

Abstract
Two metal-free, formal [2 + 2 + 2] cycloaddition strategies for the construction of polycyclic pyridine derivatives are described that proceed via pericyclic cascade mechanisms featuring the participation of unactivated cyano groups as enophile and dienophile cycloaddition partners.
The nitrile functional group rarely participates as an enophile or dienophile in Alder ene and Diels-Alder cycloadditions.i,ii Herein we describe two formal [2 + 2 + 2] strategies for the synthesis of substituted pyridines that proceed via pericyclic cascade processes involving the unusual reaction of unactivated nitriles as 2-π cycloaddition components. As outlined in Scheme 1, the first strategy begins with a propargylic ene reactioniii which is followed by an intramolecular Diels-Alder reaction in which an unactivated cyano group functions as the dienophile. When the initial propargylic ene step is blocked (e.g., by substitution at the appropriate propargylic carbon), then a second cascade sequence is operative which leads to the same pyridine products. This alternate pathway begins with an intramolecular propargylic ene reaction in which an unactivated cyano group serves as the enophilic π-bond. To our knowledge, the participation of an unactivated cyano group in a thermal ene reaction is unprecedented.iv,v The resulting allenylimine then functions as a 1-azadiene in an intramolecular hetero Diels-Alder reaction vi leading (after tautomerization) to the isolated pyridine product. Overall, these transformations provide strategies for achieving metal-free, formal [2 + 2 + 2] cycloadditionsvii that complement the well established transition-metal-catalyzed methodologyviii for the synthesis of this important heterocyclic ring system.ix
Scheme 1.

Synthesis of Pyridines via Pericyclic Cascade Strategies
Tables 1 and 2 present examples of transformations leading to pyridines that proceed via the first pathway outlined in Scheme 1. x In this pathway the first step involves a propargylic ene reaction in which the alkyne rather than cyano group functions as the enophile component. The observations summarized below suggest that this pathway is dominant even when both pathways are possible.
Table 1.
Formal [2 + 2 + 2] Cycloadditions via Propargylic Ene/Cyano Diels-Alder Reactions
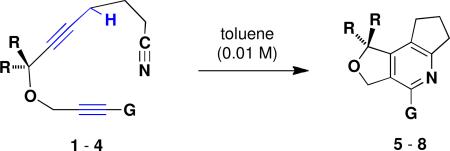 | ||||||
|---|---|---|---|---|---|---|
| entry | R | G | conditions | product | yield (%)a | |
| 1 | 1 | H | H | 160 °C, 21 h | 5 | 71 |
| 2 | 2 | CH3 | H | reflux, 66 h | 6 | 96 |
| 3 | 2 | CH3 | H | 140 °C, 18 h | 6 | 55 |
| 4 | 3 | H | C≡CSiMe3 | reflux, 24 h | 7 | 30 |
| 5 | 4 | H | C≡CCH2OSit-BuMe2 | reflux, 46 h | 8 | 37 |
Isolated yield of products purified by chromatography.
Table 2.
Formal [2 + 2 + 2] Cycloadditions via Propargylic Ene/Cyano Diels-Alder Reactions
 | ||||||
|---|---|---|---|---|---|---|
| entry | R | G | conditions | product | yield (%)a | |
| 1 | 9 | Et | C≡CSi(i-Pr)3 | 200 °C, 16 h | 12 | 51 |
| 2 | 10 | CO2Et | C≡CSi(i-Pr)3 | 200 °C, 19 h | 13 | 46 |
| 3 | 11 | CO2Et | H | 200 °C, 48 h | – | 0 |
Isolated yield of products purified by chromatography.
In the case of substrate 2, substitution of two methyl groups at one propargylic carbon precludes cyano ene reaction as the first step in the cascade so that it is pathway A that must be operative. Comparison of the relative rates of reactions of substrates lacking gem-dimethyl substitution suggest that these transformations also proceed via the propargylic ene/cyano Diels-Alder mechanism. For example, activation of the terminal alkyne with an alkynyl substituent leads to a significant increase in the rate of disappearance of starting material (entries 1 and 4). This observation is consistent with an initial propargylic ene step, and would not be expected if the first step in the cascade were the cyano ene reaction. A similar trend is observed in the case of substrates 10 and 11 (Table 2) where the unactivated alkyne 11 (G = H) failed to react even at 200 °C and most of the starting material was recovered unchanged.
The following competition experiment provides further evidence for the greater enophilicity of alkynes compared to cyano groups in the propargylic ene reaction. Heating 1 equiv each of 14, 15, and N-methylmaleimide (NMM) at 160 °C led to the isolation of 16 in 86% yield and the recovery of most of nitrile 15 unchanged (eq 1). Thus, propargylic ene reaction of 14 (but not 15) takes place under these conditions, generating a vinylallene that in this case is trapped in an intermolecular [4 + 2] cycloaddition with NMM to afford 16. In summary, when both pathways in Scheme 1 are available, pathway A is favored. The relative facility of the cyano Diels-Alder reaction involving unactivated nitriles in this pathway is attributed to the entropic advantages associated with intramolecular cycloadditions and the high reactivity of the s-cis vinylallene as a Diels-Alder diene.xi
 |
(1) |
Interestingly, when the preferred pathway A is blocked, an alternate cascade sequence takes place that begins with a propargylic cyano ene reaction and which leads to the formation of the same type of pyridine products. Table 3 presents transformations involving substrates with amide, ester, and ketone tethers in which the position of the carbonyl group precludes the propargylic ene reaction with the alkyne participating as the enophile. Substrates incorporating gem-dimethyl substitution react at lower temperature, presumably due to the gem-dialkyl effect.xii The reaction of nitriles 17 and 19 also proceed with the formation of fewer byproducts, since in these cases an α-proton is not available for tautomerization of the intermediate allenylimine prior to trapping in the hetero Diels-Alder step. Thermolysis of alkynone 21 failed to produce the desired pyridine and instead furnished the product of an “ynone” type [4 + 2] cycloadditionxiii in high yield (eq 2).
 |
(2) |
Table 3.
Formal [2 + 2 + 2] Cycloadditions via Propargylic Cyano Ene/Azadiene Diels-Alder Reactions
 | ||||||
|---|---|---|---|---|---|---|
| entry | Z | R | conditions | product | yield (%)a | |
| 1 | 17 | MeN | CH3 | 115 °C, 41 h | 22 | 74 |
| 2 | 18 | MeN | H | 160 °C, 36 h | 23 | 58 |
| 3 | 19 | O | CH3 | 150 °C, 20 h | 24 | 62 |
| 4 | 20 | O | H | 210 °C, 6 h | 25 | 35 |
| 5 | 21 | CH2 | CH3 | 170 °C, 14 h | – | 0b |
Isolated yield of products purified by chromatography.
Ynone type [4 + 2] cycloadduct 27 was obtained in 91% yield (eq 2).
Further examples of the formation of pyridines via this alternate cascade pathway were observed when alkynylimine 28 was heated in toluene for several hours at 150 °C (Scheme 2). The tricyclic pyridine product 30 was obtained in 69% yield, and the yield improved to 86% when the reaction was carried out at higher dilution. This transformation can be explained as involving initial E/Z isomerization of the imine followed by sequential propargylic cyano ene and azadiene hetero Diels-Alder reactions as outlined in the lower pathway in Scheme 1. Consistent with this mechanism is the observation that the rate of disappearance of starting material was unchanged when a homologous compound bearing an additional methylene between the alkyne groups was subjected to thermolysis. The rate of the cyano ene reaction would not be expected to be affected in this case, while the rate of the hetero Diels-Alder reaction should be significantly slower. In fact, no pyridine products were obtained in the reaction of the homologous system, presumably because trapping of the reactive allenylimine intermediate is not competitive with dimerization and other alternative reaction pathways.
Scheme 2.

Formal [2 + 2 + 2] Cycloadditions via Propargylic Cyano Ene/Azadiene Diels-Alder Reactions
Schemes 3 and 4 provide further evidence in support of the proposal that the formation of the pyridine products in Table 3 begins with an intramolecular cyano ene reaction. Scheme 3 illustrates an alternate route to pyridines involving an inter-rather than intramolecular hetero Diels-Alder reaction. In this two-step approach, the intermediate allenylimine is trapped by an electron-rich alkenexiv to afford an enamine (34) which isisolated and then converted to the desired pyridine upon exposure to acid.
Scheme 3.

Stepwise [2 + 2 + 2] Cycloaddition via Propargylic Cyano Ene and Intermolecular Addition of Vinyl Ether
Scheme 4.

Evidence for Propargylic Cyano Ene Reaction: Isolation of Enamine 36
Further evidence for the cyano ene pathway was obtained when the thermolysis of nitrile 17 (Table 3, entry 1) was interrupted after only 30 min, allowing isolation of the unstable enamine 36 (Scheme 4). This compound is believed to form via tautomerization of the intermediate allenylimine 37 involved in the formation of pyridine 22. Further heating transforms 36 to 22, suggesting that 36 and 37 undergo interconversion at elevated temperature.
Throughout this study particular attention was focused on reactions of substrates with alkynyl G groups. These substituents were found not only to function as excellent activating groups for the propargylic ene and azadiene Diels-Alder reactions, but also can serve as synthetic equivalents for a variety of substituents on the pyridine ring. For example, hydration and hydrogenation of formal [2 + 2 + 2] cycloadducts 7 and 8 furnished 38 and 39, respectively, in good yield (eq 3-4).
 |
(3) |
 |
(4) |
In summary, we have described two formal [2 + 2 + 2] cycloaddition strategies for the construction of polycyclic pyridine derivatives that proceed via unusual pericyclic cascade mechanisms featuring the participation of unactivated cyano groups as enophile and dienophile cycloaddition partners. Further studies are underway in our laboratory aimed at developing additional synthetically useful variants of this [2 + 2 + 2] strategy for the synthesis of pyridines.
Supplementary Material
Acknowledgment
We thank the National Institutes of Health (GM 28273), Merck Research Laboratories, and Boehringer Ingelheim Pharmaceuticals for generous financial support. T. S. was supported in part by a Research Fellowship of the Japan Society for the Promotion of Science (JSPS) for Young Scientists. We thank Dr. Michiko Sasaki for carrying out the competition experiment described in eq 1.
Footnotes
Supporting Information Available: Experimental procedures, characterization data, and 1H and 13C NMR spectra for all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- i.Cycloadditions of unactivated nitriles generally require harsh conditions and few examples have been reported. See Janz GJ. In: 1,4-Cycloaddition Reactions. Hamer J, editor. Academic Press; New York: 1967. pp. 97–125. Boger DL, Weinreb SM. Hetero Diels-Alder Methodology in Organic Synthesis. Academic Press; San Diego: 1987. pp. 146–150.
- ii.For examples of [4 + 2] cycloadditions involving activated nitriles such as arylsulfonyl cyanides, see McClure CK, Link JS. J. Org. Chem. 2003;68:8256. doi: 10.1021/jo034961i.; Hussain I, Yawer MA, Lalk M, Lindequist’ U, Villinger A, Fischer C, Langer P. Bioorg. Med. Chem. 2008;16:9898. doi: 10.1016/j.bmc.2008.10.033. and references cited therein.
- iii.Few ene reactions involving propargylic rather than allylic hydrogen atoms have been reported previously. See Oppolzer W, Pfenninger E, Keller J. Helv. Chim. Acta. 1973;56:1807. Shea KJ, Burke LD, England WP. Tetrahedron Lett. 1988;29:407. Jayanth TT, Jeganmohan M, Cheng M-J, Chu S-Y, Cheng C-H. J. Am. Chem. Soc. 2006;128:2232. doi: 10.1021/ja058418q. Altable M, Filippone S, Martín-Domenech A, Güell M, Solà M, Martín N. Org. Lett. 2006;8:5959. doi: 10.1021/ol062353u. González I, Pla-Quintana A, Roglans A, Dachs A, Solà M, Parella T, Farjas J, Roura P, Lloveras V, Vidal-Gancedo J. Chem. Commun. 2010;46:2944. doi: 10.1039/b926497c. Robinson JM, Sakai T, Okano K, Kitawaki T, Danheiser RL. J. Am. Chem. Soc. 2010;132:11039. doi: 10.1021/ja1053829. (g) For a report of the isolation of dimers believed to be derived from the product of a propargylic ene reaction, see Peña D, Pérez D, Guitián E, Castedo L. Eur. J. Org. Chem. 2003:1238. doi: 10.1021/jo000535a.
- iv.Hamana has reported BCl3–promoted ene reactions of trichloroacetonitrile and chloroacetonitrile; see Hamana H, Sugasawa T. Chem. Lett. 1985:575.
- v.Murakami has recently reported intramolecular ene reactions of acyl nitriles promoted by carboxylic acids, see Shimizu H, Murakami M. Synlett. 2008:1817.
- vi.For reviews discussing 1-azadiene Diels-Alder reactions, see Behforouz M, Ahmadian M. Tetrahedron. 2000;56:5259. Buonora P, Olsen J-C, Oh T. Tetrahedron. 2001;57:6099. Groenendaal B, Ruijter E, Orru RVA. Chem. Commun. 2008:5474. doi: 10.1039/b809206k.
- vii.For related metal-free, formal [2 + 2 + 2] cycloadditions leading to carbocyclic systems, see ref. 3f and references cited therein.
- viii.Reviews: Varela JA, Saá C. Chem. Rev. 2003;103:3787. doi: 10.1021/cr030677f. Heller B, Hapke M. Chem. Soc. Rev. 2007;36:1085. doi: 10.1039/b607877j. Varela JA, Saá C. Synlett. 2008:2571.
- ix.For a review of methods for the de novo synthesis of pyridines, see Henry GD. Tetrahedron. 2004;60:6043.
- x.For full details on the synthesis of cycloaddition substrates, see the Supporting Information.
- xi.For a review of Diels-Alder reactions of vinylallenes, see Murakami M, Matsuda T. Cycloadditions of Allenes. In: Krause N, Hashmi ASK, editors. Modern Allene Chemistry. Vol. 2. Wiley-VCH; Weinheim: 2004. pp. 727–815.
- xii.Jung ME, Piizi G. Chem. Rev. 2005;105:1735. doi: 10.1021/cr940337h. [DOI] [PubMed] [Google Scholar]
- xiii.Wills MSB, Danheiser RL. J. Am. Chem. Soc. 1998;120:9378. [Google Scholar]
- xiv.Attempts to trap the intermediate allenylimine with the TMS enol ether derivative of cyclohexanone and with electron-deficient alkenes and alkynes were unsuccessful. Reaction with enamines proceeded in low yield.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.