

Abstract
Cellular autofluorescence, though ubiquitous when imaging cells and tissues, is often assumed to be small in comparison to the signal of interest. Uniform estimates of autofluorescence intensity obtained from separate control specimens are commonly employed to correct for autofluorescence. While these may be sufficient for high signal-to-background applications, improvements in detector and probe technologies and introduction of spectral imaging microscopes have increased the sensitivity of fluorescence imaging methods, exposing the possibility of effectively probing the low signal-to-background regime. With spectral imaging, reliable monitoring of signals near or even below the noise levels of the microscope is possible if compensation for autofluorescence and background signals can be performed accurately. We demonstrate the importance of accurate autofluorescence modeling and the utility of spectral imaging and multivariate analysis methods using a case study focusing on fluorescence confocal spectral imaging of host-pathogen interactions. In this application fluorescent proteins are produced when Francisella novicida invade host macrophage cells. The resulting analyte signal is spectrally overlapped and typically weaker than the cellular autofluorescence. In addition to discussing the advantages of spectral imaging for following pathogen invasion, we present the spectral properties and cellular origin of macrophage autofluorescence.
Keywords: spectral imaging, fluorescence imaging, Francisella, autofluorescence, spectral crosstalk, multivariate curve resolution, macrophage, fluorescent proteins
Introduction
Fluorescence microscopy is a powerful tool for imaging proteins and their interactions in cells. As bioscience researchers look deeper into the details of cellular machinery, there is an increasing need to monitor lower and lower levels of protein expression. The fundamental limit on sensitivity in the fluorescence microscope is often not the signal-to-noise ratio, but rather the signal-to-background ratio. This background signal arises from spectrally interfering species such as additional labels used in a multicolor experiment, cell growth media, and/or cellular autofluorescence that emit photons in the same spectral range as the analyte of interest, thus contaminating the signal. This phenomenon is commonly referred to as spectral crosstalk or spectral bleed through (SBT). For intense analyte signals, low levels of background interference can be tolerated, but as the analyte signal approaches the detection limit of the microscope as it often does in realistic experiments, accurate measurement and compensation for contaminants are required. Correction for the contaminating fluorescence in fluorescence microscopy has been demonstrated through the use of independent control images and can require up to seven independent images for fluorescence resonance energy transfer based measurements (Periasamy & Day, 2005). In practice the use of control images is tedious and often the fluorescent properties of the interferrant are highly variable and may change with different experimental conditions leading to insufficient background correction. Correcting for cellular autofluorescence can be particularly problematic because of its nonuniform intensity distribution within and between cells, as well as across cell lines.
The development of spectral imaging technology and accompanying spectral unmixing algorithms have promise to alleviate this limitation (Dickinson et al., 2001; Schultz et al., 2001; Keshava & Mustard, 2002; Zimmerman et al., 2003; Timlin et al., 2004; Xu & Rice, 2009). Spectral imaging systems use the entire spectral signature of a fluorescent species to generate images rather than the univariate intensity used in traditional fluorescence microscopy. This provides a sensitivity and specificity advantage if the spectra of all the fluorescing species in an image are accurately modeled using multivariate image analysis capable of mathematically separating the spectral contributions from one another. Spectral imaging and subsequent spectral unmixing have been shown to be effective in removing the effects of autofluorescence for biomedical applications (Mansfield et al., 2005; Nieman et al., 2006; Xu & Rice, 2009). We have previously demonstrated the utility of spectral imaging for removing contaminating fluorescence in DNA microarrays (Martinez et al., 2003; Timlin et al., 2005) and addressed the importance of modeling all the fluorescent species in multicolor imaging of brain tissue (Timlin et al., 2006). Recently, Haaland and coworkers have applied spectral imaging and multivariate curve resolution (MCR) for following dynamic processes in cells and demonstrated quantitative analyses without concern for SBT (Haaland et al., 2007; Davis et al., 2009).
In our current application we are interested in visualizing low levels of protein expression and protein-protein interactions to improve our understanding of virulence mechanisms in the potential biowarfare agent Francisella tularensis. For these studies we are using F. tularensis subspecies novicida strain U112 (hereafter F. novicida) that displays reduced virulence in humans but full virulence in mice. The virulence mechanism of F. tularensis has been recently shown to include a novel Type VI secretion system (T6SS) that appears to be conserved in all subspecies (de Bruin et al., 2007). Our efforts aim to develop advanced molecular biology strategies and low-light imaging methodologies to permit us to assess spatial and temporal patterns of F. novicida virulence proteins during host cell infection. We have chosen for our initial targets the virulence proteins IglA and IglB. These proteins are conserved in all subspecies of F. tularensis and are thought to play a structural role in the formation of the T6SS. For us to provide simultaneous quantification of multiple fluorescent proteins, we have employed a custom built confocal spectral microscope (Sinclair et al., 2006) and employed optimized MCR algorithms to separate the highly overlapped spectra. MCR is a versatile technique for resolving multiway data based on assuming a linear additive relationship between each signal component in the sample (Lawton & Sylvestre, 1971). MCR is thus a desirable approach for performing spectral unmixing on spatial and temporal datasets from chemical systems where Beer’s Law applies. For a review of MCR for spectroscopic applications, please refer to the work of Schoonover and coworkers (2003).
In the case of F. tularensis infection, the resulting fluorescence intensities of the biological processes of interest have weak signals, unfortunately often weaker than the native host cell autofluorescence emission, thus it is critical that we fully understand the impact of autofluorescence on our measurements. The spectral imaging and MCR approach we present in this article provides a tool not only for investigating autofluorescence but also for eliminating its effect on imaging of virulence protein interactions—the goal in this application. Specifically, we demonstrate the use of spectral imaging and MCR to monitor weakly expressed fluorescent Francisella virulence proteins in highly autofluorescent macrophage cells and provide evidence clarifying the cellular origin of inhomogeneously distributed autofluorescence observed in living macrophage cells. These methods have widespread utility in other applications in cell biology where a weak emission needs to be monitored accurately amidst interfering signals.
Methodology
Strains and Growth Conditions
Francisella tularensis subspecies novicida strain Utah 112 (BEI Resources NR-13) was used to generate all transformants in this study. Unless otherwise specified, bacteria were grown in tryptic soy broth (BBL 211768) or tryptic soy agar (BBL 211043) supplemented with 0.1% cysteine (TSBC and TSAC). When required, kanamycin (10 μg/mL) was used. Single bacterial colony isolates were grown in TSBC to mid-log phase. Sterile glycerol was added to 20%, and 0.5 mL aliquots were frozen and stored at −80°C. Freshly-thawed aliquots were used for all infection experiments and were titered by plating dilutions on TSAC plates and counting colonies.
The murine macrophage-like cell line RAW264.7 (ATCC TIB-71) was used as the host for F. novicida infections in these studies. RAW264.7 cells were grown in nontreated culture flasks (Nunc 156800) in “RAWGM1” DMEM (ATCC 30-2002) supplemented with 10% heat-inactivated fetal bovine serium (Gemini Bio-Products 100-500), 20 mM HEPES (Invitrogen 15630080), and 2 mM L-glutamine (Invitrogen 25030081) at 37°C, 5% CO2. For live cell imaging healthy RAW macrophages were either exposed to a 10 nM aqueous solution of LysoTracker Red DND99 (Invitrogen L7526) for 10 min (for correlation of lysosomal markers with autofluorescence) or not (control cells) and placed on microscope slides in media with a cover slip applied. Spectral imaging was performed immediately. Alternatively, for fixed cell imaging cells were infected with F. novicida and fixed as described below.
Fluorescent Proteins
The Francisella shuttle plasmid pFNLTP6 and pFNLTP6-groE-GFP were gifts from Thomas Zahrt of the Medical College of Wisconsin. A cassette containing the F. novicida U112 IglA and IglB genes was synthesized de novo by Epoch Biolabs, Inc., and was cloned into the KpnI/BamHI sites of pFNLTP6. For this study, the mTangerine and mHoneydew (Shaner et al., 2004) genes were synthesized de novo following codon optimization for expression in Francisella. In-frame fusions of mHoneydew at the amino terminus of IglA and mTangerine at the amino terminus of IglB were generated using standard cloning techniques.
Bacterial Transformation
Plasmid DNA was introduced into F. novicida U112 by chemical transformation as previously described (Ludu et al., 2007). Bacteria were first streaked onto day-old TSAC plates supplemented with 0.4% glucose and then transferred to TSBC media with 0.4% glucose and were grown to mid- to late-log phase. Cells were pelleted at 4300 rpm for 15 min and resuspended in 2 mL transformation buffer. Ten microliters of plasmid was mixed with 500 μL cell suspension and incubated at 37°C for 20 min without shaking. After addition of 1 mL TSBC/glucose, tubes were shaken for 2 h at 37°C at 200 rpm. Fifty to 200 μL of mix was plated on fresh TSAC/glucose selection plates. Colonies appeared after several days incubation at 37°C.
Infection of RAW264.7 Cells with Bacteria
RAW264.7 cells were infected with F. novicida U112 transformants as follows: 105 RAW264.7 cells were seeded per well into four well chamber slides or chambered coverglasses (NNI 154917, 155382) in RAWGM1 one day prior to infection. Bacteria were opsonized in 10% mouse complement serum (Innovative Research IMS-COMPL) for 30 min at 37°C for 1 h immediately prior to use. Cells were infected with F. novicida U112 transformants at an input ratio of 10–50 bacteria per cell in 0.5 mL RAWGM1 per well. Chamber slides were centrifuged at 300 g for 5 min at 4°C to promote bacterial adherence to the cells. Following an ingestion incubation of 1 h at 37°C, 5% CO2, adherent cells were gently washed three times with RAWGM; a final 1 mL of RAWGM1 was added per well. Samples were fixed at various times postinfection in 4% paraformaldehyde for 30 min, followed by three PBS washes.Chamber slides were prepared for microscopy by mounting #1.5 glass coverslips in ProLong Gold Antifade reagent (Invitrogen P36934) and sealed.
Spectral Image Acquisition
Spectral image data were collected using a scanning confocal hyperspectral microscope (Sinclair et al., 2006). In this system, fluorescence emission is excited by a continuous wave 488 nm laser and collected over a spectral range of 500 to 800 nm from each voxel using a prism-based spectrometer and the first four rows of a 512 × 512 electron multiplying charge-coupled device (EMCCD) (Andor, Inc., Upper Water-town, SD, USA; the remaining rows are used as spectrum transfer buffers). Three-dimensional scanning is achieved by software integrated motion of a single galvanometer (Cambridge Technology, Inc., Lexington, MA, USA) and a Z-axis piezo stage (Physik Instrumente, Karlsruhe, Germany) coupled to a linear motor driven XY stage (Aerotech, Pittsburgh, PA, USA), providing hybrid-triggered readout (Hing & Muller, 2003) for a maximum sustained data collection rate of 8.3 Mbits/s at a spectral resolution <3 nm. In the described experiments, high-resolution imaging was performed using an infinity corrected 60× objective (Nikon, PlanApo VC NA = 1.40), corresponding to an in-plane voxel dimension of 0.12 μm per side. Spectral arrays of 204 × 204 points were collected at a scan rate of 4,167 spectra per second, for an overall single image collection time of ~10 s. Daily instrument calibration was performed using accepted methods for hyperspectral imaging to ensure reliable microscope performance. Prior to data collection the laser power was measured at the back aperture of the objective using an optical power meter and adjusted to 150 μW, which remained stable over the course of the measurements. The emission wavelength axis was calibrated using the spectral lines from a hollow-cathode Kr+ lamp. A fluorescent polymer standard (Chroma, Inc., Bellows Falls, VT, USA) was imaged daily as an intensity standard for centering the spectral emission on the confocal pinhole. Finally, dark images were collected at regular intervals during data collection to correct for instrument offset and dark current signals originating from the EMCCD.
Data Pretreatment
Prior to analysis all spectral image data were preprocessed to enable more accurate MCR results. The preprocessing is described in complete detail for hyperspectral confocal fluorescence images by Jones and coworkers (2008). In brief, the spectra were first corrected for cosmic events by replacing an affected voxel with the spectrum from a neighboring voxel. Second, dark current contributions were removed using the first eigenvector from principal component analysis (PCA) of a dark current image. Next, an automated offset correction was performed using a region of the spectrum that is within a cut-off filter and thus contains only offset signal. Following this the data were weighted for both Poisson and read noise arising from the EMCCD as described by Jones et al. (2008). Composite images consisting of individual images of the same type were generated for subsequent analysis (i.e., one “super-image” containing all the images of unlabeled living cells, another contained all images of macrophages 7.5 h postinfection with IglB::mTangerine F. novicida, etc.).
MCR Analysis
Following preprocessing, analysis of the composite image was performed using MCR to obtain the pure spectral components and corresponding intensities of the image data. MCR is an iterative numerical analysis technique based on a constrained alternating least-squares algorithm. Details of the MCR algorithm and implementation of rigorous constraints used in this implementation are published elsewhere (Lawton & Sylvestre, 1971; Bro & DeJong, 1997; Tauler et al., 1995; Haaland et al., 2003; Kotula et al., 2003; Van Benthem & Keenan, 2004) and will not be repeated here. We have previously shown the utility of MCR when combined with hyperspectral fluorescence imaging for identifying and quantifying multiple overlapping fluorophores in complex biological samples (Timlin et al., 2005; Sutherland et al., 2007; Vermaas et al., 2008). Although we used our in-house developed MCR algorithms and software that have excellent speed and accuracy with an easy-to-use interface to perform the analyses for this article, MCR software is commercially available from several chemometric software sources (i.e., Camo, Inc., Woodbridge, NJ, USA, see http://www.camo.com/), or refer to Roma Tauler’s website to download an MCR toolbox for Matlab (http://www.ub.edu/mcr/welcome.html).
For the analyses of hyperspectral images of autofluorescence, exogenously introduced LysoTracker Red fluorescence, and virulence protein expression presented in this article, MCR was performed on all pixels in the “super-image” that contained signal at any wavelength >3× the dark current variation. By restricting our analysis to pixels over this threshold, we effectively apply a filter that highlights only the pixels within a cell for spectral analysis. PCA was applied to the resulting image data to determine the number of non-noise-related spectral components present (Golub & Reinsch, 1970). MCR analysis was then initiated with random number-based spectral estimates with spectral and concentration nonnegativity constraints. Within the MCR algorithm, the spectral estimates were normalized to unit length to ensure algorithm stability. PCA of the weighted spectral residuals was performed to confirm the adequacy of the fit to the model of the image data. The resulting spectral estimates are unweighted. Once good spectral estimates were obtained from MCR, the spectral components were rescaled to a maximum emission of 1 and used to perform a final classical least-squares analysis of the image data to obtain the corresponding intensities of each component (Haaland et al., 1985). This was done to facilitate direct visual comparison of the relative intensities of the components and their intensities across different images. All preprocessing and analysis steps were performed using dual-processor desktop computers and custom code written in Matlab (Mathworks, Inc.) or C/C++.
Spatial Correlation of Autofluorescence with LysoTracker Red
The colocalization coefficients of the autofluorescence and LysoTracker Red emission were calculated using the method of Manders and coworkers (1993). MCR component concentrations generated from imaging data from living macrophage cells stained with LysoTracker Red were used for this analysis. Briefly, the independent colocalization coefficients Mauto and MLyso were computed by summing the individual component intensities from voxels where the other component was present and normalizing by the sum of all of the image intensities for that component. The resulting colocalization coefficients are on a scale of 0 to 1, removing the ambiguity involved in negative correlation values. This method is desirable because the independent metrics are insensitive to varying component intensities, photobleaching, detection efficiencies, and the relative number of the objects of each component in the images. As above this analysis was implemented using Matlab (Mathworks, Inc., Natwick, MA, USA) on a dual processor desktop computer.
Results
The pure component spectral estimates from MCR analyses of hyperspectral images containing F. novicida expressing fluorescent proteins [mTangerine::IglB, mHoneydew::IglA, green fluorescent protein (GFP)] and typical macrophage autofluorescence are shown in Figure 1. These spectra were obtained from multiple images from separate experiments of F. novicida challenged cells and are normalized to maximum intensity and overlaid for comparison. It is clear that the macrophage autofluorescence is spectrally overlapped with these fluorescent proteins and would confound attempts to visualize protein localization patterns without the extra dimension of spectral information. It should also be noted that, although not the subject of this article, the fluorescent proteins themselves are spectrally overlapped and contribute spectral crosstalk with each other, requiring spectral imaging to use these in a multicolor experiment.
Figure 1.
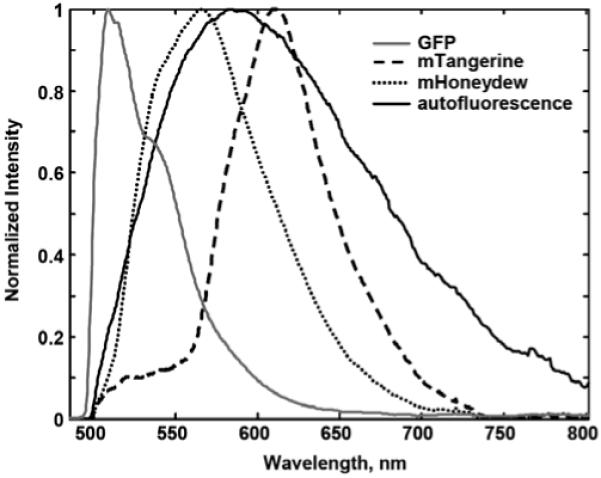
MCR extracted spectral signatures of F. novicida fluorescent proteins (solid gray trace = GFP, dotted black trace = mHoneydew::IglA, dashed black trace = mTangerine::IglB) and macrophage autofluorescence (solid black trace) demonstrating high degree of spectral overlap. Spectra have been normalized to a maximum peak = 1. 84 × 67 mm (300 × 300 dpi).
Typical concentration maps corresponding to macrophage autofluorescence and GFP expression following an overnight infection of RAW cells with F. novicida containing nonprotein targeted GFP expressed off of the GroE promoter (PgroE gfp) (Maier et al., 2004) are shown in Figures 2C and 2D, respectively. For comparison, the total intensity image is shown in Figure 2A and corresponds more closely to what might result from a filter-based microscope equipped with a GFP filter. Focusing on the two areas indicated by the arrows in Figure 2 illustrates a common problem. Without information from the spectra of the fluorescent species, one is forced to look for patterns in this total intensity image that resemble expectations of infecting bacteria. The areas indicated by the arrows in Figure 2A appear very similar in size, morphology, and brightness and as such would likely be assigned to invading bacteria. However, a look at the raw spectral data from these two voxels clearly shows these are not the same fluorescent species (Fig. 2B inset). While one of the arrows does point to an area of GFP expression, the other points to an area of more concentrated macrophage autofluorescence. The MCR analysis of the hyperspectral image data cube successfully separates the spectrally overlapped data (Fig. 2B) into its two underlying fluorescent components and independent concentration maps are calculated for each species shown as the solid lines in Figure 1 (macrophage autofluorescence, Fig. 2C; GFP, Fig. 2D). These maps provide a clearer understanding of the spatial distribution of the fluorescent protein versus macrophage autofluorescence and can be compared in the color overlay format (color overlay of Fig. 2C–E). Visual inspection of the spectral residuals from the MCR model confirms that no additional fluorescent spectral components were present in the images.
Figure 2.

Spatial distribution of groE::GFP expression in F. novicida infected cells. Cells were fixed following infection and prior to imaging. A: Total summed intensity image, equivalent to GFP filter-based fluorescence image. B: Raw spectral data from each voxel in the confocal image. Inset: Spectral data from the voxels indicated by the arrows. C, D: MCR extracted concentration maps of macrophage autofluorescence (C) and groE::GFP expression (D). E: Color overlay of C and D. Color available online only. Arrows indicate areas of interest and are in the same position in each panel. Scale bar = 5 μm. 52 × 42 mm (300 × 300 dpi).
A more comprehensive assessment of the spectral properties and emission intensity of living and fixed RAW macrophage autofluorescence is shown in Figure 3. The autofluorescence spectra identified from fixed and living RAW macrophages (Fig. 3A) were indistinguishable leading to the conclusion that the same chemical species comprise the autofluorescence in both conditions. A likely chemical candidate for the observed autofluorescence signal is the redox cofactor, flavin adenine dinucleotide, which is ubiquitous in the mitochondria of eukaryotic cells, excitable by our 488 nm laser, and known to have broad fluorescence emission over the spectral range observed (Andersson et al., 1998). Though the autofluorescence was found to arise from the same chemical constituent, the spatial arrangement and average intensities are clearly different between fixed and living cells (Fig. 3B,C). Live RAW macrophages exhibit patchy areas of autofluorescence while fixed cells showed a more uniform cytoplasmic distribution and about twice the autofluorescence intensity relative to live cells as indicated by the relative intensity of the images. To address the hypothesis that autofluorescence originates from acidifying organelles in RAW macrophages, experiments were performed with LysoTracker Red, which is known to accumulate in the acidic organelles of cells after 5–30 min of exposure. The colocalization coefficients of the autofluorescence and LysoTracker Red components were calculated. The fluorescence spectrum of LysoTracker Red is shown in Figure 3A and is readily detected at the concentration described (despite low excitation efficiency at 488 nm) along with cellular autofluorescence. Visual observation of the spectra indicate the two species are highly correlated spatially (each voxel contains contributions from both autofluorescence and LysoTracker Red). MCR successfully separates the LysoTracker Red spectrum from autofluorescence even amidst these high levels of spatial colocalization because the two differ in relative intensity from voxel to voxel. The MCR maps show similar patchy signals from autofluorescence and LysoTracker Red (Fig. 3C,D). The spatial correlation between autofluorescence and LysoTracker Red was quantified from a dataset consisting of 12 images of living macrophages stained with LysoTracker Red. The average colocalization coefficient for the autofluorescence component (with the LysoTracker Red component) Mauto was found to be 0.94 ± 0.03, whereas the average colocalization coefficient for the LysoTracker Red component (with the macrophage autofluorescence component) MLyso was found to be 0.96 ± 0.03.
Figure 3.
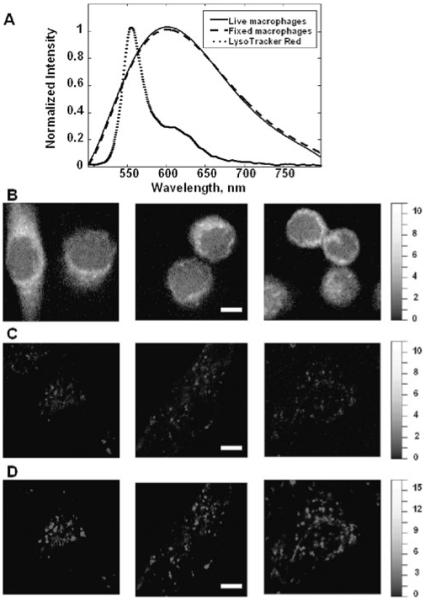
Comparison of autofluorescence properties in living and fixed macrophage cells. A: Spectral signatures recovered from MCR analysis of hyperspectral imaging data. B: MCR extracted concentration map of macrophage autofluorescence in fixed cells. C: MCR extracted concentration map of macrophage autofluorescence in a separate set of living cells. D: MCR extracted concentration map of LysoTracker Red fluorescence from the same cells shown in C. The spectra in A were derived from images in B, C, and D. Scale bar = 5 μm. Intensity scales have been adjusted for display purposes. 61 × 84 mm (300 × 300 dpi).
The relative intensity of macrophage autofluorescence compared to F. novicida protein expression at two different time points after infection is shown in Figure 4. These results are part of a larger experiment designed to follow the levels of F. novicida protein expression in space and time. For this experiment it is important to note that although the relative 488 nm excitation efficiencies differ significantly for the two fluorescence constructs, when the quantum efficiencies and molar absorptivities of the chromophores are considered (Shaner et al., 2004), the total fluorescence emission for mTangerine::IglB is actually ~10% greater than that of mHoneydew::IglA. The additional consideration of the relative detector efficiency at the emission wavelengths of the two fluorescent constructs results in nearly identical number of detected photons of mTangerine::IglB and mHoneydew::IglA (emission from mTangerine::IglB was calculated to be 0–5% greater than mHoneydew::IglA), allowing qualitative comparison of the relative expression levels of these two virulence proteins. Accurate modeling of macrophage autofluorescence and subsequent removal of its contribution from the fluorescent protein images is necessary because of the low levels of protein fluorescence. For example, at the 7.5 h time point, the average intensity levels of IglA and IglB protein expression are of the same magnitude as the autofluorescence. At 20 h postinfection, the average IglA protein expression intensity has increased to levels approximately two to three times that of the autofluorescence and is relatively uniformly distributed throughout the cell. In contrast, at the 20 h time point, the IglB protein expression is on average still on the same order of magnitude as the autofluorescence, but shows small, tight foci of higher concentration. The different spatial and temporal expression patterns observed in these two proteins are hypothesized to play a key role in the organism’s virulence, and a complete study of the biological significance of the distinct spatiotemporal immune response will be the subject of a future publication. Table 1 contains a summary of the image analysis statistics at these two time points. Although only one cell is shown for each of the conditions in Figure 4, the values reported in Table 1 were generated from a larger set containing separate observations of five cells for all time points and proteins except mHoneydew expression at t = 20 h, which was generated from observations of three cells. It should be noted that the Igl and autofluorescence areas reported in Table 1 are not expected to sum to the total cell area as these species are not mutually exclusive in their location.
Figure 4.
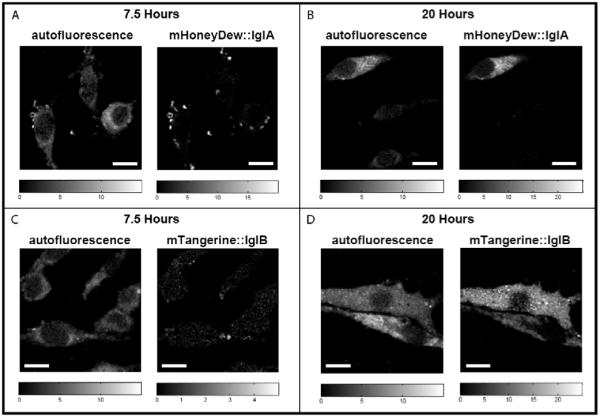
Spatial distribution of mHoneydew::IglA (A, B) and mTangerine::IglB (C, D) expression in F. novicida infected macrophage cells at 7.5 h (A, C) and 20 h (B, D) time points postinfection. Cells were fixed following infection and prior to imaging. Images shown are MCR extracted concentration maps. Scale bar = 5 μm. 101 × 71 mm (300 × 300 dpi).
Table 1.
Summary of the Image Analysis Statistics
| Cell Area (μm2) |
AF Area (μm2) |
Igl Area (μm2) |
% Cell Area with Igl Expression |
Mean AF Intensity (a.u.) |
Mean Igl Intensity (a.u.) |
Image Characteristics |
|
|---|---|---|---|---|---|---|---|
| mTangerine::IglB 7 h | 33.1 | 9.2 | 0.2 | 0.3 | 4 ± 0.4 | 3.8 ± 0.7 | 1–5 individual, isolated bacteria per cell |
| mHoneydew::IglA 7 h | 44.0 | 13.9 | 2.0 | 4.9 | 4.9 ± 0.9 | 11.8 ± 2.7 | 1–5 individual, isolated bacteria per cell |
| mTangerine::IglB 20 h | 82.0 | 51.4 | 54.3 | 61.7 | 5.9 ± 1.1 | 7.8 ± 2.1 | Cells are fully engulfed with bacteria. |
| mHoneydew::IglA 20 h | 54.6 | 28.7 | 29.9 | 51.9 | 9.8 ± 3.4 | 16.6 ± 6.2 | Cells are fully engulfed with bacteria. |
Discussion
The importance of spectral imaging for visualizing low levels of fluorescent virulence protein emission in the presence of highly overlapped cellular autofluorescence is illustrated in Figures 2 and 4. When SBT occurs, a microscopist might attempt to select a different label or a different wavelength region; however, cell and tissue autofluorescence is broad enough to interfere significantly with any of the visible-emitting fluorescent proteins. One exception is the far red fluorescent proteins, though these are often not desirable for imaging because of their tendency to form dimers, low quantum yield, photoinstability, and slow maturation time (Heikel et al., 2001; Shaner et al., 2004). Spectral imaging coupled with MCR allows the ability to extract the spectra of the underlying fluorescent species in these spectrally overlapped images of host-pathogen interactions (without a need to know their spectral features a priori) and is capable of resolving all four of the spectra shown in Figure 1 from the same image. The MCR algorithm assumes a linear additive relationship (Beer’s Law), and the use of constraints (spectral and concentration nonnegativity, etc.) ensures realistic solutions. For two components to be identified as separate species by MCR, they must vary in spectral shape and/or spatial location throughout the image. Using iterative constrained alternating least squares, MCR results in pure spectral components and a corresponding concentration map (component image) for each of the independently varying fluorescent species in the image. The pure component spectra can be thought of as “what” is present in the image, while the concentration maps tell “where” and “how much” of each species is present. Investigation of the spectral residuals provides confidence that the spectral model is valid for new images. If additional species (like an additional autofluorescence spectrum) were present and unaccounted for in the model, large spectral residuals would be observed, indicating the need to add another component to the model. MCR models developed for these images of virulence protein expression resulted in the autofluorescence spectra shown in Figure 1 as well as additional spectra as appropriate to the experiment (i.e., if the unlabeled cells were imaged just the autofluorescence spectrum was used, but if cells plus bacteria were labeled with GFP were imaged then both the GFP and autofluorescence spectra were used to create the model). All models explained >99.8% of the variance in the images. To achieve the accurate models of weak protein fluorescence shown in Figure 4, it was necessary to weight the data for the effects of both Poisson and detector read noise.
Both of the autofluorescence patterns observed in Figure 3 are difficult at best to correct for with traditional background estimation and subtraction methods based on separate control samples. The live cell autofluorescence is even more problematic because it is highly variable within a cell and could not be represented accurately by any single numerical value. In addition, without spectral imaging it would not be possible to determine the location of either the mHoneydew- or mTangerine-labeled bacteria in a living cell because of this patchy, contaminating autofluorescence that is brighter than the protein expression. This would prevent imaging studies from visualizing interactions in the important early stages of infection. At the much later time points, once the bacteria have replicated within the host, the expression intensity levels do increase. At these higher expression levels, it is still important to accurately model the macrophage autofluorescence as Table 1 shows. Consider the IglB expression at 20 h post-infection. Due to the degree of spectral overlap, even at the late time points a filter-based microscope would see these two species as contributing about equally to the signal detected.
It was unexpected that the living cells and fixed cells possess the same spectral signature for autofluorescence. This indicates the same molecular moieties are contributing to the spectrum. The differences in spatial localization and intensity of macrophage autofluorescence shown in Figure 3 are most likely due to the loss of membrane integrity in the fixation process. During fixation organelle membranes become disrupted and the autofluorescent species can leak out, filling the cytoplasm more uniformly. The results may not necessarily be the same if a different cellular system were interrogated or if a different fixation protocol were used. For example, glutaraldehyde fixation is thought to generate new fluorescent species.
Although macrophage autofluorescence is often considered an inconvenience in cell-based imaging applications, it does have a benefit when using a spectral imaging microscope—the autofluorescence signal can be used to generate contrast and “label” acidifing organelles, such as lysosomes and mitochondria, reducing the need for an additional label in an already crowded spectral space. This is readily demonstrated by the strong correlation between autofluorescence and emission from exogenously introduced LysoTracker Red.
Conclusions
In this work we have investigated the spectral properties of macrophage autofluorescence in living and fixed cells using a confocal spectral imaging microscope and MCR to perform the spectral unmixing. We have demonstrated the importance of accurate measurement of macrophage autofluorescence for our specific application following Igl protein expression during F. novicida infection. The relative protein expression levels are of the same magnitude of the autofluorescence at early time points (<10 h post infection) and can even have similar spatial distribution (particularly in living cells), making it impossible to interpret images accurately without the use of spectral imaging and an unmixing analysis to separate the overlapped spectra into their underlying fluorescent species. By developing an accurate model for the autofluorescence, the images of the analytes can be effectively corrected for the autofluorescence contribution. The combination of spectral imaging and MCR employed in this work are widely applicable to many imaging applications where spectral bleed through leads to contaminating signal. In this study we employed the combination of a custom hyperspectral confocal microscope and in-house written MCR software, both available by license or partnership with Sandia National Laboratories. In recent years, however, spectral imaging microscopes have become available from most major microscope vendors and several commercial chemometrics software packages include MCR algorithms. Thus, with adequate understanding of the noise characteristics of the microscope and the trade-off between the available spectral resolution and the resulting discerning power, the methods detailed in this article are achievable in the broader imaging community.
Acknowledgments
The following reagent was obtained through the National Institutes of Health (NIH) Biodefense and Emerging Infections Research Resources Repository, National Institute of Allergy and Infectious Diseases, NIH: F. novicida Type Strain, Strain Utah 112, NR-13. The authors wish to thank Roger Tsien for the fruit fluorescent protein plasmids, Thomas Zhart for the Francisella shuttle plasmids, Bryan Carson and Amanda Carroll-Portillo for helpful discussion about macrophage autofluorescence and cell biology, and the following people for algorithm and software development (listed alphabetically): David M. Haaland, Paul G. Kotula, Michael R. Keenan, Tony Ohlhausen, Greg Poulter, Christopher L. Stork, and Mark H. Van Benthem. This work was supported in part by the Laboratory Directed Research and Development program at Sandia National Laboratories and by the NIH through the NIH Director’s New Innovator Award Program, 1-DP2-OD006673-01 (J.A.T.). Sandia is a multiprogram laboratory operated by Sandia Corporation, a Lockheed Martin Company, for the U.S. Department of Energy under Contract DE-ACO4-94AL85000.
References
- Andersson H, Baechi T, Hoechl M, Richter C. Autofluorescence in living cells. J Microsc. 1998;191:1–7. doi: 10.1046/j.1365-2818.1998.00347.x. [DOI] [PubMed] [Google Scholar]
- Bro R, DeJong SA. A fast non-negativity constrained least squares algorithm. J Chemom. 1997;11:393–401. [Google Scholar]
- Davis RW, Arango DC, Jones HDT, Van Benthem MH, Haaland DM, Brozik SM, Sinclair MB. Antimicrobial peptide interactions with silica bead supported bilayers and E. coli: Buforin II, magainin II, and arenicin. J Pept Sci. 2009;15:511–522. doi: 10.1002/psc.1152. [DOI] [PubMed] [Google Scholar]
- de Bruin OM, Ludu JS, Nano FE. The Francisella pathogenicity island protein IglA localizes to the bacterial cytoplasm and is needed for intracellular growth. BMC Microbiol. 2007;7:1471. doi: 10.1186/1471-2180-7-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickinson ME, Bearman GH, Tille S, Lansford R, Fraser SE. Multi-spectral imaging and linear unmixing add a whole new dimension to laser scanning fluorescence microscopy. Biotechniques. 2001;31:1272–1278. doi: 10.2144/01316bt01. [DOI] [PubMed] [Google Scholar]
- Golub GH, Reinsch C. Singular value decomposition and least squares solutions. Numer Math. 1970;14:403–420. [Google Scholar]
- Haaland DM, Easterling RG, Vopicka DA. Multivariate least-squares methods applied to the quantitative spectral analysis of multicomponent samples. Appl Spectrosc. 1985;39:73–83. [Google Scholar]
- Haaland DM, Jones HDT, Sinclair MB, Carson B, Branda C, Poschet JF, Rebeil R, Tian B, Liu P, Brasier AR. Hyperspectral confocal fluorescence imaging of cells. In: Brown CD, Druy MA, Coates JP, editors. Next-Generation Spectroscopic Technologies. Vol. 6765. SPIE; Boston, MA: 2007. [Google Scholar]
- Haaland DM, Timlin JA, Sinclair MB, Van Benthem MH, Martinez MJ, Aragon AD, Werner-Washburne M. Multivariate curve resolution for hyperspectral image analysis: Applications to microarray technology. In: Levenson RM, Bearman GH, Mahadevan-Jansen A, editors. Spectral Imaging: Instrumentation, Applications, and Analysis. SPIE; San Jose, CA: 2003. [Google Scholar]
- Heikel AA, Hess ST, Baird GS, Tsien RY, Webb WW. Molecular spectroscopy and dynamics of intrinsically fluorescent proteins: Coral red (dsRed) and yellow (Citrine) Proc Nat Acad Sci USA. 2001;97:11996–12001. doi: 10.1073/pnas.97.22.11996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hing P, Muller HW. CCD cameras simplify biology. Biophotonics. 2003;9:52–58. [Google Scholar]
- Jones HDT, Haaland DM, Sinclair MB, Melgaard DK, Van Benthem MH, Pedroso MC. Weighting hyperspectral image data for improved multivariate curve resolution results. J Chemom. 2008;22:482–490. [Google Scholar]
- Keshava N, Mustard JF. Spectral unmixing. IEEE Signal Proc Mag. 2002;19:44–57. [Google Scholar]
- Kotula PG, Keenan MR, Michael JR. Automated analysis of SEM X-Ray spectral images: A powerful new microanalysis tool. Microsc Microanal. 2003;9:1–17. doi: 10.1017/S1431927603030058. [DOI] [PubMed] [Google Scholar]
- Lawton WH, Sylvestre EA. Self modeling curve resolution. Technometrics. 1971;13:617–633. [Google Scholar]
- Ludu JS, Nix EB, Duplantis BN, de Bruin OM, Gallagher LA, Hawley LM, Nano FE. Genetic elements for selection, deletion mutagenesis and complementation in Francisella spp. FEMS Microbiol Lett. 2007;278:86–93. doi: 10.1111/j.1574-6968.2007.00979.x. [DOI] [PubMed] [Google Scholar]
- Maier TM, Havig A, Casey M, Nano FE, Frank DW, Zahrt TC. Construction and characterization of a highly efficient Francisella shuttle plasmid. Appl Environ Microbiol. 2004;70:7511–7519. doi: 10.1128/AEM.70.12.7511-7519.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manders EMM, Verbeek FJ, Aten JA. Measurement of co-localization of objects in dual-color confocal images. J Microsc. 1993;169:375–382. doi: 10.1111/j.1365-2818.1993.tb03313.x. [DOI] [PubMed] [Google Scholar]
- Mansfield JR, Gossage KW, Hoyt CC, Levenson RM. Autofluorescence removal, multiplexing, and automated analysis methods for in-vivo fluorescence imaging. J Biomed Opt. 2005;10:41207. doi: 10.1117/1.2032458. [DOI] [PubMed] [Google Scholar]
- Martinez MJ, Aragon AD, Rodriguez AL, Weber JM, Timlin JA, Sinclair MB, Haaland DM, Werner-Washburne M. Identification and removal of contaminating fluorescence from commercial and in-house printed DNA microarrays. Nucleic Acids Res. 2003;31:e18. doi: 10.1093/nar/gng018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieman LT, Sinclair MB, Timlin JA, Jones HDT, Haaland DM. In: Kovacevic J, Meijering E, editors. Hyperspectral imaging system for quantitative identification and discrimination of fluorescent labels in the presence of autofluorescence; IEEE International Symposium on Biomedical Imaging; Arlington, VA: IEEE. 2006.pp. 1703–1706. [Google Scholar]
- Periasamy A, Day RN. Molecular Imaging. Oxford University Press; New York: 2005. [Google Scholar]
- Schoonover JR, Marx R, Zhang SL. Multivariate curve resolution in the analysis of vibrational spectroscopy data files. Appl Spectrosc. 2003;57:154A–170A. doi: 10.1366/000370203321666461. [DOI] [PubMed] [Google Scholar]
- Schultz RA, Nielsen T, Zavaleta JR, Ruch R, Wyatt R, Garner H. Hyperspectral imaging: A novel approach for microscopic analysis. Cytometry. 2001;43:239–247. doi: 10.1002/1097-0320(20010401)43:4<239::aid-cyto1056>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- Shaner NC, Campbell RE, Steinbach PA, Giepmans BN, Palmer AE, Tsien RY. Improved monomeric red, orange, and yellow fluorescent proteins derived from Dicosoma sp. red fluorescent protein. Nat Biotechnol. 2004;22:1567–1572. doi: 10.1038/nbt1037. [DOI] [PubMed] [Google Scholar]
- Sinclair MB, Haaland DM, Timlin JA, Jones HDT. Hyperspectral confocal microscope. Appl Opt. 2006;45:6283–6291. doi: 10.1364/ao.45.006283. [DOI] [PubMed] [Google Scholar]
- Sutherland V, Timlin JA, Nieman LT, Guzowski JF, Chawla MK, Roysam B, Worley PF, McNaughton BL, Sinclair MB, Barnes CA. Advanced imaging of multiple mRNAs in brain tissue using a custom hyperspectral imager and multivariate curve resolution. J Neurosci Meth. 2007;160:144–148. doi: 10.1016/j.jneumeth.2006.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tauler R, Smilde A, Kowalski B. Selectivity, local rank, three-way data analysis and ambiguity in multivariate curve resolution. J Chemom. 1995;9:31–58. [Google Scholar]
- Timlin JA, Haaland DM, Sinclair MB, Aragon AD, Martinez MJ, Werner-Washburne M. Hyperspectral microarray scanning: Impact and reliability of gene expression data. BMC Genomics. 2005;6:72. doi: 10.1186/1471-2164-6-72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timlin JA, Nieman LT, Jones HDT, Sinclair MB, Haaland DM, Guzowski JF. Imaging multiple endogenous and exogenous fluorescent species in cells and tissues. In: Farkas DL, Nicolau DV, Leif RC, editors. Imaging, Manipulation, and Analysis of Biomolecules, Cells, and Tissues IV. SPIE; San Jose, CA: 2006. [Google Scholar]
- Timlin JA, Sinclair MB, Haaland DM, Martinez MJ, Manginell M, Brozik SM, Guzowski JF, Werner-Washburne M. In: Leahy RM, Roux C, editors. Hyperspectral imaging of biological targets: The difference a high resolution spectral dimension and multivariate analysis can make; IEEE International Symposium on Biomedical Imaging; Arlington, VA: IEEE. 2004.pp. 1529–1532. [Google Scholar]
- Van Benthem MH, Keenan MR. Fast algorithm for the solution of large scale non-negativity constrained least squares problems. J Chemom. 2004;18:441–450. [Google Scholar]
- Vermaas WFJ, Timlin JA, Jones HDT, Sinclair MB, Nieman LT, Hamad SW, Melgaard DK, Haaland DM. In vivo hyperspectral confocal fluorescence imaging to determine pigment localization and distribution in cyanobacterial cells. Proc Natl Acad Sci USA. 2008;105:4050–4055. doi: 10.1073/pnas.0708090105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H, Rice BW. In-vivo fluorescence imaging with a multivariate curve resolution spectral unmixing technique. J Biomed Opt. 2009;14:064011. doi: 10.1117/1.3258838. [DOI] [PubMed] [Google Scholar]
- Zimmerman T, Rietdorf J, Pepperkok R. Spectral imaging and its applications in live cell microscopy. FEBS Lett. 2003;546:87–92. doi: 10.1016/s0014-5793(03)00521-0. [DOI] [PubMed] [Google Scholar]