

Abstract
Concise asymmetric total syntheses of vindoline (1) and vindorosine (2) are detailed based on a unique intramolecular [4+2]/[3+2] cycloaddition cascade of 1,3,4-oxadiazoles inspired by the natural product structures. A chiral substituent on the tether linking the dienophile and oxadiazole was used to control the facial selectivity of the initiating Diels–Alder reaction and set the absolute stereochemistry of the remaining six stereocenters in the cascade cycloadduct. This key reaction introduced three rings and four C–C bonds central to the pentacyclic ring system setting all six stereocenters and introducing essentially all the functionality found in the natural products in a single step. Implementation of the approach for the synthesis of 1 and 2 required the development of a ring expansion reaction to provide a 6-membered ring suitably functionalized for introduction of the Δ6,7-double bond found in the core structure of the natural products. Two unique approaches were developed that defined our use of a protected hydroxymethyl group as the substituent that controls the stereochemical course of the cycloaddition cascade. In the course of these studies, several analogues of vindoline were prepared containing deep-seated structural changes presently accessible only by total synthesis. These analogues, bearing key modifications at C6–C8, were incorporated into vinblastine analogues and used to probe the unusual importance (100-fold) and define the potential role of the vinblastine Δ6,7-double bond.
Introduction
Vindoline (1),1,2 a major alkaloid of Cantharanthus roseus, constitutes the more complex lower half of vinblastine (4)2–5 and serves as both a biosynthetic3,4 and synthetic6,7 precursor to this important natural product (Figure 1). Vinblastine (4) and vincristine (5) represent the most widely recognized members of the vinca alkaloids as a result of their clinical use as antitumor drugs. Originally isolated in trace quantities from Cantharanthus roseus (L.) G. Don,3 their biological properties were among the first to be shown to arise from inhibition of microtubule formation and mitosis that today is still regarded as one of the more successful drug targets for the treatment of cancer.3–5 We previously reported the development of a concise total synthesis of (−)- and ent-(+)-vindoline8–10 enlisting a powerful intramolecular tandem [4+2]/[3+2] cycloaddition cascade of 1,3,4-oxadiazoles with resolution of a key intermediate,11 its extension to the preparation of a series of related natural products including vindorosine (2),10,12 and the subsequent development of a biomimetic Fe(III)-promoted coupling with catharanthine (3) for their single step incorporation into total syntheses of vinblastine, vincristine, and related natural products.6f
Figure 1.
Natural product structures.
Herein, we report full details13 of the development of asymmetric total syntheses of vindoline (1) and the related natural product vindorosine (2) based on an additional implementation of the tandem [4+2]/[3+2] cycloaddition reaction in which the tether linking the initiating dienophile and oxadiazole bears a chiral substituent that sets absolute stereochemistry of the remaining six stereocenters in the cascade cycloadducts (Figure 2).
Figure 2.

Key elements of the initial synthetic strategy.
Relative to our early work,10 the dienophile linking tether was reduced in length by one carbon insuring that the initiating Diels–Alder reaction could be conducted under milder conditions11 than previously observed. More significantly, this change permitted the effective control of the facial selectivity of the initiating Diels–Alder reaction and subsequent transmission of the attached substituent stereochemistry throughout the newly constructed pentacyclic ring system that was not observed with a longer dienophile tether.10 The approach required that the activating acyl chain carbonyl now reside in the dipolarophile tether and that the initiating Diels–Alder reaction of the cycloaddition cascade afford a fused 5-membered ring. A subsequent ring expansion to provide the unsaturated 6-membered ring found in the core structure of the natural products was anticipated to be accomplished by using a hydroxymethyl side chain substituent that, upon alcohol activation for displacement, would undergo aziridinium ion formation and subsequent nucleophilic attack to provide a more stable 6- versus 5-membered ring suitably functionalized for introduction of the Δ6,7-double bond. As detailed herein, this rearrangement is subject to stereoelectronic control for both the aziridinium ion formation as well as its subsequent cleavage by nucleophilic attack, the latter of which also proved to be subject to kinetic and thermodynamic control of the regioselectivity. Investigations into these issues provided a unique and remarkably concise approach to the total synthesis of the natural products, clarified an important stereochemical requirement for the final regioselective elimination, and ultimately led to the development of an additional second unanticipated approach to the key ring expansion.
Results and Discussion
Cycloaddition substrate preparation and examination of the cycloaddition cascade
The most important question addressed in initial studies was the stereochemical fate of the key cycloaddition cascade. In prior studies, we observed significant differences in the rate and facility of the reaction that was dependent on the stereochemistry of the initiating dienophile. Although both (Z)- and (E)-enol ethers proved successful at initiating the cycloaddition cascade, the reaction rate for the (Z)-isomer, providing directly the correct C4 stereochemistry, was considerably slower than the corresponding (E)-isomer, the latter of which required an inversion of the cycloadduct C4 sterochemistry for use in the targeted synthesis.10 Accordingly, substrates bearing both the (Z)-enol ether illustrated in Figure 2 as well as the corresponding (E)-enol ether were prepared and examined (Scheme 1). The side chain chirality was set using aspartic acid as the starting material (both enantiomers prepared, natural enantiomer series shown). Teoc protection of D-H2N-Asp(OBn)-OH (6, quant.) followed by mixed anhydride formation (i-BuOCOCl, NMM, DME, −15 °C, 15 min) and its reduction (NaBH4, H2O, −20 °C) provided the primary alcohol 8 (91%), which was protected as its MOM ether 9 (MOMCl, i-Pr2NEt, CH2Cl2, 25 °C, 2 h, 84%).14 Benzyl ester hydrogenolysis (H2, 10% Pd/C, THF, 25 °C, 0.5 h), coupling of the crude carboxylic acid with N,O-dimethylhydroxylamine (1.2 equiv EDCI, DMAP, i-Pr2NEt, CH2Cl2, 0–25 °C, 16 h, 94% from 9), and reaction of the Weinreb amide 1015,16 with EtMgBr (3 equiv, 3 equiv of CeCl3, THF, 0 °C, 1 h, quant.) cleanly provided the ethyl ketone 11. Notably, the reaction of the Grignard reagent with a substrate closely related to 10 (–OTBS vs –OMOM) in the absence of CeCl3 was considerably less effective (67% vs quant.).17 Wittig olefination with Ph3P=CHOBn18 provided a 1:1 mixture of the separable (E) and (Z) enol ethers 12, which were independently carried through the subsequent 3–4 steps for oxadiazole formation and the preparation of (E)- and (Z)-15. Thus, their Teoc deprotection (Bu4NF) and treatment of the crude amines with carbonyldiimidazole afforded (Z)- and (E)-13 (86–100%) that were converted to the oxadiazole precursors 14 by treatment with methyl oxalylhydrazide19 in the presence of HOAc. Cyclization to form the corresponding oxadiazoles 15 was mediated by TsCl and Et3N (CH2Cl2, 94–97%). Coupling of (Z)-15 and (E)-15 with 2-(1-methylindol-3-yl)acetic acid (16) provided the substrates (Z)-17 and (E)-17, respectively, with which the initial examination of the cycloaddition cascade was conducted. Although they lack the aryl methoxy substituent required for the total synthesis of vindoline, they are substrates that lead to vindorosine and were judged to be ideal surrogates for detailed examination of the key cycloaddition reaction.
Scheme 1.

As detailed in early studies, trisubstitution on the dienophile typically precludes [4+2] cycloaddition initiation of the reaction cascade.11 The exception to this generalization is the use of trisubstituted olefins bearing an electron-donating substituent to activate the tethered dienophile for participation in an inverse electron demand Diels–Alder reaction with the electron-deficient 1,3,4-oxadiazole. The additional shortening of the dienophile tether length from four to three atoms was expected to further enhance this reactivity leading to a cycloaddition cascade that would proceed under milder conditions than previously observed. Thus, the electron-rich dienophiles of (Z)-17 and (E)-17 were judged to be well suited for initiation of the cycloaddition cascade enhancing the intrinsic reactivity of the dienophile, reinforcing the [4+2] cycloaddition regioselectivity dictated by the dienophile tether, and introducing the C4 alkoxy substituent. The distinction in the two substrates is that (Z)-17 permits the direct introduction of the naturally occurring C4 stereochemistry, whereas (E)-17 provides the C4 isomer requiring a subsequent inversion of configuration at this center. Cyclization of (E)-17 proceeded cleanly and effectively providing essentially or predominantly a single diastereomer 20 in superb conversions (72%, xylene, 145–150 °C, 10 h) and only small amounts (0–13%) of a second diastereomer were occasionally isolated (Scheme 2). Notably, the temperature needed to initiate the cycloaddition cascade is lower (145 vs 180 °C) and the reaction time required for complete reaction is shorter (10 vs 24 h) than those observed with substrates bearing a longer dienophile tether.10 In fact, (E)-17 possessed sufficient reactivity that cycloaddition was observed even in refluxing toluene (110 °C), albeit requiring longer reaction times (e.g., 50% 20 at 66 h). Diastereoselective reductive cleavage of the oxido bridge in 20 was effected by treatment with NaCNBH3 (2 equiv, 20% HOAc/i-PrOH, 0–25 °C, 40 min, 94%) in a reaction that proceeds by acid-catalyzed generation of an acyliminium ion flanked by two quaternary centers that is subsequently reduced by hydride addition to the less hindered convex face, and provided 21 whose structure and stereochemistry were confirmed in a single crystal X-ray structure determination.20 Following initial studies in which 20 was characterized and as we scaled up the reaction sequence, it proved most convenient to run the cycloaddition reaction and subsequent reductive oxido bridge cleavage without the intermediate purification of 20, providing 21 directly in good overall conversions (70% for the two steps). By contrast and consistent with expectations,10 the tandem cycloaddition of (Z)-17 proved more challenging to implement. After considerable exploration, (Z)-17 was found to provide the desired cycloadduct 18 as the major diastereomer (6–10:1 dr) when warmed in o-Cl2C6H4 (140 °C, 20 h, 51–57%). Like substrate (E)-17, the reaction is initiated at the lower reaction temperatures (140 °C), but satisfactory conversions required more dilute reaction concentrations (0.1 mM vs 1–5 mM) to avoid competitive intermolecular reactions in the slower 1,3-dipolar cycloaddition. Because the immediate cycloadduct 18 proved somewhat unstable to the conditions of chromatographic purification, the crude product was typically subjected to reductive oxido bridge cleavage (NaCNBH3, HCl, THF/i-PrOH, 0 °C, 1 min, 99%) providing 19 directly in yields up to 61% for the two step conversion. In this case, the corresponding C18 methoxy N,O-ketal was isolated if the reaction was conducted in MeOH (vs i-PrOH) resulting from oxido bridge cleavage and solvent trap of the iminium ion without reduction and more extended reaction times or higher reaction temperatures can lead to increasing amounts of MOM ether deprotection (0–10%) under the mildly acidic conditions employed.
Scheme 2.

The reaction cascade is initiated by [4+2] cycloaddition of the 1,3,4-oxadiazole with the tethered electron-rich enol ether whose reactivity and regioselectivity are matched to react with the electron-deficient oxadiazole in an inverse electron demand Diels–Alder reaction (Figure 2). Loss of N2 from the initial cycloadduct provides a carbonyl ylide, which undergoes a subsequent 1,3-dipolar cycloaddition with the tethered indole.21 The diene and dienophile substituents complement and reinforce the [4+2] cycloaddition regioselectivity dictated by the linking tether, the intermediate 1,3-dipole is stabilized by the complementary substitution at the dipole termini, and the intrinsic regioselectivity of the attached dipolarophile (indole) reinforces the [3+2] cycloaddition regioselectivity that is set by its linking chain. The chiral substituent on the dienophile tether effectively controls the facial selectivity of the initiating [4+2] cycloaddition reaction preferring that the protected hydroxylmethyl group at C7 and the C5 ethyl group reside trans to one another on the newly formed 5-membered ring avoiding a cis pseudodiaxial-1,3-interaction on the sterically more congested concave face of the transition state leading to the initial [4+2] cycloadduct. This establishes the absolute stereochemistry at C5 which in turn is transmitted throughout the cascade cycloadduct where the remaining relative stereochemistry is controlled by a combination of the dienophile geometry (C4 and C5 stereochemistry) and an endo indole [3+2] cycloaddition sterically directed to the face opposite the newly formed 5-membered ring.10–12 The minor diastereomer derived from the cycloaddition of (E)-17 that was occasionally observed and isolated (>7–15:1) is derived from exo indole [3+2] cycloaddition on the face of the 1,3-dipole opposite the newly formed 5-membered ring (C2/C11 diastereomer) indicating that the facial selectivity for the initiating Diels-Alder reaction is >20:1 (detection limits), whereas the minor diastereomer isolated from the cycloaddition of (Z)-17 (6–10:1) is derived from the alternative [4+2] cycloaddition facial selectivity and has the C7 protected hydoxymethyl group and C5 ethyl cis to one another. The latter minor diastereomer as well as 18 both are recovered unchanged when independently resubjected to thermal reaction conditions as pure samples (175 °C, 8 h), whereas the former pure minor diastereomer isolated from the reaction of (E)-17 slowly converts to 20 (80–98%, whereas pure 20 is recovered unchanged) indicating that, in selected instances, the 1,3-dipolar cycloaddition may be thermally reversible, partitioning to the thermodynamically more stable reaction product (ΔE = >4.9kcal/mol for 20, MM2). Significantly, this latter observation accounts for the fact that this minor diastereomer was not always observed, having been funneled on to the thermodynamically more stable product because of the reaction conditions employed. With recognition of this unique ability to funnel this minor diastereomer into the desired diastereomer 20 under thermal conditions, the reaction conditions employed for (E)-17 were adjusted to first promote the cycloaddition cascade (125–140 °C, 8 h, xylene), then warmed at a higher temperature of 175 °C (8 h) to promote the further in situ conversion of the minor diastereomer to 20 (74% overall following oxido bridge cleavage).
Although the stereochemical assignments were firmly established by X-ray analysis of various intermediates described herein including 21,20 detailed spectroscopic analysis of the cycloaddition products 18 and 20, as well as their minor diastereomers, confirmed the assignments distinguishing the two series. The cycloadduct 18 exhibited diagnostic NOE’s of C5-Et with C4-H, C6-Hα, C7-H and C13-H and of C4-H with N1-Me placing them all on the α-face of the structure, as well as between C2-H/C10-Hβ, C6-Hα/C7-H and C6-Hβ/C19-H2. Its minor diastereomer, enantiomeric in structure with 18 except for the C7 stereochemistry, exhibited diagnostic NOE’s of C5-Et with C4-H, C6-Hβ, C19-H2 and C13-H and of C4-H with N1-Me placing them all on the β-face of the structure, as well as between C2-H/C10-Hα, C6-Hα/C7-H, and C6-Hβ/C19-H2. The cycloadduct 20 exhibited diagnostic NOE’s of C5-Et with C4-OBn, C6-Hα, C7-H, and C13-H and failed to show a C4-H/N1-Me NOE. Instead, C4-H exhibited an NOE with C6-Hβ and the expected NOE’s of C6-Hβ/C19-H2 and C6-Hα/C7-H were observed. Its minor diastereomer, epimeric only at C2 and C11, displayed diagnostic NOE’s of C5-Et with C2-H and C10-Hα and no longer displayed an NOE with C13-H. Otherwise, the diagnostic NOE’s of 20 characterizing the C3–C7 stereochemistry were also observed with this minor diastereomer (C5-Et with C4-OBn, C6-Hα, and C7-H, C4-H/C6-Hβ, C6Hβ/C19-H2, and C6-Hα/C7-H).
Asymmetric total synthesis of vindorosine
With 19 and 20 in hand, their conversion to vindorosine was explored as a prelude to efforts on vindoline itself. Vindorosine (2)22 is among the most highly functionalized and stereochemically rich natural products within the family of alkaloids isolated from the Madagascan periwinkle (Catharanthus roseus (L.) G. Don). It is identical in structure to vindoline with the exception that it lacks the C16 methoxy substituent. As a consequence, it has been the subject of a series of beautiful and historically important total syntheses.10,23
The two cycloadducts 19 and 21 were converted to the same key intermediate 24 by hydrogenolysis of benzyl ether 19 to liberate the free alcohol or benzyl ether hydrogenolysis (86%), oxidation of the free alcohol 22 to the ketone 23 (IBX, DMSO, 25 °C, 3.5 h, 78%) and diastereoselective ketone reduction (LiAl(OtBu)3H, THF, 0 °C, 12 h, 90%, 10:1 dr) from the less hindered convex face for 21 (Scheme 3).24,25 Subsequent C4 alcohol acetylation (Ac2O, DMAP, pyridine, 25 °C, 1 h, 98%) provided 25. Consequently, the routes to vindorosine from either 19 or 21 converged requiring only the additional two steps of oxidation and reduction for the inversion of the C4 stereochemistry for utilization of 21.
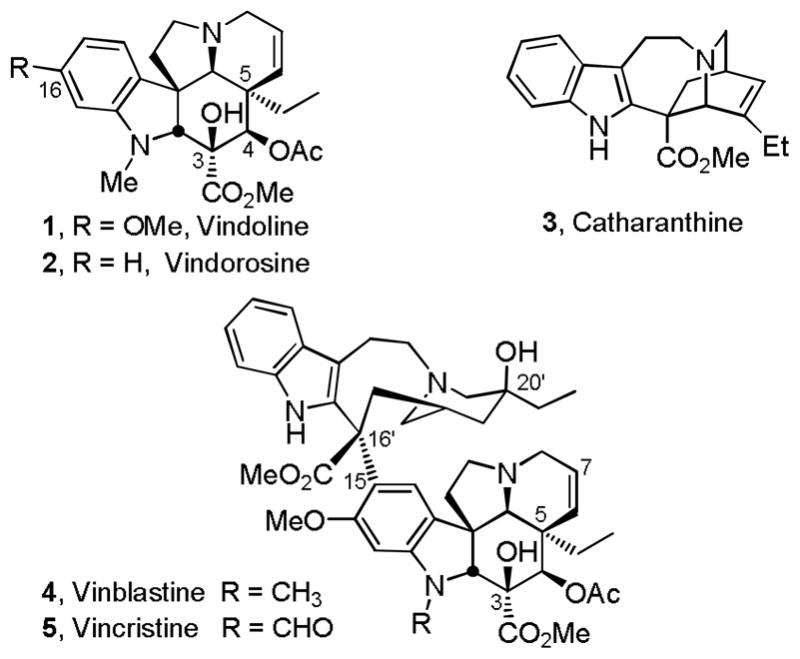
Scheme 3.

Several approaches for the conversion of 25 to vindorosine were examined and the first of the most concise routes that emerged from the efforts is summarized in Scheme 3.24 Reductive removal of the lactam carbonyl was most directly accomplished by O-methylation (MeOTf, 2,6-di-t-butylpyridine, CH2Cl2, 25 °C, 2 h) followed by NaBH4 reduction of the intermediate methoxyiminium ion (MeOH, 25 °C, 5 min, 89%) to provide 26, although alternative approaches involving thiolactam formation and reduction also proved effective. Having liberated the basic amine and following MOM ether deprotection of 26 (HCl, MeOH, 25 °C, 4 h, 92%) to liberate the primary alcohol 27, the ring expansion by way of alcohol activation and aziridinium ion formation was examined. Disappointingly albeit not surprising, all efforts to effect the ring expansion upon activation of the alcohol 27 by way of conversion to the tosylate 32 (TsCl, DMAP, Et3N, CH2Cl2, 25 °C, 16 h, 91%), chloride 33 (Ph3P–CCl4, Et3N, MeCN, 80 °C, 30 min, 90%), or bromide 34 (Ph3P–CBr4, Et3N, MeCN, 25 °C, 30 min, 96%) failed to lead to aziridinium ion formation (equation 1). Instead and only upon exposure to forcing conditions, such derivatives simply led to elimination and subsequent N,O-ketal formation to provide 35 whose structure and stereochemistry were confirmed by X-ray analysis26 (equation 1). Protection of the C3 tertiary alcohol prevented the N,O-ketal formation, but still led to elimination and not aziridinium ion formation. Clearly, the geometrical constraints imposed by the fused 6,5,5-ring system, which place the nitrogen lone pair on the convex face of the molecule opposite the hydroxymethyl substituent, are sufficient to preclude its participation in the requisite stereoelectronically-controlled displacement reaction for aziridinium ion formation.27
 |
(1) |
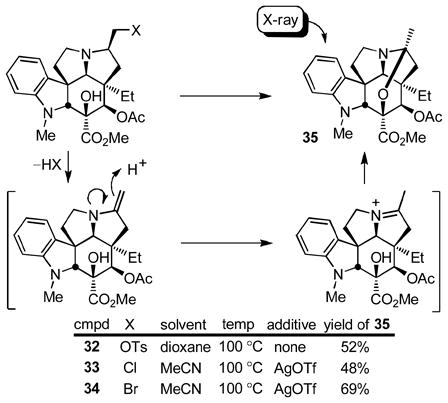
Although such an elimination reaction might be used to effect the required ring expansion and its potential was briefly examined (e.g., NBS, H2O–THF), a more attractive approach emerged as we began examining methods for the inversion of stereochemistry at C7 in order to place the hydroxymethyl substituent on the same face of the molecule as the nitrogen lone pair. Since this entails isomerization of the C7 substituent from the congested concave face of the molecule to its less hindered convex face, oxidation of the alcohol 27 to the corresponding aldehyde and its epimerization were examined. Swern oxidation (CH2Cl2, 1 h, 80%) provided an unstable α-aminoaldehyde that not only rapidly epimerized, but was also especially prone to hydrate and enol formation. Moreover, we found that simply exposing the crude aldehyde to silica gel in the presence of Et3N (EtOAc) led to clean conversion to the stable N,O-ketal 28 (80%), equation 2.
 |
(2) |
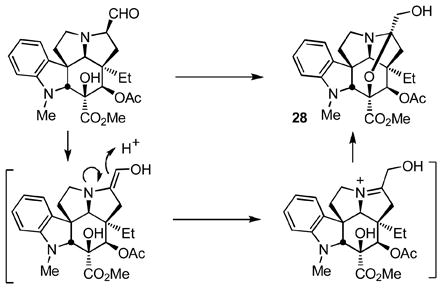
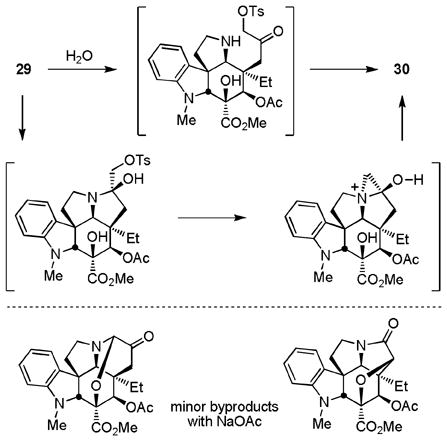
Activation of the primary alcohol for displacement (TsCl, DMAP, Et3N, CH2Cl2, 25 °C, 16 h, 87%) followed by treatment of tosylate 29 with mild base in the presence of water (HOCO2NH4, EtOH–H2O, 50 °C, 24 h)28 led to clean ring expansion to provide the 6-membered ketone 30 (76%, Scheme 2). Originally examined using NaOAc28 (THF–H2O, reflux, 36 h, 65%), the conversion of 29 to 30 improved using ammonium hydrogen carbonate (ammonium bicarbonate) and the two significant byproducts (equation 3) were no longer observed. Although several mechanistic possibilities can be envisioned for this transformation, some of which proceed through an aziridinium ion,27,28 it is most simply and formally represented as hydrolysis of the N,O-ketal to release a reactive α-tosyloxymethyl ketone followed by its intramolecular N-alkylation to provide the 6-membered ketone 30 (equation 3). This acyclic intermediate was not detected in the reaction mixture and could not be trapped with the inclusion of Boc2O, but its intermediacy would nicely account for the generation of the first of the two byproducts by base-catalyzed oxidative elimination of p-toluenesulfenic acid to produce the corresponding aldehyde. Final conversion of 30 to vindorosine required diastereoselective reduction (L-selectride, THF, −78 °C, 0.5 h, 93%)12c from the less hindered, convex face of the ketone to provide alcohol 31, which we have previously converted to the natural product (74%)10,12 enlisting a regioselective elimination reaction that occurs upon Mitsunobu activation of the secondary alcohol in the absence of added nucleophiles (Scheme 3). The approach, which was explored further with the total synthesis of vindoline, provided vindorosine in 11 or 13 steps starting from the cycloaddition precursors (Z)- or (E)-17, respectively.
 |
(3) |
As the work progressed, we continued to pursue the use of a well-precedented ring expansion reaction proceeding via an intermediate aziridinium ion.27 With the recognition that this requires inversion of the C7 stereochemistry and that a C7 aldehyde epimerization from the congested concave to convex face of the molecule is facile, we examined its potential with intermediates in which the α-amino group is still a part of the adjacent amide anticipating that this would permit the generation of an intrinsically less electrophilic aldehyde (Scheme 4). Acid-catalyzed MOM deprotection of 25 (HCl, H2O–MeOH, 25 °C, 7 h, 93%) and oxidation of the primary alcohol 3629 (IBX, DMSO, 40 °C, 3.5 h) provided the aldehyde 37 that underwent smooth and complete (>99%) epimerization to the isomeric aldehyde 38 upon exposure to silica gel (EtOAc) in the presence of Et3N (Scheme 4). Subsequent reduction of the aldehyde 38 with NaBH4 (MeOH–THF, 0 °C, 0.5 h) provided the alcohol 39 (90% from 36, 3-steps) that was reprotected as its MOM ether 40 (MOMCl, i-Pr2NEt, CH2Cl2, 25 °C, 4 h, 95%). Although a bit lengthy, the first 3 steps of this sequence as well as the epimerization could be conducted without the purification or storage of intermediates providing 39 in ca. 80% yield overall from 25 (Scheme 4). Reductive removal of the lactam carbonyl by O-methylation (MeOTf, CH2Cl2, 25 °C, 2 h) and NaBH4 treatment of the resulting methoxyiminium ion (HCl, MeOH, 25 °C, 1 h, 70%) and subsequent MOM ether deprotection of 41 (HCl, H2O–MeOH, 25 °C, 1 h, 97%) provided the primary alcohol 42, isomeric at C7 with the primary alcohol 27.
Scheme 4.

Unlike 27, activation of the primary alcohol of 42 for displacement led to aziridinium ion formation that could be kinetically trapped with nucleophiles to provide products resulting from attack on the least hindered center without ring expansion or thermodynamically trapped to provide the more stable, ring expanded products derived from attack on the more substituted center (equation 4). The latter reaction leading to ring expansion requires a nucleophile capable of reversible aziridinium ion formation and was effectively observed in reactions leading to the chlorides 43 or 44. Thus, treatment of 42 with Ph3P–CCl4 (Et3N, MeCN, 25 °C, 30 min) at room temperature provided the primary chloride 43 (72%) that on warming (MeCN, 70 °C, 2 h) smoothly converted to the ring expanded secondary chloride 44 (91%) whose structure and stereochemistry were confirmed by X-ray analysis.30 Alternatively, the addition of NaI (5 equiv) to a solution containing the primary chloride 43 generated in situ at room temperature and warming the mixture at 70 °C (MeCN, 1 h) cleanly provided the ring expanded secondary iodide 45 (72–80%) whose structure and stereochemistry were also confirmed by X-ray analysis (Scheme 4).31 Kuehne,32 and earlier Hammer,33 both describe a similar kinetic versus thermodynamic preference for aziridinium ring opening reactions, although Kuehne has also observed a reversal of this intrinsic preference32a and the propensity for even kinetic ring expansion has been generalized with simpler systems.27 Interestingly and although not investigated in detail, a single effort to prepare either the corresponding primary bromide (Ph3P–CBr4) or iodide (Ph3P, I2, imidazole) from 42 provided the desired products by TLC and LCMS, but both reverted back to 42 upon aqueous workup suggesting they possess a reactivity for aziridinium ion formation that might preclude their simple isolation. Thus, the chloride addition to such an intermediate aziridinium ion is reversible, and yet sufficiently stable that it can provide two regioisomeric products whose generation is condition and temperature dependent.
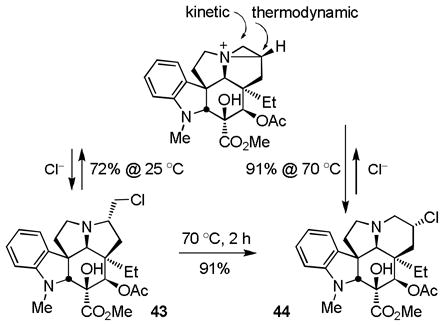 |
(4) |
The final stages of the conversion of 44 or 45 to vindorosine also provided a series of important observations. To date, neither the chloride 44 nor the more reactive iodide 45 could be eliminated directly upon base treatment to provide vindorosine, although this was not extensively examined. Rather, and representative of the reactions observed upon use of the more forcing conditions, treatment of iodide 45 with DBU (toluene, 120 °C, 3 h) provided 3526 (>90%, equation 5) in a reaction that apparently first undergoes ring contraction to the corresponding primary iodide, base-catalyzed elimination of HI, and subsequent N,O-ketal formation (see equation 1). Similarly, efforts to displace the iodide with typical oxygen nucleophiles resulted in ring contraction in reactions that also proceed through aziridinium ion formation and subsequent irreversible kinetic nucleophilic addition to its least substituted center (equation 5). An especially attractive approach combining both the ring expansion reaction with a subsequent in situ oxidation34 was also initially explored at this stage and entailed the exposure of 43 or 45 to a modified Kornblum oxidation35 with DMSO as the oxygen nucleophile, providing products capable of in situ oxidation to the corresponding carbonyl compound. Although treatment of 43 with AgBF4/DMSO was not productive, treatment of 45 (10 equiv AgBF4, DMSO, 25 °C, 9 h) provided 28 (49%) derived from the ring contracted aldehyde and the corresponding primary alcohol 42 (19%) indicative of kinetic DMSO addition to the less hindered carbon of the intermediate aziridinium ion followed by eliminative oxidation or solvolysis (equation 5). As highlighted and developed below, the successful implementation of this approach would require a DMSO equivalent capable of reversible aziridinium ion formation and slower in situ oxidation (elimination). Unsure whether this might be practical to implement and avoiding the complicating reactions derived from an intermediate aziridinium ion for the time being, treatment of iodide 45 with Bu3SnH in the presence of TEMPO provided the TEMPO adducts 46 (93%, 2:1α:β) as a mixture of diastereomers (Scheme 4). Reductive cleavage of the β-diastereomer of the TEMPO adduct 46 (Zn, HOAc/THF/H2O 3:1:1, 60 °C, 3 h, 77%) provided the key alcohol 31 that we previously converted to vindorosine. Additionally, the isomeric C7 α-alcohol derived from the α-diastereomer of the TEMPO adduct or the diastereomeric mixture of alcohols were oxidized to the ketone 30 (IBX, DMSO) and converted to vindorosine in two steps by diastereoselective reduction and regioselective elimination of the resulting alcohol 31 (L-selectride; Ph3P–DEAD) as detailed in Scheme 3. Significant in these studies, the additional key observation was made that efforts to eliminate a C7 leaving group bearing the α-stereochemistry preferentially and nonproductively form the aziridinium ion, whereas those bearing the β-stereochemistry (e.g., 31) are stereoelectronically precluded from forming an analogous aziridinium ion and productively participate in a regioselective elimination reaction to provide vindorosine.
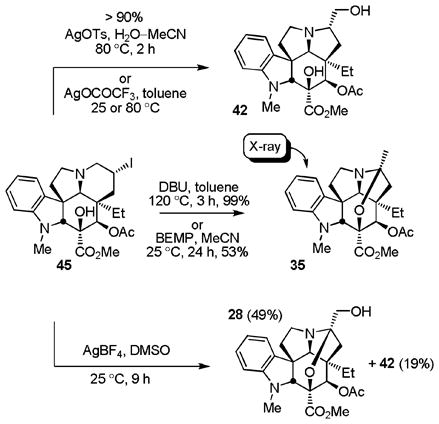 |
(5) |
As informative as these latter studies were, this second approach to vindorosine still suffered in length, requiring a cumbersome deprotection/reprotection/deprotection of the MOM ether in the sequence required for inversion of the hydroxymethyl side chain stereochemistry, and the final conversions to the key secondary alcohol 31 were still disappointing. Thus, after establishing the viability of the approach and following its implementation in a synthesis of vindoline, we reexamined the two limiting features of the approach. First, conditions were developed that avoid the MOM ether reprotection/deprotection of the epimerized alcohol and streamlined the preparation of the secondary iodide 45. Thus, direct conversion of the primary alcohol 39 to the corresponding primary chloride 47 (polymer-bound Ph3P, CCl4, 25 °C, 12 h, 98%) followed by reductive removal of the lactam carbonyl (MeOTf; NaBH4) provided the sensitive primary chloride 43 avoiding the intermediate primary alcohol MOM ether protection/deprotection (Scheme 5). Direct treatment of 43, without purification, with NaI (MeCN, 70 °C, 30 min) provided the ring expanded secondary iodide 45 in good overall conversions (59%, for 3-steps from 47). Next, we examined modifications on the Kornblum oxidation that would permit O-nucleophile displacement of the iodide, albeit with the potential for reversible aziridinium ion formation with a reagent that would necessarily undergo a slower eliminative oxidation reaction to generate the ketone 30 directly. Meeting these requirements, treatment of 45 with 4-(dimethylamino)pyridine N-oxide (DMAPO)36 under conditions that first entail aziridinium ion formation and kinetic DMAPO addition to the less substituted carbon (50 °C, 2 h, EtCN; LCMS detection of intermediate) and its subsequent partitioning to the thermodynamically more stable ring-expanded 6-membered ring addition product (120 °C, 2 h, EtCN; LCMS detection of rearrangement product), followed by treatment of the reaction mixture with base (1.5 equiv BEMP,37 25 °C, 3 h) to promote the oxidative elimination of DMAP provided the ketone 30 in a single step and in excellent conversions (78%) given the complexity of the reaction. The use of DBU36 in place of BEMP was effective, but required higher reaction temperatures (80 vs 25 °C), and little or no desired product was observed without exposure of the mixture to the higher reaction temperatures (50 vs 120 °C) despite the disappearance of 45. Efforts to convert the primary chloride 43 directly to 30 under these conditions were not as effective. However, if the conversion was conducted with KI (1.5 equiv) as an additive (3 equiv of DMAPO, 40 °C, 2 h; 120 °C, 1 h, EtCN) followed by treatment with base (3 equiv of BEMP, 25 C, 3 h), the direct conversion of 43 to 30 was achieved in comparable overall yields (48%). Interestingly, the use of the more common and soluble NaI in place of KI simply led to unproductive and preferential consumption of DMAPO. With these modifications and following diastereoselective ketone reduction (L-selectride) and alcohol elimination (Ph3P–DEAD) to provide vindorosine, this second route employing a ring expansion reaction proceeding through an aziridinium ion required 12 or 14 steps from the cycloaddition substrates (Z)- and (E)-17, respectively,
Scheme 5.

Asymmetric total synthesis of vindoline
Coupling of both (Z)-15 and (E)-15 with 2-(6-methoxy-1-methylindol-3-yl)acetic acid (48)38 provided the cycloaddition substrates (Z)-49 and (E)-49 for extension of the studies to the total synthesis of vindoline. Both substrates participated in the cycloaddition cascade (140 to 150 °C, 10–16 h) in a fashion analogous to 17 with the (E)-isomer again providing higher yields and a better diastereoselectivity than the (Z)-isomer (Scheme 6). In both instances, the initial cascade cycloadducts 50 and 52 were isolated and characterized, but most conveniently subjected to reductive oxido bridge cleavage prior to purification providing 51 and 53 directly, and X-ray analysis39 of the primary alcohols derived from both 51 and 53 confirmed the structural and stereochemical assignments. Unlike the studies with 19 and 21, a significant competitive indole-initiated [4+2] cycloaddition was also now observed, especially with (Z)-49, lowering the effective two-step conversions to 51 (41% vs 61% for 19) or 53 (55% vs 70–74% for 21).
Scheme 6.

With notable exceptions to accommodate the more nucleophilic aryl system derived from the added C16 methoxy group, most other aspects of the two approaches developed for the synthesis of vindorosine translated nicely to the synthesis of vindoline. Occasionally and with the benefit of further studies, the conversions for some steps were improved considerably. The two cycloadducts 51 and 53 were converted to the same key intermediate 56 by hydrogenolysis of benzyl ether 51 to liberate the free alcohol or benzyl ether hydrogenolysis (91%), oxidation of the free alcohol 54 to the ketone 55 (DMP, pyridine–CH2Cl2 1:4, 0 °C, 3 h, 76%) and diastereoselective ketone reduction (LiAl(OtBu)3H, THF, 0 °C, 10 h, 87%) from the less hindered convex face for 53 (Scheme 7). In the conversion of 54 to 55, IBX (vs DMP) proved less effective, and the optimized reduction of ketone 55 (87%) was established to be highly diastereoselective (30:1 dr). Subsequent C4 alcohol acetylation (Ac2O, DMAP, pyridine, 95%) provided 57. Thus, the routes to vindoline using either 51 or 53 converged requiring only the additional two steps of oxidation (76%) and reduction (87%) for the inversion of the C4 stereochemistry for use of 53, the product of the more effective of the cascade cycloaddition reactions.
Scheme 7.

O-Methylation and reductive removal of the lactam carbonyl (MeOTf, 2,6-di-t-butylpyridine, CH2Cl2, 25 °C, 2 h; NaBH4, MeOH, 25 °C, 5 min, 96%) followed by MOM ether deprotection (HCl, MeOH, 25 °C, 16 h) liberated the primary alcohol 59 (85% for two steps from 57, Scheme 7). Oxidation of 59 (3 equiv of SO3–pyr, 3 equiv of Et3N, CH2Cl2–DMSO 5:1, 25 °C, 0.5 h) followed by exposure of the crude aldehyde to silica gel in the presence of Et3N (EtOAc) cleanly afforded the N,O-ketal 60 (85%). Formation of the primary tosylate 61 (TsCl, DMAP, Et3N, CH2Cl2, 25 °C, 16 h, 93%) and it subjection to the ring expansion reaction conditions developed herein (HOCO2NH4, EtOH–H2O, 50 °C, 24 h) provided the key 6-membered ring ketone 62 (78%) in conversions that represent an improvement over that originally reported (NaOAc, dioxane–H2O, 70 °C, 61%)13. Diastereoselective reduction of 62 (L-selectride, THF, −78 °C, 0.5 h, 91%)12c provided the penultimate secondary alcohol 63 (91%), which turn underwent regioselective elimination10 to provide vindoline (1) upon Mitsunobu activation in the absence of added nucleophiles.
In an analogous fashion and in a second route to vindoline utilizing the aziridinium ion-based ring expansion reaction (Scheme 8), MOM ether deprotection of 57 (HCl, MeOH, 25 °C, 2 h, 84%) followed by oxidation of the primary alcohol 64 to the corresponding aldehyde (1.2 equiv DMP, pyridine–CH2Cl2 1:4, 0 °C, 1 h), its clean and complete epimerization to the C7 epimeric aldehyde (SiO2, 2% Et3N–EtOAc, 25 °C), and subsequent reduction (NaBH4, MeOH–THF, 0 °C, 0.5 h) provided the C7 epimeric primary alcohol 65 (82% from 64). MOM ether protection of 65 (98%), reductive removal of the lactam carbonyl (MeOTf, CH2Cl2, 25 °C, 2 h; NaBH4, HCl–MeOH, 0 °C, 0.5 h, 71%), and MOM ether deprotection (HCl, 85%) provided the primary alcohol 68 poised for the key ring expansion reaction. Treatment of 68 with Ph3P–CCl4 for in situ generation of the primary chloride followed by addition of NaI (10 equiv) and warming the solution at 70 °C (0.5 h) provided 69 (76%) resulting from reversible aziridinium ion formation partitioning to the thermodynamically more stable secondary iodide. Finally, treatment of 69 with Bu3SnH in the presence of TEMPO followed by reduction of the TEMPO adducts 70 (Zn, HOAc, THF/H2O 2:1, 25 °C, 2 h, 78–99%) provided the penultimate secondary alcohol 63, as well as the C7 epimeric alcohol 71, the former of which was previously converted to vindoline (Ph3P–DEAD, THF, 25 °C, 75%). As detailed below, both 71 and 73 (equation 6) available only through implementation of this approach, as well 63, were incorporated into vinblastine analogues.
Scheme 8.

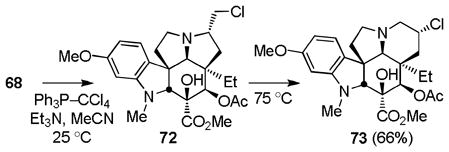 |
(6) |
The final improvement on this approach is detailed in Scheme 9 and follows advancements first developed with vindorosine altering the order of the steps to avoid the primary alcohol deprotection/reprotection and implementing the unique ring expansion–Kornblum oxidation. Thus, following inversion of the C7 stereochemistry, conversion of the primary alcohol 65 to the primary chloride 74 (91%) and reductive removal of the lactam carbonyl provided the chloride 72 that was immediately subjected to ring expansion treatment with NaI to provide 69 (69% from 74). Modified Kornblum oxidation of 69 under conditions that lead to aziridinium ion formation and that thermally partition the initially formed aminooxonium addition product on to the thermodynamically more stable 6-membered ring adduct provided the ketone 62 (66%) that was previously converted to vindoline.
Scheme 9.

Key vinblastine analogues
One of the most surprising observations made in studies with an initial series of key analogues of vinblastine6f was the impact of removing the 6,7-double bond in the vindoline subunit. The compound Δ6,7-dihydrovinblastine was found to be ca. 100-fold less active (cytotoxic) than vinblastine indicating that this region of the natural product has a pronounced impact on its properties. An examination of the recent X-ray crystal structure of vinblastine bound to tubulin40 does not reveal an obvious pronounced stabilizing interaction with the olefin that might account for the difference. As a result, this impact potentially could be derived from either steric or conformational features resulting from the presence of the double bond, or arises from an effect that the olefin might have on the pKa of the allylic amine in 1. Several key vindoline analogues prepared herein contain subtle (63, 71, and 73) or more deep-seated changes (59 and 68) in this C6–C8 region of vindoline with which we could now probe the origin of this effect. Each contains a heteroatom β to the basic amine modulating its pKa in a manner potentially analogous to the 6,7-double bond, and 59 and 68 incorporate the additional deep-seated change of replacing the olefin-containing 6-membered ring with a saturated 5-membered ring bearing either an α or β oriented C7 hydroxymethyl substituent. These five compounds were coupled with catharanthine (3) using the single-step biomimetic Fe(III)-promoted coupling and subsequent free radical oxidation6f to afford the corresponding vinblastine analogues without optimization (Scheme 10). In addition to providing the vinblastine analogues shown, the corresponding epimeric C20′ leurosidine analogues (76, 79, 82, and 85) were generated in the now characteristic ca. 2:1 β:α diastereoselectivity for the introduction of the C20′ alcohol and the intermediate anhydrovinblastines (77, 80, 83, 86, and 89) were also isolated in yields ranging from 17–22%. Interestingly and for presently unknown reasons, the couplings and in situ oxidation of the analogues 59 and 68 containing the ring contracted 5-membered rings did not proceed nearly as well as typical substrates.
Scheme 10.

The vinblastine analogues bearing the key changes in the structure of 1 were used to further assess the importance and potential role of the 6,7-double bond in the cellular activity of the natural product (cytotoxic activity against L1210 murine leukemia and HCT116 colon cancer cell lines). Additionally, the analogues were examined for their susceptibility to multidrug resistance (MDR), resulting from overexpression and drug efflux by Pgp, using a well-established companion vinblastine resistant cell line (HCT116/VM46).41 Key elements of the results of these studies are summarized in Figure 4.
Figure 4.

Cytotoxic activity.
As observed in prior studies, the vinblastine analogues proved more active than the corresponding anhydrovinblastine analogues (ca 10-fold), which in turn were more active than the corresponding leurosidine derivatives (not shown, see Supporting Information). Although the vinblastine analogues containing the vindoline saturated and C7 substituted 6-membered rings (75, 78, and 81) were found to approach the activity of Δ6,7-dihydrovinblastine (90),42 they remained at least 100-fold less potent than vinblastine. Thus, the C7 substitutions in 75 and 81 did not significantly affect the activity adversely relative to 90, but they also did not make up for the loss in activity observed with removal of the 6,7-double bond, whereas 78 experienced a further 10-fold loss in activity. Notably, analogue 81 possesses the opportunity to generate an aziridinium ion capable of reacting with tubulin, yet its cellular activity did not reflect an enhancement in activity that would be expected to accompany such an alkylation. Finally, the vinblastine analogues 84 and 87 incorporating the vindoline subunits containing the 5-membered rings bearing a hydroxymethyl substituent were even 10-fold less potent and ca. 1000-fold less active than vinblastine. Clearly, the C6–C8 region of vinblastine including its 6,7-double bond has a surprisingly large impact on the properties of 1, and plays a previously unappreciated major role in establishing its functional activity. Additional studies further probing this effect are in progress and will be disclosed in due time.43
Conclusions
Full details of the development of two concise asymmetric total syntheses of vindorosine and vindoline are disclosed enlisting an intramolecular [4+2]/[3+2] cycloaddition cascade of 1,3,4-oxadiazoles in which a chiral substituent on the tether linking the dienophile and oxadiazole was used to control the facial selectivity of the initiating Diels–Alder reaction and set the absolute stereochemistry of the remaining six stereocenters in the cascade cycloadduct. Implementation of the approach for the synthesis of 1 and 2 required that the initiating Diels–Alder reaction of the cycloaddition cascade afford a fused 5-membered ring and the development of a subsequent ring expansion reaction to provide a 6-membered ring suitably functionalized for introduction of the 6,7-double bond found in the core structure of the natural products. Two unique approaches were developed for the key ring expansion that defined our use of a protected hydroxymethyl group as the substituent used to control the stereochemical course of the cycloaddition cascade. In the course of these studies, several analogues of vindoline were prepared containing deep-seated structural changes presently accessible only by total synthesis. These analogues, bearing key modifications at C6–C8, were incorporated into vinblastine analogues and used to probe the importance (100-fold) and further define the potential role of the vinblastine 6,7-double bond.
Supplementary Material
Figure 3.

Structure of minor diastereomers and diagnostic NOEs.
Acknowledgments
We gratefully acknowledge the financial support of the National Institutes of Health (CA042056 and CA115526) and the Skaggs Institute for Chemical Biology. We wish to thank Dr. Raj Chadha for the X-ray structures disclosed herein, William M. Robertson for the cytotoxic assays, and we are especially grateful to Dr. Paul Hellier of Pierre Fabre for the generous gift of catharanthine. D.K. is a Skaggs Fellow. Dedicated to Professor Raymond Funk in honor of his sixtieth birthday.
Footnotes
Supporting Information Available. Full experimental details and compound characterizations are provided. This material is available free of charge via the internet at http://pubs.acs.org.
References
- 1.Gorman M, Neuss N, Biemann K. J Am Chem Soc. 1962;84:1058. [Google Scholar]
- 2.Moncrief JW, Lipscomb WN. J Am Chem Soc. 1965;84:4963. doi: 10.1021/ja00949a056. [DOI] [PubMed] [Google Scholar]
- 3.Noble RL, Beer CT, Cutts JH. Ann N Y Acad Sci. 1958;76:882. doi: 10.1111/j.1749-6632.1958.tb54906.x.Noble RL. Lloydia. 1964;27:280.Svoboda GH, Nuess N, Gorman M. J Am Pharm Assoc Sci Ed. 1959;48:659. doi: 10.1002/jps.3030481115.For related, naturally occurring vinca alkaloids: see Leurosidine: Svoboda GH. Lloydia. 1961;24:173.Deoxyvinblastine: De Bruyn A, Sleechecker J, De Jonghe JP, Hannart J. Bull Soc Chim Belg. 1983;92:485.Neuss N, Gorman M, Cone NJ, Huckstep LL. Tetrahedron Lett. 1968:783.N-Desmethylvinblastine: Simonds R, De Bruyn A, De Taeye L, Verzele M, De Pauw C. Planta Med. 1984;50:274. doi: 10.1055/s-2007-969701.Desacetoxyvinblastine: Neuss N, Barnes AJ, Jr, Huckstep LL. Experientia. 1975;31:18. doi: 10.1007/BF01924654.Desacetylvinblastine: Svoboda GH, Barnes AJ., Jr J Pharm Sci. 1964;53:1227. doi: 10.1002/jps.2600531023.Anhydrovinblastine: Goodbody AE, Watson CD, Chapple CCS, Vukovic J, Misawa M. Phytochem. 1988;27:1713.
- 4.Review: Neuss N, Neuss MN. In: The Alkaloids. Brossi A, Suffness M, editors. Vol. 37. Academic; San Diego: 1990. p. 229.
- 5.Reviews: Pearce HL. In: The Alkaloids. Brossi A, Suffness M, editors. Vol. 37. Academic; San Diego: 1990. p. 145.Borman LS, Kuehne ME. In: The Alkaloids. Brossi A, Suffness M, editors. Vol. 37. Academic; San Diego: 1990. p. 133.
- 6.(a) Mangeney P, Andriamialisoa RZ, Langlois N, Langlois Y, Potier P. J Am Chem Soc. 1979;101:2243. doi: 10.1021/jo01336a006. [DOI] [PubMed] [Google Scholar]; (b) Kutney JP, Choi LSL, Nakano J, Tsukamoto H, McHugh M, Boulet CA. Heterocycles. 1988;27:1845. [Google Scholar]; (c) Kuehne ME, Matson PA, Bornmann WG. J Org Chem. 1991;56:513. [Google Scholar]; Bornmann WG, Kuehne ME. J Org Chem. 1992;57:1752. [Google Scholar]; Kuehne ME, Zebovitz TC, Bornmann WG, Marko I. J Org Chem. 1987;52:4340. [Google Scholar]; (d) Magnus P, Mendoza JS, Stamford A, Ladlow M, Willis P. J Am Chem Soc. 1992;114:10232. [Google Scholar]; (e) Yokoshima S, Ueda T, Kobayashi S, Sato A, Kuboyama T, Tokuyama H, Fukuyama T. J Am Chem Soc. 2002;124:2137. doi: 10.1021/ja0177049. [DOI] [PubMed] [Google Scholar]; Kuboyama T, Yokoshima S, Tokuyama H, Fukuyama T. Proc Natl Acad Sci USA. 2004;101:11966. doi: 10.1073/pnas.0401323101. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Ishikawa H, Colby DA, Boger DL. J Am Chem Soc. 2008;130:420. doi: 10.1021/ja078192m. [DOI] [PMC free article] [PubMed] [Google Scholar]; Ishikawa H, Colby DA, Seto S, Va P, Tam A, Kakei H, Rayl TJ, Hwang I, Boger DL. J Am Chem Soc. 2009;131:4904. doi: 10.1021/ja809842b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Review: Kuehne ME, Marko I. In: The Alkaloids. Brossi A, Suffness M, editors. Vol. 37. Academic; San Diego: 1990. p. 77.
- 8.Racemic total syntheses: Ando M, Büchi G, Ohnuma T. J Am Chem Soc. 1975;97:6880.Kutney JP, Bunzli-Trepp U, Chan KK, De Souza JP, Fujise Y, Honda T, Katsube J, Klein FK, Leutwiler A, Morehead S, Rohr M, Worth BR. J Am Chem Soc. 1978;100:4220.Andriamialisoa RZ, Langlois N, Langlois Y. J Org Chem. 1985;50:961. doi: 10.1021/jo01336a006.Ban Y, Sekine Y, Oishi T. Tetrahedron Lett. 1978;2:151.Takano S, Shishido K, Sato M, Yuta K, Ogasawara K. J Chem Soc, Chem Commun. 1978:943.Takano S, Shishido K, Matsuzaka J, Sato M, Ogasawara K. Heterocycles. 1979;13:307.Danieli B, Lesma G, Palmisano G, Riva R. J Chem Soc, Chem Commun. 1984:909.Danieli B, Lesma G, Palmisano G, Riva R. J Chem Soc, Perkin Trans. 1987;1:155.Zhou S, Bommeziijn S, Murphy JA. Org Lett. 2002;4:443. doi: 10.1021/ol0171618.
- 9.Enantioselective total syntheses: Feldman PL, Rapoport H. J Am Chem Soc. 1987;109:1603.Kuehne ME, Podhorez DE, Mulamba T, Bornmann WG. J Org Chem. 1987;52:347.Cardwell K, Hewitt B, Ladlow M, Magnus P. J Am Chem Soc. 1988;110:2242.Kobayashi S, Ueda T, Fukuyama T. Synlett. 2000:883.
- 10.(a) Ishikawa H, Elliott GI, Velcicky J, Choi Y, Boger DL. J Am Chem Soc. 2006;128:10596. doi: 10.1021/ja061256t. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Choi Y, Ishikawa H, Velcicky J, Elliott GI, Miller MM, Boger DL. Org Lett. 2005;7:4539. doi: 10.1021/ol051975x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wilkie GD, Elliott GI, Blagg BSJ, Wolkenberg SE, Soenen DB, Miller MM, Pollack S, Boger DL. J Am Chem Soc. 2002;124:11292. doi: 10.1021/ja027533n.Elliott GI, Fuchs JR, Blagg BSJ, Ishikawa H, Tao H, Yuan Z, Boger DL. J Am Chem Soc. 2006;128:10589. doi: 10.1021/ja0612549.For reviews of heterocyclic azadiene cycloaddition reactions, see: Boger DL. Tetrahedron. 1983;39:2869.Boger DL. Chem Rev. 1986;86:781.Boger DL, Weinreb SM. Hetero Diels–Alder Methodology in Organic Synthesis. Academic; San Diego: 1987.
- 12.(a) Elliott GI, Velcicky J, Ishikawa H, Li Y, Boger DL. Angew Chem Int Ed. 2006;45:620. doi: 10.1002/anie.200503024. [DOI] [PubMed] [Google Scholar]; (b) Yuan Z, Ishikawa H, Boger DL. Org Lett. 2005;7:741. doi: 10.1021/ol050017s. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Ishikawa H, Boger DL. Heterocycles. 2007;72:95. [Google Scholar]; (d) Campbell EL, Zuhl AM, Liu CM, Boger DL. J Am Chem Soc. 2010;132:3009. doi: 10.1021/ja908819q. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Va P, Campbell EL, Robertson WM, Boger DL. J Am Chem Soc. 2010;132:8489. doi: 10.1021/ja1027748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kato D, Sasaki Y, Boger DL. J Am Chem Soc. 2010;132:3685. doi: 10.1021/ja910695e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rodriguez M, Llinares M, Doulut S, Heitz A, Martinez J. Tetrahedron Lett. 1991;32:923. [Google Scholar]
- 15.Nahm S, Weinreb SM. Tetrahedron Lett. 1981;22:3815. [Google Scholar]
- 16.Compound 10 was also prepared in a single operation (47%) from L-TeocHN-Ser(OMOM)-OH by treatment with (1) i-BuOCOCl, Et3N, THF; (2) CH2N2, THF; (3) PhCO2Ag, MeNH(OMe), Et3N, THF. This avoids the use of an unnatural amino acid precursor for the natural enantiomer series.
- 17.Imamoto T, Takiyama N, Nakamura K, Hatajima T, Kamiya Y. J Am Chem Soc. 1989;111:4392. [Google Scholar]
- 18.Tam TF, Fraser–Reid B. J Chem Soc, Chem Commun. 1980:556. [Google Scholar]
- 19.Christl M, Lanzendoerfer U, Groetsch MM, Ditterich E, Hegmann J. Chem Ber. 1990;123:2031. [Google Scholar]
- 20.The structure and stereochemistry of 21 were established by X-ray (CCDC 758511) and conducted on the unnatural enantiomer series.
- 21.Padwa A, Price AT. J Org Chem. 1998;63:556. doi: 10.1021/jo971424n. [DOI] [PubMed] [Google Scholar]; 1995;60:6258. [Google Scholar]
- 22.Mosa BK, Trojanek J. Collect Czech Chem Commun. 1963;28:1427. [Google Scholar]
- 23.Buchi G, Matsumoto KE, Nishimura H. J Am Chem Soc. 1971;93:3299. doi: 10.1021/ja00742a042.Kuehne ME, Podhorez DE, Mulamba T, Bornmann WG. J Org Chem. 1987;52:347.Elliott GI, Velcicky J, Ishikawa H, Li Y, Boger DL. Angew Chem Int Ed. 2006;45:620. doi: 10.1002/anie.200503024.Formal syntheses: Takano S, Shishido K, Sato M, Ogasawara K. Heterocycles. 1977;6:1699.Veenstra SJ, Speckamp WN. J Am Chem Soc. 1981;103:1645.Natsume M, Utsunomiya I. Chem Pharm Bull. 1984;32:2477.Andriamialisoa RZ, Langlois N, Langlois Y. J Org Chem. 1985;50:961. doi: 10.1021/jo01336a006.Winkler JD, Scott RD, Williard PG. J Am Chem Soc. 1990;112:8971.Heurenx N, Wouters J, Marko IE. Org Lett. 2005;7:5245. doi: 10.1021/ol0521127.
- 24.Often times the work was conducted with only the natural or unnatural enantiomer series (see Supporting Information), but this is represented herein depicting only the natural enantiomer series for the sake of clarity.
- 25.Reduction of 23 with NaBH4 predominately provided the epimeric C4 alcohol 22.
- 26.The structure and stereochemistry of 35 (CCDC 780504) were confirmed by X-ray analysis conducted in the unnatural enantiomer series.
- 27.Cossey J, Pardo DG. Chemtracts: Org Chem. 2002;15:579. [Google Scholar]
- 28.(a) Frehel D, Badorc A, Pereillo JM, Maffrand JM. J Heterocyclic Chem. 1985;22:1011. [Google Scholar]; (b) Kavadias G, Velkof S, Belleau B. Can J Chem. 1979;57:1861, 1866. [Google Scholar]; (c) Takeda M, Inoue H, Konda M, Saito S, Kugita H. J Org Chem. 1972;37:2677. [Google Scholar]; (d) Hoffman RV, Jankowski BC, Carr CS, Duesler EN. J Org Chem. 1986;51:130. [Google Scholar]
- 29.The structure and stereochemistry of 36 (CCDC 759386) were confirmed by X-ray analysis conducted in the unnatural enantiomer series.
- 30.The structure and stereochemistry of 44 (CCDC 759385) were confirmed by X-ray analysis conducted in the unnatural enantiomer series.
- 31.The structure and stereochemistry of 45 (CCDC 759387) were confirmed by X-ray analysis conducted in the natural enantiomer series.
- 32.(a) Kuehne ME, Okuniewicz FJ, Kirkemo CL, Bohnert JC. J Org Chem. 1982;47:1335. [Google Scholar]; (b) Kuehne ME, Podhorez DE. J Org Chem. 1985;50:924. [Google Scholar]
- 33.Hammer CF, Heller SR, Craig JH. Tetraherdon Lett. 1972;28:239. [Google Scholar]
- 34.D’hooghe M, Baele J, Contreras J, Boelens M, De Kimpe N. Tetrahedron Lett. 2008;49:6039. [Google Scholar]
- 35.Kornblum N, Jones WJ, Anderson GJ. J Am Chem Soc. 1959;81:4113. [Google Scholar]; Ganem B, Boeckman RK., Jr Tetrahedron Lett. 1974;15:917. [Google Scholar]; Lemal DM, Fry AJ. J Org Chem. 1964;29:1673. [Google Scholar]; Dave P, Byun HS, Engel R. Synth Commun. 1986;16:1343. [Google Scholar]
- 36.Mukaiyama S, Inanaga J, Yamaguchi M. Bull Chem Soc Jpn. 1981;54:2221. [Google Scholar]
- 37.Schwesinger R. Chimia. 1985;39:269. [Google Scholar]
- 38.Feldman PL, Rapoport H. Synthesis. 1986:735. [Google Scholar]; Drost KJ, Jones RJ, Cava MP. J Org Chem. 1989;54:5985. [Google Scholar]
- 39.The structure and stereochemistry of 53 were confirmed by X-ray analysis conducted the free alcohol derived by MOM ether deprotection (HCl, MeOH, 0–25 °C, 1.5 h, 66%) in the natural enantiomer series (CCDC 758510). Similarly, the structure and stereochemistry of 51 were confirmed by X-ray analysis conducted the free alcohol derived by MOM ether deprotection (HCl, MeOH, 25 °C, 16 h, quant.) in the natural enantiomer series (CCDC 780505).
- 40.Gigant B, Wang C, Ravelli RBG, Roussi F, Steinmetz MO, Curmi PA, Sobel A, Knossow M. Nature. 2005;435:519. doi: 10.1038/nature03566. [DOI] [PubMed] [Google Scholar]
- 41.(a) Dumontet C, Sikic BI. J Clin Oncol. 1999;17:1061. doi: 10.1200/JCO.1999.17.3.1061. [DOI] [PubMed] [Google Scholar]; (b) Kavallaris M, Verrills NM, Hill BT. Drug Res Updates. 2001;4:392. doi: 10.1054/drup.2002.0230. [DOI] [PubMed] [Google Scholar]
- 42.For an earlier characterization of 90, see: Neuss N, Barnes AJ, Huckstep LL. Experientia. 1975;31:18. doi: 10.1007/BF01924654.Barnett CJ, Cullinan GJ, Gerzon K, Hoying RC, Jones WE, Nevlon WM, Poore GA, Robison RL, Sweeney MJ, Todd GC, Dyke RW, Nelson RL. J Med Chem. 1978;21:88. doi: 10.1021/jm00199a016.
- 43.Abbreviations: Ac = acetyl; BEMP = 2-tert-butylimino-2-diethylamino-1,3-dimethylperhydro-1,3,2-diazaphosphorine; Boc = tert-butyloxycarbonyl; Bn = benzyl; CDI = 1,1′-carbonyldiimidazole; DBU = 1,8-diazabicyclo[5.4.0]undec-7-ene; DEAD = diethyl azodicarboxylate; DMAP = 4-(dimethylamino)pyridine; DMAPO = 4-(dimethylamino)pyridine N-oxide; DME = dimethoxyethane; DMF = N,N-dimethylformamide; DMP = Dess–Martin periodinane; DMSO = dimethylsulfoxide; EDCI = 1-[3-(dimethylamino)propyl]-3-ethylcarbodiimide hydrochloride; Fe2(ox)3 = iron(III) oxalate; IBX = 2-iodoxybenzoic acid; KHMDS = potassium bis(trimethylsilyl)amide; MOM = methoxymethyl; NBS = N-bromosuccinimide; Pyr = pyridine; NMM = N-methylmorpholine; TEMPO = 2,2,6,6-tetramethypiperidine-1-oxyl; Teoc = 2-(trimethylsilyl)ethoxycarbonyl; Teoc–O S u = 1-[2-(trimethyl-silyl)ethoxycarbonyloxy]pyrrolidin-2,5-dione; THF = tetrahydrofuran; Tf = trifluoromethanesulfonyl; Ts = p-toluenesulfonyl.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.