days after immunization, naïve mice were sacrificed, and splenocytes coincubated in the presence or absence of SIINFEKL. Pulsed cells were incubated in the presence of high levels of CFSE, while nonpulsed cells were incubated with low levels of CFSE. After administration of CFSE-high and CFSE-low splenocytes to immunized mice, the specific cytotoxic activity against the SIINFEKL peptide was measured by an in vivo killing assay. The data represent the mean percentage values from triplicate samples.
In addition to measuring the in vivo cytotoxicity, we also measured by ELISPOT the number of IFN-γ-producing cells specific for the SIINFEKL peptide induced by immunization. Thus, 7 days after immunization, splenic cells were restimulated in vitro in the presence or absence of the SIINFEKL peptide. The number of IFN-γ spots was counted to evaluate the frequency of SIINFEKL-specific cytotoxic T cells. The ELISPOT assay revealed IFN-γ-producing OVA-specific cells from mice injected with OVA-NP (129.25 ± 56.20 spot-forming cells [SFC]/106 cells compared with 27.5 ± 11.1 SFC/106 cells in the absence of peptide, P < 0.05) (Fig. 4). In contrast, IFN-γ-producing splenocytes specific for the SIINFEKL peptide were not detected in mice immunized with NPs or OVA alone.
FIG. 4.

In vivo induction of OVA-specific IFN-γ-secreting CD8+ T cells. Mice were immunized with OVA, poly(anhydride) NPs, and OVA plus NP and 8 days after immunization sacrificed. Splenocytes were cultured in triplicate in the presence of SIINFEKL or culture medium alone (negative control). Each bar represents the mean value of SFC/106 cells.
Nanoparticle-induced TLRs.
In order to understand the adjuvant capacities shown by the poly(anhydride) NPs, their ability to trigger different TLR responses was measured. As shown in Fig. 5, nanoparticles significantly activated hTLR2-, hTLR4-, and hTLR5-expressing cell lines. In addition the hTLR3 cell line was also weakly activated. The TLR− cell line was not stimulated at all. This cell line does not express any TLR but still has an NF-κβ-inducible promoter. This result confirmed that all measured OD values were strictly a consequence of TLR stimulation.
FIG. 5.

Effects of nanoparticles on the activation of TLR signaling. Bars represent engagement to TLR2, TLR3, TLR4, TLR5, TLR7, TLR8, and TLR9 after incubation with positive controls and poly(anhydride) NPs. Names inside the bars of positive controls represent the specific agonist for each TLR used in the study: PAM2 (100 ng/ml) for TLR2, poly(I:C) (100 ng/ml) for TLR3, E. coli K12 LPS (1 μg/ml) for TLR4, flagellin (1 μg/ml) for TLR5, R848 (10 μg/ml) for TLR7 and TLR8, and ODN 2006 (10 μg) for TLR9. A TLR nonexpressing recombinant cell line is also included (TLR−). Results are given in OD values.
Immunization and protection studies of mice against Salmonella serovar Enteritidis.
Ten days after the intraperitoneal administration of poly(anhydride) NPs, splenocytes showed a basal expression of IFN-γ (20 pg/ml). Restimulation with an antigenic complex containing LPS (HE, from S. Enteritidis) resulted in a significative (P > 0.001) increase in detected IFN-γ (577 pg/ml). Nonimmunized animals showed no IFN-γ release before restimulation and a very small amount (40 pg/ml) after incubation with HE containing LPS. When IL-4 production was measured, a very weak basal secretion was detected for restimulated splenocytes in both the inoculated and control groups (10 pg/ml).
Nanoparticle inoculation elicited a 30% protection against lethal intraperitoneal challenge with S. Enteritidis (1.6 × 102 CFU) (Fig. 6). In addition, the end survival date was significantly delayed (P < 0.001) from day 7 for the control group until day 23 for the treated animals.
FIG. 6.

Comparative protection against the virulent serovar of S. Enteritidis. Groups of 10 BALB/c mice were i.p. immunized with poly(anhydride) NPs (♦) or PBS (○). Ten days later, mice were challenged i.p. with 1.6 × 102 CFU of the virulent strain 3934. Data are expressed as percentage of survivals after challenge.
DISCUSSION
The incorporation of antigens into poly(anhydride) NPs has been demonstrated to enhance the immune responses in terms of antibody production and overall protection levels (16, 17, 35). This fact may be explained by the implemented possibilities that NPs render to the antigen: controlled release from the vehicle and chemotaxis for APC recruitment. Besides, Gomez et al. reported that after oral administration, the bioadhesive nature of the polymer enhanced the interaction of poly(anhydride) NPs with the gut mucosa (1, 16, 21). In addition, the ability of nanoparticles to partially erode allows a controlled release of the antigens that could favor their sustained exposition to the immune system, increasing the efficiency of vaccination. In fact, we observed the formation of an antigen depository after intradermal vaccination that increased antigen residence time that, in turn, was translated into higher responses (I. Tamayo, C. Gamazo, J. deSouza, and J. M. Irache, unpublished results). Moreover, these NPs allow the adhesion of antigens and ligands to their outer shells, creating high-density antigen surfaces that increase the possibilities of antigen recognition and/or capture by the APCs. Based on the ability of NPs to induce potent immune responses, our hypothesis was that NPs are able to trigger determined elements of the immune system. Explicitly, due to their particulate nature, we hypothesized that NPs would interact with APCs, specifically DCs, through PRRs, including TLR.
Of the receptors (PRR) that sense the plethora of “danger” signals, TLRs represent one of the most prominent groups. Not in vain, TLR engagement leads to DC activation and promotes T-cell priming and acquired immunity. In addition, it also activates microbicidal effects and inflammation in cells of the innate immune system. It is well established that some pathogen products, PAMPs, are the main natural actors involved in the activation of DCs through PRR, resulting in a defined maturation phenotype responsible for driving a polarized Th1 or Th2 response. Thus, DCs are capable of integrating signals from pathogens, cytokines, and T cells, leading to the generation of an adaptive immune response of the appropriate class (44). However, it has become clear that PRR may sense nonmicrobial “patterns” (45), including polysaccharides from plants (43), dietary fatty acid lipids (27), and some xenobiotic products used in pharmacotherapy (31). These include synthetic ligands of TLR7, such as imiquimod, R848, and loxoribine (2), but the TLR family is also critical for the recognition of certain endogenous molecules (24). Indeed, TLR2 can be activated by Hsp-60, -70, or -96 or by high-mobility group protein B1 (HMGB1), whereas TLR4 can be triggered by Hsp22, -60, -70, or -96, fibrinogen, HMGB1, hyaluronan fragments, or by spliced exon encoding the type III repeat extra domain A (EDA) from fibronectin as recently described (26).
When the potential of NPs to act as TLR ligands was screened, we observed the high ability to stimulate TLR2 and, to a lesser extent, TLR3, TLR4, and TLR5 signaling. In general, the hydrophobicity of a PAMP is a crucial structural parameter for receptor specificity (6). Therefore, we can assume that NP polymer hydrophobicity is in the base of its TLR agonist capacity. However, some specificity should drive the specific activation; in fact, NP-mediated activation was a consequence exclusively of TLR ligation as demonstrated by the absence of TLR−-cell line activation.
TLR2 appears to contain several epitopes that may interact with ligands, potentially suggesting that different regions within the molecule might contribute to the recognition of specific agonists (30). Thus, TLR2 seems to be highly promiscuous, able to recognize the most diverse set of nonrelated TLR2 agonists, most of them glycolipids, lipopeptides, or glycosyl-phosphatidylinositol (GPI)-anchored structures (58). Even some other polymers used in nanotechnology, such as alginate polymers, have been reported to be potent immune-stimulating agents in eliciting cytokine production by monocytes through TLR (14). The authors of this report suggested that short oligosaccharides from alginates serve as agonists for TLR2 and TLR4. Some alginate-derived neoglycolipids have also been described as potent ligands for TLR4 (60). It is noteworthy that many of the structurally defined nonbacterial agonists for TLR2 share a diacylglycerol group for which TLR2 has a structurally defined hydrophobic binding pocket (52). However, from the strict structural point of view, many of the PAMPs published for TLR2 have almost nothing in common. It has been also suggested that the promiscuity of TLR2 is linked to the fact that TLR2 interacts with other receptors, such as TLR1, TLR6, CD36, or dectin-1 (61). However, our results did not show TLR1 or TLR6 activation, rather a TLR4 activation. Agonists for TLR4 may be related to TLR2 agonists since both may require acylated saturated fatty acids in their molecules. Thus, some kind of promiscuity has been related also to TLR4, with a growing list of ligands (23). Similarly, TLR5 can signal as a homodimer, but it can also build a heterodimer with TLR4, which results in the activation of an alternative downstream signaling pathway (32).
Napolitani et al. (34) identified a “combinatorial code” by which DCs promote Th1 responses, describing that synergistic TLR stimulation is probably the rule. So, our hypothesis is that the NP is able to bind to APC via proximal TLR, leading to the assembly of membrane signaling complexes and the recruitment of the cytoplasmic protein MyD88, which triggers the activation of the transcription factor NF-κB, which induces the expression of TH1-derived cytokine genes (20).
To evaluate this hypothesis of the effect of TLR synergy on Th1 priming, we stimulated DCs with NP and tested their capacity to trigger CD86 and CD54 costimulatory molecule expression. Results demonstrated not only the high expression of these costimulatory molecules but also the secretion of Th1-derived cytokines from DCs. One of the most important characteristics that place DCs as key players is their ability to decode and integrate danger signals that are transported in a mature state to the T-cell areas, initiating an immune response (42). As a result of an effective interaction, the DCs undergo a maturation process. Thus, the analysis of the so-called signal 3, with regard to the treatment with poly(anhydride) NPs, revealed a significant increase of TNF-α expression that followed a dose-response pattern. Additionally, the detected levels of IL-12 were also significantly increased, as a consequence of coincubation with NPs. IL-12 from DCs is a key marker in the innate responses that drive Th1 polarization (55). It is, then, plausible to suppose that detection of both cytokines endorses the hypothesis of TLR stimulation. Nevertheless, to address speculations about beneficial effects of TLR signaling, a more detailed examination, including one of a TLR knockout system (2, 4, 5), remains to be performed.
After confirming positive in vitro DC maturation and cytokine expression, we analyzed the ability of NPs to induce an in vivo cellular response. Studies performed with the model OVA-peptide SIINFEKL showed that the coadministration of empty NPs with OVA resulted in the induction of cytotoxic T cells specific for target cells displaying OVA. Similarly, ELISPOT assays revealed the presence of IFN-γ-producing cells in response to stimulation with the SIINFEKL peptide in splenocytes from mice immunized with NPs and OVA, indicating that NPs may be considered an adjuvant for the induction of CD8+ T-cell immune responses.
All together, these results agree with previous observations that suggested the potent immunostimulating nature of these NPs. Estevan et al. (13) described that inoculation of mice with empty NPs induced a significant level of protection against a challenge with Salmonella enterica serovar Abortusovis. In fact, we performed an experiment with empty nanoparticles in order to test their ability to induce a protective response against a lethal challenge with Salmonella serovar Enteritidis. Results demonstrate a level of protection time compatible with the activation immune response, as discussed above, with a significant delay in the mean survival date of treated mice from day 7 until day 23 postchallenge. For the particular case of Salmonella infections, the first stages postimmunization were shown to be nonspecific even when immunized with live vaccines (53). In addition, as a facultative intracellular pathogen, T cell-mediated immunity is required for clearance of the pathogen. The pattern of cytokines released after NP inoculation is consistent with the hypothesis that NPs activate an innate response mediated by PRRs, enough to control the growth of bacteria until the establishment of the proper T cell.
Classically, nanoparticles are being exploited as antigen delivery systems, but a more active role may be behind their adjuvant properties. The results described here suggest an active interaction of NP with DCs that will render multiple stimuli mediated by PRRs, which might allow a more effective response. Taken together, our results shed light on how NPs act as active Th1 adjuvants in immunoprophylaxis and immunotherapy (10, 16, 17, 35). More information on NP recognition, intracellular trafficking, and processing in DCs would help us to establish a greater degree of accuracy in this matter.
Acknowledgments
This work was supported by Instituto de Salud Carlos III, Spain (FI08/00432), and Departamento de Salud, Gobierno de Navarra, Spain (2118/2007).
Footnotes
Published ahead of print on 14 July 2010.
REFERENCES
- 1.Agüeros, M., P. Areses, M. A. Campanero, H. Salman, G. Quincoces, I. Penuelas, and J. M. Irache. 2009. Bioadhesive properties and biodistribution of cyclodextrin-poly(anhydride) nanoparticles. Eur. J. Pharm. Sci. 37:231-240. [DOI] [PubMed] [Google Scholar]
- 2.Akira, S., and H. Hemmi. 2003. Recognition of pathogen-associated molecular patterns by TLR family. Immunol. Lett. 85:85-95. [DOI] [PubMed] [Google Scholar]
- 3.Arbós, P., M. Wirth, M. A. Arangoa, F. Gabor, and J. M. Irache. 2002. Gantrez AN as a new polymer for the preparation of ligand-nanoparticle conjugates. J. Control Release 83:321-330. [DOI] [PubMed] [Google Scholar]
- 4.Baums, C. G., C. Kock, A. Beineke, K. Bennecke, R. Goethe, C. Schroder, K. H. Waldmann, and P. Valentin-Weigand. 2009. Streptococcus suis bacterin and subunit vaccine immunogenicities and protective efficacies against serotypes 2 and 9. Clin. Vaccine Immunol. 16:200-208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bertholet, S., Y. Goto, L. Carter, A. Bhatia, R. F. Howard, D. Carter, R. N. Coler, T. S. Vedvick, and S. G. Reed. 2009. Optimized subunit vaccine protects against experimental leishmaniasis. Vaccine 27:7036-7045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Beutler, B. 2003. Not “molecular patterns” but molecules. Immunity 19:155-156. [DOI] [PubMed] [Google Scholar]
- 7.Bungener, L., F. Geeraedts, V. W. Ter, J. Medema, J. Wilschut, and A. Huckriede. 2008. Alum boosts TH2-type antibody responses to whole-inactivated virus influenza vaccine in mice but does not confer superior protection. Vaccine 26:2350-2359. [DOI] [PubMed] [Google Scholar]
- 8.Champion, C. I., V. A. Kickhoefer, G. Liu, R. J. Moniz, A. S. Freed, L. L. Bergmann, D. Vaccari, S. Raval-Fernandes, A. M. Chan, L. H. Rome, and K. A. Kelly. 2009. A vault nanoparticle vaccine induces protective mucosal immunity. PLoS One 4:e5409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chopra, A., T. S. Kim, I. Sullivan, D. Martinez, and E. P. Cohen. 2006. Treatment of squamous carcinoma in mice with a vaccine enriched for cells that induce immunity to squamous carcinoma-a new vaccination strategy. Int. J. Cancer 119:339-348. [DOI] [PubMed] [Google Scholar]
- 10.Da Costa Martins, R., J. M. Irache, J. M. Blasco, M. P. Munoz, C. M. Marin, M. Jesus Grillo, M. Jesus De Miguel, M. Barberan, and C. Gamazo. 2010. Evaluation of particulate acellular vaccines against Brucella ovis infection in rams. Vaccine 28:3038-3046. [DOI] [PubMed] [Google Scholar]
- 11.Da Silva, C. A., D. Hartl, W. Liu, C. G. Lee, and J. A. Elias. 2008. TLR-2 and IL-17A in chitin-induced macrophage activation and acute inflammation. J. Immunol. 181:4279-4286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Del Giudice, G. 2003. Vaccination strategies. An overview. Vaccine 21(Suppl. 2):S83-S88. [DOI] [PubMed] [Google Scholar]
- 13.Estevan, M., J. M. Irache, M. J. Grillo, J. M. Blasco, and C. Gamazo. 2006. Encapsulation of antigenic extracts of Salmonella enterica serovar Abortusovis into polymeric systems and efficacy as vaccines in mice. Vet. Microbiol. 118:124-132. [DOI] [PubMed] [Google Scholar]
- 14.Flo, T. H., L. Ryan, E. Latz, O. Takeuchi, B. G. Monks, E. Lien, O. Halaas, S. Akira, G. Skjak-Braek, D. T. Golenbock, and T. Espevik. 2002. Involvement of toll-like receptor (TLR) 2 and TLR4 in cell activation by mannuronic acid polymers. J. Biol. Chem. 277:35489-35495. [DOI] [PubMed] [Google Scholar]
- 15.Galindo-Rodriguez, S. A., E. Allemann, H. Fessi, and E. Doelker. 2005. Polymeric nanoparticles for oral delivery of drugs and vaccines: a critical evaluation of in vivo studies. Crit. Rev. Ther. Drug Carrier Syst. 22:419-464. [DOI] [PubMed] [Google Scholar]
- 16.Gómez, S., C. Gamazo, B. S. Roman, M. Ferrer, M. L. Sanz, and J. M. Irache. 2007. Gantrez AN nanoparticles as an adjuvant for oral immunotherapy with allergens. Vaccine 25:5263-5271. [DOI] [PubMed] [Google Scholar]
- 17.Gómez, S., C. Gamazo, R. B. San, C. Vauthier, M. Ferrer, and J. M. Irachel. 2006. Development of a novel vaccine delivery system based on Gantrez nanoparticles. J. Nanosci. Nanotechnol. 6:3283-3289. [DOI] [PubMed] [Google Scholar]
- 18.Green, S. J. 2008. Clinical development of TLR agonists as adjuvants: “post-alum adjuvant” candidates may reach beyond their intended purpose. Clin. Pharmacol. Ther. 83:813-814. [DOI] [PubMed] [Google Scholar]
- 19.Guy, B. 2007. The perfect mix: recent progress in adjuvant research. Nat. Rev. Microbiol. 5:505-517. [DOI] [PubMed] [Google Scholar]
- 20.Hirata, N., Y. Yanagawa, T. Ebihara, T. Seya, S. Uematsu, S. Akira, F. Hayashi, K. Iwabuchi, and K. Onoe. 2008. Selective synergy in anti-inflammatory cytokine production upon cooperated signaling via TLR4 and TLR2 in murine conventional dendritic cells. Mol. Immunol. 45:2734-2742. [DOI] [PubMed] [Google Scholar]
- 21.Irache, J. M., H. H. Salman, S. Gomez, S. Espuelas, and C. Gamazo. 2009. Poly(anhydride) nanoparticles as adjuvants for mucosal vaccination. Front. Biosci. 2:876-890. [DOI] [PubMed] [Google Scholar]
- 22.Jasani, B., H. Navabi, and M. Adams. 2009. Ampligen: a potential Toll-like 3 receptor adjuvant for immunotherapy of cancer. Vaccine 27:3401-3404. [DOI] [PubMed] [Google Scholar]
- 23.Jenkins, S. J., J. P. Hewitson, S. Ferret-Bernard, and A. P. Mountford. 2005. Schistosome larvae stimulate macrophage cytokine production through TLR4-dependent and -independent pathways. Int. Immunol. 17:1409-1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kanzler, H., F. J. Barrat, E. M. Hessel, and R. L. Coffman. 2007. Therapeutic targeting of innate immunity with Toll-like receptor agonists and antagonists. Nat. Med. 13:552-559. [DOI] [PubMed] [Google Scholar]
- 25.Lambert, G., E. Fattal, H. Pinto-Alphandary, A. Gulik, and P. Couvreur. 2000. Polyisobutylcyanoacrylate nanocapsules containing an aqueous core as a novel colloidal carrier for the delivery of oligonucleotides. Pharm. Res. 17:707-714. [DOI] [PubMed] [Google Scholar]
- 26.Lasarte, J. J., N. Casares, M. Gorraiz, S. Hervas-Stubbs, L. Arribillaga, C. Mansilla, M. Durantez, D. Llopiz, P. Sarobe, F. Borras-Cuesta, J. Prieto, and C. Leclerc. 2007. The extra domain A from fibronectin targets antigens to TLR4-expressing cells and induces cytotoxic T cell responses in vivo. J. Immunol. 178:748-756. [DOI] [PubMed] [Google Scholar]
- 27.Lee, J. Y., A. Plakidas, W. H. Lee, A. Heikkinen, P. Chanmugam, G. Bray, and D. H. Hwang. 2003. Differential modulation of Toll-like receptors by fatty acids: preferential inhibition by n-3 polyunsaturated fatty acids. J. Lipid Res. 44:479-486. [DOI] [PubMed] [Google Scholar]
- 28.Lindblad, E. B. 2004. Aluminium compounds for use in vaccines. Immunol. Cell Biol. 82:497-505. [DOI] [PubMed] [Google Scholar]
- 29.Mann, J. F., R. Acevedo, J. D. Campo, O. Perez, and V. A. Ferro. 2009. Delivery systems: a vaccine strategy for overcoming mucosal tolerance? Expert Rev. Vaccines 8:103-112. [DOI] [PubMed] [Google Scholar]
- 30.Meng, G., A. Grabiec, M. Vallon, B. Ebe, S. Hampel, W. Bessler, H. Wagner, and C. J. Kirschning. 2003. Cellular recognition of tri-/di-palmitoylated peptides is independent from a domain encompassing the N-terminal seven leucine-rich repeat (LRR)/LRR-like motifs of TLR2. J. Biol. Chem. 278:39822-39829. [DOI] [PubMed] [Google Scholar]
- 31.Miyake, K. 2007. Innate immune sensing of pathogens and danger signals by cell surface Toll-like receptors. Semin. Immunol. 19:3-10. doi: 10.1016/j.smim.2006.12.002. [DOI] [PubMed] [Google Scholar]
- 32.Mizel, S. B., A. N. Honko, M. A. Moors, P. S. Smith, and A. P. West. 2003. Induction of macrophage nitric oxide production by Gram-negative flagellin involves signaling via heteromeric Toll-like receptor 5/Toll-like receptor 4 complexes. J. Immunol. 170:6217-6223. [DOI] [PubMed] [Google Scholar]
- 33.Najar, H. M., and J. P. Dutz. 2007. Topical TLR9 agonists induce more efficient cross-presentation of injected protein antigen than parenteral TLR9 agonists do. Eur. J. Immunol. 37:2242-2256. [DOI] [PubMed] [Google Scholar]
- 34.Napolitani, G., A. Rinaldi, F. Bertoni, F. Sallusto, and A. Lanzavecchia. 2005. Selected Toll-like receptor agonist combinations synergistically trigger a T helper type 1-polarizing program in dendritic cells. Nat. Immunol. 6:769-776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ochoa, J., J. M. Irache, I. Tamayo, A. Walz, V. G. DelVecchio, and C. Gamazo. 2007. Protective immunity of biodegradable nanoparticle-based vaccine against an experimental challenge with Salmonella Enteritidis in mice. Vaccine 25:4410-4419. [DOI] [PubMed] [Google Scholar]
- 36.Ochoa-Repáraz, J., B. Sesma, M. Alvarez, R. M. Jesus, J. M. Irache, and C. Gamazo. 2004. Humoral immune response in hens naturally infected with Salmonella Enteritidis against outer membrane proteins and other surface structural antigens. Vet. Res. 35:291-298. [DOI] [PubMed] [Google Scholar]
- 37.O'Hagan, D. T. 2007. MF59 is a safe and potent vaccine adjuvant that enhances protection against influenza virus infection. Expert Rev. Vaccines 6:699-710. [DOI] [PubMed] [Google Scholar]
- 38.O'Hagan, D. T., A. Wack, and A. Podda. 2007. MF59 is a safe and potent vaccine adjuvant for flu vaccines in humans: what did we learn during its development? Clin. Pharmacol. Ther. 82:740-744. [DOI] [PubMed] [Google Scholar]
- 39.Pashine, A., N. M. Valiante, and J. B. Ulmer. 2005. Targeting the innate immune response with improved vaccine adjuvants. Nat. Med. 11:S63-S68. [DOI] [PubMed] [Google Scholar]
- 40.Perrie, Y., A. R. Mohammed, D. J. Kirby, S. E. McNeil, and V. W. Bramwell. 2008. Vaccine adjuvant systems: enhancing the efficacy of sub-unit protein antigens. Int. J. Pharm. 364:272-280. [DOI] [PubMed] [Google Scholar]
- 41.Petrovsky, N., and J. C. Aguilar. 2004. Vaccine adjuvants: current state and future trends. Immunol. Cell Biol. 82:488-496. [DOI] [PubMed] [Google Scholar]
- 42.Pulendran, B. 2004. Modulating vaccine responses with dendritic cells and Toll-like receptors. Immunol. Rev. 199:227-250. [DOI] [PubMed] [Google Scholar]
- 43.Raghu, R., D. Sharma, R. Ramakrishnan, S. Khanam, G. J. Chintalwar, and K. B. Sainis. 2009. Molecular events in the activation of B cells and macrophages by a non-microbial TLR4 agonist, G1-4A from Tinospora cordifolia. Immunol. Lett. 123:60-71. [DOI] [PubMed] [Google Scholar]
- 44.Reis e Sousa, C. 2004. Activation of dendritic cells: translating innate into adaptive immunity. Curr. Opin. Immunol. 16:21-25. doi: 10.1016/j.coi.2003.11.007. [DOI] [PubMed] [Google Scholar]
- 45.Rifkin, I. R., E. A. Leadbetter, L. Busconi, G. Viglianti, and A. Marshak-Rothstein. 2005. Toll-like receptors, endogenous ligands, and systemic autoimmune disease. Immunol. Rev. 204:27-42. [DOI] [PubMed] [Google Scholar]
- 46.Rowe, R. C., P. J. Sheskey, and S. C. Owen. 2005. Handbook of pharmaceutical excipients. Pharmaceutical Press and American Pharmacist Association, London, United Kingdom.
- 47.Salman, H. H., C. Gamazo, M. A. Campanero, and J. M. Irache. 2005. Salmonella-like bioadhesive nanoparticles. J. Control Release 106:1-13. [DOI] [PubMed] [Google Scholar]
- 48.Salman, H. H., C. Gamazo, M. A. Campanero, and J. M. Irache. 2006. Bioadhesive mannosylated nanoparticles for oral drug delivery. J. Nanosci. Nanotechnol. 6:3203-3209. [DOI] [PubMed] [Google Scholar]
- 49.Salman, H. H., J. M. Irache, and C. Gamazo. 2009. Immunoadjuvant capacity of flagellin and mannosamine-coated poly(anhydride) nanoparticles in oral vaccination. Vaccine 27:4784-4790. doi: 10.1016/j.vaccine.2009.05.091. [DOI] [PubMed] [Google Scholar]
- 50.Sharp, F. A., D. Ruane, B. Claass, E. Creagh, J. Harris, P. Malyala, M. Singh, D. T. O'hagan, V. Petrilli, J. Tschopp, L. A. O'Neill, and E. C. Lavelle. 2009. Uptake of particulate vaccine adjuvants by dendritic cells activates the NALP3 inflammasome. Proc. Natl. Acad. Sci. U. S. A. 106:870-875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Szymczakiewicz-Multanowska, A., N. Groth, R. Bugarini, M. Lattanzi, D. Casula, A. Hilbert, T. Tsai, and A. Podda. 2009. Safety and immunogenicity of a novel influenza subunit vaccine produced in mammalian cell culture. J. Infect. Dis. 200:841-848. [DOI] [PubMed] [Google Scholar]
- 52.Tapping, R. I. 2009. Innate immune sensing and activation of cell surface Toll-like receptors. Semin. Immunol. 21:175-184. doi: 10.1016/j.smim.2009.05.003. [DOI] [PubMed] [Google Scholar]
- 53.Tite, J. P., G. Dougan, and S. N. Chatfield. 1991. The involvement of tumor necrosis factor in immunity to Salmonella infection. J. Immunol. 147:3161-3164. [PubMed] [Google Scholar]
- 54.Toyota-Hanatani, Y., M. Inoue, T. Ekawa, H. Ohta, S. Igimi, and E. Baba. 2008. Importance of the major Fli C antigenic site of Salmonella enteritidis as a subunit vaccine antigen. Vaccine 26:4135-4137. [DOI] [PubMed] [Google Scholar]
- 55.Trinchieri, G. 2003. Interleukin-12 and the regulation of innate resistance and adaptive immunity. Nat. Rev. Immunol. 3:133-146. doi: 10.1038/nri1001. [DOI] [PubMed] [Google Scholar]
- 56.Trinchieri, G., and A. Sher. 2007. Cooperation of Toll-like receptor signals in innate immune defence. Nat. Rev. Immunol. 7:179-190. [DOI] [PubMed] [Google Scholar]
- 57.Valensi, J. P., J. R. Carlson, and G. A. Van Nest. 1994. Systemic cytokine profiles in BALB/c mice immunized with trivalent influenza vaccine containing MF59 oil emulsion and other advanced adjuvants. J. Immunol. 153:4029-4039. [PubMed] [Google Scholar]
- 58.Wetzler, L. M. 2003. The role of Toll-like receptor 2 in microbial disease and immunity. Vaccine 21(Suppl. 2):S55-S60. [DOI] [PubMed] [Google Scholar]
- 59.Wischke, C., J. Zimmermann, B. Wessinger, A. Schendler, H. H. Borchert, J. H. Peters, T. Nesselhut, and D. R. Lorenzen. 2009. Poly(I:C) coated PLGA microparticles induce dendritic cell maturation. Int. J. Pharm. 365:61-68. [DOI] [PubMed] [Google Scholar]
- 60.Xu, R., and Z. H. Jiang. 2008. Synthesis of beta-(1→4)-oligo-d-mannuronic acid neoglycolipids. Carbohydr. Res. 343:7-17. [DOI] [PubMed] [Google Scholar]
- 61.Yadav, M., and J. S. Schorey. 2006. The beta-glucan receptor dectin-1 functions together with TLR2 to mediate macrophage activation by mycobacteria. Blood 108:3168-3175. [DOI] [PMC free article] [PubMed] [Google Scholar]