

Abstract
In recent years, techniques have been developed and perfected for high-throughput identification of proteins and their accurate partial sequencing by shotgun nano-liquid chromatography-tandem mass spectrometry (nano-LC-MS/MS), making it feasible to assess global protein expression profiles in organisms with sequenced genomes. We implemented comprehensive proteomics to assess the expressed portion of the genome of Methylobacillus flagellatus during methylotrophic growth. We detected a total of 1,671 proteins (64% of the inferred proteome), including all the predicted essential proteins. Nonrandom patterns observed with the nondetectable proteins appeared to correspond to silent genomic islands, as inferred through functional profiling and genome localization. The protein contents in methylamine- and methanol-grown cells showed a significant overlap, confirming the commonality of methylotrophic metabolism downstream of the primary oxidation reactions. The new insights into methylotrophy include detection of proteins for the N-methylglutamate methylamine oxidation pathway that appears to be auxiliary and detection of two alternative enzymes for both the 6-phosphogluconate dehydrogenase reaction (GndA and GndB) and the formate dehydrogenase reaction (FDH1 and FDH4). Mutant analysis revealed that GndA and FDH4 are crucial for the organism's fitness, while GndB and FDH1 are auxiliary.
Methylotrophy is the ability of some microorganisms to grow on compounds containing no carbon-carbon bonds (C1 compounds) as sole sources of carbon and energy. This type of metabolism requires special enzyme systems and pathways that accomplish three principal metabolic goals: (i) energy-producing primary oxidation of a C1 substrate to formaldehyde or methyl transfer to produce methyl-tetrahydrofolate (H4F), (ii) energy-producing oxidation of formaldehyde or methyl-H4F to CO2, and (iii) energy-consuming assimilation of formaldehyde, methylene-H4F, and/or CO2 into biomass (1, 22). In the course of microbial evolution, multiple pathways for each step have been assembled, and, theoretically, any combination of these should enable methylotrophy (9). Moreover, some organisms possess multiple pathways for certain metabolic tasks. For example, methane can be oxidized to methanol by either soluble or particulate methane monooxygenase, and many methanotrophs possess both enzymes (25). The presence of multiple formate dehydrogenases is typical among methylotrophs (10). Some methylotrophs, such as Methylococcus capsulatus, encode multiple pathways for C1 assimilation (37). Methylotrophs employing the ribulose monophosphate (RuMP) cycle for formaldehyde assimilation can also oxidize formaldehyde via this pathway by employing a single additional reaction, catalyzed by 6-phosphogluconate dehydrogenase (Gnd) (1, 22). In addition, most RuMP cycle methylotrophs also possess a linear pathway for formaldehyde oxidation employing tetrahydromethanopterin (H4MPT) as a cofactor (35).
Methylobacillus flagellatus is a typical representative of RuMP cycle methylotrophs. It has a very limited substrate repertoire, growing robustly only on methanol or methylamine (16). Genomic analysis has revealed specific lesions in pathways for utilization of multicarbon compounds, confirming that, indeed, this organism must rely exclusively on methylotrophy to sustain its growth (11). At the same time, redundant methylotrophy pathways have been deduced from the genome. For example, in addition to the bona fide methanol dehydrogenase, four homologs of the large subunit are encoded (11). Besides the well-characterized methylamine dehydrogenase, an alternative system for methylamine oxidation is encoded, consisting of N-methylglutamate synthase and N-methylglutamate dehydrogenase (N-methylglutamate pathway) (21). Both cyclic and linear pathways for formaldehyde oxidation are encoded, along with two formate dehydrogenases. In addition, multiple terminal cytochrome oxidases are encoded (11).
It has been assumed that metabolism of both methanol and methylamine, with the exception of the respective specific primary oxidation step, is carried out by M. flagellatus (and other methylotrophs) in exactly the same fashion (1, 22). However, this assumption has not been experimentally tested. The relative contributions of each of the redundant pathways encoded in the genome (for example, linear versus cyclic oxidation of formaldehyde) also remained unclear (7). These questions can be assessed through analysis of the expressed portion of the genome under specific growth conditions and especially precisely through a comprehensive analysis of the protein content in the cell (proteome) (3).
In recent years, techniques have been developed and perfected for high-throughput identification of proteins and their accurate partial sequencing by shotgun nano-liquid chromatography-tandem mass spectrometry (nanoLC-MS/MS), making it feasible to assess relatively complete global protein expression profiles in prokaryotic organisms with sequenced genomes (2, 17). In this study, we employed comprehensive proteomics of M. flagellatus to determine what portion of the encoded proteome is detectable during methylotrophic growth under defined laboratory conditions and to obtain new insights into the physiology of the organism through this analysis.
MATERIALS AND METHODS
Cultivation, sample preparation, and high-performance liquid chromatography (HPLC) prefractionation.
For the proteome analysis, cultures of M. flagellatus KT were grown in 250 ml of minimal mineral medium [15 mM K2HPO4, 16 mM NaH2PO4, 0.8 mM MgSO4, 4 mM (NH4)2SO4, and 1× Vishniac trace elements (34)] supplemented with either 2% methanol or 0.4% methylamine (wt/vol) at 37°C to optical densities at 600 nm (OD600) of 0.6 ± 0.05. Cells were harvested by centrifugation at 4,500 × g for 7 min at 4°C. The pellets were resuspended in cooled 20 mM Tris HCl buffer (pH 8.0) and centrifuged again at 4,500 × g for 7 min and again for 4 min to remove all of the supernatant. The pellets were immediately frozen in liquid nitrogen and stored at −80°C until further use.
Lysis was carried out as described previously (3), except that the resuspension buffer was 15 mM Tris HCl (pH 8.5)-5 mM dithiothreitol-0.01% Rapigest (Waters, Milford, MA) and the boiling time was 5 min. Digestion was carried out as described previously (3), except that after alkylation, a second reduction was carried out with 10 mM dithiothreitol for 30 min at 37°C and the pellet wash protocol was 250 μl of the following: once with 0.5% trifluoroacetic acid (TFA) and 5% acetonitrile, once with 0.5% TFA and 25% acetonitrile, and once with 0.5% TFA and 50% acetonitrile. Suspensions after the first two washes were centrifuged for 5 min at 8,500 rpm in a tabletop centrifuge (Eppendorf, Westbury, NY), and the supernatant was collected. The suspension after the final wash was centrifuged for 10 min at 14,000 rpm, and the supernatant was collected. All the supernatants were pooled, and the pH was adjusted to 2.5 with TFA if necessary. After the supernatants were frozen, the volume of the samples was reduced to 150 μl by lyophilization. HPLC prefractionation was carried out as described previously (3) and five fractions were collected.
Proteomics.
The proteomics experiments were carried out essentially as previously described (3, 4). Detailed proteomics methods are presented in File S1 in the supplemental material.
Bioinformatics.
Gene product annotations, cluster of orthologous gene (COG) assignments, and transmembrane domain and signal peptide predictions were downloaded from the Joint Genome Institute (JGI) IMG/M interface (23) (Taxon Object ID 637000165) and loaded into a MySQL database. Similarly, the log2 sums of spectral counts for each protein under methanol and methylamine growth conditions were loaded into the MySQL database, using a text file extracted from the FileMaker database (see File S2 in the supplemental material). The gene product annotations and proteomics data sets were joined on open reading frame (ORF) identity such that each protein detected in proteomics mapped back to a gene and its associated product annotations.
For each predicted gene, we calculated the fraction of neighboring gene products undetected in proteomics (shown by the arrow color in File S3 in the supplemental material). A neighboring gene was one that ended within 2.5 kb of the start of the focus ORF or one that started within 2.5 kb of the end of the focus ORF. A product was undetected if the log2 sum of spectral counts from both the methanol and methylamine experiments was zero.
In File 3 in the supplemental material, the genes were plotted from the start base to the end base with an arrow depicting the direction of the coding strand (left to right, +; right to left, −). The color of the arrows was scaled, with the mean fraction of neighboring gene products scaled from black to red, representing detected to undetected, respectively. Above each arrow is a portion of the product annotation from JGI. Below the arrows are bars indicating COG assignments made by JGI (if available) colored by COG class (see the legend on the first page of File S3 in the supplemental material).
Mutant generation and phenotypic analysis.
Null mutants in the following enzymatic systems were generated: the N-methylglutamate pathway (MgdC); the oxidative branch of the RuMP pathway, represented by two functional homologs, GndA and GndB; and two alternative formate dehydrogenase systems, the NAD-linked formate dehydrogenase (Fdh1A) and the novel formate dehydrogenase whose electron transfer partners remain unknown (Fdh4A). The gndA insertion mutant was generated using the previously described donor construct based on the pAYC61 suicide vector (5). Mutations in mgdC, gndB, fdh1A, and fdh4A were generated using the previously described suicide vector pCM184, essentially as previously described (24). The double-crossover nature of the mutants was validated using diagnostic PCR tests, essentially as previously described (7). To generate unmarked mutations, a recombinase-expressing vector was introduced into the mutants, and the kanamycin resistance gene was specifically excised at the cre-lox sites (24). The primers used to amplify the respective chromosomal regions and the resulting plasmids and mutant strains are listed in File S4 in the supplemental material. The phenotypes of the mutants were tested on methanol- and methylamine-supplemented plates and in methanol- and methylamine-supplemented liquid media. Growth curves were monitored in 50-ml cultures grown in 250-ml flasks with rotary shaking as 250 rpm, at 37°C. Optical density was measured at 600 nm. Both methanol and methylamine were supplied at 60 mM.
RESULTS AND DISCUSSION
Proteome coverage and statistics.
A total of 627,843 peptide mass spectra were matched to the 2,759 proteins inferred from the M. flagellatus genome (11). For all biological replicates, we qualitatively identified 1,671 proteins. Specifically, 1,384 and 1,494 proteins were detected in the two methanol replicates and 1,349 and 1,498 proteins were detected in the two methylamine replicates. The two sets of biological replicates were highly reproducible quantitatively, as determined from correlation analysis using linear regression, with coefficients of determination (r2) of 0.999 for the methanol samples and 0.999 for the methylamine samples. The overlaps between the methanol and methylamine biological replicates were also very similar due to the small number of genes with significantly altered protein abundances identified when the two sets of growth conditions were compared (r2 = 0.993 to 0.994). A subset of 143 genes in the genome is duplicated, thus encoding identical proteins (11), of which 90 were detected in this study. Only one copy of each gene was considered in calculating the qualitative proteome coverage at 63.9%. A comprehensive list of all identified proteins along with abundance and protein intensity ratios is shown in File S2 in the supplemental material. When statistically compared, of the 1,671 detectable proteins, 191 (11.4%) were more represented in the methylamine cultures and 81 (less than 5%) were more represented in the methanol cultures, suggesting that the organism indeed requires relatively minor adjustments for the switch between the two substrates. The proteins with the highest levels of detection were common to both growth conditions, including some of the methylotrophy enzymes, with few exceptions (Table 1).
TABLE 1.
Most abundant proteins detected in this study in methanol- and methylamine-grown Methylobacillus flagellatus
| Mfla identifier (protein name) | Protein/function | Spectral counta |
|||
|---|---|---|---|---|---|
| Total (log2) | MA/Me ratio (log2) | MA (log2) | Me (log2) | ||
| Mfla_0364 (GroEL) | Chaperonin | 14.103 | −0.095 | 13.061 | 13.144 |
| Mfla_2453 | Hypothetical protein | 13.880 | 4.446 | 13.800 | 9.662 |
| Mfla_0265 | Translation elongation factor | 13.746 | −0.769 | 12.327 | 13.069 |
| Mfla_0535 | Oxidoreductase | 13.359 | −0.721 | 11.953 | 12.675 |
| Mfla_0751 (Hsp70) | Heat shock protein | 12.978 | 0.368 | 12.166 | 11.762 |
| Mfla_2044 (MxaF) | MDH large subunitb | 12.931 | 0.527 | 12.179 | 11.631 |
| Mfla_1578 | OmpA/MotB precursor | 12.915 | −0.093 | 11.873 | 11.956 |
| Mfla_0250 (Hps) | Hexulosephosphate synthaseb | 12.910 | −0.274 | 11.768 | 12.040 |
| Mfla_0016 | DNA-binding protein | 12.772 | −1.500 | 10.829 | 12.337 |
| Mfla_2249 (Tkt) | Transketolaseb | 12.657 | −0.044 | 11.651 | 11.663 |
| Mfla_0548 (MauB) | MADH large subunitb | 12.602 | 6.529 | 12.568 | 6.092 |
| Mfla_2744 | ATP synthase F1 beta subunit | 12.428 | −0.567 | 11.115 | 11.685 |
| Mfla_1654 (Hps) | Hexulosephosphate synthaseb | 12.329 | −0.548 | 11.036 | 11.572 |
| Mfla_0122 | TonB-dependent receptor | 12.040 | −0.251 | 10.944 | 11.130 |
| Mfla_2338 | Outer membrane protein | 11.875 | −1.075 | 10.317 | 11.276 |
| Mfla_0272 | RNA polymerase beta subunit | 11.777 | −0.050 | 10.761 | 10.793 |
| Mfla_2140 | Cold shock protein | 11.774 | 0.044 | 10.894 | 10.644 |
| Mfla_0927/1071 | Ribosomal protein S1 | 11.762 | −0.094 | 10.718 | 10.805 |
| Mfla_2746 | ATP synthase F1 alpha subunit | 11.738 | −0.346 | 10.562 | 10.895 |
| Mfla_1655 (Tal) | Transaldolaseb | 11.700 | −0.642 | 10.396 | 10.957 |
| Mfla_1170 | AAA ATPase | 11.691 | −0.011 | 10.684 | 10.698 |
| Mfla_0273 | RNA polymerase beta′ subunit | 11.672 | −0.093 | 10.631 | 10.712 |
| Mfla_1529 | Elongation factor Ts | 11.578 | −0.824 | 10.156 | 10.904 |
| Mfla_0269 | Ribosomal protein L1 | 11.562 | −0.367 | 10.362 | 10.737 |
| Mfla_1986 | Flagellin | 11.505 | 0.383 | 10.684 | 10.301 |
MA, methylamine; Me, methanol; MDH, methanol dehydrogenase; MADH, methylamine dehydrogenase.
Protein with known function in methylotrophy.
Of the 2,616 different proteins predicted from genome analysis exclusive of gene duplications, we detected 1,671 proteins at different levels of abundance (see File S2 in the supplemental material). There are a number of possible reasons to explain the failure to detect the remaining 945 proteins, as follows. (i) These proteins may be false negatives in the proteome analysis. This is especially true for proteins that are difficult to detect using proteomics techniques, such as proteins with significant stretches of transmembrane regions or small proteins having small numbers of tryptic peptides (38, 39). (ii) Some predicted proteins are not real, resulting from genome overinterpretation (putative proteins translated from noncoding regions, often annotated as hypothetical, including small hypothetical proteins). (iii) Finally, some proteins must be true negatives, i.e., they are not expressed under the growth conditions used in the experiment or they are expressed at such low levels that they are not detected and thus of doubtful biological relevance. This third category of proteins is of biological importance since it represents proteins that are not essential under a given set of experimental conditions. Thus, we examined the nondetected portion of the inferred proteome in detail.
We compared the distributions of the detected and nondetected proteins among different functional categories. For this analysis, we leveraged the COG (clusters of orthologous genes) assignments and transmembrane helices and signaling peptide predictions by the Integrated Microbial Genomes (IMG) interface (23). We determined that, indeed, among the nondetected predicted proteins, 265 were hypothetical proteins with no COG assignments (see File S5 in the supplemental material). In contrast, a much smaller proportion of predicted hypothetical proteins were found among the detected proteins in both methanol and methylamine samples (Fig. 1), and these are now validated as true proteins, though their functions are as yet unknown.
FIG. 1.

Pie charts depicting proportions of proteins with assigned functional categories (not shown) versus proportions of hypothetical proteins in proteins translated from the genomic sequence (A), nondetected proteins (B), and detected proteins (C).
Of the remaining undetected proteins, 266 were predicted to contain transmembrane helices and 180 were predicted to contain signal peptides, and therefore these might be expected to be underrepresented. The undetected proteins with COG assignments were sorted into functional categories (see File S5 in the supplemental material) and manually analyzed. Most of these appeared to have nonessential functions, such as transport, secretion, regulation, etc., and many belonged to COG categories with unknown functions or a general function prediction only, consistent with proteins that could be unexpressed. At the same time, most of the proteins encoding predicted essential functions were detected (see below). Exceptions, as expected, were represented by proteins containing large numbers of transmembrane helices (>50% of the protein length).
In order to determine whether the expressed genes corresponding to the detected proteins were randomly distributed over the chromosome or belonged to putative transcriptional units, we visualized the locations of proteins on the chromosome in color (see File S3 in the supplemental material). We found that the genes corresponding to detected and nondetected proteins tended to form clusters (genomic islands), suggesting nonrandom distribution and potential coregulation. We incorporated the nonrandom factors described above into this analysis and plotted the corresponding genome regions for selected nondetected islands (Fig. 2). As can be seen, some genomic islands are rich in proteins that would be difficult to detect, such as the secretory proteins in the Mfla_1333-to-Mfla_1353 region. As such, these genomic islands may very well be expressed despite the large number of undetected proteins. However, many of the islands contain a large number of readily detectable proteins, suggesting that their absence is neither random nor a biochemical artifact. These genomic islands are likely truly silent, i.e., not expressed under a given set of environmental conditions. To assign a statistical probability to this conclusion, we tested a null hypothesis of detecting or not detecting a product of a given gene being independent of detecting or not detecting the products of neighboring genes. A G test showed that for a given gene with a nondetected product, the likelihood of neighbors with nondetected products was higher than expected, given the ratio of detected versus nondetected proteins (a P value of 3E−57). Overall, these analyses suggest that we were able to detect most of the proteins that correspond to genes expressed during methylotrophic growth, thus defining a comprehensive methylotrophy proteome.
FIG. 2.

Examples of major types of nonexpressed gene islands. Genes corresponding to small proteins (<10 kDa) or proteins with substantial predicted transmembrane regions (>50% of protein's length) are shown in red, and genes corresponding to proteins with readily detectable characteristics are shown in black.
Central metabolism.
M. flagellatus exhibits high methanol dehydrogenase (MDH) activity during growth on both methanol and methylamine (20). Accordingly, the large subunit of MDH (MxaF, Mfla_2044) was one of the most frequently detected proteins in both methanol- and methylamine-grown cultures (Table 1; see File S6 in the supplemental material). Peptides were also detected for the small subunit, the associated cytochrome c, and all the remaining MDH accessory proteins (Mfla_2034 to Mfla_2043) (11), as well as all the PQQ biosynthesis proteins (Mfla_0734, Mfla_0735, and Mfla_1680 to Mfla_1683), except for PqqA (Mfla_0021). Three copies of pqqA are present in the genome (of which only one is translated in the current genome annotation [11]). The translated PqqA polypeptide contains only 23 amino acid residues. Its tryptic digestion should result in two peptides, neither of which were matched with the spectra collected in this work. While no transcriptomic analysis has been conducted using M. flagellatus, the pqqA gene has been shown to be highly transcribed in other organisms (27, 33). One explanation for this pattern is offered by a hypothesis that the peptide translated from pqqA serves as a precursor in synthesis of PQQ (27, 29, 33). However, if this were the case, stoichiometric rather than catalytic amounts of the polypeptide would be required (29, 33). The lack of PqqA spectra in our databases may point toward an alternative possibility, that pqqA may code for a small RNA and not a peptide. In addition to the bona fide MDH, four homologs of the large subunit of MDH, MxaF, are encoded in the genome (Mfla_344, Mfla_1451, Mfla_1717, and Mfla_2314). Of these, three were detected at low spectral counts and one was not detected (see File S6 in the supplemental material).
In the methylamine-grown culture, some of the most differentially expressed proteins were the ones involved in the methylamine dehydrogenase (MADH) function, while none or very few peptides of these proteins were observed in the methanol-grown culture (see File S6 in the supplemental material). In terms of spectral counts, the two subunits of MADH (Mfla_0548 and Mfla_0551) and the associated electron acceptor azurin (Mfla_0556) were most abundant, along with MauD (Mfla_0550), whose exact role in the MADH reaction remains unknown. MauD has also been previously detected at high spectral counts in methylamine-grown Methylotenera mobilis (4). The MADH system has been previously demonstrated to be essential for growth on methylamine (14, 15). However, the recently described genes for an alternative methylamine oxidation enzyme system, the one involving an N-methylglutamate as an intermediate, have also been recognized on the chromosome of M. flagellatus (Mfla_0452 to Mfla_0459) (21). Proteins involved in this pathway, like MADH proteins, were detected almost exclusively in the methylamine culture (see File S6 in the supplemental material), suggesting that the pathway is functional and is induced during growth on methylamine. However, in terms of spectral counts, the proteins involved in this pathway were less abundant. One of the most frequently detected proteins in the methylamine cultures was a protein of unknown function (Mfla_2453) (Table 1) that is predicted to possess an outer membrane channel motif, and this protein could potentially be a porin. Prior to experimental work with this protein, we can only speculate that it could be involved in transport of either methylamine or a methylamine-specific product, such as ammonium, across the cell membrane.
All of the enzymes involved in the assimilatory RuMP cycle (detailed in reference 11 and File S6 in the supplemental material) were detected at high spectral counts in both methanol- and methylamine-grown cultures. Two genes for hexulosephosphate synthase (Mfla_0250 and Mfla_1654), the key assimilatory enzyme, have been previously identified, one as part of the large C1 cluster and one inserted into a histidine biosynthesis gene cluster (11). Both were detected at high spectral counts, the latter having a somewhat higher count (Table 1). Of the two isoenzymes of 6-phosphogluconate dehydrogenase, the enzyme involved in the dissimilatory RuMP cycle (cyclic oxidation), the NAD-linked enzyme (GndA, Mfla_0918/1062) was detected at much higher spectral counts than the NADP-linked enzyme (GndB, Mfla_2599). Surprisingly, the latter was statistically overexpressed in methylamine-grown cells (see File S6 in the supplemental material), potentially suggesting a special role for this enzyme in methylamine metabolism. Interestingly, the previously analyzed methylamine utilizer, Methylotenera mobilis, possesses (and expresses during growth on methylamine) only GndB, not GndA (4).
All the known proteins involved in the H4MPT-linked formaldehyde oxidation pathway were detected (Mfla_1579 to Mfla_1582, Mfla_1646 to Mfla_1652, Mfla_1656 to Mfla_1662) along with proteins for two formate dehydrogenases, FDH1 (Mfla_0718 to Mfla_0720) and FDH4 (Mfla_0338), named after the homologous enzymes formerly characterized in Methylobacterium extorquens AM1 (8) that, together with the H4MPT-linked pathway, form the linear formaldehyde oxidation path (Fig. 3). Interestingly, the NAD-linked FDH (FDH1, Mfla_718 to Mfla_722) was detected only in methylamine-grown cells, while FDH4 (Mfla_433) was detected under both conditions. Fae, formaldehyde-activating enzyme, is the first enzyme in the linear formaldehyde oxidation path, and it is known to be highly expressed in methylotrophs (36). We previously identified two bona fide fae genes (Mfla_1652 and Mfla_2543) and two distant homologs named fae2 (Mfla_2524) and fae3 (Mfla_2364) whose functions remain enigmatic (11). All four of the fae proteins were detected. The Fae encoded in the large C1 gene cluster (Mfla_1652) was found at the highest spectral counts, followed by Fae2, Fae3, and Fae (Mfla_2543). These data suggest that Fae2 and Fae3 may have separate and important functions in C1 metabolism.
FIG. 3.
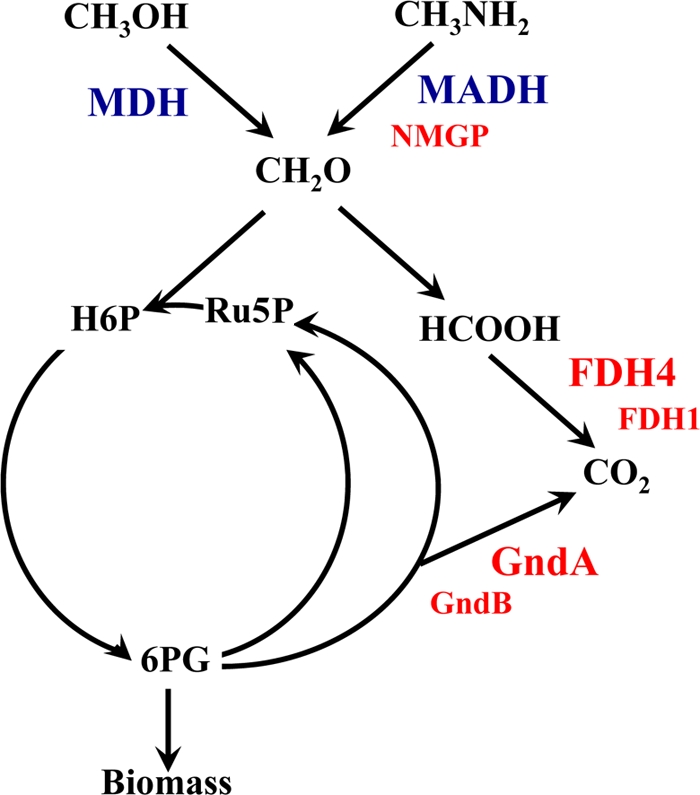
Schematic representation of central metabolism of M. flagellatus. MDH, methanol dehydrogenase; MADH, methylamine dehydrogenase; NMGP, N-methylglutamate pathway; GndA and GndB, NAD-linked and NADP-linked 6-phosphogluconate dehydrogenases, respectively; FDH1 and FDH4, alternative formate dehydrogenases; H6P, hexulose 6-phosphate; Ru5P, ribulose 5-phosphate. Enzyme systems mutated in this work are shown in red. Essential enzymes are shown in large font, and auxiliary enzymes are shown in small font.
All of the enzymes that were previously implicated in C3 interconversions (11) were detected, as well as all of the enzymes involved in the incomplete tricarboxylic cycle (see File S6 in the supplemental material).
Essential functions and secondary metabolism.
We detected proteins representing all of the essential functions, such as transcription, translation, ribosomal structure, and biosynthesis of vitamins and cofactors, with one exception. Two proteins involved in the biosynthesis of cobalamine were not detected and appeared to be part of a silent operon (Mfla_94 to Mfla_105). This fits with the current understanding of methylotrophic metabolism of organisms utilizing the RuMP cycle, which does not involve cobalamine-dependent reactions (1, 22).
Of the nonessential functions, we detected most of the proteins involved in flagellum biosynthesis and function, all encoded by a single gene cluster (Mfla_1928 to Mfla_1988). We found that both exopolysaccharide synthesis gene clusters predicted from the genome sequence (Mfla_1268 to Mfla_1280 and Mfla_2007 to Mfla_2031) were expressed during methylotrophic growth. Although they are proposed as nonessential for energy generation during methylotrophic growth (31, 32), we found most of the proteins composing the NADH dehydrogenase complex (Mfla_2048 to Mfla_2061). Interestingly, some of the genes associated with the CRISPR element (Mfla_0601 to Mfla_0607) were expressed, suggesting that cells maintain active phage defense mechanisms (28) even when grown as axenic cultures.
Insights from the silent gene islands.
We carefully analyzed the silent genes and gene islands and manually sorted them into functional categories (see Files S3 and S5 in the supplemental material). One goal of this analysis was to confirm that no essential functions were encoded by these genes, and another goal was to detect genes and pathways that are not expressed during growth on methanol or methylamine but could potentially indicate the existence of alternative modes of metabolism and/or alternative growth substrates. From these analyses, many silent genes and gene islands were predicted to encode transport, secretion, and efflux functions (Fig. 2; see File S5 in the supplemental material). Another major functional category of silent genes included genes belonging to prophage sequences and other insertional elements, such as integrons (Mfla_403 to Mfla_410) (26) or putative integrons (Mfla_2544 to Mfla_2549). Two silent gene islands encoded proteins with homology to adhesins and hemagglutinins, proteins typically synthesized and excreted by pathogenic bacteria. Other silent gene islands encoded metabolic traits that were not essential for growth in the laboratory (nitrate transport and metabolism, Mfla_0315 to Mfla_0331; cobalamin biosynthesis, Mfla_0095 to Mfla_0105, etc.). The only clue toward identifying potential alternative growth substrates was the gene cluster predicted to encode transport and degradation of alkanesulfonates (Mfla_1534 to Mfla_1566) previously described for serine cycle methylotrophs (19). A few other silent gene islands encoded predicted transporter peptides, along with predicted oxidoreductases that may be indicative of other potential alternative substrates. However, these substrates cannot be deduced based on general function prediction and remain unknown.
Metabolic insights from mutant analysis.
The results of the proteomic analysis provided new insights into methylotrophic metabolism of M. flagellatus. The MADH system has been a hallmark of methylamine metabolism in methylotrophs, including M. flagellatus, having been demonstrated to be essential for methylamine utilization (5, 14, 15, 30). However, the finding of an alternative system involving N-methylglutamate as an intermediate specifically expressed during growth on methylamine raised a question of whether this additional system contributed to growth on methylamine. The role of the linear pathway for oxidizing formaldehyde has been approached experimentally before (7, 18). However, while high activities for the enzymes of the H4MPT pathway have been measured, mutants in these enzymes revealed only very subtle phenotypes, suggesting a minor role in formaldehyde oxidation. Formate dehydrogenase activity was reported only in cells of M. flagellatus (and other RuMP methylotrophs) when grown in a chemostat at low dilution rates (6, 12, 13), consistent with the nonessential role of the linear pathway. However, here we detected all the components of the linear pathway, including the two alternative formate dehydrogenases. Of these, the NAD-linked enzyme (Mfla_0718 to Mfla_0720) was detected only in methylamine cells, while the second enzyme (Mfla_0338) was detected under both conditions. The latter enzyme is a homolog of a novel formate dehydrogenase, FDH4, that was recently identified and investigated in M. extorquens (8). This enzyme remains enigmatic, with no cofactors or electron acceptors identified and no assay existing for its activity. Mutants of M. extorquens defective in this enzyme reveal a dramatic phenotype: during growth in liquid, they start accumulating formate in growth medium, which eventually results in growth arrest and lysis (8). The fdh4A gene was found to be acid inducible, suggesting a potential involvement in the acid stress response. In addition, experiments on overexpression of fdh4A and an adjacent gene named fdh4B prompted hypotheses of additional, potentially regulatory functions that remain poorly understood (8). We were interested in testing the roles of the alternative formate dehydrogenases in M. flagellatus to determine whether this step of the linear pathway is essential, whether they were redundant, and whether the dramatic phenotype observed for the fdh4A mutant in M. extorquens will be observed in the case of M. flagellatus.
Proteomics revealed the presence of both GndA (Mfla_0918/Mfla_1062) and GndB (Mfla_2599), the latter seeming to be upregulated in the methylamine-grown cells. We have previously failed to obtain GndA mutants (7). However, in light of the presence of GndB, along with the enzymes for the linear pathway, it appeared necessary to retest the relative contributions of these enzymes to formaldehyde oxidation.
To address the questions posed above, we constructed mutants with deficiencies in the N-methylglutamate pathway (mgdC, mfla_0454), GndA (gndA, mfla_0918/mfla_1062), GndB (gndB, mfla_2599), FDH1 (fdh1A, mfla_0720), and FDH4 (fdh4A, mfla_0338). A simplified scheme of the metabolism of M. flagellatus featuring these reactions is depicted in Fig. 3. Double-crossover (null) mutants were obtained using all five genes. In the case of GndA, double-crossover mutants were obtained only when selected for on methylamine, not methanol, plates.
Tests on plates indicated that mutants with mutations in only two genes, gndA and fhd4A, showed reduced growth on both methanol and methylamine. Tests in liquid cultures confirmed these observations. Mutants with mutations in mgdC, gndB, and fdh1A grew at the same rate and to the same optical density as wild-type cells on both methanol and methylamine, while cells with mutations in gndA and fdh4A showed growth defects (Fig. 4 shows results from two representative experiments out of a total of nine experiments). A dramatic phenotype was manifested by the GndA mutant, which essentially could not grow on methanol in liquid culture. On methylamine, the mutant revealed a long (8- to 10-h) lag phase, after which it grew at a rate close to the wild-type rate (approximately 3-h doubling time). Such a phenotype agrees with the elevated levels of GndB on methylamine (see File S7 in the supplemental material), suggesting that GndB, after a critical threshold concentration is reached for the enzyme, can act in place of GndA and that the change in the ratio of NADH (primarily produced by GndA) to NADPH (primarily produced by GndB) does not affect the metabolism of M. flagellatus in a dramatic way during growth on methylamine.
FIG. 4.

Growth of wild-type and mutant M. flagellatus on methanol and methylamine. Results from two representative experiments are shown (experiment 1, A and B; experiment 2, C and D). Data are OD600 values plotted against time (hours).
The Fdh4A mutant displayed reduced growth on both methanol and methylamine in terms of having a lag period of 8 to 10 h, before onset of growth. In addition, this mutant revealed patterns of stochastic behavior, i.e., sometimes the strain would not grow in liquid culture beyond a few cell divisions (Fig. 4A, C, and D) and sometimes it would (Fig. 4B). This type of behavior may reflect subtle differences in microelement content of the media that may have occurred between different experiments or an involvement in a bistable switch type of network. While a very interesting phenomenon, differentiating between these possibilities is beyond the scope of this work.
Overall, the phenotypes of the mutants indicated that, while both cyclic and linear formaldehyde oxidation pathways are functionally present in M. flagellatus and both involve alternative solutions (GndA versus GndB and FDH1 versus FDH4), their functions are likely not redundant. GndA appears to have a more important function than GndB, and FDH4 appears to have a more important function than FDH1.
To ultimately answer the question of whether either the linear or the cyclic pathway alone can support methylotrophic growth of M. flagellatus, we attempted to generate mutants deficient in different combinations of alternative Gnd and FDH enzymes, as follows: GndA/GndB, Fdh1A/Fdh4A, GndA/Fdh1A, GndA/Fdh4A, GndB/Fdh4A, and GndB/Fdh1A. Of all these combinations, we were able to select only double mutants deficient in GndB/Fdh1A, with selection on either methanol or methylamine. No strains containing the other combinations of mutations were obtained. This result fits well with the phenotypes of the single mutants. It appears from these results that both linear and cyclic pathways for oxidation of formaldehyde, and specifically the formate oxidation step, are required. While no visible phenotypes were observed for GndB or FDH1 mutants under laboratory growth conditions, GndB at least can replace the metabolically important GndA under certain circumstances, indicating that these systems likely play a role in the organism's fitness.
Conclusions.
The comprehensive proteomics approach implemented here revealed that during methylotrophic growth, M. flagellatus expresses up to 64% of its proteome, including all the inferred methylotrophy proteins and all the inferred essential proteins, as well as proteins involved in some auxiliary functions such as motility, polysaccharide biosynthesis, and phage defense. Many of the nondetectable proteins appear to correspond to silent genomic islands, and a portion of the nonexpressed proteins may represent artifacts of genome annotation.
Protein contents in methylamine- and methanol-grown cells significantly overlap. The newly detected N-methylglutamate pathway is functionally expressed during growth on methylamine but appears to play an auxiliary role, while the linear pathway for formaldehyde oxidation and especially the formate oxidation step appear important for the organism's fitness.
Supplementary Material
Acknowledgments
This research was supported by the National Science Foundation as part of the Microbial Observatories program (MCB-0604269).
We thank Qiangwei Xia for discussions and help with the protein abundance ratio calculations and Fred Taub for the FileMaker database.
Footnotes
Published ahead of print on 16 July 2010.
Supplemental material for this article may be found at http://jb.asm.org/.
REFERENCES
- 1.Anthony, C. 1982. The biochemistry of methylotrophs. Academic Press, London, United Kingdom.
- 2.Armengaud, J. 2009. A perfect genome annotation is within reach with the proteomics and genomics alliance. Curr. Opin. Microbiol. 12:292-300. [DOI] [PubMed] [Google Scholar]
- 3.Bosch, G., E. Skovran, Q. Xia, J. A. Miller, M. E. Lidstrom, and M. Hackett. 2008. Comprehensive proteomics of Methylobacterium extorquens AM1 metabolism under single carbon and non-methylotrophic conditions. Proteomics 8:3494-3505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bosch, G., T. Wang, E. Latypova, M. G. Kalyuzhnaya, M. Hackett, and L. Chistoserdova. 2009. Insights into the physiology of Methylotenera mobilis as revealed by metagenome-based shotgun proteomic analysis. Microbiology 155:1103-1110. [DOI] [PubMed] [Google Scholar]
- 5.Chistoserdov, A. Y., L. V. Chistoserdova, W. S. McIntire, and M. E. Lidstrom. 1994. Genetic organization of the mau cluster in Methylobacterium extorquens AM1: complete nucleotide sequence and generation and characteristics of mau mutants. J. Bacteriol. 176:4052-4065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chistoserdova, L. V., A. Y. Chistoserdov, N. L. Schklyar, M. V. Baev, and Y. D. Tsygankov. 1991. Oxidative and assimilative enzyme activities in continuous cultures of the obligate methylotroph Methylobacillus flagellatum. Antonie Van Leeuwenhoek 60:101-107. [DOI] [PubMed] [Google Scholar]
- 7.Chistoserdova, L., L. Gomelsky, J. A. Vorholt, M. Gomelsky, Y. D. Tsygankov, R. K. Thauer, M. E. Lidstrom. 2000. Analysis of two formaldehyde oxidation pathways in Methylobacillus flagellatus KT, a ribulose monophosphate cycle methylotroph. Microbiology 146:233-238. [DOI] [PubMed] [Google Scholar]
- 8.Chistoserdova, L., G. J. Crowther, J. A. Vorholt, E. Skovran, J. C. Portais, and M. E. Lidstrom. 2007. Identification of a fourth formate dehydrogenase in Methylobacterium extorquens AM1 and confirmation of the essential role of formate oxidation in methylotrophy. J. Bacteriol. 189:9076-9081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chistoserdova, L., M. G. Kalyuzhnaya, and M. E. Lidstrom. 2005. C1 transfer modules: from genomics to ecology. ASM News 71:521-528. [Google Scholar]
- 10.Chistoserdova, L., M. G. Kalyuzhnaya, and M. E. Lidstrom. 2009. The expanding world of methylotrophic metabolism. Annu. Rev. Microbiol. 63:477-499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chistoserdova, L., A. Lapidus, C. Han, L. Goodwin, L. Saunders, T. Brettin, R. Tapia, P. Gilna, S. Lucas, P. M. Richardson, and M. E. Lidstrom. 2007. The genome of Methylobacillus flagellatus, the molecular basis for obligate methylotrophy, and the polyphyletic origin of methylotrophy. J. Bacteriol. 189:4020-4027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chu, I. M., and E. T. Papoutsakis. 1987. Growth dynamics of a methylotroph (Methylomonas L3) in continuous cultures. I. Fast transients induced by methanol pulses and methanol accumulation. Biotechnol. Bioeng. 29:55-64. [DOI] [PubMed] [Google Scholar]
- 13.Chu, I. M., and E. T. Papoutsakis. 1987. Growth dynamics of a methylotroph (Methylomonas L3) in continuous cultures. II. Growth inhibition and comparison against an unstructured model. Biotechnol. Bioeng. 29:65-71. [DOI] [PubMed] [Google Scholar]
- 14.Gak, E. R., A. Y. Chistoserdov, and M. E. Lidstrom. 1995. Cloning, sequencing, and mutation of a gene for azurin in Methylobacillus flagellatum KT. J. Bacteriol. 177:4575-4578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gak, E. R., Y. D. Tsygankov, and A. Y. Chistoserdov. 1997. Organization of methylamine utilization genes (mau) in “Methylobacillus flagellatum” KT and analysis of mau mutants. Microbiology 143:1827-1835. [DOI] [PubMed] [Google Scholar]
- 16.Govorukhina, N. I., L. V. Kletsova, Y. D. Tsygankov, Y. A. Trotsenko, and A. I. Netrusov. 1987. Characteristics of a new obligate methylotroph. Microbiologiya 56:849-854. [Google Scholar]
- 17.Gupta, N., J. Benhamida, V. Bhargava, D. Goodman, E. Kain, I. Kerman, N. Nguyen, N. Ollikainen, J. Rodriguez, J. Wang, M. S. Lipton, M. Romine, V. Bafna, R. D. Smith, and P. A. Pevzner. 2008. Comparative proteogenomics: combining mass spectrometry and comparative genomics to analyze multiple genomes. Genome Res. 18:1133-1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kalyuzhnaya, M. G., N. Korotkova, G. J. Crowther, C. J. Marx, M. E. Lidstrom, and L. Chistoserdova. 2005. Analysis of gene islands involved in methanopterin-linked C1 transfer reactions reveals new functions and provides evolutionary insights. J. Bacteriol. 187:4607-4614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kelly, D. P., and J. C. Murrell. 1999. Microbial metabolism of methanesulfonic acid. Arch. Microbiol. 172:341-348. [DOI] [PubMed] [Google Scholar]
- 20.Kletsova, L. V., N. I. Govorukhina, Y. D. Tsygankov, and Y. A. Trosenko. 1987. Metabolism of the obligate methylotroph Methylobacillus flagellatum. Microbiologiya 56:901-906. [Google Scholar]
- 21.Latypova, E., S. Yang, Y. S. Wang, T. Wang, T. A. Chavkin, M. Hackett, H. Schäfer, and M. G. Kalyuzhnaya. 2010. Genetics of the glutamate-mediated methylamine utilization pathway in the facultative methylotrophic beta-proteobacterium Methyloversatilis universalis FAM5. Mol. Microbiol. 75:426-439. [DOI] [PubMed] [Google Scholar]
- 22.Lidstrom, M. E. 2006. Aerobic methylotrophic prokaryotes, p. 618-634. In A. Balows, H. G. Truper, M. Dworkin, W. Harder, and K. H. Schleifer (ed.), The prokaryotes. Springer Verlag, New York, NY.
- 23.Markowitz, V. M., E. Szeto, K. Palaniappan, Y. Grechkin, K. Chu, I. M. Chen, I. Dubchak, I. Anderson, A. Lykidis, K. Mavromatis, N. N. Ivanova, and N. C. Kyrpides. 2008. The integrated microbial genomes (IMG) system in 2007: data content and analysis tool extensions. Nucleic Acids Res. 36:D528-D533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Marx, C. J., and M. E. Lidstron. 2002. Broad-host-range cre-lox system for antibiotic marker recycling in gram-negative bacteria. Biotechniques 33:1062-1067. [DOI] [PubMed] [Google Scholar]
- 25.McDonald, I. R., L. Bodrossy, Y. Chen, and J. C. Murrell. 2008. Molecular ecology techniques for the study of aerobic methanotrophs. Appl. Environ. Microbiol. 74:1305-1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nemergut, D. R., M. S. Robeson, R. F. Kysela, A. P. Martin, S. K. Schmidt, and R. Knight. 2008. Insights and inferences about integron evolution from genomic data. BMC Genomics 9:261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ramamoorthi, R., and M. E. Lidstrom. 1995. Transcriptional analysis of pqqD and study of the regulation of pyrroloquinoline quinone biosynthesis in Methylobacterium extorquens AM1. J. Bacteriol. 177:206-211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sorek, R., V. Kunin, and P. Hugenholtz. 2008. CRISPR—a widespread system that provides acquired resistance against phages in bacteria and archaea. Nat. Rev. Microbiol. 6:181-186. [DOI] [PubMed] [Google Scholar]
- 29.Toyama, H., and M. E. Lidstrom. 1998. pqqA is not required for biosynthesis of pyrroloquinoline quinone in Methylobacterium extorquens AM1. Microbiology 144:183-191. [DOI] [PubMed] [Google Scholar]
- 30.Van der Palen, C. J., D. J. Slotboom, L. Jongejan, W. N. Reijnders, N. Harms, J. A. Duine, and R. J. van Spanning. 1995. Mutational analysis of mau genes involved in methylamine metabolism in Paracoccus denitrificans. Eur. J. Biochem. 230:860-871. [DOI] [PubMed] [Google Scholar]
- 31.Van Dien, S. J., and M. E. Lidstrom. 2002. Stoichiometric model for evaluating the metabolic capabilities of the facultative methylotroph Methylobacterium extorquens AM1, with application to reconstruction of C(3) and C(4) metabolism. Biotechnol. Bioeng. 78:296-312. [DOI] [PubMed] [Google Scholar]
- 32.Van Dien, S. J., Y. Okubo, M. T. Hough, N. Korotkova, T. Taitano, and M. E. Lidstrom. 2003. Reconstruction of C(3) and C(4) metabolism in Methylobacterium extorquens AM1 using transposon mutagenesis. Microbiology 149:601-609. [DOI] [PubMed] [Google Scholar]
- 33.Velterop, J. S., E. Sellink, J. J. Meulenberg, S. David, I. Bulder, and P. W. Postma. 1995. Synthesis of pyrroloquinoline quinone in vivo and in vitro and detection of an intermediate in the biosynthetic pathway. J. Bacteriol. 177:5088-5098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vishniac, W., and M. Santer. 1957. The thiobacilli. Bacteriol. Rev. 21:195-213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vorholt, J. A., L. Chistoserdova, S. M. Stolyar, M. E. Lidstrom, and R. K. Thauer. 1999. Distribution of tetrahydromethanopterin-dependent enzymes in methylotrophic bacteria and phylogeny of methenyl tetrahydromethanopterin cyclohydrolases. J. Bacteriol. 181:5750-5757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vorholt, J. A., C. J. Marx, M. E. Lidstrom, and R. K. Thauer. 2000. Novel formaldehyde-activating enzyme in Methylobacterium extorquens AM1 required for growth on methanol. J. Bacteriol. 182:6645-6650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ward, N., O. Larsen, J. Sakwa, L. Bruseth, H. Khouri, A. S. Durkin, G. Dimitrov, L. Jiang, D. Scanlan, K. H. Kang, M. Lewis, K. E. Neslon, B. Methé, M. Wu, J. F. Heidelberg, I. T. Paulsen, D. Fouts, J. Ravel, H. Tettelin, Q. Ren, T. Read, R. T. DeBoy, R. Seshadri, S. L. Salzberg, H. B. Jensen, N. K. Birkelenad, W. C. Nelson, R. J. Dodson, S. H. Grindhaug, I. Holt, I. Eidhammer, I. Jonasen, S. Vanaken, T. Utterback, T. V. Feldblyum, C. M. Fraser, J. R. Lillehaug, and J. A. Eisen. 2004. Genomic insights into methanotrophy: the complete genome sequence of Methylococcus capsulatus (Bath). PLoS Biol. 2:e303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Washburn, M. P., R. Ulaszek, C. Deciu, D. M. Schieltz, and J. R. Yates III. 2002. Analysis of quantitative proteomic data generated via multidimensional protein identification technology. Anal. Chem. 74:1650-1657. [DOI] [PubMed] [Google Scholar]
- 39.Washburn, M. P., D. Wolters, and J. R. Yates III. 2001. Large-scale analysis of the yeast proteome by multidimensional protein identification technology. Nat. Biotechnol. 19:242-247. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.