

Abstract
Metallo-β-lactamases (MBLs) are important enzymatic factors in resistance to β-lactam antibiotics that show important structural and functional heterogeneity. BJP-1 is a subclass B3 MBL determinant produced by Bradyrhizobium japonicum that exhibits interesting properties. BJP-1, like CAU-1 of Caulobacter vibrioides, overall poorly recognizes β-lactam substrates and shows an unusual substrate profile compared to other MBLs. In order to understand the structural basis of these properties, the crystal structure of BJP-1 was obtained at 1.4-Å resolution. This revealed significant differences in the conformation and locations of the active-site loops, determining a rather narrow active site and the presence of a unique N-terminal helix bearing Phe-31, whose side chain binds in the active site and represents an obstacle for β-lactam substrate binding. In order to probe the potential of sulfonamides (known to inhibit various zinc-dependent enzymes) to bind in the active sites of MBLs, the structure of BJP-1 in complex with 4-nitrobenzenesulfonamide was also obtained (at 1.33-Å resolution), thereby revealing the mode of interaction of these molecules in MBLs. Interestingly, sulfonamide binding resulted in the displacement of the side chain of Phe-31 from its hydrophobic binding pocket, where the benzene ring of the molecule is now found. These data further highlight the structural diversity shown by MBLs but also provide interesting insights in the structure-function relationships of these enzymes. More importantly, we provided the first structural observation of MBL interaction with sulfonamides, which might represent an interesting scaffold for the design of MBL inhibitors.
β-Lactamases are bacterial enzymes that confer resistance to β-lactam antibiotics, the most widely used family of anti-infective agents, by hydrolyzing the amide bond of the β-lactam ring and thus bear significant clinical relevance (32, 41, 43). Two structurally and mechanistically distinct families of β-lactamases are known: the active-site serine enzymes (Ambler's classes A, C, and D), acting via an acylation-deacylation mechanism, and the metallo-β-lactamases (MBLs) (Ambler's class B), which require zinc in their active sites and whose catalytic mechanism is less well understood, although several hypotheses have been provided (4, 13, 21, 33). On the basis of sequence homology, three subclasses that also differ by the natures and positions of the residues that constitute the metal binding site(s) have been defined (44).
From a structural standpoint, MBLs share a unique fold that was first identified when the structure of the BcII enzyme from Bacillus cereus was solved; this fold consists of a β-sandwich flanked by α-helices, the metal center being located at the interface of two roughly symmetrical domains (9). Subsequently, many proteins encoded by the genomes of many organisms (from Archaea to mammalians) appeared to share the same typical fold and were grouped in the so-called MBL superfamily, which contains zinc hydrolases that might exhibit a wide variety of enzymatic and/or cellular functions (e.g., DNA repair, RNA maturation, cell detoxification, metabolism, degradation of pesticides and organophosphates, and quorum sensing) (4, 14).
From a functional standpoint, MBLs are characterized by strong hydrolytic activity against carbapenems, the most recent broad-spectrum β-lactams, which are often used as last-resort drugs, largely accounting for the clinical relevance of these enzymes (44). In addition, and due to their different catalytic mechanism with respect to that of serine β-lactamases, MBLs are not susceptible to any of the commercially available β-lactamase inactivators (e.g., clavulanate) used in β-lactam/β-lactamase deactivator combinations (44). Besides the acquired MBLs, which are currently being disseminated in many important opportunistic pathogens (such as Enterobacteriaceae, Pseudomonas, and Acinetobacter spp.), many of these enzymes have been found to be encoded by the genomes of some microorganisms of limited or no clinical relevance (e.g., Caulobacter vibrioides). These endogenous enzymes could represent interesting models for investigating the evolutionary relationships between MBLs and the other members of the MBL superfamily and their structure-activity relationships and for identifying broad-spectrum MBL inhibitors, whose development would address an increasingly important clinical need (16, 42, 49, 50).
BJP-1, the endogenous subclass B3 MBL from Bradyrhizobium japonicum, a bacterium widely used in agriculture whose genome was recently released, was previously identified and characterized following a postgenomic study of MBL determinants present in the genomes of related bacteria belonging to the order Rhizobiales (27, 49). This enzyme exhibits many interesting functional features, e.g., an overall low affinity for β-lactam compounds, a situation similar to that for CAU-1 and CAR-1, two enzymes that were identified by means of a postgenomic approach. It has been hypothesized that CAU-1 and CAR-1 might represent interesting evolutive intermediates of MBLs or might even be examples of catalytic promiscuity, their primary function possibly being different from antibiotic resistance (16, 50). In addition, the catalytic efficiency of BJP-1 for the hydrolysis of β-lactam compounds was significantly lower than those of the other subclass B3 MBLs, such as L1 and GOB-1. Finally, and by contrast with other MBLs, BJP-1 was poorly susceptible to metal chelators, likely reflecting differences in the affinities of zinc ions for their respective binding sites.
In order to provide a rationale for the above-mentioned unique features of BJP-1, we determined the crystal structures of the native BJP-1 and compared them to the available structures of other MBLs. In addition, to probe the potential for the development of broad-spectrum MBL inhibitors, we also obtained the structure of BJP-1 in complex with a simple sulfonamide compound, which is known to inhibit several Zn-dependent enzymes, like carbonic anhydrase and carboxypeptidase (26, 39).
MATERIALS AND METHODS
BJP-1 purification and crystallization.
Purified BJP-1 was obtained using the production and purification protocol described previously (49). Crystallization screening was performed immediately after purification of the enzyme. BJP-1 was concentrated to 10 mg/ml, and the purification buffer was changed to 0.1 M Tris-HCl (pH 8.5) using a Microcon 10-kDa-cutoff ultrafiltration device (Millipore, Bedford, MA). The crystallization trials were performed using the sitting-drop method (96-well CrystalEX plates; Corning) (6). The drops consisted of 2 μl protein solution and 2 μl reservoir solution equilibrated at room temperature (20°C) against a reservoir volume of 100 μl. The initial screens tested were Crystal Screen, Crystal Screen 2, and Grid Screen Ammonium Sulfate (Hampton Research, Aliso Viejo, CA). Initially, small, ill-formed crystals were obtained in the drops under conditions 17 and 22 of Crystal Screen (0.2 M Li2SO4·H2O, 0.1 M Tris-HCl [pH 8.5], and 30% [wt/vol] polyethylene glycol 4000 [PEG 4000] and 0.2 M sodium acetate trihydrate, 0.1 M Tris-HCl [pH 8.5], and 30% [wt/vol] PEG 4000, respectively). However, since these crystals were not suitable for X-ray diffraction analysis, further optimization of the initial crystallization conditions was accomplished by changing various crystallization parameters. Optimization of crystallization conditions was performed at room temperature by using a 24-well sitting-drop plate sealed with clear sealing tape (Cryschem plate; Hampton Research), 2 and 4 μl of protein solution, and 2 μl of reservoir solution equilibrated against 700 and 800 μl reservoir solution. Crystallization trials were performed either in the presence or absence of 5 mM ZnCl2, with protein concentrations ranging from 5 to 15 mg/ml, PEG 4000 concentrations ranging from 25 to 45% (wt/vol), and ammonium or sodium acetate and lithium sulfate concentrations ranging from 0.2 to 0.5 M. The best crystals were grown using 30 to 35% PEG 4000, 0.5 M sodium acetate at pH 8.5, a protein concentration of 10 mg/ml, a 4-μl drop volume, and an 800-μl reservoir volume. BJP-1 crystals grew in a few weeks as clustered parallelepipedons to an average size of about 100 μm. Crystals of the BJP-1 complex with 4-nitrobenzenesulfonamide (NBSA) were obtained by soaking the BJP-1 crystals in the crystallization solution, to which was added 60 mM NBSA dissolved in dimethyl sulfoxide (DMSO) and diluted in a 1:10 ratio with PEG 400 to ensure mixing in the crystallization solution and, at the same time, cryoprotection. Data collection was performed after 3 h of soaking in the above solution.
Diffraction data collection, structure solution, and refinement.
A single-wavelength anomalous diffraction (SAD) data set to 1.4-Å resolution was collected at the Zn edge (λ = 1.2813 Å) on a native BJP-1 crystal at the European Synchrotron Radiation Facility (ESRF) ID29 beamline using an ADSC Quantum 315R detector. Diffraction data on the BJP-1-NBSA complex were collected at the ESRF ID14-1 beamline (λ = 0.9340 Å) to 1.33-Å resolution using an ADSC Quantum 210 detector.
The data were processed using the program MOSFLM (31) and scaled using the program SCALA (18). The data collection parameters and data reduction statistics are reported in Table 1. The structure was solved using the anomalous data with the program SHARP/AutoSHARP (55), which provided the initial phases based on the zinc substructure. Part of the model was then built into the experimental electron density using the ARP/wARP suite (37) and manually completed by using XtalView (34) and Coot (17). The refinement was carried out with REFMAC5 (38) from the Collaborative Computational Project 4 (CCP4) suite (11) using translation/libration/screw (TLS) parameterization (56, 57). Only the zinc ions were refined anisotropically. In between the refinement cycles, the models were subjected to manual rebuilding. Water molecules were added using the standard procedures within the ARP/wARP suite (37).
TABLE 1.
Data collection and refinement statistics
| Parameter | Valuea for: |
|
|---|---|---|
| Native BJP-1 | BJP-1-NBSA complex | |
| X-ray source | ESRF ID29 | ESRF ID14-1 |
| Wavelength (Å) | 1.2813 | 0.9340 |
| Data collection temp (K) | 100 | 100 |
| Space group | P1 | P1 |
| Cell dimensions (Å) | a = 42.50, b = 44.77, c = 76.97, α = 78.92, β = 89.51, γ = 61.91 | a = 42.33, b = 44.76, c = 76.62, α = 78.73, β = 89.43, γ = 61.99 |
| No. of subunits/asymmetric units | 2 | 2 |
| Matthews coefficient (Å3 Da−1) | 2.10 | 2.03 |
| Solvent content (%) | 41.57 | 39.44 |
| Resolution limits (Å) | 38.50-1.40 (1.48-1.40) | 22.25-1.33 (1.40-1.33) |
| No. of reflections measured | 326,935 (38,207) | 391,600 (55,084) |
| No. of unique reflections | 87,611 (10,821) | 102,968 (14,679) |
| Completeness (%) | 91.0 (77.2) | 91.8 (89.4) |
| Rmergeb (%) | 6.0 (33.6) | 6.0 (15.4) |
| Multiplicity | 3.7 (3.5) | 3.8 (3.8) |
| I/σI | 7.1 (1.8) | 14.8 (6.9) |
| Wilson B factor (Å2) | 14.05 | 9.61 |
| Ranomc (%) | 5.8 (24.6) | |
| Anomalous completeness (%) | 85.0 (67.7) | |
| Phasing powerd (acentric) | 1.22 | |
| RCullise (acentric) | 0.772 | |
| Figure of meritf (acentric) | 0.32 | |
| Rcrystb (%) | 16.21 (34.10) | 12.79 (13.10) |
| Rfreeb (%) | 19.35 (36.70) | 17.34 (18.10) |
| No. of protein atoms | 4,174 | 4,053 |
| No. of metal ions | 6 | 8 |
| No. of NBSA atoms | 26 | |
| No. of other ligand atoms | 7 | |
| No. of water molecules | 496 | 690 |
| Avg B factor (Å2) | 12.1 | 8.43 |
| RMSD bond length (Å) | 0.026 | 0.024 |
| RMSD bond angle (°) | 2.204 | 2.056 |
The data in parentheses refer to the highest-resolution shell.
Rmerge = Σh Σi|li,h − lh|/ΣhΣili,h × 100·Rcryst (Rfree) = Σh||Fh,obs|−|Fh,calc||/Σh|Fh,obs|×100.
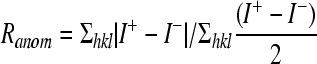 .
.
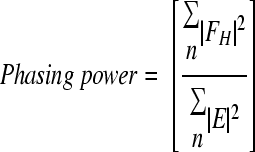 with
with 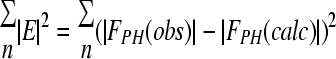 .
.
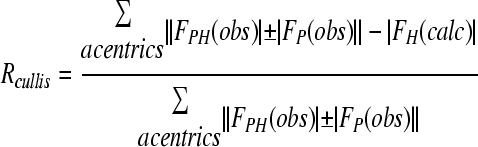 .
.
Figure of merit =m = 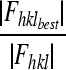 with
with 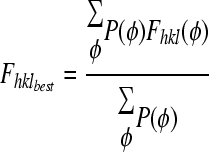 .
.
The structure of the BJP-1-NBSA complex was solved by molecular replacement with MOLREP (53), using the native structure as model, and refined by the same procedures described for the native enzyme except that, due to the higher resolution of the BJP-1-NBSA data set, all atoms have been refined with anisotropic temperature factors. The stereochemical quality of both refined models was assessed using the program PROCHECK (30). Figures were made using PyMol (http://www.pymol.org/) or CCP4 mg (40) software. Secondary structure assignment was performed using the program STRIDE (22).
Protein data bank accession numbers.
The coordinates and structure factors of the native BJP-1 and its complex with NBSA have been deposited at the Protein Data Bank under codes 3LVZ and 3M8T, respectively.
RESULTS AND DISCUSSION
Structure of BJP-1 and its complex with NBSA.
BJP-1 crystallized in the P1 space group (Table 1), and the diffraction data set allowed the structure of the enzyme at 1.40-Å resolution, the highest resolution obtained for subclass B3 MBLs, to be obtained. After refinement, the final model of the native BJP-1 consisted of two independent BJP-1 molecules in the crystal cell (residues 23 to 294 for chain A and 22 to 294 for chain B, with reference to SwissProt entry Q89GW5), six zinc ions, and 496 water molecules (Table 1; Fig. 1). NSBA-BJP-1 complex crystals produced an even better data set than native crystals, and the structure could be refined up to 1.33-Å resolution (Table 1). Similar to the native structure, the final model consisted of two independent BJP-1-NBSA complexes in the crystal cell (residues 32 to 292 for chains A and B), eight zinc ions, 1 DMSO molecule, and 690 water molecules (Table 1).
FIG. 1.

(a) Stereo views of the two independent subunits present in the P1 cells of both native and inhibited BJP-1s are shown as cartoons highlighting the tertiary structure of the enzyme. The active-site dinuclear zinc cluster is represented as gray spheres, while the accessory zinc ion is shown as a pale green sphere. (b) Topology diagram for BJP-1, where helices are represented as cylinders (H1 to H7 are α-helices; E1 to E5 are 310-helices) and β-strands as arrows.
The secondary structure of BJP-1 (PDB code, 3LVZ) is shown in Fig. 1; it has the following composition: β-strand, 22.7%; α-helix, 27.3%; helix 310, 4.9%; loops, 45.1%. The overall structure and topology of BJP-1 (Fig. 1b) displayed the main features of class B β-lactamases, consisting of a β sandwich composed of two facing β-sheets (one formed by seven β-strands and the other by five β-strands) surrounded by seven α-helices. Both β-sheets (A and B in Fig. 1b) are mixed β-sheets composed of β-strands in parallel and antiparallel conformations.
The two independent molecules in both the native and complex structures were almost identical, as reflected by the very low root mean square deviation (RMSD) values obtained upon superimposition of the two subunits (0.074 and 0.090 Å), and displayed the same backbone conformation in all parts of the structure. However, the final electron density maps showed different lengths of the polypeptide depending on the disorder present at the amino and carboxy termini of the molecules. The maximum span shown by the structures is from Thr-22 to Lys-294 in subunit B of the native enzyme crystal, but it should be pointed out that in this case, the 3 terminal residues are affected by higher disorder than the analogous portions of the other molecules. The quality of both native and complex structures was overall very good, and, due to the high resolution of the data, the positioning of the model atoms could be made with high accuracy (Table 1) and also multiple conformations for various residues could be observed. Three amino acids, Asp-84, Gly-195, and Ser-221 (using the standard numbering scheme for class B β-lactamases, which will be used henceforth for comparison with other MBLs [23, 24]), were found to adopt strained backbone conformations and were indicated as outliers in the Ramachandran plot. Asp-84 was observed in such a strained conformation in all reported MBL structures and interacted with the residue at position 115 (preceding the zinc ligand His-116), and its probable role in enzyme structure and active-site organization has been previously proposed (9, 12, 25). However, our structure also showed residues 195 and 221 in strained conformations and might also indicate the potential importance of these residues. Indeed, Gly-195 showed dihedral angles that are unusual for Gly residues and is located immediately before invariant zinc-coordinating His-196. Overall, Gly-195 is well conserved in MBLs and might be important for the correct positioning of His-196. Finally, Ser-221 is conserved in all subclass B3 enzymes and replaces Cys-221, found in subclass B1 and B2 enzymes. This residue has also been observed in disallowed regions of the Ramachandran plot, both in the native and in the complex structures. In the BJP-1-NBSA complex structure, the electron density map allowed for modeling Ser-221 in two conformations, likely due to the partial occupancy of the inhibitor, indicating that inhibitor binding might induce a conformational change of this residue.
Comparison with other MBLs.
The comparison of BJP-1 with the other B3 subclass β-lactamases of known structure, L1 from Stenotrophomonas maltophilia (1SML) and FEZ-1 from Fluoribacter gormanii (1K07) showed that, despite the 34% and 37% sequence identities, the structures shared very close backbone conformations, resulting in RMSDs of 1.60 Å and 1.25 Å, respectively, by superimposition on the common Cα atoms (213 and 202, respectively) (Fig. 2). However, some relevant structural differences were also found (Fig. 2). BJP-1, like FEZ-1, lacks the 12-residue N-terminal unstructured peptide present in L1 and shows a nicely structured amino-terminal α-helix, which is not present in the other two enzymes. Interestingly, this helix (H1) covers the active site, from which it is displaced upon the binding of the NBSA inhibitor (Fig. 2; see below for details).
FIG. 2.

(a) Stereo view of a least-squares superimposition of BJP-1 (red ribbon) with L1 (blue ribbon) and FEZ-1 (green ribbon), highlighting the structural differences between the three class B3 β-lactamases of known structure. The location of the active site common to the three enzymes is shown by the gray spheres representing the Zn(II) ions (shown for BJP-1 only). The N-terminal α-helix unique to the native BJP-1 structure is orange. The position of the Phe-31 side chain in the native enzyme is shown by red sticks. The 4-nitrobenzenesulfonamide (NBSA) molecule found in the BJP-1 complex is shown as cyan sticks. (b) Stereo view of a superimposition of the native (red ribbon) and NBSA-bound (yellow ribbon) BJP-1 structures showing the overlap in the positions occupied by the Phe-31 side chain (red) and the NBSA molecule (cyan).
In addition, major displacements occurred in the carboxy-terminal region, where helices H5 and H6 are linked by a large loop to the terminal helix H7 (Fig. 1 and 2). Here, both FEZ-1 and BJP-1 have insertions that made the backbone conformations differ by more than 10 Å with respect to that of L1. FEZ-1 differed from BJP-1 by more that 3.4 Å, on average, in this region. Other significant differences are located on the long loop (residues 218 to 239) linking β11B to helix H5, where BJP-1 adopts a conformation that is different from those of FEZ-1 and L1 and that is relevant to the shape and dimensions of the active site (see below for details). Finally BJP-1 differs from the other two enzymes in the long loop linking helix H4 to the β7A. It is worth noting that all three structural elements involved in variable parts of the structure surround the active-site cavity, where the metal ions are bound. The disulfide bridge present in both FEZ-1 and L1 at the same position, between the C-terminal helix and the loop linking H5 to β12B, is missing in BJP-1, where a different disulfide bridge occurs between Cys-200 and Cys-220 (residues 181 and 201 in the BJP-1 numbering), as hypothesized previously (49). A further difference presented by BJP-1 is the presence of an additional metal binding site located between helix H5 and the B β-sheet, where a zinc ion from the crystallization buffer is bound, with partial occupancy, to Glu-176, His-190, and Lys-250 and to the C-terminal carboxylate of Lys-321 from an adjacent molecule, completing a tetrahedral coordination polyhedron. This observation provides a further interesting example of the structural role played by metal ions in facilitating protein crystallization by stabilizing intermolecular interactions (6).
Active-site and metal coordination.
The active site of BJP-1 is located in a narrow cleft located above the β-sheets, contoured by the amino-terminal helix (H1), by the loops H4-E3 and β11B-H5, and defined by loop β5A-H3 and by the loops β9B-β10B and β12B-E4, which contain all the zinc ligands: His-116, His-118, and His-196 (forming the so-called H3 site) and Asp-120, His-121, and His-263 (forming the so-called DHH site) (Fig. 3 a).
FIG. 3.

Stereo views of the active site of native BJP-1 and BJP-1 in complex with NBSA. (a) Active-site structure of native BJP-1 with the residues involved in zinc binding represented as green sticks. A 2Fo-Fc electron density map computed with phases from the refined model is superimposed as a blue grid contoured at 1.5 σ. The residues forming the hydrophobic pocket hosting the side chain of the N-terminal Phe-31 are shown as magenta sticks. (b) Active-site structure of NBSA-inhibited BJP-1. The picture shows the same residues reported in panel a with the same color code. A 2Fo-Fc electron density map computed with phases from the refined model is superimposed as a blue grid contoured at 1.5 σ. The NBSA inhibitor bridges the two zinc ions with the sulfonamide nitrogen, while the two sulfone oxygens asymmetrically bind the zinc ions. The inhibitor phenyl ring replaces the Phe-31 side chain of the native enzyme.
The electron density of the dinuclear zinc site shows that the coordination environment of the two Zn(II) ions is completed by a hydroxide anion (OHbr) bridging the two zinc ions, similar to other MBL structures. The anionic nature of the bridging ligand is indicated by the short observed coordination distances to the metal ions (Fig. 3a; Table 2). The two subunits differ slightly in the binding of OHbr, indicating some flexibility of the metal site: in subunit A, OHbr is asymmetric and located significantly closer to Zn1 (as already observed for L1 β-lactamase [47, 52]), while in subunit B the two OHbr-Zn distances are almost identical within experimental errors.
TABLE 2.
Zinc coordination distances
| Zn atom | Atom or molecule | Zn distancea (Å) for: |
|||
|---|---|---|---|---|---|
| Native BJP-1, subunit: |
NBSA-BJP-1 complex, subunit: |
||||
| A | B | A | B | ||
| Zn1 | His-116 Nɛ | 2.06 | 2.08 | 2.03 | 2.04 |
| His-118 Nδ | 2.08 | 2.11 | 2.06 | 2.05 | |
| His-196 Nɛ | 2.03 | 1.97 | 2.04 | 2.06 | |
| OHbr | 1.94 | 1.98 | |||
| N NBSAb | 1.83 | 1.94 | |||
| O NBSA | 2.86 | 2.87 | |||
| Water | 3.25 | 3.22 | 3.14 | 3.38 | |
| Zn2 | 3.45 | 3.43 | 3.42 | 3.44 | |
| Zn2 | Asp-120 Oδ2 | 2.06 | 2.09 | 2.02 | 2.01 |
| His-121 Nɛ | 2.07 | 2.08 | 2.04 | 2.05 | |
| His-263 Nɛ | 2.05 | 2.09 | 2.04 | 2.05 | |
| OHbr | 2.06 | 1.96 | |||
| N NBSA | 1.92 | 1.86 | |||
| O NBSA | 3.28 | 3.24 | |||
| Water | 2.53 | 2.45 | 2.78 | 3.05 | |
| Zn3 | Glu-176 Oɛ2 | 2.00 | 1.96 | 2.04 | 2.03 |
| His-190 Nδ | 2.06 | 2.10 | 2.02 | 2.02 | |
| Lys-250 Nζ | 1.98 | 2.00 | 2.00 | 1.97 | |
| Lys-321 O (symmetrical) | 1.99 | 2.03 | 2.06 | 2.01 | |
Estimated SD on distances were below 0.06 and 0.04 Å for the native and complex structures, respectively. τ = (β − α)/60, where α and β are the basal diagonal angles at Zn with β > α(1); (β = Asp-120 Oδ1-Zn-Water angle; α = His-263 Nɛ-Zn-OHbr angle, in this case). τ for subunit A = 0.29; τ for subunit B = 0.30.
The N atom of NBSA replaces the OHbr atom in the NBSA-BJP-1 complex bridging the two Zn ions.
Zn1 in the H3 site adopts a distorted tetrahedral geometry, while Zn2 coordination in the DHH site is better described by a square pyramidal geometry, as a water molecule, loosely bound to Zn2 at an average of 2.50 Å, acts as a fifth zinc ligand. The base of the pyramid is constituted by Asp-120 Oδ1, His-263 Nɛ2, a water molecule (water), and OHbr, and His-121 Nɛ2 is the apex.
The square pyramidal geometry for Zn2 is established by the values of the stereochemical τ parameter (Table 2, footnote a), which depend on the angular values of the base diagonal (Asp-120 Oδ1-Zn2-water angle and His-263 Nɛ2-Zn2-OHbr angle, in this case). τ has been introduced to describe the geometry of five-coordinated metal coordination compounds, with τ of 0 for an ideal square pyramidal geometry and τ of 1 for an ideal trigonal bipyramidal geometry (1).
Although the geometry for Zn2 in dinuclear B1 and B3 β-lactamases has usually been described as trigonal bipyramidal (13, 19), the calculation of the τ value using the available structures of other MBLs also indicates that, in these cases, the geometry of Zn2 would be better described by a square pyramid. Indeed, the τ values for L1 and the dizinc BcII (PDB codes, 1SML and 1BC2, respectively) range from 0.27 to 0.43.
Binding of sulfonamide inhibitor in BJP-1.
In the complex structure, the NBSA ligand replaces the bridging hydroxide anion between the two Zn(II) ions, as shown in Fig. 3b. The ligand has been modeled with the amide nitrogen as the bridging ligand, due to the higher Lewis base properties and in analogy with the established binding of primary sulfonamides to zinc enzymes, like carbonic anhydrase (2, 7, 10, 26, 51, 54) and carboxypeptidase (39), or model compounds (28). The very short Zn-N-NBSA distances (Table 2) indicate that the species bound is the anionic nitrogen of the sulfonamide, deprotonated by binding to the dinuclear zinc center (3, 28), while the two sulfone oxygens behave as weaker zinc ligands. The binding of NBSA to BJP-1 is, to our knowledge, the first example of a sulfonamide bound to a dinuclear metal center in proteins. The binding of the inhibitor changes the coordination number and geometry for Zn1 toward a distorted trigonal bipyramid by adding the O2 atom of NBSA to the Zn(II) coordination sphere (τ = 0.70 for both subunits). On the other hand, the sulfone O1 atom of NBSA remains at a longer distance from Zn2; this, however, also results in a change toward a trigonal bipyramidal geometry (τ = 0.51 to 0.61 for the two subunits). The NBSA O1 and O2 atoms are at H-bond distance from two or three water molecules, depending on the subunit. The NBSA nitro group was not involved in any H bond, but the nitrobenzene moiety was firmly held in its binding site by contacts with Trp-39, Met-87, Leu-119, Leu-162, and Leu-226 side chains (residues 34, 73, 104, 145, and 207 in the BJP-1 numbering), which form a hydrophobic pocket within the active-site cavity. In the native enzyme, this hydrophobic pocket was occupied by the side chain of Phe-31 (residue 26 in the BJP-1 numbering), which was displaced by NBSA upon binding. This also affected the whole N-terminal helix, which moved away in the BJP-1-NBSA complex. In that structure, only a diffuse electron density corresponding to the N-terminal helix could be observed, reflecting the overall disorder of this part of the protein and preventing its proper modeling (these residues are missing from the final BJP-1-NBSA complex structure).
Comparison of the two independent molecules of the complex by least-squares superimposition shows that the binding mode of NBSA is the same in both subunits. The comparison by least-squares superposition with the native enzyme shows that, while the Zn1-Zn2 distance remains essentially the same (Table 2), Zn2 moves by a significant 0.36 Å upon the binding of the inhibitor, indicating a higher flexibility of the DHH metal binding site and suggesting that this site might have a somewhat lower affinity for zinc in BJP-1 than the H3 site.
Significance for substrate profile and broad-spectrum inhibition of MBLs.
The peculiar functional properties of BJP-1, i.e., overall low affinities for β-lactam substrates and the absence of interaction with carboxypenicillins (Table 3) could possibly be explained by analyzing the structural features of the native and inhibited forms and by comparing them with the available subclass B3 MBL structures. Although the metal binding sites of the various enzymes have essentially the same shape, the BJP-1 active site is significantly narrower due to the presence of the N-terminal helix (H1). Moreover, the side chain of residue Phe-31 occupies a position in which the peptide bond present in most β-lactam substrates (part of the C6/C7 substituent, also known as the R1 substituent) is expected to bind, as revealed by the superimposition of the hydrolyzed moxalactam-L1 complex structure (2AIO) with that of the native BJP-1 (48). Although the NBSA complex shows that this residue could adopt a different conformation and make room for an inhibitor and/or substrate, this likely requires a conformational change of the N-terminal helix, which might represent a kinetically and thermodynamically limiting factor for the binding of β-lactam substrates; this would be consistent with the overall high Km values observed for BJP-1 with most antibiotics (49).
TABLE 3.
Kinetic properties of some subclass B3 metallo-β-lactamases with relevant β-lactam compoundsa
| Substrate | BJP-1 |
L1 |
FEZ-1 |
GOB-1 |
||||
|---|---|---|---|---|---|---|---|---|
| Km (μM) | kcat/Km (M−1·s−1) | Km (μM) | kcat/Km (M−1·s−1) | Km (μM) | kcat/Km (M−1·s−1) | Km (μM) | kcat/Km (M−1·s−1) | |
| Ampicillin | 670 | 1.9 × 104 | 40 | 4.4 × 106 | >5,000d | 1.1 × 104 | —e | — |
| Piperacillin | 700 | 6.7 × 104 | 20 | 7.0 × 106 | 4,200 | 1.2 × 104 | 170 | 1.7 × 106 |
| Ticarcillin | NDb | NHc | 140 | 2.7 × 106 | >5,000d | 1.3 × 104 | 150 | 5.2 × 105 |
| Cephalothin | 230 | 5.8 × 105 | 8.9 | 9.2 × 106 | 120 | 2.5 × 106 | 24 | 6.7 × 105 |
| Ceftazidime | >700d | 4.3 × 103 | 145 | 2.0 × 105 | >1,000d | 4.0 × 103 | 71 | 7.6 × 105 |
| Imipenem | 260 | 6.0 × 104 | 90 | 7.3 × 105 | >1,000d | 2.0 × 105 | 60 | 6.6 × 105 |
| Meropenem | 190 | 8.3 × 105 | 10 | 4.5 × 106 | 85 | 5.0 × 105 | 5 | 5.3 × 106 |
The lack of interaction with carboxypenicillins (e.g., ticarcillin) likely depends on the nature and position of the residues constituting loop H5-β11 (Fig. 2b). In the BJP-1 structure, the side chain of Leu-226 protrudes into the active-site cavity and determines a narrowing of the cleft where the carboxy group of the C6 substituent of ticarcillin would possibly bind (48). In the hydrolyzed moxalactam-L1 complex (moxalactam also has a carboxy group in its C7 substituent), the corresponding loop is located 4 Å away from the active site (compared to BJP-1), creating a much larger cavity, able to accommodate bulkier substrates and consistent with the broader substrate profile exhibited by this enzyme.
The binding of the sulfonamide group observed in the BJP-1-NBSA complex represents the first structural observation of such a molecule bound to a class B β-lactamase, indicating that the sulfonamide group is able to bind dinuclear MBLs by either competing with the substrate β-lactam group for binding to the Zn(II) ion or displacing both the OHbr nucleophile and the Zn2-bound water molecule, which appear to play an active role in catalysis (15, 46). NBSA, one of the simplest model aromatic sulfonamides known to bind to other zinc enzymes (29), was able to inhibit BJP-1 in solution with a Ki of 100 μM. Although the analysis of NBSA binding shown by the crystal structures would apparently suggest a stronger inhibition effect, the rather high Ki value reflects a rather low gain in complexation energy between the free and inhibited BJP-1s, likely due to the presence of the Phe-31 side chain in the pocket where the inhibitor binds. Thus, it is expected that other MBLs, whose active sites are not similarly hindered, would be more efficiently inhibited by NBSA.
In conclusion, the NBSA-BJP-1 complex structure potentially provides the structural basis for the rational design of sulfonamide-based MBL inhibitors. The crystal structure of the B1 MBL VIM-2 in complex with a mercaptocarboxylate inhibitor (PDB entry 2YZ3) shows the thiolate sulfur of the inhibitor bridging the two Zn(II) ions in a binding mode that resembles that of NBSA in BJP-1, suggesting that sulfonamide-based inhibitors might also bind to subclass B1 MBLs (58). Very recently, sulfonamide compounds have been found to behave as competitive inhibitors of the clinically relevant subclass B1 MBL VIM-2 (36). The structure of the BJP-1-NBSA complex provides the structural rationale for the mechanism of binding and inhibition of these compounds with MBLs and might represent valuable information to design new MBL inhibitors based on the sulfonamide scaffold. Interestingly, many sulfonamide molecules are currently used as successful and safe drugs in human therapy to treat a wide variety of pathologies (e.g., HIV infection, migraine, and glaucoma) (51). This also demonstrates that the specificity for different enzyme targets could be reasonably achieved by varying the chemical structure of a common scaffold. Furthermore, the ability of sulfonamides to bind mononuclear zinc enzymes like carbonic anhydrase and carboxypeptidase suggests that this chemical determinant might be exploited to also inhibit subclass B1 and B2 enzymes, independently of their zinc content. In conclusion, we believe that it is worth pursuing the study of sulfonamide-based molecules to develop specific and broad-spectrum inhibitors potentially active on the three subclasses of metallo-β-lactamases.
Acknowledgments
We acknowledge the European Synchrotron Radiation Facility (ESRF; Grenoble, France) for having provided access to the ID14-1 and ID29 beamlines and the ESRF staff for excellent assistance.
This work was funded in part by grants from the European Union and by the Italian Ministero dell'Istruzione, Università e Ricerca (MIUR) (contract numbers HPRN-CT-2002-00264 and 2005061894_004). S.M. also acknowledges the contribution of MIUR (PRIN2007 2007M5MWM9_001 research grant). V.C. was the recipient of a postdoctoral fellowship from the Italian CIRMPP consortium.
Footnotes
Published ahead of print on 9 August 2010.
REFERENCES
- 1.Addison, A. W., T. Nageswara Rao, J. Reedijk, J. van Rijn, and G. C. Verschoor. 1984. Synthesis, structure, and spectroscopic properties of copper(II) compounds containing nitrogen-sulphur donor ligands; the crystal and molecular structure of aqua[1,7-bis(N-methylbenzimidazol-2-yl)-2,6-dithiaheptane]copper(II) perchlorate. J. Chem. Soc. Dalton Trans. 7:1349-1356. [Google Scholar]
- 2.Alterio, V., M. Hilvo, A. Di Fiore, C. T. Supuran, P. Pan, S. Parkkila, A. Scaloni, J. Pastorek, S. Pastorekova, C. Pedone, A. Scozzafava, S. M. Monti, and G. De Simone. 2009. Crystal structure of the catalytic domain of the tumor-associated human carbonic anhydrase IX. Proc. Natl. Acad. Sci. U. S. A. 106:16233-16238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Basolo, F., and R. G. Pearson. 1967. Acid-base properties of complex ions, p. 31-33. In Mechanisms of inorganic reactions. Wiley, New York, NY.
- 4.Bebrone, C. 2007. Metallo-β-lactamases (classification, activity, genetic organization, structure, zinc coordination) and their superfamily. Biochem. Pharmacol. 74:1686-1701. [DOI] [PubMed] [Google Scholar]
- 5.Bellais, S., D. Aubert, T. Naas, and P. Nordmann. 2000. Molecular and biochemical heterogeneity of class B carbapenem-hydrolyzing β-lactamases in Chryseobacterium meningosepticum. Antimicrob. Agents Chemother. 44:1878-1886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Benvenuti, M., and S. Mangani. 2007. Crystallization of soluble proteins in vapor diffusion for x-ray crystallography. Nat. Protoc. 2:1633-1651. [DOI] [PubMed] [Google Scholar]
- 7.Boriack, P. A., D. W. Christianson, J. Kingery-Wood, and G. M. Whitesides. 1995. Secondary interactions significantly removed from the sulfonamide binding pocket of carbonic anhydrase II influence inhibitor binding constants. J. Med. Chem. 38:2286-2291. [DOI] [PubMed] [Google Scholar]
- 8.Carenbauer, A. L., J. D. Garrity, G. Periyannan, R. B. Yates, and M. W. Crowder. 2002. Probing substrate binding to metallo-β-lactamase L1 from Stenotrophomonas maltophilia by using site-directed mutagenesis. BMC Biochem. 3:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Carfi, A., S. Pares, E. Duee, M. Galleni, C. Duez, J. M. Frere, and O. Dideberg. 1995. The 3-D structure of a zinc metallo-β-lactamase from Bacillus cereus reveals a new type of protein fold. EMBO J. 14:4914-4921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chakravarty, S., and K. K. Kannan. 1994. Drug-protein interactions. Refined structures of three sulfonamide drug complexes of human carbonic anhydrase I enzyme. J. Mol. Biol. 243:298-309. [DOI] [PubMed] [Google Scholar]
- 11.Collaborative Computational Project, N4. 2008. The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 50:760-763. [DOI] [PubMed] [Google Scholar]
- 12.Concha, N. O., B. A. Rasmussen, K. Bush, and O. Herzberg. 1996. Crystal structure of the wide-spectrum binuclear zinc β-lactamase from Bacteroides fragilis. Structure 4:823-836. [DOI] [PubMed] [Google Scholar]
- 13.Crowder, M. W., J. Spencer, and A. J. Vila. 2006. Metallo-β-lactamases: novel weaponry for antibiotic resistance in bacteria. Acc. Chem. Res. 39:721-728. [DOI] [PubMed] [Google Scholar]
- 14.Daiyasu, H., K. Osaka, Y. Ishino, and H. Toh. 2001. Expansion of the zinc metallo-hydrolase family of the β-lactamase fold. FEBS Lett. 503:1-6. [DOI] [PubMed] [Google Scholar]
- 15.Dal Peraro, M., A. J. Vila, P. Carloni, and M. L. Klein. 2007. Role of zinc content on the catalytic efficiency of B1 metallo β-lactamases. J. Am. Chem. Soc. 129:2808-2816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Docquier, J. D., F. Pantanella, F. Giuliani, M. C. Thaller, G. Amicosante, M. Galleni, J. M. Frere, K. Bush, and G. M. Rossolini. 2002. CAU-1, a subclass B3 metallo-β-lactamase of low substrate affinity encoded by an ortholog present in the Caulobacter crescentus chromosome. Antimicrob. Agents Chemother. 46:1823-1830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Emsley, P., and K. Cowtan. 2004. Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60:2126-2132. [DOI] [PubMed] [Google Scholar]
- 18.Evans, P. R. 1997. SCALA, continuous scaling program. Joint CCP4 ESF-EACBM Newsl. 33:22-24. [Google Scholar]
- 19.Fabiane, S. M., M. K. Sohi, T. Wan, D. J. Payne, J. H. Bateson, T. Mitchell, and B. J. Sutton. 1998. Crystal structure of the zinc-dependent β-lactamase from Bacillus cereus at 1.9 Å resolution: binuclear active site with features of a mononuclear enzyme. Biochemistry 37:12404-12411. [DOI] [PubMed] [Google Scholar]
- 20.Felici, A., and G. Amicosante. 1995. Kinetic analysis of extension of substrate specificity with Xanthomonas maltophilia, Aeromonas hydrophila, and Bacillus cereus metallo-β-lactamases. Antimicrob. Agents Chemother. 39:192-199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fisher, J. F., S. O. Meroueh, and S. Mobashery. 2005. Bacterial resistance to β-lactam antibiotics: compelling opportunism, compelling opportunity. Chem. Rev. 105:395-424. [DOI] [PubMed] [Google Scholar]
- 22.Frishman, D., and P. Argos. 1995. Knowledge-based protein secondary structure assignment. Proteins 23:566-579. [DOI] [PubMed] [Google Scholar]
- 23.Galleni, M., J. Lamotte-Brasseur, G. M. Rossolini, J. Spencer, O. Dideberg, and J. M. Frere. 2001. Standard numbering scheme for class B β-lactamases. Antimicrob. Agents Chemother. 45:660-663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Garau, G., I. Garcia-Saez, C. Bebrone, C. Anne, P. Mercuri, M. Galleni, J. M. Frere, and O. Dideberg. 2004. Update of the standard numbering scheme for class B β-lactamases. Antimicrob. Agents Chemother. 48:2347-2349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Garcia-Saez, I., P. S. Mercuri, C. Papamicael, R. Kahn, J. M. Frere, M. Galleni, G. M. Rossolini, and O. Dideberg. 2003. Three-dimensional structure of FEZ-1, a monomeric subclass B3 metallo-β-lactamase from Fluoribacter gormanii, in native form and in complex with D-captopril. J. Mol. Biol. 325:651-660. [DOI] [PubMed] [Google Scholar]
- 26.Hakansson, K., and A. Liljas. 1994. The structure of a complex between carbonic anhydrase II and a new inhibitor, trifluoromethane sulphonamide. FEBS Lett. 350:319-322. [DOI] [PubMed] [Google Scholar]
- 27.Kaneko, T., Y. Nakamura, S. Sato, K. Minamisawa, T. Uchiumi, S. Sasamoto, A. Watanabe, K. Idesawa, M. Iriguchi, K. Kawashima, M. Kohara, M. Matsumoto, S. Shimpo, H. Tsuruoka, T. Wada, M. Yamada, and S. Tabata. 2002. Complete genomic sequence of nitrogen-fixing symbiotic bacterium Bradyrhizobium japonicum USDA110. DNA Res. 9:189-197. [DOI] [PubMed] [Google Scholar]
- 28.Kimura, E., and E. Kikuta. 2000. Why zinc in zinc enzymes? From biological roles to DNA base-selective recognition. J. Biol. Inorg. Chem. 5:139-155. [DOI] [PubMed] [Google Scholar]
- 29.Koike, T., E. Kimura, I. Nakamura, Y. Hashimoto, and M. Shiro. 1992. The first anionic sulfonamide-binding zinc(II) complexes with a macrocyclic triamine: chemical verification of the sulfonamide inhibition of carbonic anhydrase. J. Am. Chem. Soc. 114:7338-7345. [Google Scholar]
- 30.Laskowski, R. A., M. W. Macarthur, D. S. Moss, and J. E. Thornton. 1993. PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 26:283-291. [Google Scholar]
- 31.Leslie, A. G. W. 2006. The integration of macromolecular diffraction data. Acta Crystallogr. D Biol. Crystallogr. 62:48-57. [DOI] [PubMed] [Google Scholar]
- 32.Livermore, D. M. 2009. Has the era of untreatable infections arrived? J. Antimicrob. Chemother. 64(Suppl. 1):i29-i36. [DOI] [PubMed] [Google Scholar]
- 33.Matagne, A., A. Dubus, M. Galleni, and J. M. Frere. 1999. The β-lactamase cycle: a tale of selective pressure and bacterial ingenuity. Nat. Prod. Rep. 16:1-19. [DOI] [PubMed] [Google Scholar]
- 34.McRee, D. E. 1999. XtalView/Xfit—a versatile program for manipulating atomic coordinates and electron density. J. Struct. Biol. 125:156-165. [DOI] [PubMed] [Google Scholar]
- 35.Mercuri, P. S., F. Bouillenne, L. Boschi, J. Lamotte-Brasseur, G. Amicosante, B. Devreese, J. Van Beeumen, J. M. Frere, G. M. Rossolini, and M. Galleni. 2001. Biochemical characterization of the FEZ-1 metallo-β-lactamase of Legionella gormanii ATCC 33297T produced in Escherichia coli. Antimicrob. Agents Chemother. 45:1254-1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Minond, D., S. A. Saldanha, P. Subramaniam, M. Spaargaren, T. Spicer, J. R. Fotsing, T. Weide, V. V. Fokin, K. B. Sharpless, M. Galleni, C. Bebrone, P. Lassaux, and P. Hodder. 2009. Inhibitors of VIM-2 by screening pharmacologically active and click-chemistry compound libraries. Bioorg. Med. Chem. 17:5027-5037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Morris, R. J., A. Perrakis, and V. S. Lamzin. 2003. ARP/wARP and automatic interpretation of protein electron density maps. Methods Enzymol. 374:229-244. [DOI] [PubMed] [Google Scholar]
- 38.Murshudov, G. N., A. A. Vagin, and E. J. Dodson. 1997. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 53:240-255. [DOI] [PubMed] [Google Scholar]
- 39.Park, J. D., D. H. Kim, S. J. Kim, J. R. Woo, and S. E. Ryu. 2002. Sulfamide-based inhibitors for carboxypeptidase A. Novel type transition state analogue inhibitors for zinc proteases. J. Med. Chem. 45:5295-5302. [DOI] [PubMed] [Google Scholar]
- 40.Potterton, E., S. McNicholas, E. Krissinel, K. Cowtan, and M. Noble. 2002. The CCP4 molecular-graphics project. Acta Crystallogr. D Biol. Crystallogr. 58:1955-1957. [DOI] [PubMed] [Google Scholar]
- 41.Rice, L. B. 2009. The clinical consequences of antimicrobial resistance. Curr. Opin. Microbiol. 12:476-481. [DOI] [PubMed] [Google Scholar]
- 42.Rossolini, G. M., M. A. Condemi, F. Pantanella, J. D. Docquier, G. Amicosante, and M. C. Thaller. 2001. Metallo-β-lactamase producers in environmental microbiota: new molecular class B enzyme in Janthinobacterium lividum. Antimicrob. Agents Chemother. 45:837-844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rossolini, G. M., and J. D. Docquier. 2006. New β-lactamases: a paradigm for the rapid response of bacterial evolution in the clinical setting. Future Microbiol. 1:295-308. [DOI] [PubMed] [Google Scholar]
- 44.Rossolini, G. M., and J. D. Docquier. 2007. Class B β-lactamases, p. 115-144. In R. A. Bonomo and M. E. Tolmasky (ed.), Enzyme-mediated resistance to antibiotics: mechanisms, dissemination and prospects for inhibition. ASM Press, Washington, DC.
- 45.Simm, A. M., C. S. Higgins, A. L. Carenbauer, M. W. Crowder, J. H. Bateson, P. M. Bennett, A. R. Clarke, S. E. Halford, and T. R. Walsh. 2002. Characterization of monomeric L1 metallo-β-lactamase and the role of the N-terminal extension in negative cooperativity and antibiotic hydrolysis. J. Biol. Chem. 277:24744-24752. [DOI] [PubMed] [Google Scholar]
- 46.Simona, F., A. Magistrato, M. Dal Peraro, A. Cavalli, A. J. Vila, and P. Carloni. 2009. Common mechanistic features among metallo-β-lactamases: a computational study of Aeromonas hydrophila CphA enzyme. J. Biol. Chem. 284:28164-28171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Spencer, J., A. R. Clarke, and T. R. Walsh. 2001. Novel mechanism of hydrolysis of therapeutic β-lactams by Stenotrophomonas maltophilia L1 metallo-β-lactamase. J. Biol. Chem. 276:33638-33644. [DOI] [PubMed] [Google Scholar]
- 48.Spencer, J., J. Read, R. B. Sessions, S. Howell, G. M. Blackburn, and S. J. Gamblin. 2005. Antibiotic recognition by binuclear metallo-β-lactamases revealed by X-ray crystallography. J. Am. Chem. Soc. 127:14439-14444. [DOI] [PubMed] [Google Scholar]
- 49.Stoczko, M., J. M. Frere, G. M. Rossolini, and J. D. Docquier. 2006. Postgenomic scan of metallo-β-lactamase homologues in rhizobacteria: identification and characterization of BJP-1, a subclass B3 ortholog from Bradyrhizobium japonicum. Antimicrob. Agents Chemother. 50:1973-1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Stoczko, M., J. M. Frere, G. M. Rossolini, and J. D. Docquier. 2008. Functional diversity among metallo-β-lactamases: characterization of the CAR-1 enzyme of Erwinia carotovora. Antimicrob. Agents Chemother. 52:2473-2479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Supuran, C. T., and J. Y. Winum. 2009. Drug design of zinc-enzyme inhibitors: functional, structural, and disease applications. John Wiley & Sons, Hoboken, NJ.
- 52.Ullah, J. H., T. R. Walsh, I. A. Taylor, D. C. Emery, C. S. Verma, S. J. Gamblin, and J. Spencer. 1998. The crystal structure of the L1 metallo-β-lactamase from Stenotrophomonas maltophilia at 1.7 Å resolution. J. Mol. Biol. 284:125-136. [DOI] [PubMed] [Google Scholar]
- 53.Vagin, A., and A. Teplyakov. 1997. MOLREP: an automated program for molecular replacement. J. Appl. Crystallogr. 30:1022-1025. [Google Scholar]
- 54.Vidgren, J., A. Liljas, and N. P. C. Walker. 1990. Refined structure of the acetazolamide complex of human carbonic anhydrase-II at 1.9 Å. Int. J. Biol. Macromol. 12:342-344. [DOI] [PubMed] [Google Scholar]
- 55.Vonrhein, C., E. Blanc, P. Roversi, and G. Bricogne. 2007. Automated structure solution with autoSHARP. Methods Mol. Biol. 364:215-230. [DOI] [PubMed] [Google Scholar]
- 56.Winn, M. D., M. N. Isupov, and G. N. Murshudov. 2001. Use of TLS parameters to model anisotropic displacements in macromolecular refinement. Acta Crystallogr. D Biol. Crystallogr. 57:122-133. [DOI] [PubMed] [Google Scholar]
- 57.Winn, M. D., G. N. Murshudov, and M. Z. Papiz. 2003. Macromolecular TLS refinement in REFMAC at moderate resolutions. Methods Enzymol. 374:300-321. [DOI] [PubMed] [Google Scholar]
- 58.Yamaguchi, Y., W. Jin, K. Matsunaga, S. Ikemizu, Y. Yamagata, J. Wachino, N. Shibata, Y. Arakawa, and H. Kurosaki. 2007. Crystallographic investigation of the inhibition mode of a VIM-2 metallo-β-lactamase from Pseudomonas aeruginosa by a mercaptocarboxylate inhibitor. J. Med. Chem. 50:6647-6653. [DOI] [PubMed] [Google Scholar]