

Abstract
Riboswitches are elements of mRNA that regulate gene expression by undergoing structural changes upon binding of small ligands. Although the structures of several riboswitches have been solved with their ligands bound, the ligand-free states of only a few riboswitches have been characterized. The ligand-free state is as important for the functionality of the riboswitch as the ligand-bound form, but the ligand-free state is often a partially folded structure of the RNA with conformational heterogeneity that makes it particularly challenging to study. Here, we present models of the ligand-free state of a thiamine pyrophosphate (TPP) riboswitch that are derived from a combination of complementary experimental and computational modeling approaches. We obtain a global picture of the molecule using small angle X-ray scattering (SAXS) data and use an RNA structure modeling software, MC-SYM, to fit local structural details to this data on an atomic scale. We have used two different approaches to obtain these models. Our first approach develops a model of the RNA from the structures of its constituent junction fragments in isolation. The second approach treats the RNA as a single entity, without bias from the structure of its individual constituents. We find that both approaches give similar models for the ligand-free form, but the ligand-bound models differ for the two approaches, and only the models from the second approach agree with the ligand-bound structure known previously from X-ray crystallography. Our models provide a picture of the conformational changes that may occur in the riboswitch upon binding its ligand. Our results also demonstrate the power of combining experimental SAXS data with theoretical structure prediction tools in the determination of RNA structures beyond riboswitches.
INTRODUCTION
Since the first discovery of catalytic RNA in the early 1980s, RNA research has exploded to reveal a myriad of non-protein coding RNAs that perform functions such as catalysis, recognition, and regulation previously considered exclusive to proteins. The functional versatility demonstrated by RNA is often coupled to their ability to form diverse three-dimensional structures without which many RNAs would not attain their specific biological activities 1. This knowledge lends itself to questions about how a functional RNA chooses a particular structure, what drives the folding of an RNA sequence to that native structure, and what alternative conformations, active or inactive, are accessible to the RNA.
Riboswitches are functional RNA molecules that provide a protein-free means of regulating gene expression 2; 3; 4; 5; 6. Riboswitches are elements of mRNA that adopt alternative conformations upon binding specific small metabolites, thereby turning on or off target genes downstream in the mRNA. The ability to recognize and bind small metabolites with high specificity and the use of conformational rearrangements to be effective biological switches indicates a high degree of structural sophistication in riboswitches that makes them attractive targets for RNA structure and folding studies.
The regulation performed by a riboswitch relies on the interplay of its ligand-bound and free forms, which usually involves switching between alternative conformations or secondary structures by the riboswitch. X-ray crystallography has elucidated the structures of a number of riboswitches in their ligand-bound form 7; 8; 9; 10; 11; 12; 13. However, these structures do not provide information about the unliganded states of the riboswitch. In some cases, such as the lysine and glmS riboswitch, nearly identical structures have been observed irrespective of ligand binding 7; 14. Such an observation is consistent with an energy landscape of conformations that can be biased by conditions, such as high concentrations of Mg2+, to allow population of fully ‘folded’ states. However it has not been ascertained whether these folded states are accessible to the riboswitch in vivo in the absence of the ligand. In general, it appears that the ligand-free state of a riboswitch represents a partially folded RNA structure, and that binding of the ligand leads to global changes in architecture or local structural organization of the riboswitch to varying degrees, as revealed by chemical probing, small angle X-ray scattering, NMR, and dual trap optical tweezers experiments 10; 15; 16; 17; 18. In some cases, the ligand-free state may be heterogeneous and therefore difficult to crystallize. Hence, the characterization of the ligand-free structure of a riboswitch presents an alluring challenge that will enhance our understanding of both riboswitches specifically and RNA folding and structure in a broader context.
The thiamine pyrophosphate (TPP) riboswitch regulates genes that code for proteins involved in the thiamine biosynthetic pathway. Crystal structures have been obtained for the TPP riboswitch thiM from E.coli and thiC from A.thaliana bound to TPP 11; 19. The structures of the riboswitch for the different organisms show a high degree of similarity, folded into the same overall ‘tuning-fork’ conformation, with the ligand bound between the two prongs in an extended conformation (Figure 1B). The secondary structure of the riboswitch consists of five base-paired helical stems, P1 to P5, connected by non-base-paired junction regions (Figure 1A). The architecture of the riboswitch is built around a three-way junction with helix P1 branching into two stacked helices in each branch, P2 and P3 on one side, and P4 and P5 on the other, with the two branches arranged in a parallel fashion. The P2/P3 stack forms the “pyrimidine sensor helix”, where the thiamine moiety of TPP is inserted into a pocket formed by J2/3 junction, and the P4/P5 stack is identified as the “pyrophosphate sensor helix”, where junction J4/5 forms a hydrogen-bonding network with the pyrophosphate end of the ligand 19. The negative pyrophosphate moiety is accommodated via a divalent ion in the crystal structure 20 Additionally, tertiary interactions are observed between loop L5 and helix P3, which stabilize the structure of the RNA.
Figure 1.
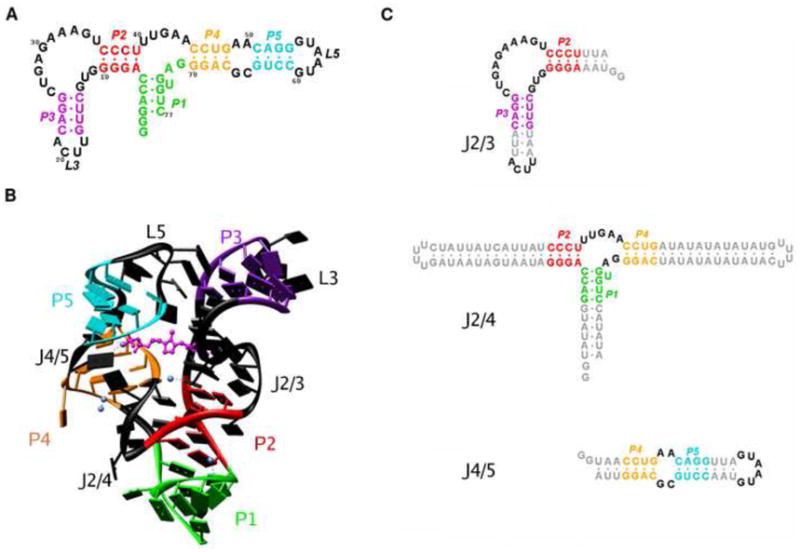
The aptamer domain of the thiamine pyrophosphate riboswitch from the thiC gene in A. thaliana examined in this study. (A) Secondary structure diagram of the riboswitch construct. The five base-paired helices are labeled P1 – P5 and are each given a unique color: P1 (green), P2 (red), P3 (violet), P4 (yellow), P5 (cyan). The non base-paired regions – junctions and loops – are shown in black. (B) Structure of the TPP riboswitch aptamer as characterized previously by X-ray crystallography. The color scheme is as defined in (A). The ligand TPP is shown in magenta in a ball-and-stick representation. Mg2+ ions found in the crystal structure are represented as blue spheres. (C) Constructs of the three junctions in the structure of the aptamer. The junctions are defined by the adjoining helices: J2/3 between P2 and P3 (left), J2/4 between P1, P2 and P4 (middle), and J4/5 between P4 and P5.
The global conformation of the riboswitch in the absence of TPP remains elusive. We have used computational modeling in conjunction with small angle X-ray scattering (SAXS) experiments 21; 22 to develop a model for the overall structure of the riboswitch in its unliganded form. Specifically, we use two contrasting approaches, a ‘divide-and-conquer’ approach, and a ‘global’ approach, to independently obtain candidate structures for the TPP-free state. At present, no single algorithm exists that enables the modeling of a tertiary structure based only on the sequence of the RNA and the low-resolution structural envelope obtained from SAXS data. Our results demonstrate the utility of combining computational structure modeling of RNA with a global experimental approach such as SAXS in order to overcome some of the challenges encountered in probing partially folded RNA structures with the conventional methods of X-ray crystallography and NMR. Our models provide a stepping stone for further analysis of the unliganded riboswitch structure. In addition, a comparison of our results from the two approaches provides important clues about the modeling of RNA structure.
RESULTS AND DISCUSSION
We use small-angle X-ray scattering (SAXS) to obtain structural information about the different conformations of the thiC aptamer domain from A.thaliana in varying ligand and Mg2+ concentrations. Being a solution technique, SAXS is particularly well suited to probe unfolded and partially folded structures, compared to higher resolution techniques such as X-ray crystallography and NMR spectroscopy 22; 23; 24.
SAXS has also been increasingly utilized for monitoring ion and ligand-dependent conformational changes in macromolecules in solution 16; 25; 26; 27; 28; 29; 30; 31; 32. We use our SAXS data in conjunction with a structure prediction algorithm, MC-SYM. Our approach allows us to combine the global structure information contained in SAXS with the local RNA structural information used in computational modeling to obtain low-resolution atomic models of the TPP riboswitch aptamer in the absence of TPP. We also obtain models for the ligand-bound state for the riboswitch, which we compare to the crystal structure to test the validity of our approach.
Three state model of the TPP riboswitch aptamer
We obtained SAXS data for the aptamer domain of the TPP riboswitch with varying ion and ligand concentrations (Figure 2). The data are shown in Kratky representation, q2 · I, where the scattering intensity I is weighted by the square of the momentum transfer q. This representation has been shown to be particularly useful for monitoring conformational changes in proteins and nucleic acids 23. Molecules that are folded and compact show a characteristic peak in a Kratky plot, whereas completely denatured polymers, which have a random and extended conformation, assume a linear rise at large q.
Figure 2.
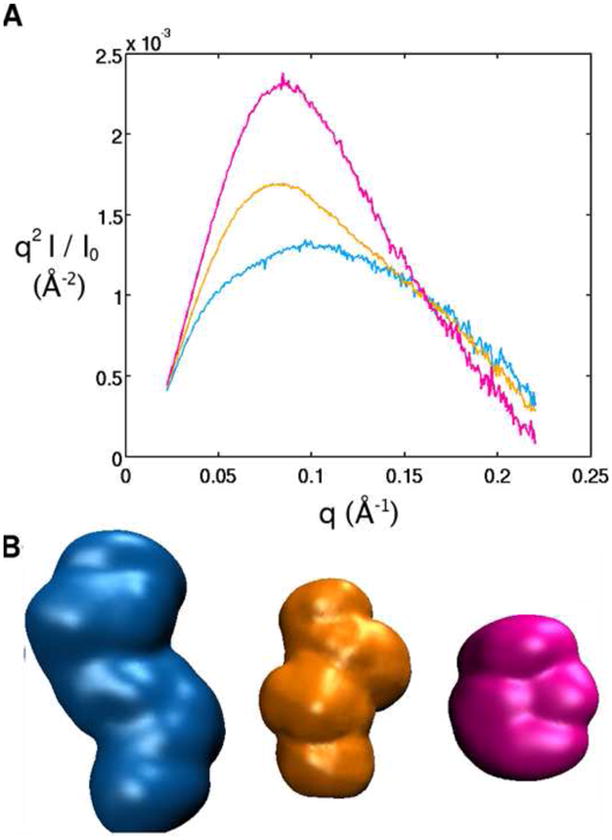
Small Angle X-ray Scattering (SAXS) data for the structure of the TPP riboswitch aptamer in the absence of Mg 2+ and TPP (blue) defined as the ‘unfolded’ state, in 10 mM Mg2+ but no TPP (orange) defined as the ‘intermediate’ state, and in 10 mM Mg2+ and 10 mM TPP (pink) defined as the ‘folded’ state. Data is also shown for the denatured riboswitch in 7 M urea (grey). (A) SAXS profiles in Kratky representation (q2I(q) vs. q for the full scattering profile) in the different solution conditions. (B) Low resolution bead models for the TPP riboswitch in its unfolded, intermediate and folded state, obtained from the SAXS profiles in (A). See Materials and Methods for details on the structure reconstruction.
The SAXS data reveal four distinct scattering profiles for the RNA in different solution conditions, indicating the existence of at least four conformational states (Figure 2A). In the presence of low concentrations of monovalent cations without any divalent cations or TPP, the SAXS profile assumes a plateau shape without any single peak, consistent with an ‘unfolded state’ composed of a heterogeneous ensemble of extended conformations. The radius of gyration (Rg) in this state is found to be 34 ± 1 Å from Guinier analysis (Methods) 33. The riboswitch is not completely denatured in the unfolded state, as seen by a comparison with the data for the RNA in 7 M urea, and the difference presumably arises from the presence of secondary structure in the absence of urea.
Upon addition of millimolar concentrations of Mg2+ to the unfolded state, the Kratky plot becomes significantly different, with the appearance of a shoulder in the profile and the beginning of the formation of a peak. The intermediate state is fully populated at these concentrations of Mg2+, as further addition of Mg2+ no longer produces changes in the scattering profile (data not shown). The radius of gyration also decreases to 28 ± 1 Å, indicating the population of a more compact ‘intermediate state’. Electrostatic relaxation of the riboswitch likely contributes to the structural rearrangement responsible for the observed change in the shape of the SAXS profile and the Rg. The divalent ions screen the negatively charged sugar-phosphate backbone of the RNA, allowing it to come together to adopt a stable global fold 34; 35; 36; 37; 38.
The riboswitch undergoes further compaction when both Mg2+ and TPP are present, as seen from the large peak in the SAXS profile, characteristic of globular structures, and the decreased Rg of 22 ± 1 Å (see Supplementary Information, and Figure S1 for a description of minor discrepancies observed in measurements of ligand-bound state). In this ‘folded’ state, the RNA binds the TPP ligand, and tertiary contacts are formed that hold the RNA in this compact conformation 19. From the different SAXS profiles in the presence and absence of TPP it is evident that although the RNA becomes more compact when transitioning to the intermediate state from the unfolded state, it is still distinct from the bound state. These observations suggest that the RNA may be partially folded without the ligand, and hence, an ensemble of conformers may still be present in the intermediate state. Our focus is to develop a model that represents an average of the ensemble present in the partially folded ligand-free state of the RNA. We also obtain models for the folded state of the RNA, and we compare these models to the crystal structure of the ligand-bound state of this riboswitch.
For visualization of the changes brought about in the RNA as a function of Mg2+ and TPP, we also obtained low-resolution bead models from the experimental SAXS profiles, shown in Figure 2B (Methods). The Rg and low-resolution envelopes, in agreement with the qualitative conclusion from the peaks observed in the SAXS profiles, demonstrate that the RNA becomes compact upon addition of Mg2+ and assumes an even tighter conformation when TPP is bound.
Bead models have previously been successfully employed in structural studies for proteins and RNA 16; 39; 40; 41; 42. Although these low-resolution models are useful for visualizing overall changes in the RNA, they do not provide any link between the three-dimensional structure of the RNA and its primary sequence; the placing of structural elements of the RNA within the envelope remains elusive. Our methods are aimed at building models of the TPP riboswitch that go beyond these low resolution-envelopes and elucidate the global conformations of the RNA.
Two approaches to obtain models of the ligand-free and bound riboswitch
We build on the approaches taken before using FRET, electrophoretic gel mobility, chemical probing, transient electric birefringence, NMR and most recently SAXS, to characterize the global conformation of RNA molecules 43; 44; 45; 46; 47; 48; 49; 50. Drawing on the hierarchical folding characteristic of RNA, such that regions of local secondary structure are typically stably formed under conditions not yet conducive to tertiary structure formation, we modeled the structure of the TPP riboswitch as rigid base-paired helices connected by unpaired junction regions 51. These junction regions govern the overall arrangement of the helices in the global fold observed for the RNA.
We used two approaches, a ‘divide-and-conquer’ approach and a ‘global’ approach, to orient all the helices in the RNA with respect to one another to obtain structural models of the ligand-free state of the TPP riboswitch aptamer. The divide-and-conquer approach has been used previously to obtain models for a number of RNA systems, where the structure of the RNA is obtained by solving the structures of smaller constituent fragments of the RNA through experimental and computational means and then reconstructing the whole structure by conjoining of these fragments 44; 52; 53. In contrast, in the global approach we test models of the structure of the RNA as a whole, independent of experimental agreement of the fragment structures in isolation. Finally, we compare the ligand-free models from the two approaches with one another and the ligand-bound models with the crystal structure, and address the potential origins of observed differences. These comparisons allow us to probe the assumptions underlying each approach and elements that may work in concert to impart a unique fold on a given RNA sequence in a particular divalent ion environment.
The divide-and-conquer approach
Helical junctions have been recognized to play important roles in the organization of RNA structure, allowing helical regions to form compact domains via stacking interactions or parallel packing, and bridging distant regions of the RNA to allow formation of tertiary contacts 54. We draw on previous methodologies used to obtain structural models for RNA systems such as the VS ribozyme 53, to isolate and study the separate junction components of the RNA independently, and to then reassemble the structure of the RNA from these components 44.
We divided the TPP riboswitch into three fragments for this purpose (Figure 1C): two two-way junctions, J2/3 with adjacent helices P2 and P3, and J4/5 with adjacent helices P4 and P5, and one three-way junction J2/4 linking helices P1, P2 and P4. Structure prediction is expected to be more effective for the smaller junction fragments as opposed to the whole RNA, as the conformational space that needs to be sampled for each construct is reduced and there is less chance of obscuring SAXS contributions via compensating conformational changes that lead to the same or similar SAXS patterns. Several algorithms are available that predict three-dimensional structures of RNA for a given sequence 55; 56; 57 and we chose MC-SYM for its fragment-based approach to obtain a library of possible structures of the three junction constructs for comparison to experiments.
We obtained two hundred models of each of the three junctions using MC-SYM. SAXS profiles were then computed theoretically for each of the two hundred structures predicted by MC-SYM for the three junctions using CRYSOL 58. We also obtained experimental SAXS data for the junctions to compare with the theoretical predictions. Constructs of the three fragments of the TPP riboswitch were prepared for SAXS experiments, in which the junction core of each of the fragments was preserved, and the helices were extended to stabilize their structure in the absence of the rest of the RNA (Methods). Experimental SAXS profiles for each of these junctions were obtained in the presence of 10 mM Mg2+. The theoretical profiles were compared to the experimental data for each junction with a least squares fitting criterion (see Methods), which resulted in three best-fitting models for each of the three junctions. These best-fitting structures were then reassembled together in all possible combinations using MC-SYM to obtain twenty-seven structural models for the whole riboswitch (Methods).
Solution structures of the isolated junctions of TPP riboswitch aptamer
Junction J4/5 is a symmetric junction with single strands, two nucleotides in length, connecting helices P4 and P5 on either side. The three models for J4/5 that give the best fits to the experimental SAXS profiles also show good agreement with the low resolution envelope computed for this junction from the SAXS data. All models share similar structures with helices P4 and P5 in an almost stacked conformation, with a slight kink between the helices at the junction region, so that the helices subtend an angle slightly less than 180° (Figure 3).
Figure 3.
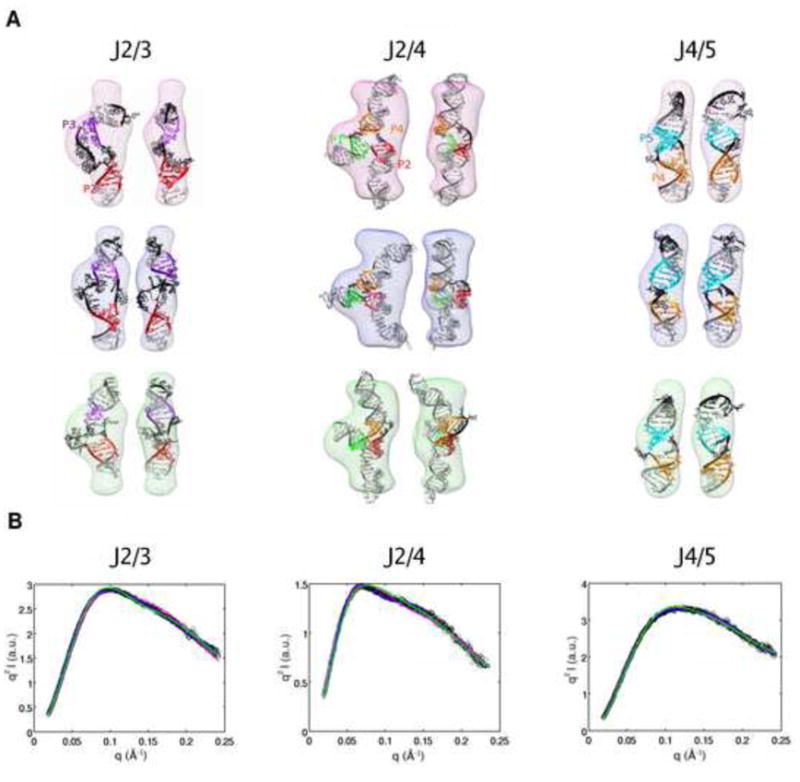
Fitting of the three highest-ranking models of the isolated junctions of the TPP riboswitch to experimental data: J2/3 (left column), J2/4 (middle column), J4/5 (right row). Models for each of the junctions were generated using MC-SYM. (A) The three highest-ranking models for the junctions overlaid on the bead models calculated for the experimental profiles shown in (B). The coloring of the helices is identical to Figure 1. The bead models are colored differently to indicate models ranked 1 to 3: rank 1 (pink), rank 2 (blue), rank 3 (green). Two views of each model are shown side by side, with a rotation of 90° about the z-axis, in the same colored bead models. (B) Experimental SAXS profiles of the isolated junctions in Kratky representation overlaid on the predicted SAXS profiles from the three highest-ranking models of the junctions, as determined from a least-squares fitting routine (Methods). The black circles represent the experimental data, and the solid lines show the predicted profiles from the models. The solid lines are colored the same as the bead model to identify the model that gives that profile.
For junction J2/3, the best fitting models again show a stacked conformation for helices P2 and P3 with a slight bend at the junction. This junction is asymmetric, with a single-stranded region of three nucleotides connecting one side of helices P2 and P3, and a ten nucleotide long single-stranded segment connecting the other side. The junction appears to accommodate the stacked conformation of the helices despite its asymmetry by keeping the shorter single stranded segment stretched out, and forming a bulge in the longer connecting segment. The structure of the bulge shows some variability in the three best fitting models and is likely not well constrained by the SAXS data (Figure 3).
The two two-way junctions are connected via the three-way junction J2/4, which organizes the overall architecture of the RNA. This junction connects helix P1 to P2 continuously with no intervening single-stranded nucleotides. Helix P2 is connected to P4 through a 5-nucleotide single-stranded segment, and helix P4 is connected back to P1 via two single-stranded nucleotides. The best-fitting models for the three-way junction show varying inter-helical angles, with the largest angle consistently subtended between P2 and P4 (Figure 3). The angles between P1 and P4 and between P1 and P2 vary in the models, with P1 bent more towards P2 in two of the models, and P1 forming the smallest angle with P4 in the remaining model.
The calculated SAXS profiles for the best-fitting models of each junction are similar (Figure 3), and cannot be used to distinguish structural details, such as small changes in inter-helical angle and structures of kinks between helices, that vary between the models. This is expected, since SAXS is a low-resolution technique. However, the best-fitting profiles do result in similar overall conformations of the junctions, and this similarity provides confidence in the global structures of the isolated junctions that we choose for the divide-and-conquer approach.
Reconstituted ligand-free structure of TPP riboswitch from the divide-and-conquer approach
There are twenty-seven total models of the whole riboswitch from all possible combinations of the three best-fitting structures of the three junctions. The SAXS profile for these models were computed and compared to the experimental SAXS data for the whole riboswitch in divalent ion in the absence of TPP. These structures were then ranked according to their fit to the experimental data (Methods).
The highest-ranking model of the riboswitch, reconstituted from the divide-and-conquer approach, was obtained from a combination of highest-ranking models 2, 1 and 1 of J2/3, J2/4 and J4/5 respectively (Figures 3, 4). We observed good agreement between this model of the riboswitch, and the SAXS data, for both the experimental scattering profile (Figure 5A) and the low-resolution envelope generated from this profile (Figure 4A). This agreement suggests that the highest-ranking model provides an adequate representation of the average structure of the conformers present in the ligand-free state of the riboswitch.
Figure 4.
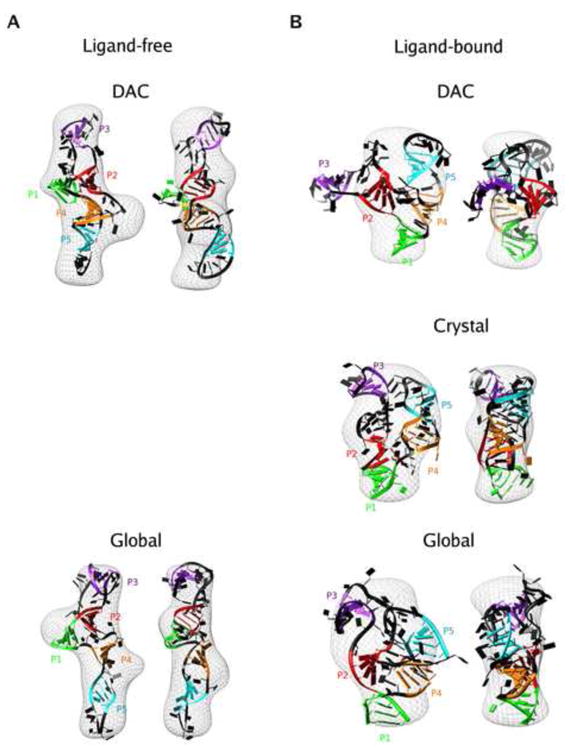
Models of TPP riboswitch aptamer in ligand-free (left column) and ligand-bound (right column) states. Models were generated using MC-SYM as described in Methods. (A) Models for the ligand-free state superimposed on the bead models obtained from the experimental profile in excess Mg2+. The two models shown are derived from the divide-and-conquer (DAC) approach (top) and the global approach (bottom). Two views are shown for each model, with a rotation of 90° about the z-axis between the views. (B) The structures for the ligand bound state superimposed on experimental SAXS electron density map obtained in the presence of excess Mg2+ and TPP. The different structures are the model obtained for the bound state using the global approach(bottom) and the crystal structure for the bound state (middle) The coloring of the helices follows the convention defined in figure 1.
Figure 5.
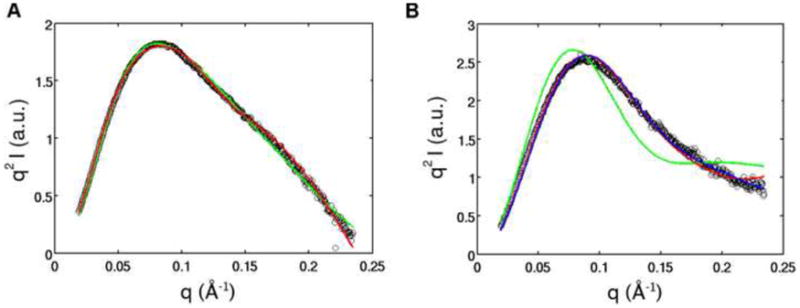
Comparison of SAXS profiles of the models of the ligand-bound and free state from the divide-and-conquer (DAC) and global approach, the crystal structure for the ligand bound state, and experimental data. Experimental SAXS profiles for TPP aptamer in 10 mM Mg2+ only (left) and 10 mM Mg2+ and 10 mM TPP (right) overlaid on theoretical profiles predicted for each of these states using CRYSOL. Black circles indicate the experimental data. The color of the line represents the different approaches used to derive the models: divide-and-conquer (green), global (red). The profile predicted for the crystal structure is shown in blue.
The model shows an extended conformation compared to the structure observed by X-ray crystallography for its ligand-bound counterpart (Figure 4). The most striking difference between the two states is the relative position of the P2/P3 and P4/P5 stacked helices. In the ligand-bound state, the crystal structure shows these helices arranged parallel to one another, held together by TPP. This allows loop-helix interactions to occur between L5, the loop that caps P5, and helix P3. In the absence of TPP, our model shows these helices pried apart from one another, with an angle of almost 120° between them, thereby assuming an ‘open’ form, compared to the ligand-bound crystal structure (Figure 4). This positioning of helical elements suggests a lack of pre-organization in the structure of this riboswitch prior to the binding of TPP.
Ligand-bound structure of TPP riboswitch from divide-and-conquer approach
The theoretical SAXS profiles of the twenty-seven models obtained from the divide-and-conquer approach were also compared to the experimental SAXS profile for the TPP riboswitch in the presence of both Mg2+ and TPP. The models were now ranked according to their fit to the experimental data for the TPP-bound state of the riboswitch (Methods).
In the case of the ligand-bound state, even the highest-ranking model did not give a good fit to the experimental SAXS profile or to the low-resolution electron density map (Figures 4B and 5B). The Kratky representation of the SAXS profile of this model shows a more defined peak than that of the divide-and-conquer ligand-free model, indicating that the ligand-bound model is more compact. This observation is confirmed by a closer look at the ligand-bound model, which shows the P2/P3 and P4/P5 stalks subtending an angle of almost 90°, compared to the ligand-free model in which this angle is ~ 120°.
Although comparisons to the experimental SAXS profiles for the ligand-free and ligand-bound states of the riboswitch rank the models correctly in terms of compactness, the ligand-bound model is unable to capture the parallel arrangement of P2/P3 and P4/P5 observed in the crystal structure. This stems from the library of J2/4 structures used in the divide-and-conquer approach in which all the structures of this junction have the helices P2 and P4 wide apart.
Presumably a junction with a given sequence is biased towards a certain structure that is inherent to that sequence. When probed experimentally in isolation, the junction reflects this inherent structure. However, when the junction is placed within the parent RNA structure, it may not retain its inherent structure, due to the presence and influence of other elements of the RNA. Most simply, changes can arise due to steric clashes that can sometimes be avoided by local rearrangements. Furthermore, tertiary interactions observed between L5/P3 in the crystal structure of the riboswitch may enforce a different junction conformation than that which is most stable in isolation; for example a more parallel arrangement of P2 and P4 is observed in the ligand-bound crystal structure. The divide-and-conquer approach is blind to the interactions between the smaller fragments of the RNA, and in cases where these interactions play an important role in the folding of the parent structure, the divide-and-conquer approach will not suffice to give a good representative model.
The global approach
The divide-and-conquer approach operates with an underlying assumption that the structures of the isolated junctions remain the same when these junctions are reincorporated into their parent structure. In contrast, the global approach treats the RNA as one unit, where the sum of the conformations adopted by the smaller fragments in isolation does not necessarily constitute the overall structure. Therefore, in the global approach, models of the structures of the constituent fragments are not discriminated against based on the lack of their agreement with experimental data for these fragments in isolation.
The assembly of the models of the whole TPP riboswitch is again performed with MC-SYM, and in this case, we allow all 200 models of all three junctions to be put together in different combinations. This is in contrast to the divide-and-conquer approach, where we assembled only the models of the junctions that gave good agreement with the experimental data for the junctions in isolation. We generated a total of 40,000 candidate structures of the whole riboswitch. Theoretical SAXS profiles were computed for all of the 40,000 models of the whole riboswitch, and these profiles were tested against experimental SAXS data for the RNA (Methods).
Ligand-free structure of TPP riboswitch from global approach
The highest-ranking 38 models from the global approach gave p-values of >0.95 when tested against experimental data for the ligand-free state of the riboswitch (Figure S3A). Considerable variations exist between these 38 models from the global approach, with an average RMSD of ~12 Å between the different models. The differences arise from the twist of helices with respect to one another, and from small changes in inter-helical angles, which cannot be differentiated using SAXS (Figure S3B). In addition, we are using one-dimensional SAXS profiles to represent three-dimensional models, and the mapping of the profiles to the structures cannot be unique. However, these models all have the same overall conformation, with helical branches P2/P3 and P4/P5 separated by an average angle of 120° ± 15°.
One representative model from this set of thirty-eight is shown separately for clarity (Figures 4A and 5A). The extended conformation of the ligand-free state is common to both the divide-and-conquer and the global approach (Figure 4A). This observation reinforces the conclusions that the ligand-free state of the TPP riboswitch has an open form and that binding of TPP is required to bring the structure to the compact conformation observed in the crystal structure.
Ligand-bound structure obtained from global approach
For the ligand-bound state of the riboswitch, the top 27 ranked models produced good fits to the experimental data, as determined by a chi-squared goodness of fit test (Figure S4). There was more variation between these models than between the set of thirty-eight models isolated for the ligand-free state, as indicated by a higher average RMSD of ~17 Å between the models (Figure S4).
One example shown for clarity gives a sense of the level of structural detail that can be discriminated from a SAXS profile (Figure 4B). The theoretical profiles from the crystal structure and the model give similar fits to the experimental SAXS data (Figure 5B). The overall conformation of the model is similar to the crystal structure, in which helices P2/P3 and P4/P5 come together in an almost parallel fashion (See Supplementary Information Figure S5 for cautionary note). However, higher resolution details, such as the orientation of the individual residues, helical twists and structures of the non-base paired regions are different between the two structures. A superimposition of all twenty-seven models also shows that the global conformation is conserved, but detailed structural differences are present (Figure S4B). Although higher resolution structural details beyond global conformations cannot be accessed using SAXS, the combination of global information content from SAXS and atomic models of the RNA obtained from MC-SYM provide a powerful tool for obtaining possible structures of the overall conformation of the RNA molecules.
The global approach does not discriminate against junctions based on their structures in isolation, and therefore includes a larger pool of candidates of the junction fragments, which are then reassembled into the parent structure. This leads to different ligand-bound models from the divide-and-conquer and global approaches, but similar models for the ligand-free state (Figure 4). The divide-and-conquer approach can be adequate for modeling RNA structures, in the absence of inter-fragment interactions in the form of tertiary contacts or steric clashes. For the ligand-free state, the structures of the isolated junctions appear to be preserved in the riboswitch, such that the inclusion of junction structures, which stray from their isolated conformations, does not change the overall conformation of the models that fit the data well. However, in the ligand-bound state, tertiary interactions exist between P3 and L5, which hold P2/P3 and P4/P5 in a parallel configuration in the crystal structure. This conformation is rendered inaccessible to the divide-and-conquer approach, because the tertiary contact perturbs the structure of the junctions away from their inherently favored conformations. Inclusion of a larger population of junction structures in the global approach partially removes this problem and gives better representative models.
In addition, the pyrimidine and pyrophosphate moieties of TPP are recognized and bound separately by the two junctions J2/3 and J4/5, and these interactions also enforce the tuning-fork conformation of the bound state. MC-SYM does not account for the effects of ligand binding on RNA structure and since the ligand has an important effect on the folding energy landscape of this riboswitch, MC-SYM is not expected to capture the bound state of the RNA. This limitation of MC-SYM can lead to under-sampling of the bound conformation, and demonstrates that modeling the association of RNA with ligands and proteins by structure prediction software remains a challenge. Conversely, the SAXS data for the whole riboswitch in the presence of TPP accounts for the presence of the ligand. The use of this data for isolating models of the bound state with the global approach therefore helps in identifying the correct global conformation from all the other models.
CONCLUSIONS
Riboswitches have garnered much excitement as an RNA-only mechanism of gene expression regulation. However, much remains to be learned about riboswitches, and characterization of the ligand-free states of these RNA switches is one of the gaps that needs to be filled 59. We have presented a structural model of the ligand-free state of the TPP riboswitch aptamer. Previous studies using chemical probing and fluorescence spectroscopy indicated that this riboswitch undergoes significant structural modulation upon binding of the ligand, and our results provide a picture of the global rearrangement that occurs once TPP binds 11; 19; 60. Our model shows an open state for the TPP riboswitch that leaves both the binding sites for the thiamine and pyrophosphate moieties accessible to the ligand for binding. As the two functional groups of TPP are recognized by two distal regions of the RNA, TPP binding bridges the gap between these regions, and ‘staples’ the RNA together into its compact bound conformation.
Our model complements an earlier study that revealed this riboswitch controls splicing 61. It was proposed that nucleotides in the P4/P5 stem of the riboswitch interact with the nucleotides adjacent to the splice site, rendering the splice site inaccessible for binding by the spliceosome. TPP binding brings the P4/P5 stem away from the splice site, thereby allowing splicing to occur. Our model indicates that in the TPP-free state, P4/P5 and P2/P3 are splayed wide apart, and in this conformation the P4/P5 stem is available for interaction with alternative nucleotides in the splice site.
The limitations of the divide-and-conquer approach from our results demonstrate that the absence of information about RNA interactions with proteins and other ligands restricts the predictive abilities of RNA structure modeling algorithms. Our approach obtains information about these interactions lacking in MC-SYM from SAXS and hence partially alleviates some problems that remain in RNA structure determination. SAXS is a low-resolution technique and structures with variations in details such as helical twist give very similar SAXS profiles, as seen for the best fitting models of the individual junctions and the models derived from the global approach. Additionally, SAXS reports on the ensemble measurement, and the models described in our results represent low-resolution ‘average’ structures that are consistent with our data. Further refinement of these average models, and the building of physically and experimentally consistent ensembles of structures are future challenges that need to be addressed. Nevertheless, our results provide a first step towards developing a methodology that will advance our ability to model and predict RNA structures that are especially difficult to solve with currently available methods.
MATERIALS AND METHODS
RNA synthesis
The TPP riboswitch aptamer domain and the junction constructs were prepared by in vitro transcription with T7 RNA polymerase. Templates for the constructs were synthesized by assembly PCR, using synthetic DNA oligonucleotides purchased from IDT DNA technologies, Coralville, IA. All the constructs were purified by electrophoresis on a 10% acrylamide gel in the presence of 7 M urea.
We used the same construct as Thore et al. to study the full riboswitch aptamer 19. To study the individual component junctions, we used constructs where the core of the junctions were preserved, with the helical arms extended to seven base pairs each for helices P2, P3, P4 and P5 with tetra-U loops introduced at the ends of helices P3 and P5 in junctions J2/3 and J4/5 respectively (Figure 1). The helix P1 was extended to ten basepairs (bp) and helices P2 and P4 were extended to seventeen bp with tetra-U loop caps for junction J2/4 (Figure 1). The sequences for the aptamer construct and the junction constructs are given below:
TPP aptamer: 5′–GGGACCAGGGGUGCUUGUUCACAGGCUGAGAAAGCCCUUU GAACCUGAACAGGGUAAUGCCUGCGCAGGGAGUGUC–3′
J2/3: 5′–GGUAAAGGGGUGCUUGUAAUUCAUUACAGGCUGAGAAAGUCCCUU UA–3′
J2/4: 5′–GGUAUAUGGACCAGGGAUAAUGAUAAUAGUUUUCUAUUAUCAUU AUCCCUUUGAACCUGAUAUAUAUAUAUGUUUUCAUAUAUAUAUAUCAGGG AGGUCCAUAUA–3′
J4/5: 5′–GGUAACCUGAACAGGUUAGUAAUGUAACCUGCGCAGGUUA–3′
SAXS data acquisition
SAXS data were acquired at beamline 12-ID of the Advanced Photon Source, Argonne, IL, USA. The measurements were performed at 25°C using a sample-detector distance of 2 m, a CCD detector, and a custom-made sample cell 62. The X-ray energy was set to 12 keV for all measurements.
SAXS data were collected for sample concentrations of 0.5, 1 and 2 mg/ml (corresponding to 20, 40 and 80 μM respectively). The RNA was thawed and incubated at 65°C for 10 minutes, after which it was mixed in buffer (100 mM Tris•HCl, pH 7.0), and various concentrations of MgCl2 and TPP (Sigma-Aldrich). The samples were centrifuged for 10 minutes at 13,000 g immediately before the measurement. Five sequential exposures of 0.2 s each were obtained for each sample and averaged to improve the quality of data. Profiles for identical solutions, without the RNA, were also measured and these profiles used for buffer subtraction. Scattering profiles obtained at different RNA concentrations were superimposable after normalizing by forward scattering intensity, indicating the absence of interparticle interference and aggregation effects (data not shown). We tested for possible radiation damage by comparing subsequent exposures of the same sample and no changes were detected.
SAXS data analysis
Scattering intensity was obtained as a function of momentum transfer, q, in the range 0.02 < q < 0.25 Å−1. The radius of gyration was obtained by Guinier analysis of the low scattering angle region of the profile 33. In addition, the program GNOM was used to obtain histograms of intramolecular distances, P(r), by a regularized transform of the scattering intensity 63. The maximum intramolecular distance, Dmax, which is taken as an input parameter in GNOM, was increased in steps of 2 Å until a good fit to the experimental data and a smooth non-negative P(r) function was obtained. This yielded values of Dmax for each measurement (Table 1).
Table 1.
Radius of gyration and Dmax for the TPP riboswitch under different solution conditions measured by SAXS.
| [Mg2+] (mM) | [TPP] (mM) | Rg (Å) | Dmax(Å) |
|---|---|---|---|
| 0 | 0 | 34 ± 1 | 115 ± 5 |
| 10 | 0 | 28 ± 1 | 95 ± 5 |
| 10 | 10 | 22 ± 1 | 65 ± 5 |
Ab initio 3D modeling of SAXS data
The program DAMMIN was used to reconstruct three-dimensional bead models from the SAXS data, as described earlier 16; 64. Ten independent DAMMIN runs were performed on each scattering profile, representative of an RNA state, in ‘slow’ mode, using default parameters, and input values of Dmax obtained as described above. The models were aligned and averaged using SUPCOMB and DAMAVER respectively 65; 66. The reconstructed bead models were converted to electron density maps with the program Situs 67; 68. This used real space convolution with a Gaussian kernel with a width of 6 Å and voxel spacing of 2 Å. The models were visualized in VMD and Chimera 69; 70.
Obtaining low-resolution atomic models of the aptamer and junction constructs by computational prediction
The MC-SYM web server and program were used to generate models of the junction constructs and the TPP riboswitch aptamer 55. MC-SYM harnesses a fragment-based approach to model RNA structure. A query sequence provided to MC-SYM is divided into shorter constituent sequences. MC-SYM uses known RNA structures to build a library of three-dimensional structures for the constituent sequences. These structures serve as fragments that are then assembled together via common nucleotides between the fragments in different combinations to output many candidate structures for the whole RNA. MC-SYM provides flexibility to the user to divide the query sequence into different desired constituent sequences than those defined by default. In this case, it allows the user to input models corresponding to the desired constituent sequences to build up the fragment library. Therefore, MC-SYM provides a platform where atomic models for an RNA sequence can be generated from scratch using default libraries, or user-defined libraries of known structures.
To obtain models of the junction constructs, sequences for the junctions were submitted, along with a dot-bracket rendition of the secondary structure prediction for the sequences from the crystal structure of the bound state, to the MC-SYM pipeline webpage to generate a script. The resulting MC-SYM script was submitted to the MC-SYM web-server with a modification to increase the number of models generated to 200 for each construct, and the generated models were downloaded from the web-server. For the isolated junction constructs, the default MC-SYM libraries of fragments were used.
The whole aptamer construct of the riboswitch was modeled using a locally compiled version of MC-SYM for both the divide-and-conquer and global approaches. The input script for the program was used to divide the aptamer sequence into three fragments labeled J2/4 (corresponding to residues 1 – 10, 36 – 48 and 67 – 77 in the aptamer), J2/3 (residues 10 – 36), and J4/5 (residues 48 – 67). The divide-and-conquer approach and the global approach differ in the libraries of structures of these three fragments used to assemble the whole molecule.
For the global approach, the 200 models generated for each of the junctions were used as the library of structures for the fragments (global library). The closing base pair in P2 for J2/3 (residues 10 and 36 in the aptamer) and in P4 for J4/5 (residues 48 and 67 in the aptamer) were used as common residues between J2/3 and J2/4, and J2/4 and J4/5 respectively. These were used to link all combinations of the fragments together that did not result in steric overlaps to obtain models of the whole riboswitch. This yielded 40,000 total models for the aptamer domain of the riboswitch.
In contrast to the global approach, the divide-and-conquer approach limited the libraries of the three fragments to only those models of the junctions that gave good fits to the experimental SAXS data for the isolated junction constructs (χ2 > 0.95; see next section in Methods). This included three models for each of the junctions, and all possible combinations of three models for the three fragments led to twenty-seven reconstituted models of the riboswitch from the divide-and-conquer approach. The junctions were assembled in a similar fashion as in the global approach.
Upon using only half of the models for each junction with both the global and divide-and-conquer approach, we find similar results for the reconstituted structures, indicating that 200 models for each junction are adequate for sampling the conformational space.
Fitting of models to SAXS data
The program CRYSOL was used with default input parameters to calculate predicted SAXS profiles for all the models generated using MC-SYM 58. These predicted profiles were tested against the experimental SAXS data using a two-step process to isolate the models that gave the best fits to the SAXS data. In the first step, a least-squared criterion was used to test the predicted profiles of the models. To ensure a good fit of the SAXS profiles calculated from the MC-SYM models at all scattering angles, instead of being dominated by the low-q space, we resorted to a three-part approach. A least squares test was first performed on the first third of the SAXS profile (0.02 < q < 0.1 Å−1), which contains information about the overall size and shape of the particle. The criteria for passing this test was set to ensure that the sum of the least squared difference of the normalized intensity (I/I0 where I0 is the forward scattering intensity at q = 0) between the experimental and theoretical profile was <0.1. Upon passing this test, the next third (0.1 < q < 0.18 Å−1) of the profile was tested in a similar manner, and finally, the last third (0.18 < q < 0.25 Å−1) of the profile was checked. Models that passed these criteria and gave the lowest least-squared difference in intensity for the scattering profiles were taken to the second step.
The models that passed the least-squared test were ranked based on the p-values obtained from a chi-squared goodness of fit of their calculated profile with the experimental profile. Only three models yielded a p-value of greater than 0.95 for each junction and these models were selected as the best-fitting models (Figure S3). These best fitting models for the junctions and aptamer constructs were identified to be the best representatives of the acquired SAXS data.
Supplementary Material
Acknowledgments
We thank Nathan Boyd for help with sample preparations, and Rhiju Das, Adelene Sim, Vincent B. Chu and members of the Herschlag lab for helpful discussions and comments. This research was supported by the National Institutes of Health Grant PO1 GM0066275 and a Stanford Graduate Fellowship to M.A. Use of the Advanced Photon Source was supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. W-31-109-Eng-38.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Pyle AM, Green JB. RNA folding. Curr Opin Struct Biol. 1995;5:303–10. doi: 10.1016/0959-440x(95)80091-3. [DOI] [PubMed] [Google Scholar]
- 2.Coppins RL, Hall KB, Groisman EA. The intricate world of riboswitches. Curr Opin Microbiol. 2007;10:176–81. doi: 10.1016/j.mib.2007.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nudler E, Mironov AS. The riboswitch control of bacterial metabolism. Trends Biochem Sci. 2004;29:11–7. doi: 10.1016/j.tibs.2003.11.004. [DOI] [PubMed] [Google Scholar]
- 4.Schwalbe H, Buck J, Furtig B, Noeske J, Wohnert J. Structures of RNA switches: insight into molecular recognition and tertiary structure. Angew Chem Int Ed Engl. 2007;46:1212–9. doi: 10.1002/anie.200604163. [DOI] [PubMed] [Google Scholar]
- 5.Wakeman CA, Winkler WC, Dann CE., 3rd Structural features of metabolite-sensing riboswitches. Trends Biochem Sci. 2007;32:415–24. doi: 10.1016/j.tibs.2007.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mandal M, Breaker RR. Gene regulation by riboswitches. Nat Rev Mol Cell Biol. 2004;5:451–63. doi: 10.1038/nrm1403. [DOI] [PubMed] [Google Scholar]
- 7.Garst AD, Heroux A, Rambo RP, Batey RT. Crystal structure of the lysine riboswitch regulatory mRNA element. J Biol Chem. 2008;283:22347–51. doi: 10.1074/jbc.C800120200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Batey RT, Gilbert SD, Montange RK. Structure of a natural guanine-responsive riboswitch complexed with the metabolite hypoxanthine. Nature. 2004;432:411–5. doi: 10.1038/nature03037. [DOI] [PubMed] [Google Scholar]
- 9.Montange RK, Batey RT. Structure of the S-adenosylmethionine riboswitch regulatory mRNA element. Nature. 2006;441:1172–5. doi: 10.1038/nature04819. [DOI] [PubMed] [Google Scholar]
- 10.Dann CE, 3rd, Wakeman CA, Sieling CL, Baker SC, Irnov I, Winkler WC. Structure and mechanism of a metal-sensing regulatory RNA. Cell. 2007;130:878–92. doi: 10.1016/j.cell.2007.06.051. [DOI] [PubMed] [Google Scholar]
- 11.Serganov A, Polonskaia A, Phan AT, Breaker RR, Patel DJ. Structural basis for gene regulation by a thiamine pyrophosphate-sensing riboswitch. Nature. 2006;441:1167–71. doi: 10.1038/nature04740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cochrane JC, Lipchock SV, Strobel SA. Structural investigation of the GlmS ribozyme bound to Its catalytic cofactor. Chem Biol. 2007;14:97–105. doi: 10.1016/j.chembiol.2006.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Serganov A, Huang L, Patel DJ. Coenzyme recognition and gene regulation by a flavin mononucleotide riboswitch. Nature. 2009;458:233–7. doi: 10.1038/nature07642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Klein DJ, Ferre-D’Amare AR. Structural basis of glmS ribozyme activation by glucosamine-6-phosphate. Science. 2006;313:1752–6. doi: 10.1126/science.1129666. [DOI] [PubMed] [Google Scholar]
- 15.Ottink OM, Rampersad SM, Tessari M, Zaman GJ, Heus HA, Wijmenga SS. Ligand-induced folding of the guanine-sensing riboswitch is controlled by a combined predetermined induced fit mechanism. RNA. 2007;13:2202–12. doi: 10.1261/rna.635307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lipfert J, Das R, Chu VB, Kudaravalli M, Boyd N, Herschlag D, Doniach S. Structural transitions and thermodynamics of a glycine-dependent riboswitch from Vibrio cholerae. J Mol Biol. 2007;365:1393–406. doi: 10.1016/j.jmb.2006.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Greenleaf WJ, Frieda KL, Foster DA, Woodside MT, Block SM. Direct observation of hierarchical folding in single riboswitch aptamers. Science. 2008;319:630–3. doi: 10.1126/science.1151298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Winkler WC, Nahvi A, Sudarsan N, Barrick JE, Breaker RR. An mRNA structure that controls gene expression by binding S-adenosylmethionine. Nat Struct Biol. 2003;10:701–7. doi: 10.1038/nsb967. [DOI] [PubMed] [Google Scholar]
- 19.Thore S, Leibundgut M, Ban N. Structure of the eukaryotic thiamine pyrophosphate riboswitch with its regulatory ligand. Science. 2006;312:1208–11. doi: 10.1126/science.1128451. [DOI] [PubMed] [Google Scholar]
- 20.Thore S, Frick C, Ban N. Structural basis of thiamine pyrophosphate analogues binding to the eukaryotic riboswitch. J Am Chem Soc. 2008;130:8116–7. doi: 10.1021/ja801708e. [DOI] [PubMed] [Google Scholar]
- 21.Lipfert J, Doniach S. Small-angle X-ray scattering from RNA, proteins, and protein complexes. Annu Rev Biophys Biomol Struct. 2007;36:307–27. doi: 10.1146/annurev.biophys.36.040306.132655. [DOI] [PubMed] [Google Scholar]
- 22.Koch MH, Vachette P, Svergun DI. Small-angle scattering: a view on the properties, structures and structural changes of biological macromolecules in solution. Q Rev Biophys. 2003;36:147–227. doi: 10.1017/s0033583503003871. [DOI] [PubMed] [Google Scholar]
- 23.Doniach S. Changes in biomolecular conformation seen by small angle X-ray scattering. Chem Rev. 2001;101:1763–78. doi: 10.1021/cr990071k. [DOI] [PubMed] [Google Scholar]
- 24.Lipfert J, Herschlag D, Doniach S. Riboswitch conformations revealed by small-angle X-ray scattering. Methods Mol Biol. 2009;540:141–59. doi: 10.1007/978-1-59745-558-9_11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Russell R, Millett IS, Doniach S, Herschlag D. Small angle X-ray scattering reveals a compact intermediate in RNA folding. Nat Struct Biol. 2000;7:367–70. doi: 10.1038/75132. [DOI] [PubMed] [Google Scholar]
- 26.Fang X, Littrell K, Yang XJ, Henderson SJ, Siefert S, Thiyagarajan P, Pan T, Sosnick TR. Mg2+-dependent compaction and folding of yeast tRNAPhe and the catalytic domain of the B subtilis RNase P RNA determined by small-angle X-ray scattering. Biochemistry. 2000;39:11107–13. doi: 10.1021/bi000724n. [DOI] [PubMed] [Google Scholar]
- 27.Russell R, Millett IS, Tate MW, Kwok LW, Nakatani B, Gruner SM, Mochrie SG, Pande V, Doniach S, Herschlag D, Pollack L. Rapid compaction during RNA folding. Proc Natl Acad Sci U S A. 2002;99:4266–71. doi: 10.1073/pnas.072589599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Russell R, Zhuang X, Babcock HP, Millett IS, Doniach S, Chu S, Herschlag D. Exploring the folding landscape of a structured RNA. Proc Natl Acad Sci U S A. 2002;99:155–60. doi: 10.1073/pnas.221593598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Das R, Kwok LW, Millett IS, Bai Y, Mills TT, Jacob J, Maskel GS, Seifert S, Mochrie SG, Thiyagarajan P, Doniach S, Pollack L, Herschlag D. The fastest global events in RNA folding: electrostatic relaxation and tertiary collapse of the Tetrahymena ribozyme. J Mol Biol. 2003;332:311–9. doi: 10.1016/s0022-2836(03)00854-4. [DOI] [PubMed] [Google Scholar]
- 30.Das R, Mills TT, Kwok LW, Maskel GS, Millett IS, Doniach S, Finkelstein KD, Herschlag D, Pollack L. Counterion distribution around DNA probed by solution X-ray scattering. Phys Rev Lett. 2003;90:188103. doi: 10.1103/PhysRevLett.90.188103. [DOI] [PubMed] [Google Scholar]
- 31.Takamoto K, Das R, He Q, Doniach S, Brenowitz M, Herschlag D, Chance MR. Principles of RNA compaction: insights from the equilibrium folding pathway of the P4-P6 RNA domain in monovalent cations. J Mol Biol. 2004;343:1195–206. doi: 10.1016/j.jmb.2004.08.080. [DOI] [PubMed] [Google Scholar]
- 32.Kwok LW, Shcherbakova I, Lamb JS, Park HY, Andresen K, Smith H, Brenowitz M, Pollack L. Concordant exploration of the kinetics of RNA folding from global and local perspectives. J Mol Biol. 2006;355:282–93. doi: 10.1016/j.jmb.2005.10.070. [DOI] [PubMed] [Google Scholar]
- 33.Glatter O, Kratky O. Small angle X-ray scattering. Academic; London: 1982. [DOI] [PubMed] [Google Scholar]
- 34.Woodson SA. Metal ions and RNA folding: a highly charged topic with a dynamic future. Curr Opin Chem Biol. 2005;9:104–9. doi: 10.1016/j.cbpa.2005.02.004. [DOI] [PubMed] [Google Scholar]
- 35.Draper DE. RNA folding: thermodynamic and molecular descriptions of the roles of ions. Biophys J. 2008;95:5489–95. doi: 10.1529/biophysj.108.131813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Draper DE, Grilley D, Soto AM. Ions and RNA folding. Annu Rev Biophys Biomol Struct. 2005;34:221–43. doi: 10.1146/annurev.biophys.34.040204.144511. [DOI] [PubMed] [Google Scholar]
- 37.Draper DE. A guide to ions and RNA structure. RNA. 2004;10:335–43. doi: 10.1261/rna.5205404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chu VB, Bai Y, Lipfert J, Herschlag D, Doniach S. A repulsive field: advances in the electrostatics of the ion atmosphere. Curr Opin Chem Biol. 2008;12:619–25. doi: 10.1016/j.cbpa.2008.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Aparicio R, Fischer H, Scott DJ, Verschueren KH, Kulminskaya AA, Eneiskaya EV, Neustroev KN, Craievich AF, Golubev AM, Polikarpov I. Structural insights into the beta-mannosidase from T reesei obtained by synchrotron small-angle X-ray solution scattering enhanced by X-ray crystallography. Biochemistry. 2002;41:9370–5. doi: 10.1021/bi025811p. [DOI] [PubMed] [Google Scholar]
- 40.Egea PF, Rochel N, Birck C, Vachette P, Timmins PA, Moras D. Effects of ligand binding on the association properties and conformation in solution of retinoic acid receptors RXR and RAR. J Mol Biol. 2001;307:557–76. doi: 10.1006/jmbi.2000.4409. [DOI] [PubMed] [Google Scholar]
- 41.Fujisawa T, Kostyukova A, Maeda Y. The shapes and sizes of two domains of tropomodulin, the P-end-capping protein of actin-tropomyosin. FEBS Lett. 2001;498:67–71. doi: 10.1016/s0014-5793(01)02498-x. [DOI] [PubMed] [Google Scholar]
- 42.Lipfert J, Chu VB, Bai Y, Herschlag D, Doniach S. Low resolution models for nucleic acids from small-angle X-ray scattering with applications to electrostatic modeling. J Appl Crystallogr. 2007;40:s229–s234. [Google Scholar]
- 43.Lipfert J, Ouellet J, Norman DG, Doniach S, Lilley DM. The complete VS ribozyme in solution studied by small-angle X-ray scattering. Structure. 2008;16:1357–67. doi: 10.1016/j.str.2008.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lukavsky PJ, Kim I, Otto GA, Puglisi JD. Structure of HCV IRES domain II determined by NMR. Nat Struct Biol. 2003;10:1033–8. doi: 10.1038/nsb1004. [DOI] [PubMed] [Google Scholar]
- 45.Walter F, Murchie AI, Thomson JB, Lilley DM. Structure and activity of the hairpin ribozyme in its natural junction conformation: effect of metal ions. Biochemistry. 1998;37:14195–203. doi: 10.1021/bi981513+. [DOI] [PubMed] [Google Scholar]
- 46.Murchie AI, Thomson JB, Walter F, Lilley DM. Folding of the hairpin ribozyme in its natural conformation achieves close physical proximity of the loops. Mol Cell. 1998;1:873–81. doi: 10.1016/s1097-2765(00)80086-6. [DOI] [PubMed] [Google Scholar]
- 47.Shen Z, Hagerman PJ. Conformation of the central, three-helix junction of the 5 S ribosomal RNA of Sulfolobus acidocaldarius. J Mol Biol. 1994;241:415–30. doi: 10.1006/jmbi.1994.1517. [DOI] [PubMed] [Google Scholar]
- 48.Serganov AA, Masquida B, Westhof E, Cachia C, Portier C, Garber M, Ehresmann B, Ehresmann C. The 16S rRNA binding site of Thermus thermophilus ribosomal protein S15: comparison with Escherichia coli S15, minimum site and structure. RNA. 1996;2:1124–38. [PMC free article] [PubMed] [Google Scholar]
- 49.Tuschl T, Gohlke C, Jovin TM, Westhof E, Eckstein F. A three-dimensional model for the hammerhead ribozyme based on fluorescence measurements. Science. 1994;266:785–9. doi: 10.1126/science.7973630. [DOI] [PubMed] [Google Scholar]
- 50.Bassi GS, Mollegaard NE, Murchie AI, von Kitzing E, Lilley DM. Ionic interactions and the global conformations of the hammerhead ribozyme. Nat Struct Biol. 1995;2:45–55. doi: 10.1038/nsb0195-45. [DOI] [PubMed] [Google Scholar]
- 51.Brion P, Westhof E. Hierarchy and dynamics of RNA folding. Annu Rev Biophys Biomol Struct. 1997;26:113–37. doi: 10.1146/annurev.biophys.26.1.113. [DOI] [PubMed] [Google Scholar]
- 52.Lynch SR, Gonzalez RL, Puglisi JD. Comparison of X-ray crystal structure of the 30S subunit-antibiotic complex with NMR structure of decoding site oligonucleotide-paromomycin complex. Structure. 2003;11:43–53. doi: 10.1016/s0969-2126(02)00934-6. [DOI] [PubMed] [Google Scholar]
- 53.Lafontaine DA, Norman DG, Lilley DM. The global structure of the VS ribozyme. EMBO J. 2002;21:2461–71. doi: 10.1093/emboj/21.10.2461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lilley DM. Structures of helical junctions in nucleic acids. Q Rev Biophys. 2000;33:109–59. doi: 10.1017/s0033583500003590. [DOI] [PubMed] [Google Scholar]
- 55.Parisien M, Major F. The MC-Fold and MC-Sym pipeline infers RNA structure from sequence data. Nature. 2008;452:51–5. doi: 10.1038/nature06684. [DOI] [PubMed] [Google Scholar]
- 56.Das R, Baker D. Automated de novo prediction of native-like RNA tertiary structures. Proc Natl Acad Sci U S A. 2007;104:14664–9. doi: 10.1073/pnas.0703836104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jonikas MA, Radmer RJ, Laederach A, Das R, Pearlman S, Herschlag D, Altman RB. Coarse-grained modeling of large RNA molecules with knowledge-based potentials and structural filters. RNA. 2009;15:189–99. doi: 10.1261/rna.1270809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Svergun D, Barberato C, Koch MH. CRYSOL - a program to evaluate X-ray solution scattering of biological macromolecules from atomic coordinates. J Appl Crystallogr. 1995;28:768–773. [Google Scholar]
- 59.Garst AD, Batey RT. A switch in time: Detailing the life of a riboswitch. Biochim Biophys Acta. 2009;1789:584–91. doi: 10.1016/j.bbagrm.2009.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lang K, Rieder R, Micura R. Ligand-induced folding of the thiM TPP riboswitch investigated by a structure-based fluorescence spectroscopic approach. Nucleic Acids Res. 2007;35:5370–8. doi: 10.1093/nar/gkm580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wachter A, Tunc-Ozdemir M, Grove BC, Green PJ, Shintani DK, Breaker RR. Riboswitch control of gene expression in plants by splicing and alternative 3′ end processing of mRNAs. Plant Cell. 2007;19:3437–50. doi: 10.1105/tpc.107.053645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lipfert J, Millett I, Seifert S, Doniach S. A Sample Holder for Small-Angle X-ray Scattering Static and Flow Cell Measurements. Rev Sci Inst. 2006;77:046108. [Google Scholar]
- 63.Svergun D. Determination of the regularization parameter in indirect-transform methods using perceptual criteria. J Appl Crystallogr. 1992;25:495–503. [Google Scholar]
- 64.Svergun DI. Restoring low resolution structure of biological macromolecules from solution scattering using simulated annealing. Biophys J. 1999;76:2879–86. doi: 10.1016/S0006-3495(99)77443-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kozin M, Svergun D. Automated matching of high and low resolution structural models. J Appl Crystallogr. 2001;34:33–41. [Google Scholar]
- 66.Volkov V, Svergun D. Uniqueness of ab initio shape determination in small-angle scattering. J Appl Crystallogr. 2003;36:860–864. doi: 10.1107/S0021889809000338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wriggers W, Milligan RA, McCammon JA. Situs: A package for docking crystal structures into low-resolution maps from electron microscopy. J Struct Biol. 1999;125:185–95. doi: 10.1006/jsbi.1998.4080. [DOI] [PubMed] [Google Scholar]
- 68.Wriggers W, Chacon P. Using situs for the registration of protein structures with low-resolution bead models from X-ray solution scatterin. J Appl Crystallogr. 2001;34:773–776. [Google Scholar]
- 69.Pettersen E, Goddard T, Huang C, Couch G, Greenblatt M, Meng E, Ferrin E. UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem. 2004;13:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 70.Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. J Mol Graph. 1996;14:33–8. 27–8. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.