

Abstract
Peptide dendrimers consist of a peptidyl branching core and/or covalently attached surface functional units. They show a variety of biological properties, including antiviral activity. In this study, a minilibrary of linear, dimeric, and dendrimeric peptides containing clusters of basic amino acids was evaluated for in vitro activity against human papillomaviruses (HPVs). The peptide dendrimer SB105-A10 was found to be a potent inhibitor of genital HPV types (i.e., types 16, 18, and 6) in pseudovirus-based neutralization assays. The 50% inhibitory concentration was between 2.8 and 4.2 μg/ml (0.59 and 0.88 μM), and no evidence of cytotoxicity was observed. SB105-A10 interacts with immobilized heparin and with heparan sulfates exposed on the cell surface, most likely preventing virus attachment. The findings from this study indicate SB105-A10 to be a leading candidate compound for further development as an active ingredient of a topical microbicide against HPV and other sexually transmitted viral infections.
Human papillomaviruses (HPVs) are nonenveloped, double-stranded DNA viruses belonging to the family Papillomaviridae. The 8-kb HPV genome is enclosed in a capsid shell comprising major (L1) and minor (L2) structural proteins. More than 100 HPV types have been identified so far, over 50 of which infect the genital area (21, 32). Genital HPV infections are the most common sexually transmitted infections (STIs). HPVs are highly transmissible, and most sexually active men and women will acquire an HPV infection at some time in their lives (4). Although HPV infection is transient and asymptomatic in the great majority of immunocompetent individuals, a small proportion of men and women fail to control viral infection and develop HPV-related malignancies.
HPVs are classified as “low-risk” or “high-risk” types according to their association with cervical cancer. Infections with low-risk HPV types, such as HPV-6 and -11, are rarely associated with cervical cancer but can cause benign lesions of the anogenital areas, known as condylomata acuminata or genital warts, oral papillomas, conjunctival papillomas, and low-grade squamous intraepithelial lesions of the cervix. Perinatally acquired HPV can also cause recurrent respiratory papillomatosis in infants and young children (37).
Infection with high-risk HPV types (primarily types 16, 18, 31, and 45) can cause cervical cell abnormalities that are precursors to cancer (41). HPV types 16 and 18 cause about 70% of all cases of invasive cervical cancer worldwide, with type 16 having the greatest oncogenic potential. It has been estimated that about 500,000 cases of cervical cancer and 260,000 related deaths occur each year worldwide (4).
Current treatments are ablative and directed to abnormal cells associated with HPV rather than to the virus itself; no direct anti-HPV treatment is available. The prevention of genital infection is essential for reducing genital warts, abnormal Pap tests, and cervical cancer. Male condoms cannot be recommended as a primary prevention strategy, since they provide only partial protection against HPV transmission (30, 43).
Two HPV vaccines are widely marketed internationally (52). Gardasil (Merck and Co., Inc., Whitehouse Station, NJ) is designed to protect against oncogenic HPV types 16 and 18 and low-risk HPV types 6 and 11 (58). Cervarix (GlaxoSmithKline Biologicals, Rixensart, Belgium) is designed to protect against HPV types 16 and 18 (28). Although these vaccines represent a remarkable improvement in the fight against cervical disease and other anogenital cancers, women may remain exposed to infection with high-risk HPVs that can cause cervical cancer but are not targeted by the current vaccines. Moreover, since the vaccines are relatively expensive, they may not be available to all women, especially those in developing countries.
In light of this, topical antiviral microbicides that can block the full spectrum of genital HPV infections at the portal of entry would be useful complements to prophylactic vaccines. A main objective in the development of microbicides against HPV is to block the interaction between the virion proteins and cell surface heparan sulfate proteoglycans (HSPGs) that mediate the initial attachment of HPV to the target cell (25, 35, 53). HSPGs are associated with the surfaces of many cell types and consist of a core protein with glycosaminoglycan (GAG) chains of unbranched sulfated polysaccharides known as heparan sulfates (HS), which are structurally related to heparin. Furthermore, previous studies have demonstrated that heparin and other polyanionic compounds act as HSPG antagonists, preventing the binding of HPV to the cell surface (8, 16, 39).
An ideal microbicide should also disrupt the sexual transmission of HIV and herpes simplex virus (HSV) (45, 50) as well as HPV. Notably, HPV and HSV infections are frequently found in immunocompromised HIV-positive individuals, and all three viruses use HSPGs as initial receptors for infection. Thus, polyanionic heparin-like compounds have emerged as ideal multitarget antiviral drugs directed against HPV, HIV, and HSV (50). However, despite an initial burst of promising results, to date the translation of polyanionic heparin-like compounds from molecular studies to effective clinical applications has been unsuccessful (14, 31, 55). Taken together, these considerations call for new putative microbicides possibly endowed with different mechanisms of action.
Dendrimers are large, highly branched macromolecules synthesized from a polyfunctional core and generally endowed with functional groups in the surface layer that can bind to a variety of molecular targets (5). Dendrimers are attractive as potential new therapeutics because of their ease of preparation and functionalization, stability in the presence of proteases, and ability to display multiple copies of bioactive surface groups (multivalency) for biological recognition processes (18). Accordingly, dendrimers have been studied extensively as drug delivery agents, in diagnostics, and as antitumor, antibacterial, and antiviral drugs (3). In effect, dendrimers can act as polyvalent viral inhibitors by presenting multiple contact sites on a single molecule that can efficiently prevent/disrupt those molecular interactions that mediate the interaction of viruses with the cell surface, and hence virus infection. One such example is SPL7013, a dendrimer developed by Starpharma (Melbourne, Victoria, Australia) that contains polyanionic groups which are able to attach to molecular targets present on viruses, blocking virus attachment to the cell surface and thereby preventing infection (48).
A particular subclass of dendrimers is represented by peptide dendrimers, consisting of a peptidyl branching core and/or covalently attached surface functional units (44). So far, the use of this class of dendrimers as antivirals has not received much attention compared with carbohydrate- and polyanion-derivatized dendrimers. However, their antiviral activity was recently demonstrated against human cytomegalovirus (42).
In the search for antiviral molecular antagonists of HSPGs, we screened a minilibrary of linear, dimeric, and dendrimeric peptides containing clusters of basic amino acids that could bind to the negatively charged sulfate and carboxyl groups of HS, thus inhibiting initial phases of HPV infections. The peptide dendrimer SB105-A10 emerged as a potent inhibitor of a broad spectrum of genital HPV types and exerts its action by binding to HS exposed on the cell surface.
MATERIALS AND METHODS
Cell culture.
The human cervical carcinoma cell lines SiHa, HeLa, and C33A and epithelial HL3T1 cells derived from HeLa cells were grown as monolayers in Dulbecco's modified Eagle's medium (DMEM) (Gibco-BRL, Gaithersburg, MD) supplemented with heat-inactivated 10% fetal calf serum (FCS; Gibco-BRL) and Glutamax-I (Invitrogen, Carlsbad, CA). The 293TT cell line, derived from human embryonic kidney cells transformed with the simian virus 40 (SV40) large T antigen, was cultured in the medium described above supplemented with nonessential amino acids. 293TT cells allow high levels of protein to be expressed from vectors containing the SV40 origin due to overreplication of the expression plasmid (9). Chinese hamster ovary K1 (CHO-K1) wild-type cells and HSPG-defective CHO-K1 A745 cells (provided by J. D. Esko, University of Birmingham, AL) were grown in Ham's F-12 medium (Gibco, Grand Island, NY) with 10% FCS (23). MA104 monkey kidney cells were cultured in Eagle minimal essential medium (MEM) supplemented with 10% fetal bovine serum (FBS).
Reagents.
Heparin (13.6 kDa) was obtained from Laboratori Derivati Organici Spa (Milan, Italy). Heparinase III, a glycosidase that digests the glycosaminoglycan moiety of HSPGs (22), was from Sigma-Aldrich (St. Louis, MO). Amino acids, NovaPEG Rink amide resin (0.67 g/mol), and preloaded [(Fmoc)2Lys]2-Lys-Wang acidic resin (0.1 mmol/g) were from Novabiochem (Merck Chemicals Ltd., Nottingham, United Kingdom) and ChemImpex (Wood Dale, IL). Peptide synthesis-grade N,N-dimethylformamide (DMF), N-methylpyrrolidone (NMP), trifluoroacetic acid (TFA), dichloromethane, diethyl ether, and O-(benzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium hexafluorophosphate (HBTU) were from ChemImpex (Wood Dale, IL) and Sigma-Aldrich (St. Louis, MO). All other reagents and solvents used for both synthesis and purification of peptides were purchased from Sigma-Aldrich at the highest available purity and were used with no further purification.
Peptide synthesis.
Peptides were synthesized in tetrameric, dimeric, or linear form by a standard manual solid-phase peptide Fmoc (9-fluorenylmethoxy carbonyl) strategy, working under nitrogen flow. Coupling reactions with Fmoc amino acids were activated in situ using HBTU, 1-hydroxybenzotriazole (HOBt), and diisopropylethylamine (DIPEA) (HOBt/DIPEA/HBTU ratio, 1/2/0.9). Amide peptides were synthesized on a NovaPEG Rink amide resin, while acidic multimeric peptides were synthesized on a preloaded [(Fmoc)2Lys]2-Lys-Wang acidic resin. The amidated tetrameric lysine core was synthesized on the Rink amide resin by use of an (Fmoc)2Lys-OH-protected amino acid, and the first amino acid on the core was beta-alanine. A 6-fold excess of Fmoc amino acid was employed during every coupling synthesis step, and all amino acid residues with reactive side chains were protected with the following acid-labile protecting groups: 2,2,4,6,7-pentamethyldihydro-benzofuran-5-sulfonyl for arginine, tert-butyl ether for serine, and tert-butyloxycarbonyl for lysine and tryptophan. The Fmoc group was removed by using 20% piperidine in NMP. The amino acid residues were deprotected during cleavage of the peptide from the solid support upon treatment with a TFA-triisopropylsilane-H2O solution at a ratio of 95:2.5:2.5 for 2 h. After cleavage, the solid support was removed by filtration, and the filtrate was concentrated under reduced pressure. The crude peptides were precipitated from diethyl ether, washed several times with diethyl ether, and dried under reduced pressure.
Reverse-phase high-performance liquid chromatography (RP-HPLC) peptide analyses were performed on a Jupiter Proteo analytical C12 column (4.6 × 250 mm) supplied by Phenomenex (Torrance, CA), using 0.1% TFA-H2O as solvent A and 0.1% TFA-methyl cyanide as solvent B. The column was equilibrated with a solvent A/solvent B ratio of 95 to 5 at a flow rate of 1.0 ml/min for all peptide analyses, with an increasing concentration of solvent B up to 95% over a 14-min period.
SB007-pyrE and SB105 series purification.
Purification of the SB007-pyrE and SB105 series peptides was carried out on a Jupiter Proteo semipreparative C12 column (10 × 250 mm) supplied by Phenomenex (Torrance, CA) under gradient conditions where the concentration of solvent B was raised from 5 to 95% (vol/vol) over 12 min for SB007-pyrE, SB105, SB105-A10, and SB105-dim.
Matrix-assisted laser desorption ionization-time-of-flight mass spectrometry (MALDI-TOF MS) analysis showed the following data. For SB007-pyrE, M + H calculated, 5,126.4. Found, 5,127.6 m/z. For SB105, M + H calculated, 5,195.4. Found, 5,194.9 m/z. For SB105-A10, M + H calculated, 4,682.9. Found, 4,683.7 m/z. For SB105-dim, M + H calculated, 2,577.2. Found, 2,578.4 m/z.
A labeled tetrameric peptide bearing a biotin moiety linked to the lysine core by a 30-atom pegylated spacer, called biotinylated SB105-A10, was purchased from PolyPeptide (Strasbourg, France) and had a purity of >95%.
SB056 series purification.
The purification of SB056 peptides was carried out under gradient conditions where the concentration of solvent B was raised from 5 to 95% (vol/vol) over 14 min for SB056-dim, SB056-dim5, and SB056-dim8.
SB047 and SB068-G purification.
The purification of SB047 and SB068-G peptides was carried out under gradient conditions where the concentration of solvent B was raised from 5 to 95% (vol/vol) over 14 min.
MALDI-TOF MS analysis.
The monoisotopic molecular masses of peptide compounds were determined by MALDI-TOF MS (Bruker, Germany), using sinapinic acid (SA) as the MALDI matrix for branched peptides and alpha-cyano-4-hydroxycinnamic acid (HCCA) as the MALDI matrix for linear peptides. The instruments were calibrated with peptides of known molecular mass in the range of 1,000 to 6,000 Da. SB007-pyrE M + H calculated, 5,126.4. Found, 5,127.6 m/z. SB105 M + H calculated, 5,195.4. Found, 5194.9 m/z. SB105-A10 M + H calculated, 4,682.9. Found, 4,683.7 m/z. SB105-dim M + H calculated, 2,577.2. Found, 2,578.4 m/z. SB056-dim M + H calculated, 2,693.4. Found, 2,693.8 m/z. SB056-dim5 M + H calculated, 2,891.7. Found, 2,892.8 m/z. SB056-dim8 M + H calculated, 2,975.8. Found, 2,977.2 m/z. SB047 M + H calculated, 1,237.6. Found, 1,238.8 m/z. SB068-G M + H calculated, 1,313.6. Found, 1,314.5 m/z.
HPV PsV production.
Plasmids and 293TT cells used for pseudovirus (PsV) production were kindly provided by John Schiller (National Cancer Institute, Bethesda, MD). Detailed protocols and plasmid maps for this study can be seen at http://home.ccr.cancer.gov/lco/default.asp. HPV-16, HPV-18, HPV-6, and bovine papillomavirus type 1 (BPV-1) PsVs were produced according to previously described methods (10). Briefly, 293TT cells were transfected with plasmids expressing the papillomavirus major and minor capsid proteins (L1 and L2, respectively), together with a reporter plasmid expressing the secreted alkaline phosphatase (SEAP) or green fluorescent protein (GFP), named pYSEAP or pfwB, respectively. HPV-16, HPV-6, and BPV-1 PsVs were produced using bicistronic L1/L2 expression plasmids (p16sheLL, p6sheLL, and pSheLL, respectively), and the HPV-18 PsV was produced using peL1fB and peL2bhb plasmids. Capsids were allowed to mature overnight in cell lysate; the clarified supernatant was then loaded on top of a density gradient of 27 to 33 to 39% Optiprep (Sigma-Aldrich, St. Louis, MO) at room temperature for 4 h. The material was centrifuged at 234,000 × g for 3.30 h at 16°C in an SW50.1 rotor (Beckman Coulter, Inc., Fullerton, CA) and then collected by bottom puncture of the tubes. Fractions were inspected for purity in 10% sodium dodecyl sulfate (SDS)-Tris-glycine gels, titrated on 293TT cells to test for infectivity by SEAP or GFP detection, and then pooled and frozen at −80°C until needed. The L1 protein content of PsV stocks was determined by comparison with bovine serum albumin standards in Coomassie-stained SDS-polyacrylamide gels.
SEAP-based PsV neutralization assays.
293TT cells were seeded 3 to 4 h in advance in 96-well tissue culture-treated flat-bottom plates at a density of 30,000 cells/well in 100 μl of DMEM without phenol red (Life Technologies, Inc., Gaithersburg, MD) and with 10% heat-inactivated FBS, 1% glutamate, 1% nonessential amino acids, 1% penicillin-streptomycin-amphotericin B (Fungizone), and 10 mM HEPES (neutralization buffer). To generate dose-response curves, diluted PsV stocks (80 μl/well) were placed on 96-well untreated sterile polystyrene plates (Nalge-Nunc, Roskilde, Denmark), combined with 20 μl of serially diluted peptidic compounds, and placed on ice for 1 h. The 100-μl PsV-compound mixture was transferred to the cell monolayers and incubated for 68 to 72 h at 37°C at a final concentration of PsV equal to approximately 1 ng/ml L1 (about 750 capsid equivalents of L1/cell) (8).
Following incubation, 50-μl aliquots of supernatant were clarified at 1,500 × g for 5 min. The SEAP content in the clarified supernatant was determined using a Great Escape SEAP chemiluminescence kit 2.0 (BD Clontech, Mountain View, CA) as directed by the manufacturer. Thirty minutes after the addition of the substrate, samples were read using a Wallac 1420 Victor luminometer (PerkinElmer Life and Analytical Sciences, Inc., Wellesley, MA).
The 50% inhibitory concentration (IC50) values and the 95% confidence intervals (CIs) were determined using the Prism program (GraphPad Software, San Diego, CA).
Attachment assays.
SB105-A10 was preincubated with HPV-16-SEAP PsV (30 μg/ml and 1 ng/ml L1, respectively) for 1 h at 4°C. The mixture was added to cooled 293TT cells in 96-well plates and incubated for 2 h at 4°C to ensure PsV attachment but not entry. After two gentle washes, cells were shifted to 37°C, and SEAP activity was measured in the cell culture supernatants 72 h after PsV inoculation.
Preattachment assays.
293TT cell monolayers in 96-well plates were incubated with SB105-A10 (30 μg/ml) for 2 h at 4°C. After removal of the compound and a gentle wash, HPV-16-SEAP PsV (1 ng/ml L1) was added to the cells for 2 h at 4°C. After two gentle washes, the cells were shifted to 37°C, and SEAP activity was measured in the cell culture supernatants 72 h after PsV inoculation.
Postattachment assays.
293TT cell monolayers in 96-well plates were incubated with HPV-16-SEAP PsV (1 ng/ml L1) for 2 h at 37°C, followed by two gentle washes to remove unbound virus. A fixed dose of SB105-A10 (30 μg/ml) was then added to cultures after washout of the inoculum.
GFP-based assays.
Cells were seeded in 24-well plates at a density of 1 × 105 cells/well in 400 μl of DMEM supplemented with 10% FBS. The next day, a fixed dose of SB105-A10 (30 μg/ml) was added to preplated cells together with 2 to 5 μl of PsV stock. After 44 to 52 h of incubation at 37°C, flow cytometric analysis was performed by fixing the count to 15,000 live cells and considering only highly GFP-positive cells. PsV doses were calibrated such that between 5% and 25% of cells were scored as GFP positive when no inhibitors were added. Percent inhibition was calculated by using the formula 100 × [1 − (percentage of GFP+ cells in test sample/percentage of GFP+ cells in the mock sample)].
Rotavirus infectivity assay.
Confluent MA104 cell monolayers in a 96-well plate were washed twice with MEM and then infected with 1,200 PFU of human rotavirus strain Wa (ATCC VR-2018) for 1 h at 37°C in the presence or absence of SB105-A10. Virus was preactivated with 5 μg of porcine trypsin (Sigma)/ml for 30 min at 37°C. After the adsorption period, the virus inoculum was removed, the cells were washed with MEM, and the cultures were maintained at 37°C for 16 h in medium with trypsin at 0.5 μg/ml. The infected cells were fixed and immunostained using an UltraTech HRP streptavidin-biotin detection system (Beckman Coulter).
Cell viability assay.
Cells were seeded at a density of 5 × 104/well in 24-well plates; the next day, they were treated with serially diluted peptide compounds to generate dose-response curves. After 48 or 72 h of incubation, cell viability was determined by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide method, as previously described (47). Fifty percent cytotoxic concentrations (CC50s) and 95% CIs were determined using Prism software.
Electron microscopy.
An aliquot of diluted HPV-PsV preparation was placed on a grid and air dried prior to examination. Microscopy was performed using a Philips CM10 transmission electron microscope. Micrographs were taken of random sections at different powers of magnification.
For negative staining, PsV was spotted onto carbon grids and stained with 5% ammonium molybdate (pH 7.0) containing 1% trehalose as previously described (29). Samples were examined under a Zeiss EM900 transmission electron microscope at an instrumental magnification of ×330,000.
SPR assay.
Surface plasmon resonance (SPR) measurements were performed on a BIAcore X instrument (GE Healthcare, Milwaukee, WI), using a research-grade CM3 sensor chip. The reagents 1-ethyl-3-(3-diaminopropyl)-carbodiimide hydrocloride (EDC) and N-hydroxysuccinimide (NHS) were purchased from GE Healthcare and used according to recommended protocols.
For the study of the interaction of linear, dimeric, or dendrimeric peptides with heparin, the latter was immobilized on a BIAcore sensor chip as described previously (49). Briefly, a CM3 sensor chip (GE Healthcare, Milwaukee, WI) previously activated with 50 μl of a mixture containing 0.4 M EDC and 0.1 M NHS was coated with streptavidin. Heparin was biotinylated at its reducing end and immobilized onto the streptavidin-coated sensor chip. These experimental conditions allowed the immobilization of 130 resonance units (RU), equal to 9.5 fmol/mm2 of heparin. A sensor chip coated with streptavidin alone was used to evaluate the nonspecific binding of the peptides to the sensor chip and for blank subtraction, allowing determination of the specific binding of the peptides to heparin. The various linear, dimeric, and dendrimeric peptides, in 10 mM HEPES buffer, pH 7.4, containing 150 mM NaCl, 3 mM EDTA, and 0.005% surfactant P20 (HBS-EP), were injected over the heparin or streptavidin surface for 4 min (to allow their association with immobilized molecules) and then washed until dissociation was observed. After every run, the sensor chip was regenerated by injection of glycine, pH 2.0. The Kd (dissociation constant) at equilibrium was calculated by Scatchard's analysis of the dose-response data. In particular, the Kd values were evaluated by being fitted with the proper form of Scatchard's equation for the plot of the SPR RU at equilibrium (directly proportional to the moles of bound ligand) as a function of the ligand concentration in solution. All fitting was performed by a least-square minimization procedure based on the Levemburg-Marquardt algorithm.
Cell-binding assay for biotinylated SB105-A10 peptide.
Monolayers of HL3T1, CHO-K1, and CHO-K1 A745 cells in 96-well plates were incubated for 2 h at 4°C in phosphate-buffered saline (PBS) containing 0.1 mg/ml CaCl2, 0.1 mg/ml MgCl2, and 0.1% gelatin, with increasing concentrations of biotinylated SB105-A10 peptide. At the end of incubation, cells were washed with PBS, and the amount of cell-associated biotinylated SB105-A10 peptide was determined with horseradish peroxidase-labeled streptavidin (1/5,000) and the chromogenic substrate ABTS (Kierkegaard & Perry Laboratories, Gaithersburg, MD). In some experiments, cell monolayers were washed with PBS containing 2 M NaCl, a treatment known to remove cationic polypeptides from cell surface HSPGs (57). Alternatively, HL3T1 cells were incubated with heparinase III for 2 h at 37°C or were left untreated before the binding assay. The EC50, defined as the concentration of ligand (biotinylated SB105-A10 peptide) that gives half-maximal binding values, was determined graphically from the dose-response binding curves.
RESULTS
Peptide minilibrary.
In a search for HS antagonists endowed with anti-HPV activity, we generated a minilibrary of 10 tetrameric, dimeric, linear, and/or lipidated peptide derivatives containing clusters of basic amino acids that could bind to the negatively charged sulfate and carboxyl groups of HS. The sequences of linear peptide portions anchored on the multimeric lysine core are reported in Fig. 1. The minilibrary was screened by a cell-based HPV PsV neutralization assay. Subsequently, the ability of the three hit compounds to interact with cell surface HSPGs was investigated.
FIG. 1.

Peptide structures. The peptides assayed can be classified as tetrameric, dimeric, or linear, based on their main structures. The tetrameric core consists of Lys2-Lys-beta-Ala.
Characterization of purified HPV-16 PsV.
HPV-16 was chosen as a pivotal model because it is the genotype identified most often in cervical carcinomas (4). In preliminary experiments, the quality of the HPV-16-SEAP PsV preparations was assessed by SDS-PAGE and electron microscopy analysis. As shown in Fig. 2A, a major band migrating at 55 kDa was detected by Coomassie brilliant blue staining (lane 1). The major band was confirmed to be the L1 major capsid protein by Western blot analysis with anti-L1 antibody (lane 2). The faint band that can be seen above L1 in lane 1 is probably the minor capsid protein L2, as reported previously (7, 8). No L1-reactive proteolytic degradation products were observed at molecular masses below 55 kDa. Figure 2B shows an electron micrograph of the same PsV stock. The PsV particles exhibited an average diameter of 50 to 60 nm, similar to that of an authentic HPV capsid, and appeared as individual, well-defined particles with minimal aggregation. When subjected to negative staining and observed at a higher magnification, the particles appeared to be well-assembled, icosahedral capsids (Fig. 2B, inset). Similar results were obtained with the other PsV types used in this study (data not shown).
FIG. 2.

Characterization of purified HPV-16-SEAP PsV. (A) An aliquot of purified PsV preparation was analyzed by SDS-PAGE with Coomassie brilliant blue staining (lane 1) or immunoblotting with an anti-L1 antibody (B0580; Dako Corporation, Carpinteria, CA) (lane 2). The asterisk indicates a faint band that is probably the minor capsid protein L2. (B) Electron micrograph of a purified PV preparation. Bar, 100 nm. The inset shows a negatively stained PsV capsid at a higher magnification.
HPV-neutralizing activity of linear, dimeric, or dendrimeric peptides.
To test the neutralizing activity of synthetic peptides against HPV, we used an HPV-16-SEAP PsV-based assay. The early events of a PsV infection resemble those of a natural HPV infection, since the PsV consists of a reporter plasmid encapsidated by a capsid composed of the two viral capsid proteins (L1 and L2), as in an authentic HPV capsid. After PsV binding to and entry into the cell, the reporter plasmid is transported to the nucleus for expression of the reporter gene (9). To generate dose-response curves, serial dilutions of compounds were preincubated with aliquots of HPV-16-SEAP PsV and then added to 293TT cell cultures. Inhibition of PsV-mediated delivery of the SEAP reporter plasmid was measured at 72 h postinfection by chemiluminescence analysis of the cell supernatants. As shown in Table 1, three compounds, SB007-pyrE, SB105, and SB105-A10, neutralized the HPV-16-SEAP PsV infection, with IC50s of 21.8 μg/ml (4.25 μM), 11.6 μg/ml (2.23 μM), and 2.8 μg/ml (0.59 μM), respectively. In contrast, the other compounds failed to display any significant neutralizing activity. The CC50 was >100 μg/ml for all compounds tested, indicating that the inhibitory activities observed were not due to cytotoxicity. Among the peptides tested, SB105-A10 was the most promising and was subjected to further investigations. To assess whether SB105-A10 was HPV type specific, the neutralization assays were repeated using two other HPV-SEAP PsVs (HPV-18 and HPV-6) and SEAP-BPV-1 PsV. The results shown in Table 2 demonstrate that SB105-A10 was also active on the other PsV types tested.
TABLE 1.
Inhibition of HPV-16-SEAP PsV infection of 293TT cells by synthetic peptides
| Compound | IC50(μg/ml) | 95% CI (μg/ml) | CC50 (μg/ml) |
|---|---|---|---|
| SB007-pyrE | 21.8 | 15.3-26.2 | >100 |
| SB105 | 11.6 | 9.6-13.8 | >100 |
| SB105-A10 | 2.8 | 2.1-3.8 | >100 |
| SB056-dim | >100 | >100 | |
| SB056-dim5 | >100 | >100 | |
| SB056-dim8 | >100 | >100 | |
| SB105-dim | >100 | >100 | |
| SB047 | >100 | >100 | |
| SB068-G | >100 | >100 |
TABLE 2.
Inhibition of HPV-18, HPV-6, and BPV-1 PsV infection of 293TT cells by SB105-A10
| PsV | SB105-A10 IC50 (μg/ml) | 95% CI (μg/ml) |
|---|---|---|
| HPV-18 | 2.8 | 1.6-4.9 |
| HPV-6 | 4.2 | 3.5-5.6 |
| BPV-1 | 3.9 | 3.4-4.5 |
Inhibition of HPV-16 PsV by SB105-A10 in different cell lines.
293TT cells are the preferred indicator cells for neutralization assays because high levels of the SV40 large T antigen in these cells allow for the overreplication of the SEAP reporter plasmid.
However, to verify that the inhibitory effect of SB105-A10 on HPV-16 PsV infection was not restricted to 293TT cells, we tested its activity on keratocarcinoma cell lines derived from the uterine cervix (SiHa, HeLa, and C33A), the major anatomical target for high-risk HPV infection. Unlike 293TT, these cell lines do not express the SV40 large T antigen, resulting in very low levels of SEAP protein expression. Therefore, we employed GFP as a reporter gene because it allows reliable cytometric analyses of cell types in which the reporter plasmid does not overreplicate. As reported in Table 3, the treatment with SB105-A10 (30 μg/ml) strongly neutralized the infectivity of HPV-16-GFP PsV in SiHa, HeLa, and C33A cells (inhibition equal to 89%, 86%, and 94%, respectively). Cell viability assays demonstrated that the cell lines treated with SB105-A10 revealed no evidence of cytotoxicity (CC50, >100 μg/ml).
TABLE 3.
Inhibition of HPV-16-GFP PsV infection of cervical cancer-derived cell lines by SB105-A10
| Cell line | % Inhibition by SB105-A10a | CC50 (μg/ml) |
|---|---|---|
| 293TTb | 97 | >100 |
| SiHa | 89 | >100 |
| HeLa | 86 | >100 |
| C33A | 94 | >100 |
A fixed dose of 30 mg/ml was used.
293TT is not a cervical cancer-derived cell line but is shown here as a control.
Investigation of the inhibitory mechanism of SB105-A10.
To determine at which step SB105-A10 interferes with the infection process of PsV, three types of binding assays were performed, employing HPV-16-SEAP PsV and 293TT cells.
In the “attachment assay,” SB105-A10 (30 μg/ml) was preincubated with PsV at 4°C, and then the mixture was added to cooled cells for a further 2 h of incubation at 4°C. When performed in the absence of infection inhibitors, this procedure ensures PsV attachment, but not entry, until the cells are washed twice to remove unbound PsV and the infection is then allowed to proceed at 37°C. Under these experimental conditions, SB105-A10 strongly inhibited PsV attachment to the cell surface (Fig. 3).
FIG. 3.

Investigation of inhibitory mechanism used by SB105-A10 on HPV-16-SEAP PsV infectivity on 293TT cells. Three different assays were performed: attachment assay, preattachment assay, and postattachment assay. The experimental schemes are illustrated in the corresponding panels. The results of the three assays, shown in the lower panel, are given as means ± standard deviations for triplicates.
In the “preattachment assay,” cell monolayers were incubated with SB105-A10 (30 μg/ml) for 2 h at 4°C to allow its interaction with the cell surface. After removal of unbound compound, cells were further incubated with PsV for 2 h at 4°C, washed, and shifted to 37°C for 70 h. Under these experimental conditions, SB105-A10 retained its capacity to inhibit PsV infection (Fig. 3), demonstrating that the activity of the compound depends on its interaction with the cell surface.
Finally, a “postattachment assay” was performed to evaluate whether SB105-A10 could also neutralize the infectivity of cell-bound virus. For this aim, cell monolayers were incubated with PsV for 2 h at 37°C and washed to remove unbound virus. Subsequently, SB105-A10 (30 μg/ml) was added and the cells were incubated for a further 70 h at 37°C. As shown in Fig. 3, most of the cell-bound PsV remained susceptible to inhibition by SB105-A10, demonstrating that in addition to preventing PsV attachment to cells, SB105-A10 was also able to detach PsV already bound to the cell surface.
Interaction of peptides with heparin.
Due to the presence of a stretch of basic amino acids in the sequences of the active compounds, we hypothesized that their antiviral activity could be a consequence of their binding to cell surface HSPGs.
To investigate this possibility, in a first series of experiments we evaluated the capacity of tetrameric SB105-A10, SB007-pyrE, and SB105 to bind heparin immobilized on a BIAcore sensor chip, a “cell-free” model that resembles Tat interaction with cell surface HSPGs (49). SB056-dim5 and SB047 were also included in the assay to represent inactive dimeric and linear peptides, respectively. As shown in Fig. 4A, when injected at a concentration of 300 nM, tetrameric peptides SB105-A10, SB105, and SB007-pyrE interacted with surface-immobilized heparin (approximately 40 to 70 RU bound at equilibrium), while peptides SB056-dim5 and SB047 were endowed with a very limited capacity to bind to heparin (<10 RU). In a second set of assays, we injected increasing concentrations of the peptides (from 7.5 to 300 nM) onto the heparin surface, and data from the equilibrium binding assays were used to obtain saturation curves (Fig. 4B, C, and D) and to calculate Kd values (Table 4). As expected from their very limited binding capacity (Fig. 4A), no significant Kd was calculated for peptides SB056-dim5 and SB047. In contrast, single-component binding was revealed for peptides SB007-pyrE and SB105, which interacted with heparin with similar affinities (Table 4). When assayed in the same range of concentrations, peptide SB105-A10 instead revealed a two-component binding curve, with relatively high-affinity binding occurring at low concentrations of peptide and low-affinity binding occurring at high concentrations of peptide (Fig. 4 and Table 4).
FIG. 4.
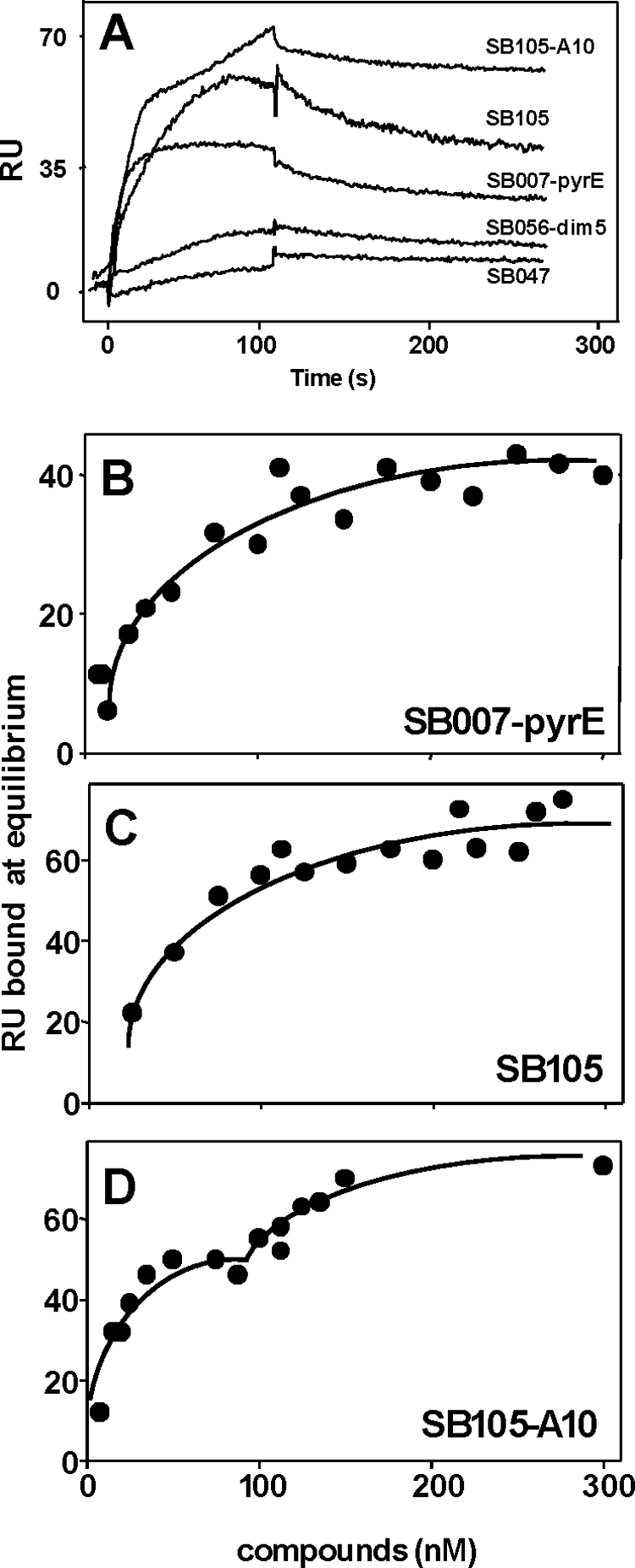
SPR analysis of HPV peptide-heparin interaction. (A) Blank-subtracted sensorgrams showing specific binding of indicated HPV peptides (assayed at 300 nM) to sensor chip-immobilized heparin. (B, C, and D) Saturation curves for the indicated HPV peptides binding to immobilized heparin. The saturation curves were obtained using the RU values at equilibrium after injection of increasing concentrations of various compounds onto the heparin surface.
TABLE 4.
Binding parameters of interactions of selected peptides with heparin by real-time SPR
| Peptide | Kd (M)a | Correlation coefficient of linear regression |
|---|---|---|
| SB007-pyrE | 46.7 × 10−9 | 0.87 |
| SB105 | 57.6 × 10−9 | 0.89 |
| SB105-A10b | 15.6 × 10−9 | 0.86 |
| 87.6 × 10−9 | 0.81 | |
| SB056-dim5 | Not determinable | Not determinable |
| SB047 | Not determinable | Not determinable |
The Kd at equilibrium was calculated by Scatchard plot analysis of the data from the saturation curves reported in Fig. 4B, C, and D.
The Kd values for the two-component binding curve of SB105-A10 are reported.
Interaction of SB105-A10 with cell surface HSPGs.
Since SB105-A10 was endowed with the strongest neutralizing activity among the heparin-binding compounds, it was subjected to further investigations. First, we evaluated the capacity of SB105-A10 to bind HSPGs expressed at the surfaces of epithelial cells by exploiting the CHO cell model. Briefly, biotinylated SB105-A10 was evaluated for the capacity to bind to the surfaces of wild-type CHO-K1 cells (which express normal levels of cell surface HSPGs) or the CHO-K1 A745 clone, which is genetically deficient in the synthesis of proteoglycans (23). As shown in Fig. 5A, biotinylated SB105-A10 bound to CHO-K1 cells in a dose-dependent manner, with an EC50 equal to 85 nM. Also, binding saturation was reached at a high concentration of the peptide (600 nM). When tested at the same doses on the CHO-K1 A745 clone, biotinylated SB105-A10 showed a significantly decreased binding capacity, indicating that HSPGs play a major role in its interaction with the cell surface. However, the presence of alternative (still uncharacterized) receptors for the peptide can be hypothesized based on the residual HSPG-independent binding that could be observed after a wash with 2 M NaCl or in HSPG-deficient CHO-K1 A745 cells. In parallel experiments, after incubation with biotinylated SB105-A10, the two cell clones were washed with 2 M NaCl, a treatment known to remove cationic polypeptides from HSPGs of the cell surface (57). As shown in Fig. 5A, a 2 M NaCl wash reduced the binding of biotinylated SB105-A10 to wild-type CHO-K1 cells, to levels comparable to those measured for the CHO-K1 A745 clone, confirming that in the former cell type, cell surface binding of SB105-A10 depends mainly on HSPGs.
FIG. 5.

Binding of biotinylated SB105-A10 to HSPGs expressed at the cell surface. (A) Monolayers of wild-type CHO-K1 or HSPG-deficient CHO-K1 A745 cells were incubated with increasing concentrations of biotinylated SB105-A10. Alternatively, they were incubated with the peptide at 5 μg/ml and then washed with PBS alone or containing 2 M NaCl. (B) Monolayers of epithelial HL3T1 cells were incubated with increasing concentrations of biotinylated SB105-A10-polyethylene glycol. In parallel experiments, they were (i) pretreated with heparinase III (15 mU/ml) and incubated with biotinylated SB105-A10 (5 μg/ml); and (ii) incubated with biotinylated SB105-A10 (5 μg/ml) in the presence of heparin (10 μg/ml), incubated with biotinylated SB105-A10 (5 μg/ml) alone, and then washed with PBS containing 2 M NaCl. At the end of incubation, the amount of biotinylated SB105-A10 bound to cells was measured. Each point is the mean ± standard error of the mean for 3 or 4 determinations performed in duplicate.
In a second set of experiments, we evaluated the binding of SB105-A10 to the surfaces of HL3T1 epithelial cells, derived from a carcinoma of the uterine cervix, a major biological target of HPV. As shown in Fig. 5B, biotinylated SB105-A10 bound to the HL3T1 cell surface in a dose-dependent way, with an EC50 equal to 53 nM, and binding saturation was reached at approximately 213 nM. Also, this interaction was partially (i) disrupted by a 2 M NaCl wash (see above), (ii) inhibited by heparin (used as an HSPG antagonist [54]), and (iii) prevented by the pretreatment of HL3T1 cells with heparinase III, a treatment that was recently demonstrated to inhibit HPV attachment in vivo (34).
The EC50 and saturation binding values obtained for the SB105-A10 peptide with both wild-type CHO and HL3T1 cells configure a low-affinity, high-capacity binding pattern, consistent with previously reported interactions of viral peptides (11) and proteins (12, 49, 47) with cell surface HSPGs. Taken together, these data demonstrate that the binding of SB105-A10 to the surfaces of two different epithelial cell types occurs mainly via HSPGs. It must be pointed out, however, that a marginal HSPG-independent binding (20 to 30% of the total) is present in both cell lines, suggesting that SB105-A10 may also bind to other receptors of the cell surface (discussed below).
SB105-A10 does not inhibit infection by human rotavirus.
Rotavirus depends on several integrins and a heat shock protein, but not on HS, for cell attachment and entry (40). Therefore, it is a useful control for investigating the specificity of the SB105-A10 mechanism of action. For this aim, we infected MA104 cells with human rotavirus strain Wa in the presence of increasing concentrations of SB105-A10. The infectivity assay showed that the peptide did not inhibit infection at any dose tested (Fig. 6). The CC50 of SB105-A10 on MA104 cells was >100 μg/ml. These data support the specificity of the SB105-A10 mechanism of action.
FIG. 6.

SB105-A10 does not inhibit the infectivity of a human rotavirus. MA104 cells grown in 96-well plates were infected for 1 h at 37°C with a human rotavirus (strain Wa) in the presence of increasing concentrations of SB105-A10. Results shown are representative of three experiments (means + standard deviations).
DISCUSSION
A wide distribution on the surfaces of eukaryotic cells and a strong interactive capacity are two features that have made HSPGs attractive adhesion molecules for all sorts of viruses during their evolution (56). Indeed, many viruses, including dengue virus (15), Japanese encephalitis virus (38), West Nile virus (38), hepatitis C virus (2), herpes simplex virus (54), foot-and-mouth disease virus (33), Sindbis virus (13), coronavirus (20), human respiratory syncytial virus (24), and HIV (46), attach to cells via cell surface HSPGs. Mucosotropic HPV also binds to cellular HSPGs, and this interaction appears to be mediated by basic domains found in the L1 and L2 viral capsid proteins (6, 35). Thus, the interaction between the basic amino acid residues of viral proteins and the negatively charged sulfated/carboxyl groups of the HS chains represents a major target for antiviral microbicides. Polyanionic heparin- or suramin-like compounds have received much attention as candidate microbicides for the prevention of viral STIs. By mimicking cellular HS, these compounds bind to virus capsids and interfere with virus attachment and entry into a target cell. Indeed, a large number of preclinical studies have shown that several polyanionic compounds can inhibit HIV, HSV, and HPV infection (50). However, three promising polyanionic compounds recently failed to pass phase III clinical trials designed to establish their efficacy in preventing HIV acquisition, making this class of compounds less attractive for further development (14, 31). An alternative strategy to prevent virus binding to cellular HSPGs is the use of compounds that interact with HS chains, thus competitively inhibiting virus attachment.
To pursue this strategy, we synthesized a minilibrary of linear, dimeric, and dendrimeric peptides containing basic amino acid residues and screened them for anti-HPV properties by exploiting a neutralization assay based on HPV PsV. The dendrimeric peptide SB105-A10 was found to be a potent, broad-spectrum inhibitor of HPV attachment that interacts with cellular HSPGs. The fact that the most efficient inhibitory compound is dendrimeric is not surprising. Cell surface HSPGs are large, complex macromolecules characterized by the presence of numerous adjacent binding sites that favor polyvalent interactions for initiating various biological processes. The multimeric structure of SB105-A10 may enable polyvalent interactions that efficiently mask the binding sites of HSPGs to pathological ligands.
The results presented in this paper indicate that binding of SB105-A10 to the cell surface, as well as its antiviral activity, is mediated mainly by HSPGs. (i) No significant binding to heparin was observed for those compounds (SB056-dim5 and SB047) which were devoid of any anti-HPV activity. In contrast, the compounds endowed with anti-HPV activity, namely, SB105-A10, SB007-pyrE, SB105, and SB105-A10, were also those endowed with the capacity to bind to heparin with similar affinities. (ii) SB105-A10 bound the surfaces of two different epithelial cell lines, with EC50s (53 to 85 nM) (Fig. 5) comparable to the Kd of its binding to heparin (15.6 to 87.6 nM) (Table 4). Also, these values are comparable to those already reported for the binding of viral proteins/peptides to heparin or HSPGs (11, 12, 49). (iii) SB105-A10 retained its capacity to inhibit HPV infection in the preattachment assay, demonstrating that its anti-HPV-16 potential is dependent on its interaction with the cell surface. (iv) SB105-A10 did not prevent infection by the human rotavirus Wa, a virus that does not exploit cellular HSPGs for attachment. (v) Genetic (CHO cell model), pharmacological (heparin and 2 M NaCl wash), and enzymatic (heparinase III) evidence directly demonstrated that the binding of SB105-A10 to different epithelial cell types occurs mainly via HSPGs. Although we have not demonstrated directly that binding of SB105-A10 to HSPGs is required for its antiviral activity, our findings support this mechanism of action. Relevant to the latter point, the binding of SB105-A10 (but not of SB007-pyrE or SB105) to heparin is biphasic, with the first binding phase occurring at low concentrations and with a relatively high affinity. This may explain, at least in part, the higher HPV inhibitory potency displayed by SB105-A10 than those of SB007-pyrE and SB105.
It must be pointed out that in both CHO and HL3T1 cells, a residual HSPG-independent binding of SB105-A10 was observed. Interestingly, it was recently shown that HPV binding per se does not specifically requires HSPGs (19, 36). Taken together, these data suggest that in addition to HSPGs, SB105-A10 can bind other receptors, pointing to an alternative mechanism of inhibition of HPV infection by the peptide.
Several competitive advantages recommend SB105-A10 for further development as an active ingredient of topical microbicides. First, its activity is not papillomavirus type restricted, since it potently inhibited infection by three common sexually transmitted HPV types causing cervical cancer and genital warts (HPV-16, HPV-18, and HPV-6). Moreover, the finding that SB105-A10 is active even against BPV-1, which is phylogenetically distant from genital HPV (21), further supports its broad-spectrum activity. Since the novel prophylactic vaccines are HPV type restricted, a microbicide against all genital HPV types could be a useful adjuvant to vaccination programs.
In addition, SB105-A10 retained its HPV-antagonist activity even when it was added to cell cultures 2 h after PsV exposure, thus exerting a postattachment inactivation similar to that previously observed with heparin and neutralizing monoclonal antibodies (17, 25). This is a valuable property for the development of a topical microbicide that can prevent HPV infection during or immediately after sexual intercourse.
Importantly, besides its anti-HPV activity, SB105-A10 also proved active against HIV-1 infection in vitro (A. Giuliani, S. Landolfo, D. Lembo, D. Gibellini, G. Pirri, L, Pizzuto, and G. Gribaudo, Italian patent application MI2009A001425). This fulfils the criteria for the development of a broad-spectrum microbicide that is effective in preventing two major sexually transmitted infections that raise the risk of cancer in seropositive individuals (1), a risk that will probably become an increasingly important complication of long-term HIV infection (27).
The therapeutic exploitation of synthetic peptides is greatly limited by the fact that their binding to natural targets occurs with affinities (Kd in the mM to μM range) that are very low compared to those of parent proteins (Kd in the pM to nM range) (26). This limit has been overcome successfully by conjugating multiple copies of peptides to carriers (26) or by synthesizing dendrimers (51), two procedures that increase the binding affinities of peptides for their natural ligands (26) almost 1,000-fold. Accordingly, the Kd value calculated for the dendrimeric peptide SB105-A10 is close to that calculated for the interaction of heparin with intact heparin-binding proteins (49).
In conclusion, our results identify SB105-A10 as a lead compound to be developed further as an active ingredient of a topical microbicide against HPV and other viral STIs. Moreover, its particular mechanism of action warrants further investigations in other research areas (e.g., respiratory syncytial virus inhibitors) where there is an unmet medical need.
Acknowledgments
This work was supported by grants from MIUR (PRIN 2008) to D.L. and from the CARIPLO Foundation, 60%, and ISS (AIDS Project) to M.R.
We are grateful to John Schiller and to Susana Pang (NCI, Bethesda, MD) for providing plasmids, cells, and technical advice for PsV production.
Footnotes
Published ahead of print on 19 July 2010.
REFERENCES
- 1.Abramowitz, L., D. Benabderrahmane, P. Ravaud, F. Walker, C. Rioux, C. Jestin, E. Bouvet, J. C. Soule, C. Leport, and X. Duval. 2007. Anal squamous intraepithelial lesions and condyloma in HIV-infected heterosexual men, homosexual men and women: prevalence and associated factors. AIDS 21:1457-1465. [DOI] [PubMed] [Google Scholar]
- 2.Barth, H., E. K. Schnober, F. Zhang, R. J. Linhardt, E. Depla, B. Boson, F. L. Cosset, A. H. Patel, H. E. Blum, and T. F. Baumert. 2006. Viral and cellular determinants of the hepatitis C virus envelope-heparan sulfate interaction. J. Virol. 80:10579-10590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Boas, U., and P. M. H. Heegaard. 2004. Dendrimers in drug research. Chem. Soc. Rev. 33:43-63. [DOI] [PubMed] [Google Scholar]
- 4.Bosch, F. X., and S. de Sanjose. 2003. Human papillomavirus and cervical cancer—burden and assessment of causality. J. Natl. Cancer Inst. Monogr. 31:3-13. [DOI] [PubMed] [Google Scholar]
- 5.Bourne, N., L. R. Stanberry, E. R. Kern, G. Holan, B. Matthews, and D. L. Bernstein. 2000. Dendrimers, a new class of candidate topical microbicides with activity against herpes simplex virus infection. Antimicrob. Agents Chemother. 44:2471-2474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bousarghin, L., A. Touze, L. Combita-Rojas, and P. Coursaget. 2003. Positively charged sequences of human papillomavirus type 16 capsid proteins are sufficient to mediate gene transfer into target cells via the heparan sulfate receptor. J. Gen. Virol. 84:157-164. [DOI] [PubMed] [Google Scholar]
- 7.Buck, C. B., N. Cheng, C. D. Thompson, D. R. Lowy, A. C. Steven, J. T. Schiller, and B. L. Trus. 2008. Arrangement of L2 within the papillomavirus capsid. J. Virol. 82:5190-5197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Buck, C. B., C. D. Thompson, J. N. Roberts, M. Muller, D. R. Lowy, and J. T. Schiller. 2006. Carrageenan is a potent inhibitor of papillomavirus infection. PLoS Pathog. 2:e69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Buck, C. B., D. V. Pastrana, D. R. Lowy, and J. T. Schiller. 2004. Efficient intracellular assembly of papillomaviral vectors. J. Virol. 78:751-757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Buck, C. B., D. V. Pastrana, D. R. Lowy, and J. T. Schiller. 2005. Generation of HPV pseudovirions using transfection and their use in neutralization assays. Methods Mol. Med. 119:445-462. [DOI] [PubMed] [Google Scholar]
- 11.Bugatti, A., P. Chiodelli, J. Rosenbluh, A. Loyter, and M. Rusnati. 2010. BSA conjugates bearing multiple copies of the basic domain of HIV-1 Tat: prototype for the development of multitarget inhibitors of extracellular Tat. Antiviral Res. 87:30-39. [DOI] [PubMed] [Google Scholar]
- 12.Bugatti, A., C. Urbinati, C. Ravelli, E. De Clercq, S. Liekens, and M. Rusnati. 2007. Heparin-mimicking sulfonic acid polymers as multitarget inhibitors of human immunodeficiency virus type 1 Tat and gp120 proteins. Antimicrob. Agents Chemother. 51:2337-2345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Byrnes, A. P., and D. E. Griffin. 1998. Binding of Sindbis virus to cell surface heparan sulfate. J. Virol. 72:7349-7356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Check, E. 2007. Scientists rethink approach to HIV gels. Nature 446:12. [DOI] [PubMed] [Google Scholar]
- 15.Chen, Y., T. Maguire, R. E. Hileman, J. R. Fromm, J. D. Esko, R. J. Linhardt, and R. M. Marks. 1997. Dengue virus infectivity depends on envelope protein binding to target cell heparan sulfate. Nat. Med. 3:866-871. [DOI] [PubMed] [Google Scholar]
- 16.Christensen, N. D., C. A. Reed, T. D. Culp, P. L. Hermonat, M. K. Howett, R. A. Anderson, and L. J. Zaneveld. 2001. Papillomavirus microbicidal activities of high-molecular-weight cellulose sulfate, dextran sulfate, and polystyrene sulfonate. Antimicrob. Agents Chemother. 45:3427-3432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Christensen, N. D., N. M. Cladel, and C. A. Reed. 1995. Postattachment neutralization of papillomaviruses by monoclonal and polyclonal antibodies. Virology 207:136-142. [DOI] [PubMed] [Google Scholar]
- 18.Cloniger, M. J. 2002. Biological applications of dendrimers. Curr. Opin. Chem. Biol. 6:742-748. [DOI] [PubMed] [Google Scholar]
- 19.Day, M. D., R. Gambhira, R. B. S. Roden, D. R. Lowy, and J. T. Schiller. 2008. Mechanisms of human papillomavirus type 16 neutralization by L2 cross-neutralizing and L1 type-specific antibodies. J. Virol. 82:4638-4646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.de Haan, C. A., Z. Li, E. te Lintelo, B. J. Bosch, B. J. Haijema, and P. J. Rottier. 2005. Murine coronavirus with an extended host range uses heparan sulfate as an entry receptor. J. Virol. 79:14451-14456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.de Villiers, E. M., C. Fauquet, T. R. Broker, H. U. Bernard, and H. zur Hausen. 2004. Classification of papillomaviruses. Virology 324:17-27. [DOI] [PubMed] [Google Scholar]
- 22.Ernst, S., R. Langer, C. L. Cooney, and R. Sasisekharan. 1995. Enzymatic degradation of glycosaminoglycans. Crit. Rev. Biochem. Mol. Biol. 30:387-444. [DOI] [PubMed] [Google Scholar]
- 23.Esko, J. D. 1991. Genetic analysis of proteoglycan structure, function and metabolism. Curr. Opin. Cell Biol. 3:805-816. [DOI] [PubMed] [Google Scholar]
- 24.Feldman, S. A., S. Audet, and J. A. Beeler. 2000. The fusion glycoprotein of human respiratory syncytial virus facilitates virus attachment and infectivity via an interaction with cellular heparan sulfate. J. Virol. 74:6442-6447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Giroglou, T., L. Florin, F. Schafer, R. E. Streeck, and M. Sapp. 2001. Human papillomavirus infection requires cell surface heparan sulfate. J. Virol. 75:1565-1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Goldfarb, D. S., J. Gariepy, G. Schoolnik, and R. D. Kornberg. 1986. Synthetic peptides as nuclear localization signals. Nature 322:641-644. [DOI] [PubMed] [Google Scholar]
- 27.Grulich, A. E., M. T. van Leeuwen, M. O. Falster, and C. M. Vajdic. 2007. Incidence of cancers in people with HIV/AIDS compared with immunosuppressed transplant recipients: a meta-analysis. Lancet 370:59-67. [DOI] [PubMed] [Google Scholar]
- 28.Harper, D. M., E. L. Franco, C. Wheeler, D. G. Ferris, D. Jenkins, A. Schuind, T. Zahaf, B. Innis, P. Naud, N. S. De Carvalho, C. M. Roteli-Martins, J. Teixeira, M. M. Blatter, A. P. Korn, W. Quint, G. Dubin, and GlaxoSmithKline HPV Vaccine Study Group. 2004. Efficacy of a bivalent L1 virus-like particle vaccine in prevention of infection with human papillomavirus types 16 and 18 in young women: a randomized controlled trial. Lancet 364:1757-1765. [DOI] [PubMed] [Google Scholar]
- 29.Harris, J. R., W. Gebauer, and J. Markl. 1995. Keyhole limpet haemocyanin: negative staining in the presence of trehalose. Micron 26:25-33. [Google Scholar]
- 30.Holmes, K. K., R. Levine, and M. Weaver. 2004. Effectiveness of condoms in preventing sexually transmitted infections. Bull. World Health Organ. 82:454-461. [PMC free article] [PubMed] [Google Scholar]
- 31.Honey, K. 2007. Microbicide trial screeches to a halt. J. Clin. Invest. 117:1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Howley, P. M., and D. R. Lowy. 2001. Papillomaviruses and their replication, p. 2197-2229. In B. N. Fields et al. (ed.), Fields virology. Lippincott-Raven, Philadelphia, PA.
- 33.Jackson, T., F. M. Ellard, R. A. Ghazaleh, S. M. Brookes, W. E. Blakemore, A. H. Corteyn, D. I. Staurt, J. W. Newman, and A. M. King. 1996. Efficient infection of cells in culture by type O foot-and-mouth disease virus requires binding to cell surface heparan sulfate. J. Virol. 70:5282-5287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Johnson, K. M., R. C. Kines, J. N. Roberts, D. R. Lowy, J. T. Schiller, and P. M. Day. 2009. Role of heparan sulfate in attachment to and infection of the murine female genital tract by human papillomavirus. J. Virol. 83:2067-2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Joyce, J. G., J. S. Tung, C. T. Przysiecki, J. C. Cook, E. D. Lehman, J. A. Sands, K. U. Jansen, and P. M. Keller. 1999. The L1 major capsid protein of human papillomavirus type 11 recombinant virus-like particles interacts with heparin and cell-surface glycosaminoglycans on human keratinocytes. J. Biol. Chem. 274:5810-5822. [DOI] [PubMed] [Google Scholar]
- 36.Kines, R. C., C. D. Thompson, D. R. Lowy, J. T. Schiller, and P. M. Day. 2009. The initial steps leading to papillomavirus infection occur on the basement membrane prior to cell surface binding. Proc. Natl. Acad. Sci. U. S. A. 106:20458-20463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lacey, C. J., C. M. Lowndes, and K. V. Shah. 2006. Burden and management of non-cancerous HPV-related conditions: HPV-6/11 disease. Vaccine 24(Suppl. 3):S35-S41. [DOI] [PubMed] [Google Scholar]
- 38.Lee, E., R. A. Hall, and M. Lobigs. 2004. Common E protein determinants for attenuation of glycosaminoglycan-binding variants of Japanese encephalitis and West Nile viruses. J. Virol. 78:8271-8280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lembo, D., M. Donalisio, M. Rusnati, A. Bugatti, M. Cornaglia, P. Cappello, M. Giovarelli, P. Oreste, and S. Landolfo. 2008. Sulfated K5 Escherichia coli polysaccharide derivatives as wide-range inhibitors of genital types of human papillomavirus. Antimicrob. Agents Chemother. 52:1374-1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.López, S., and C. F. Arias. 2004. Multistep entry of rotavirus into cells: a Versaillesque dance. Trends Microbiol. 12:271-278. [DOI] [PubMed] [Google Scholar]
- 41.Lowy, D. R., and P. M. Howley. 2001. Papillomaviruses, p. 2231-2264. In B. N. Fields et al. (ed.), Fields virology. Lippincott-Raven, Philadelphia, PA.
- 42.Luganini, A., A. Giuliani, G. Pirri, L. Pizzuto, S. Landolfo, and G. Gribaudo. 2010. Peptide-derivatized dendrimers inhibit human cytomegalovirus infection by blocking virus binding to cell surface heparan sulfate. Antiviral Res. 85:532-540. [DOI] [PubMed] [Google Scholar]
- 43.Manhart, L. E., and L. A. Koutsky. 2002. Do condoms prevent genital HPV infection, external genital warts, or cervical neoplasia? A meta-analysis. Sex. Transm. Dis. 29:725-735. [DOI] [PubMed] [Google Scholar]
- 44.Niederhafner, P., S. Jaroslav, and J. Jan. 2005. Peptide dendrimers. J. Pept. 12:757-788. [DOI] [PubMed] [Google Scholar]
- 45.Nikolic, D. S., and V. Piguet. 2010. Vaccines and microbicides preventing HIV-1, HSV-2, and HPV mucosal transmission. J. Invest. Dermatol. 130:352-361. [DOI] [PubMed] [Google Scholar]
- 46.Patel, M., M. Yanagishita, G. Roderiquez, D. C. Bou-Habib, T. Oravecz, V. C. Hascall, and M. A. Norcross. 1993. Cell-surface heparan sulfate proteoglycan mediates HIV-1 infection of T-cell lines. AIDS Res. Hum. Retroviruses 9:167-174. [DOI] [PubMed] [Google Scholar]
- 47.Pauwels, R., J. Balzarini, M. Baba, R. Snoeck, D. Schols, P. Hederwijin, J. Desmyter, and E. De Clerq. 1988. Rapid and automated tetrazolium-based colorimetric assay for the detection of anti HIV compounds. J. Virol. Methods 20:309-321. [DOI] [PubMed] [Google Scholar]
- 48.Rupp, R., S. L. Rosenthal, and L. R. Stanberry. 2007. VivaGel™ (SPL7013 gel): a candidate dendrimer-microbicide for the prevention of HIV and HSV infection. Int. J. Nanomed. 4:561-566. [PMC free article] [PubMed] [Google Scholar]
- 49.Rusnati, M., C. Urbinati, A. Caputo, L. Possati, H. Lortat-Jacob, M. Giacca, D. Ribatti, and M. Presta. 2001. Pentosan polysulfate as an inhibitor of extracellular HIV-1 Tat. J. Biol. Chem. 276:22420-22425. [DOI] [PubMed] [Google Scholar]
- 50.Rusnati, M., E. Vicenzi, M. Donalisio, P. Oreste, S. Landolfo, and D. Lembo. 2009. Sulfated K5 Escherichia coli polysaccharide derivatives: a novel class of candidate antiviral microbicides. Pharmacol. Ther. 123:310-322. [DOI] [PubMed] [Google Scholar]
- 51.Sadler, K., and J. P. Tam. 2002. Peptide dendrimers: applications and synthesis. J. Biotechnol. 90:195-229. [DOI] [PubMed] [Google Scholar]
- 52.Schiller, J. T., X. Castellsagué, L. L. Villa, and A. Hildesheim. 2008. An update of prophylactic human papillomavirus L1 virus-like particle vaccine clinical trial results. Vaccine 26(Suppl. 10):K53-K61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shafti-Keramat, S., A. Handisurya, E. Kriehuber, G. Meneguzzi, K. Slupetzky, and R. Kirnbauer. 2003. Different heparan sulfate proteoglycans serve as cellular receptors for human papillomaviruses. J. Virol. 77:13125-13135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shukla, D., and P. G. Spear. 2001. Herpesviruses and heparan sulfate: an intimate relationship in aid of viral entry. J. Clin. Invest. 108:503-510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Skoler-Karpoff, S., G. Ramjee, K. Ahmed, L. Altini, M. G. Plagianos, B. Friedland, S. Govender, A. De Kock, N. Cassim, T. Palanee, G. Dozier, R. Maguire, and P. Lahteenmaki. 2008. Efficacy of Carraguard for prevention of HIV infection in women in South Africa: a randomised, double-blind, placebo-controlled trial. Lancet 372:1977-1987. [DOI] [PubMed] [Google Scholar]
- 56.Spillmann, D. 2001. Heparan sulfate: anchor for viral intruders? Biochimie 83:811-817. [DOI] [PubMed] [Google Scholar]
- 57.Tyagi, M., M. Rusnati, M. Presta, and M. Giacca. 2001. Internalization of HIV-1 Tat requires cell surface heparan sulfate proteoglycans. J. Biol. Chem. 276:3254-3261. [DOI] [PubMed] [Google Scholar]
- 58.Villa, L. L., R. L. Costa, C. A. Petta, R. P. Andrade, K. A. Ault, A. R. Giuliano, C. M. Wheeler, L. A. Koutsky, C. Malm, M. Lehtinen, F. E. Skjeldestad, S. E. Olsson, M. Steinwall, D. R. Brown, R. J. Kurman, B. M. Ronnett, M. H. Stoler, A. Ferenczy, D. M. Harper, G. M. Tamms, J. Yu, L. Lupinacci, R. Railkar, F. J. Taddeo, K. U. Jansen, M. T. Esser, H. L. Sings, A. J. Saah, and E. Barr. 2005. Prophylactic quadrivalent human papillomavirus (types 6, 11, 16, and 18) L1 virus-like particle vaccine in young women: a randomized double-blind placebo-controlled multicentre phase II efficacy trial. Lancet Oncol. 6:271-278. [DOI] [PubMed] [Google Scholar]