

Abstract
Craniometaphyseal dysplasia is caused by mutations in ANKH (ankylosis, progressive homolog (mouse)) in the majority of cases, and all of the reported mutations are single amino acid changes. Genomic DNA from an affected patient, his biological parents and a sibling was amplified and ANKH was sequenced. The affected patient had a complex heterozygous mutation in exon 7 (c.936T>C, c.938C>G, c.942_953delTGGTTGACGGAA), predicting p.Try290Gln and p.Trp292_Glu295del. We studied the effect of the predicted mutation on the subcellular distribution of ANKH protein. Immunofluorescent labeling of COS-7 cells transduced with normal or mutant Ank (murine progressive ankylosis), showed that normal Ank localized to both the plasma membrane and cytoplasm, while mutant Ank was detected only in the cytoplasmic compartment. We propose that this craniometaphyseal dysplasia mutation causes a loss of ANKH protein expression and activity in the plasma membrane due to aberrant intracellular protein trafficking.
Keywords: Craniometaphyseal dysplasia, ANKH, deletion mutation, intracellular protein trafficking
Introduction
Craniometaphyseal dysplasia (CMD: OMIM 123000) is a rare genetic bone modeling/remodeling disorder diagnosed by early progressive hyperostosis and sclerosis of craniofacial bones, and metaphyseal widening of long bones [Beighton, 1995]. Excessive bone formation is prominent on paranasal and frontal bones, resulting in readily recognizable facial features such as a flattened nasal bridge and frontonasal bossing. Additional common radiographic findings in patients with CMD include hyperostosis of the cranial base, narrowing of the foramina, and osseous obliteration of paranasal sinuses and mastoid air cells. In addition to facial bone deformities, patients may have facial paralysis and blindness due to cranial nerve compression and hearing loss secondary to a constricted nerve or internal auditory canals [Kim et al., 2005; Sheppard et al., 2003a, b].
Gorlin et al. [1969] sub-classified familial CMD based on its inheritance pattern: milder autosomal dominant and more severe autosomal recessive forms. Craniometaphyseal dysplasia is more often inherited in an autosomal dominant pattern, with the majority of the reported cases belonging to a few large pedigrees [Carnevale et al., 1983; Nurnberg et al., 1997; Rimoin, 1969; Taylor and Sprague, 1989; Tinschert and Braun, 1998]. Six different mutations in the ANKH gene (ANKH; progressive ankylosis protein homolog) have been reported in patients with autosomal dominant CMD in nine of 11 familial and three of eight patients with sporadic CMD [Nurnberg et al., 1997; Reichenberger et al., 2001]. These mutations are predicted to alter the protein sequence by a single amino acid in exons 7-10 of the ANKH gene. ANKH is the human homolog of the mouse progressive ankylosis gene (Ank), which encodes a plasma membrane integral protein Ank involved in the transport of cytosolic inorganic pyrophosphate (PPi) into the extracellular space [Gurley et al., 2006b]. In this study, we evaluated the ANKH gene in a patient with sporadic CMD and performed in vitro analyses to assess the subcellular targeting of the ANKH protein.
Clinical Report
A 6-year-old male patient was initially evaluated in the Pediatric Plastic Surgery clinic for a progressively worsening facial deformity. He was born at full term by spontaneous vaginal delivery without prenatal or perinatal complications. His birth weight was 3.3 kg (∼35th centile) and his birth length was 45 cm (∼3rd centile). At 6 years, his height was 118 cm (∼65th centile) and his weight was 19 kg (∼25th centile); at 9 years, his height was 138 cm (∼75th centile) and his weight was 28.5 kg (∼50th centile). Clinically, he presented with a small nose, a wide and thickened bony nasal bridge, and prominent zygomatic arches (Fig 1A). Computerized tomographic (CT) scan of the skull showed multiple hyperostoses in the occipital calvarium, maxilla, mandible, and nasal bones, and bilateral obliteration of the maxillary sinus and mastoid air cells (data not shown). The patient presented with no apparent signs of middle ear dysfunction, sensorineural hearing loss, or vision loss. This was determined by tympanometry (Type A on both sides), pure tone audiometric air and bone conduction tests (0-15 dB at 250-8k Hz on both ears), speech audiometry (SRT of 8 dB on both ears), and visual acuity test (scored 20/20 for both eyes) performed at 9 years. The patient was in good overall health as well. A lateral radiograph of the patient's ankle showed mild broadening of the tibial metaphyseal plate (Fig 1B). A lateral radiograph of the skull, taken at 9 years (Fig 1C), also showed that the mineral density and thickness of the endosteal bony plate was significantly increased in the cranial vault and mandibular symphysis (Fig 1C). Dental records indicated that the patient's primary teeth were smaller than average and discolored. In a panoramic radiograph at 9 years, most of the primary teeth showed no root resorption nor delayed exfoliation, and further, the radiograph indicates that the permanent teeth were developing normally (Fig 1D). The alveolar bone showed significantly increased mineral density and a lack of typical trabecular bony architecture (Fig 1D). The patient's parents and sibling had no evident features similar to those observed in the patient, although they were not formally examined. Based on the clinical and radiographic examinations, the patient was given a clinical diagnosis of a sporadic form of craniometaphyseal dysplasia.
Figure 1.
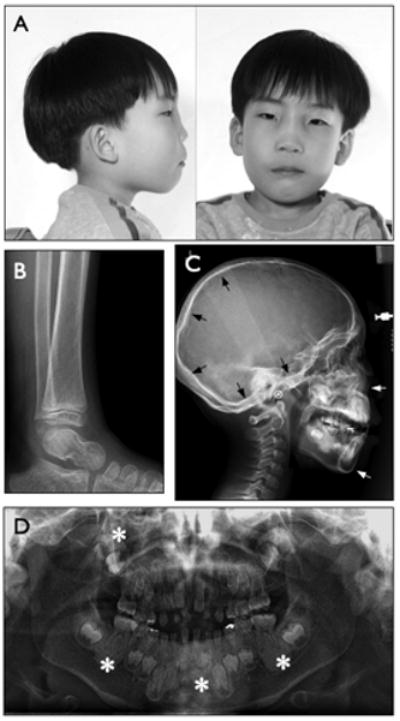
Clinical and radiographic findings from a patient diagnosed with craniometaphyseal dysplasia. A. Lateral and frontal facial photographs taken at 6 years showed thickening and broadening of the nasal bridge. B. Lateral radiograph of the ankle taken at 6 years showed mild flaring of the tibial metaphyseal plate. C. Lateral radiograph of the skull of the patient at 9 years showed hyperostosis of the cranial base, calvarial bones, facial bones, and the alveolar bone. Arrows point to increased endosteal bone density and thickness. D. Panoramic radiograph of developing dentition taken at 9 years showed delayed primary tooth root resorption and permanent tooth eruption. Asterisks point to alveolar bone and mandibular symphysis with unusually high mineral density and loss of typical trabecular bony architecture.
Methods
ANKH mutation analysis
Genomic DNA was isolated from blood samples of the patient, his parents, and his sibling. The DNA was PCR amplified and sequenced for all 12 exons of the ANKH gene. The sequencing primer sets used in this study are derived from intron sequences bordering each exon, as described in earlier studies [Nurnberg et al., 2001; Reichenberger et al., 2001] (See Supplemental Table I). To further characterize the mutation, a PCR amplicon of exon 7 was cloned into the pCR®2.1 vector (Clontech, Mountain View, CA). A total of six independent clones were selected and sequenced. As a complementary approach, a forward primer including two mutated nucleotides (5′-TACCCTGTGGGTCACATGCCACAG-3′), and a reverse primer (5′-AGGGAAGCAGGACTGAGAAGC-3′), derived from exon 7, were used to amplify genomic DNA from the patient and his parents and sibling. The amplified DNA was separated on a 1% agarose gel and visualized by ethidium bromide staining.
The STRP genotyping
The family trio was genotyped for 15 polygenic markers including one sex chromosomal marker using the AmpFISTR Identifiler PCR Amplification kit (Applied Biosystems) to confirm the biological parentage.
Antibody production
An Ank-specific antibody was raised in a rabbit against C-terminal peptide sequence TEEVTDIVEMREENE (Proteintech, Chicago, IL), purified from the antiserum and confirmed for its specificity on immunoblots.
Expression of wild type-Ank and CMD-Ank in COS-7 cells -Ank constructs
A full-length wild type Ank cDNA was cloned into pIRES2-GFP (Clontech, Mountainview, CA). The insert containing Ank-IRES-GFP was then subcloned into the pDNR-CMV donor vector (pDNR-CMV-Ank-IRES-GFP) (Clontech, Mountainview, CA). The CMD mutation reported herein was introduced to a pDNR-CMV-Ank-IRES-GFP clone to prepare the pDNR-CMV-CMD/Ank-IRES-GFP construct, using the Transformer™site-directed mutagenesis kit (Clontech). The COS-7 cell line, which does not express endogenous Ank, was transfected with either pDNR-CMV-Ank-IRES-GFP or pDNR-CMV-CMD/Ank-IRES-GFP construct, using FuGENE 6® transfection reagent (Roche, Basel, Switzerland). The transfected cells were cultured for two days and successful transfection of cells and expression of Ank protein was determined by immunoblot and by immunofluoroscent labeling. For immunofluorescent labeling of Ank protein, transfected cells were fixed in 10% formalin and incubated with the Ank-specific antibody described above and a goat anti-rabbit IgG secondary antibody tagged with FITC.
Results
The ANKH sequences for the parents and sibling of the patient were identical to the reference sequence (GenBank accession: BC014526.2, data not shown). Similarly, the sequencing data from PCR amplified genomic DNA showed that exons 1-6 and 8-12 of the patient's ANKH gene were also normal. The sequencing chromatogram of the patient's exon 7 showed double peaks. (Fig 2A). We therefore predicted that exon 7 comprised heterozygous alleles, and that one of the alleles had an insertion or a deletion. Sequencing data from several independent clones confirmed that the alleles of exon 7 were either wild-type, or contained a novel in-frame mutation (Fig 2 B,C). The mutation was complex and consisted of two substitutions (c.936T>C, c.938C>G) and one deletion (c.942_953delTGGTTGACGGAA), predicting p.Tyr290Gln and p.Trp292_Glu295del separated by one amino acid (Fig 2D). When a primer containing the mutation described above was used for genomic DNA amplification of ANKH exon 7 sequences, a discrete band of an expected size (165 bp) was produced from the patient sample only and not from the parents or sibling's samples (Fig 2E).
Figure 2.
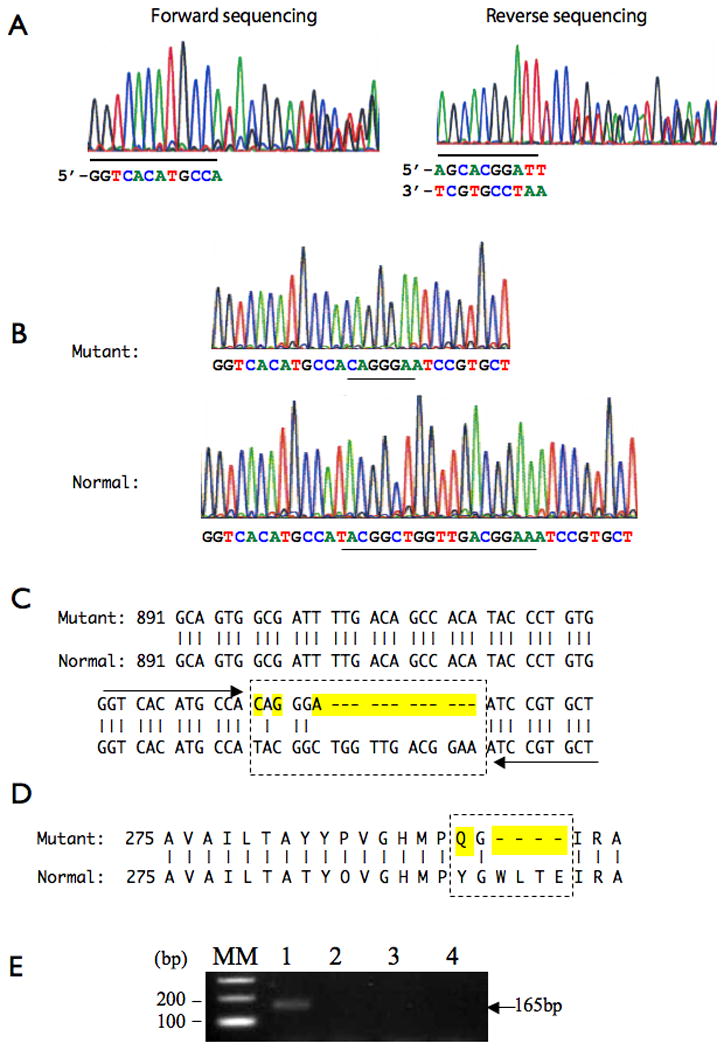
Genomic sequencing and PCR data from a patient with craniometaphyseal dysplasia. A. Exon 7 was PCR amplified from genomic DNA and sequenced using a forward and reverse primer set, derived from 5′ and 3′ intron sequences adjacent to ANKH exon 7. The regions where double picks appear are shown from both forward and reverse sequencing reactions. The sequences preceding or following the double picks are underlined. B. Clonal sequencing data showed that ANKH exon7 of the patient consists of heterologous alleles, one of which contained a mutation. C. Mutated nucleotide sequences are highlighted within a box with dotted line. Arrows indicate the sequence adjacent to the mutation as shown in Fig 2A. D. Predicted amino acid changes in the mutated allele for ANKH exon 7 are highlighted. E. PCR amplification of genomic DNA from the patient (lane1), mother (lane 2), father (lane 3) and a sibling (lane 4) with a primer set containing the CMD mutation resulted in an amplicon of predicted size (165 bp) only from the patient sample (lane 1).
To conclude that the ANKH mutation in our patient was de novo, biological parentage was verified by genotyping the family trio for 15 polygenic markers, including one sex chromosomal marker, as described in the method section. The genotyping results (See Supplemental Table II) were consistent with biological relatedness.
The mutation presented in this report predicts a loss of five amino acids, which could significantly alter protein synthesis, stability, or subcellular localization. As the first step towards understanding the biochemical basis of CMD, we investigated whether ANKH protein (ANK) containing the mutation described in this report could be synthesized by a cell. The human ANKH and murine Ank proteins are highly similar. They are the same in length and differ by only eight amino acids and all known CMD mutations occur in conserved sequences. Thus, murine Ank constructs were used in our expression study and expressed in the COS-7 cell line, which does not express endogenous Ank. The immunoblot data from transduced cell lysates showed that both mutant and normal Ank proteins were produced (Fig 3A). We next asked whether the ANKH protein containing the CMD mutation can still target the plasma membrane. Immunofluorescence microscopy of transduced cells show that wild-type Ank protein is localized in both cytoplasmic compartment and plasma membrane (Fig 3B). In contrast, mutant Ank was distributed only in the intracellular compartment and not on the plasma membrane. The poorly defined cellular outline indicates that the CMD mutation inhibits Ank protein targeting to the plasma membrane (Fig 3C).
Figure 3.
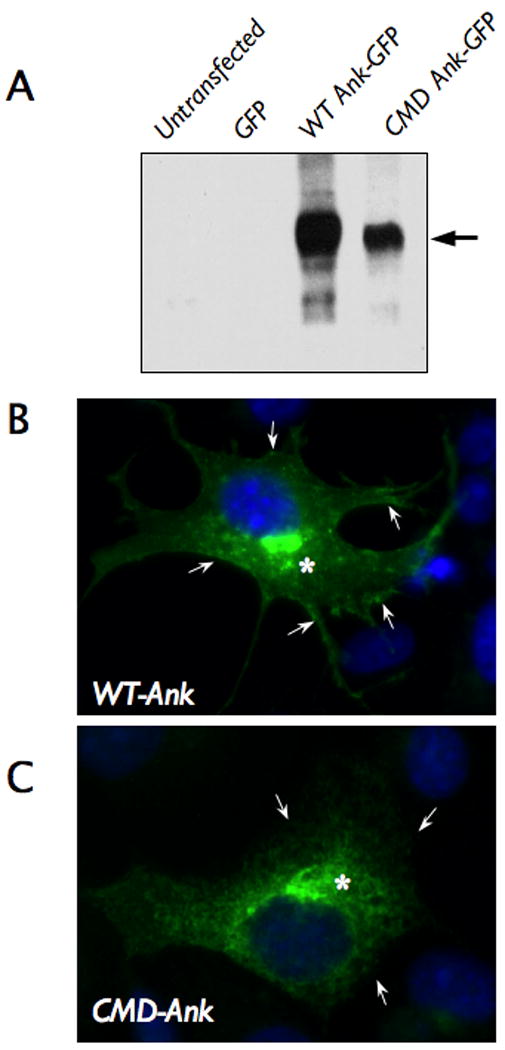
Expression of Ank protein containing the craniometaphyseal dysplasia (CMD) mutation. A. Immunoblot analysis of COS-7 cells transfected with a mammalian expression vector encoding wild type murine Ank and green fluorescent protein (GFP) (WT Ank-GFP), or Ank containing the CMD mutation and GFP (CMD Ank-GFP). Untransfected cells and the cells transfected with a vector expressing GFP alone were used as controls. B&C. Immunofluorescent labeling of cells transfected with WT-Ank or CMD-Ank construct demonstrated that WT-Ank was localized in both plasma membrane (→) and cytoplasm (*) (B), while CMD-Ank was detected only in the cytoplasmic compartment as indicated with arrows (C).
Discussion
The six mutations that cause CMD and the one reported here all occur within exons 7-10, corresponding to the regions comprising the second and third putative cytoplasmic domains from the carboxyl terminal (Table I, Fig 4) [Nurnberg et al., 2001; Reichenberger et al., 2001]. Mutations in the ANKH gene are also linked to an unrelated familial cartilage disease, chondrocalcinosis (CCAL: OMIM 118600), characterized by adult-onset of calcium pyrophosphate dihydrate deposition in the joint space [Gurley et al., 2006b; Pendleton et al., 2002]. These mutations were clustered in the first exon (-11C-T, p.Pro5Leu, p.Pro5Thr, p.Met48Thr) with the exception of a deletion mutation in the C-terminal region (p.Glu490del) (Fig 4) [Gurley et al., 2006b; Pendleton et al., 2002]. The distinct disease phenotypes of CMD and CCAL suggest that these two types of mutations may exert opposite effects on ANK activity.
Table I.
ANKH mutations in craniometaphyseal dysplasia
| Location | DNA changes** | Amino acid changes | Ref | |
|---|---|---|---|---|
| Sporadic | Exon 7 | c.942T>C | p.Trp292Arg | 1 |
| c.936T>C, c.938C>G, c.942_953delTGGTTGACGGAA | p.Tyr290Gln, p.Trp292-Glu295del | * | ||
| Intron 9 | c.1210-4A>G | p.380_V381insAla | 1 | |
| Familial | Exon 8 | c.1059T>C | p.Cys331Arg | 1 |
| Exon 9 | c.1192_1194delCCT | p.Ser375del | 1,2 | |
| c.1196_1198delCTT | p.Phe377del | 1,2 | ||
| Exon 10 | c.1233G>A | p.Gly389Arg | 1,2 |
Nurenberg et al, 2001. Nature Genet 28:37-41
Reichenberger et al, 2001. Am J. Hum. Genet 68:1321-1326
The mutation described in this report
Mutation nomenclature followed human ANKH mRNA sequences entered in GeneBank (AF274753.1)
Figure 4.
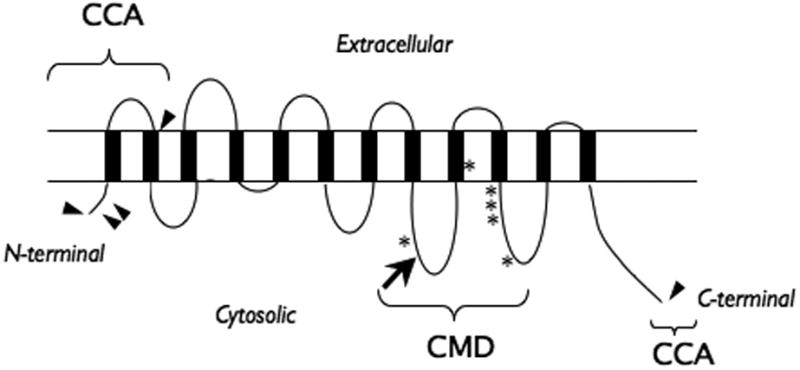
A schematic illustration of the previously described craniometaphyseal dysplasia (CMD) mutations (*), the mutation described in this study (➔) and the mutations linked to chondrocalcinosis (CCA) (▸) on a diagram predicting ANK protein with twelve transmembrane domains.
The role of ANK protein in the regulation of intracellular PPi transport across the plasma membrane has been suggested by biochemical studies that measured intracellular and extracellular PPi levels in cells expressing normal or mutant Ank proteins [Gurley et al., 2006b; Ho et al., 2000]. Over expression of wild type Ank or Ank with CCAL mutations leads to an increase in the extracellular PPi concentration and a decrease in the intracellular PPi concentration [Gurley et al., 2006b; Ho et al., 2000], offering a plausible explanation for PPi crystal formation in the joint space of patients with ANKH mutations that cause CCAL. In contrast, Ank with CMD mutations had an opposite effect on PPi levels in cultured cells [Gurley et al., 2006b]. Our immunofluorescence labeling study suggests that the reduced extracellular PPi concentration in CMD cells may be the result of an interrupted intracellular protein trafficking leading to decreased levels of ANKH targeting the plasma membrane. Extracellular PPi negatively regulates bone matrix mineralization by suppressing hydroxyapatite crystal formation to prevent excessive mineralization during bone remodeling. Therefore, a reduced extracellular PPi level may be responsible for the osteosclerosis associated with CMD mutations. Craniometaphyseal dysplasia is associated with defective bone modeling or remodeling due to reduced bone resorption [Yamamoto et al., 1993]. It is possible that a hypermineralized bone matrix is less susceptible to osteoclastic activity, favoring bone formation over bone resorption during bone remodeling. Indeed, a recently generated knock-in mouse model expressing a CMD mutation (p.Phe377del) showed reduced osteoclast activities [Chen et al., 2009]. The CMD mutations may also influence osteoblast activity. A recent in vitro study showed that over-expression of ANK stimulated osteoblast differentiation [Kirsch et al., 2009]. It is plausible that CMD mutations may slow down osteogenic differentiation, leading to an accumulation of proliferating osteogenic cell populations and ultimately an increase in bone mass. Further studies are needed to fully elucidate the roles of ANK in bone modeling/remodeling.
Autosomal dominant CMD mutations are generally regarded as loss of function mutations, which would translate to haploinsufficiency of ANK activity. However, haploinsufficiency alone does not seem to entirely account for the CMD phenotype. It has been reported that haploinsufficiency of Ank in mice (Ank +/-) does not cause abnormal skeletal phenotypes, while mice completely lacking Ank expression (Ank -/-) display skeletal characteristics similar, yet not identical, to autosomal dominant CMD phenotypes in humans [Gurley et al., 2006a]. Furthermore, heterozygous and homozygous knock-in mice harboring a CMD mutation both displayed the skeletal phenotype expected from a CMD mutation in a mutant gene dosage dependent manner and their phenotype was not identical to that of Ank null mice [Chen et al., 2009]. One possible mechanism of CMD is that mutations in the ANKH gene result in a loss of ANKH protein expression and activity in the plasma membrane, while allowing gain of aberrant function, due to mis-targeting of ANK to subcellular compartments, as seen in some other mutant proteins [Carr et al., 2007; Datta et al., 2009; Zhao et al., 2008].
In summary, we report on a novel in-frame mutation affecting multiple amino acids in exon 7 of the ANKH gene found in a young male patient clinically diagnosed with a sporadic autosomal dominant form of CMD. We also present in vitro data suggesting that the CMD mutation alters intracellular trafficking of the ANKH protein. While increasing evidence suggests the importance of ANK activity in normal and abnormal development of the skeleton, the exact mechanism by which ANK regulates the bone remodeling process remains to be clarified.
Supplementary Material
Acknowledgments
We are grateful to the family for their cooperation and permission to report on their clinical features. This study was supported in part by NIH/NIAMS grant AR50627 to H.D.N.
References
- Beighton P. Craniometaphyseal dysplasia (CMD), autosomal dominant form. J Med Genet. 1995;32:370–374. doi: 10.1136/jmg.32.5.370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carnevale A, Grether P, del Castillo V, Takenaga R, Orzechowski A. Autosomal dominant craniometaphyseal dysplasia. Clinical variability. Clin Genet. 1983;23:17–22. doi: 10.1111/j.1399-0004.1983.tb00431.x. [DOI] [PubMed] [Google Scholar]
- Carr G, Sayer JA, Simmons NL. Expression and localisation of the pyrophosphate transporter, ANK, in murine kidney cells. Cell Physiol Biochem. 2007;20:507–516. doi: 10.1159/000107534. [DOI] [PubMed] [Google Scholar]
- Chen IP, Wang CJ, Strecker S, Koczon-Jaremko B, Boskey A, Reichenberger EJ. Introduction of a Phe377del mutation in ANK creates a mouse model for craniometaphyseal dysplasia. J Bone Miner Res. 2009;24:1206–1215. doi: 10.1359/JBMR.090218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datta R, Waheed A, Bonapace G, Shah GN, Sly WS. Pathogenesis of retinitis pigmentosa associated with apoptosis-inducing mutations in carbonic anhydrase IV. Proc Natl Acad Sci U S A. 2009;106:3437–3442. doi: 10.1073/pnas.0813178106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorlin RJ, Spranger J, Koszalka MF. Genetic Craniotubular bone dysplasia and hyperostoses. A critical analysis. Birth Defects. 1969;5:17. [Google Scholar]
- Gurley KA, Chen H, Guenther C, Nguyen ET, Rountree RB, Schoor M, Kingsley DM. Mineral formation in joints caused by complete or joint-specific loss of ANK function. J Bone Miner Res. 2006a;21:1238–1247. doi: 10.1359/jbmr.060515. [DOI] [PubMed] [Google Scholar]
- Gurley KA, Reimer RJ, Kingsley DM. Biochemical and genetic analysis of ANK in arthritis and bone disease. Am J Hum Genet. 2006b;79:1017–1029. doi: 10.1086/509881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho AM, Johnson MD, Kingsley DM. Role of the mouse ank gene in control of tissue calcification and arthritis. Science. 2000;289:265–270. doi: 10.1126/science.289.5477.265. [DOI] [PubMed] [Google Scholar]
- Kim YH, Roh DH, Choi BY, Oh SH. Craniometaphyseal dysplasia. Acta Otolaryngol. 2005;125:797–800. doi: 10.1080/00016480510028474. [DOI] [PubMed] [Google Scholar]
- Kirsch T, Kim HJ, Winkles JA. Progressive ankylosis gene (ank) regulates osteoblast differentiation. Cells Tissues Organs. 2009;189:158–162. doi: 10.1159/000151725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nurnberg P, Thiele H, Chandler D, Hohne W, Cunningham ML, Ritter H, Leschik G, Uhlmann K, Mischung C, Harrop K, et al. Heterozygous mutations in ANKH, the human ortholog of the mouse progressive ankylosis gene, result in craniometaphyseal dysplasia. Nat Genet. 2001;28:37–41. doi: 10.1038/ng0501-37. [DOI] [PubMed] [Google Scholar]
- Nurnberg P, Tinschert S, Mrug M, Hampe J, Muller CR, Fuhrmann E, Braun HS, Reis A. The gene for autosomal dominant craniometaphyseal dysplasia maps to chromosome 5p and is distinct from the growth hormone-receptor gene. Am J Hum Genet. 1997;61:918–923. doi: 10.1086/514880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pendleton A, Johnson MD, Hughes A, Gurley KA, Ho AM, Doherty M, Dixey J, Gillet P, Loeuille D, McGrath R, et al. Mutations in ANKH cause chondrocalcinosis. Am J Hum Genet. 2002;71:933–940. doi: 10.1086/343054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reichenberger E, Tiziani V, Watanabe S, Park L, Ueki Y, Santanna C, Baur ST, Shiang R, Grange DK, Beighton P, et al. Autosomal dominant craniometaphyseal dysplasia is caused by mutations in the transmembrane protein ANK. Am J Hum Genet. 2001;68:1321–1326. doi: 10.1086/320612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rimoin DL, Woodruff SL, Holman BL. Craniometaphyseal dysplasia (Pyle's disease): autosomal dominant inheritance in a large kindred. Birth Defects Orig Art Ser V. 1969:9. [Google Scholar]
- Sheppard WM, Shprintzen RJ, Tatum SA, Woods CI. Craniometaphyseal dysplasia: a case report and review of medical and surgical management. Int J Pediatr Otorhinolaryngol. 2003a;67:71–77. doi: 10.1016/s0165-5876(02)00289-6. [DOI] [PubMed] [Google Scholar]
- Sheppard WM, Shprintzen RJ, Tatum SA, Woods CI. Craniometaphyseal dysplasia: a case report and review of medical and surgical management. Int J Pediatr Otorhinolaryngol. 2003b;67:687–693. doi: 10.1016/s0165-5876(03)00133-2. [DOI] [PubMed] [Google Scholar]
- Taylor DB, Sprague P. Dominant craniometaphyseal dysplasia--a family study over five generations. Australas Radiol. 1989;33:84–89. doi: 10.1111/j.1440-1673.1989.tb03242.x. [DOI] [PubMed] [Google Scholar]
- Tinschert S, Braun HS. Craniometaphyseal dysplasia in six generations of a German kindred. Am J Med Genet. 1998;77:175–181. doi: 10.1002/(sici)1096-8628(19980518)77:3<175::aid-ajmg1>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- Yamamoto T, Kurihara N, Yamaoka K, Ozono K, Okada M, Yamamoto K, Matsumoto S, Michigami T, Ono J, Okada S. Bone marrow-derived osteoclast-like cells from a patient with craniometaphyseal dysplasia lack expression of osteoclast-reactive vacuolar proton pump. J Clin Invest. 1993;91:362–367. doi: 10.1172/JCI116194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Matthies DS, Botzolakis EJ, Macdonald RL, Blakely RD, Hedera P. Hereditary spastic paraplegia-associated mutations in the NIPA1 gene and its Caenorhabditis elegans homolog trigger neural degeneration in vitro and in vivo through a gain-of-function mechanism. J Neurosci. 2008;28:13938–13951. doi: 10.1523/JNEUROSCI.4668-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.