

Abstract
Interstitial lung disease (ILD) is a challenging clinical entity associated with multiple connective tissue diseases, and is a significant cause of morbidity and mortality. Effective therapies for connective tissue disease-associated interstitial lung disease (CTD-ILD) are still lacking. Multidisciplinary clinics dedicated to the early diagnosis and improved management of patients with CTD-ILD are now being established. There is rapid progress in understanding and identifying the effector cells, the proinflammatory and profibrotic mediators, and the pathways involved in the pathogenesis of CTD-ILD. Serum biomarkers may provide new insights as risk factors for pulmonary fibrosis and as measures of disease progression. Despite these recent advances, the management of patients with CTD-ILD remains suboptimal. Further studies are therefore urgently needed to better understand these conditions, and to develop effective therapeutic interventions.
Introduction
The term interstitial lung disease (ILD) is used to describe a heterogeneous group of parenchymal lung disorders that share common radiologic, pathologic, and clinical manifestations. ILD in its various guises can be asymptomatic but detected by high-resolution computed tomography (HRCT) of the chest or by pulmonary function tests. The fibrosing forms of ILD are often incurable, and are associated with significant morbidity and mortality.
ILD is subdivided into idiopathic interstitial pneumonia, of which idiopathic pulmonary fibrosis (IPF) is one subset, and diffuse parenchymal lung diseases, which may be secondary to a variety of occupational or environmental exposures, or - as discussed in the present review - can complicate multiple rheumatic or connective tissue diseases (CTDs). These diseases include systemic sclerosis (SSc), where ILD occurs in a majority of patients, and rheumatoid arthritis (RA), polymyositis/dermatomyositis (PM/DM), Sjögren's syndrome, systemic lupus erythematosus (SLE), undifferentiated CTD, and mixed CTD, where ILD is a less frequent complication (Table 1). In addition to ILD, other forms of lung damage involving the pleura, vasculature, airways, and lymphatic tissues can complicate CTDs. These complications will not be covered in the present review.
Table 1.
Interstitial lung diseases associated with connective tissue diseases
| Rheumatic disease | Frequency of ILD (%) | Comment |
|---|---|---|
| Systemic sclerosis | 45 (clinically significant) | More common in diffuse disease; topoisomerase-1 antibodies |
| Rheumatoid arthritis | 20 to 30 | Increased risk with cigarette smoking |
| Polymyositis/dermatomyositis | 20 to 50a | More common with anti-synthetase antibodies |
| Sjögren's syndrome | Up to 25 | - |
| Systemic lupus erythematosus | 2 to 8 | Usually in patients with multisystem disease |
| Mixed connective tissue disease | 20 to 60 | - |
ILD, interstitial lung disease. aFrequency may be higher based on recent studies.
The frequency of ILD in CTDs varies based on patient selection and the methods used for detection. In general, the prevalence appears to be higher than previously thought. The clinical presentation is variable, ranging from cough to pleuritic pain and progressive shortness of breath. In some patients, ILD may be the presenting feature that predates the rheumatic disease, while in others the rheumatic symptoms predate ILD. Early recognition of pulmonary involvement in these patients is important for initiating appropriate therapy.
Multidisciplinary combined connective tissue disease-associated interstitial lung disease (CTD-ILD) clinics with rheumatologists and respiratory specialists are being established at many academic medical centers. Recent experience from one CTD-ILD clinic (at Brigham and Women's Hospital, Boston, MA, USA) indicates that, after combined evaluation by both a pulmonologist and a rheumatologist, 50% of patients referred with an initial concern for IPF or another CTD-ILD had their diagnosis changed to a CTD-ILD [1].
The underlying pathology in CTD-ILD is dominated by inflammation or fibrosis, or a combination of both with distinct radiologic and histopathologic patterns. These patterns are nonspecific interstitial pneumonia (NSIP), usual interstitial pneumonia (UIP), desquamative interstitial pneumonia, cryptogenic organizing pneumonia, diffuse alveolar damage, acute interstitial pneumonia, and lymphocytic interstitial pneumonia. Table 2 outlines the characteristic histopathologic and radiologic features of the different forms of ILD. The present review will primarily focus on the pathogenesis and treatment of SSc-associated ILD, with a brief overview of the other CTD-ILDs.
Table 2.
Characteristic histopathologic patterns and radiologic findings in the interstitium of IPF and connective tissue-associated ILD
| Disease association | Characteristic histopathologic pattern | Characteristic radiographic findings on HRCT |
|---|---|---|
| Idiopathic pulmonary fibrosis | Usual interstitial pneumonia | Peripheral and bibasilar reticulonodular opacities with honeycombing |
| Systemic sclerosis | Nonspecific interstitial pneumonia | Increased reticular markings, ground glass opacification, basilar prominence |
| Usual interstitial pneumonia | Peripheral and bibasilar reticulonodular opacities with honeycombing | |
| Rheumatoid arthritis | Usual interstitial pneumonia | Reticular changes and honeycombing |
| Nonspecific interstitial pneumonia | Ground-glass opacities with basilar prominence | |
| Polymyositis/dermatomyositis | Nonspecific interstitial pneumonia | As above |
| Usual interstitial pneumonia | As above | |
| Cryptogenic organizing pneumonia | Patchy airspace consolidation, ground glass opacities | |
| Diffuse alveolar damage | Diffuse ground glass opacities | |
| Sjögren's syndrome | Nonspecific interstitial pneumonia | As above |
| Lymphocytic interstitial pneumonia | Thin walled cysts, ground glass opacities, centrilobular nodules | |
| Systemic lupus erythematosus | Acute interstitial pneumonia | Ground glass opacities |
| Mixed connective tissue disease | Nonspecific interstitial pneumonia | Septal thickening and ground glass opacities |
HRCT, high-resolution computed tomography; ILD, interstitial lung disease; IPF, idiopathic pulmonary fibrosis.
Systemic sclerosis
SSc is characterized by tissue injury leading to excessive collagen deposition, and pulmonary disease is a leading cause of death in these patients.
Most patients with SSc have evidence of ILD by HRCT of the chest or at autopsy. Close to one-half of cases develop clinically significant ILD. In a multiethnic study, the risk for ILD in cases of SSc was greater in patients of African-American ethnicity and in those patients with more extensive skin and cardiac involvement [2]. Auto-antibody expression is a predictor of internal organ involvement, particularly lung involvement. The presence of anti-topoisomerase antibodies (Scl-70) is strongly associated with development of significant ILD, while anti-centromere antibodies appear to be protective - although patients with limited SSc are not excluded from developing ILD [2,3]. A recent European League Against Rheumatism Scleroderma Trials and Research analysis revealed in a cohort of 3,656 SSc patients that ILD was present in 53% of cases with diffuse cutaneous SSc and in 35% of cases with limited cutaneous SSc [4].
Biomarkers, although currently not available for clinical testing, may serve as indicators of disease and as predictors of progression. Serum levels of surfactant proteins A and D (SP-A and SP-D) and the glycoprotein Krebs von den lungen-6 (KL-6), produced by type II alveolar epithelial cells, are elevated in sera of patients with ILD [5]. A comparison of SP-D and KL-6 serum concentrations showed that both markers were elevated in patients with SSc-associated ILD compared with healthy controls, with SP-D being more sensitive and KL-6 more specific [5]. There is a great deal of current interest in novel biomarkers such as chitinase-like protein YKL-40, which is already shown to be useful in asthma [6].
Histologically, SSc-associated ILD is characterized by early pulmonary infiltration of inflammatory cells and subsequent fibrosis of the lung parenchyma. The most common histologic pattern seen in SSc-associated ILD is NSIP; the UIP pattern is less common. Histologically, NSIP is characterized by varying degrees of inflammation and fibrosis, with the majority of patients showing prominent inflammation. In contrast, UIP is characterized by dense patchy fibrosis with honeycombing, primarily in a subpleural distribution.
Radiologic findings associated with CTDs are summarized in Table 2. SSc-associated ILD is characterized on chest X-ray by hazy, reticular infiltrates that are prominent in the lower lobes. HRCT characteristically reveals ground glass opacities, traction bronchiectasis, and minimal honeycombing consistent with an NSIP pattern (Figure 1a). In contrast, the UIP pattern of IPF is characterized by patchy reticular opacities associated with traction bronchiectasis and honeycombing with a predominantly basal and peripheral reticular pattern (Figure 1b). The utility of HRCT to detect histologic pattern is sufficient to make the diagnosis of UIP/IPF in 50 to 60% of cases [7].
Figure 1.
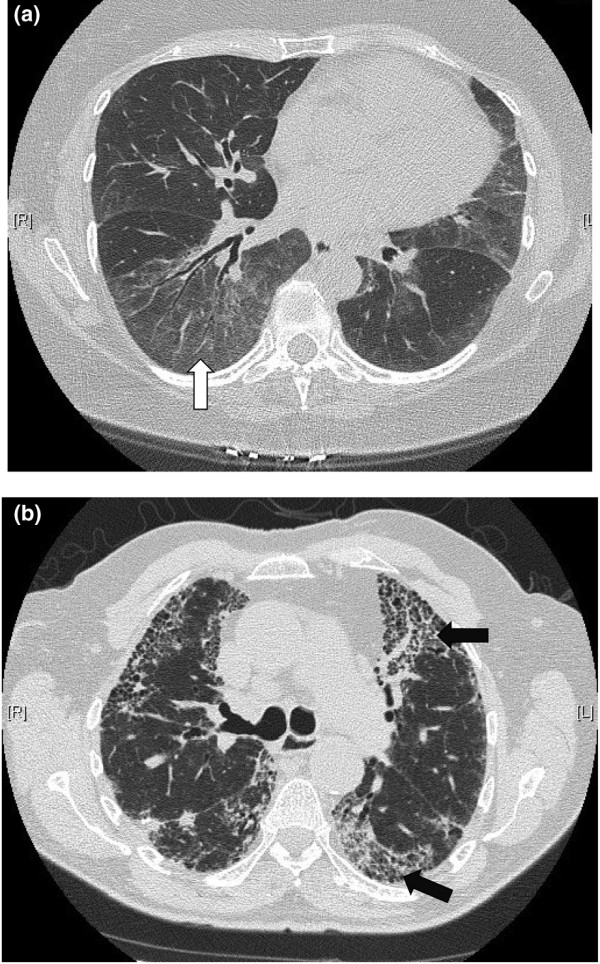
Characteristic radiographic findings on high-resolution computed tomography. High-resolution computed tomography of the chest reveals (a) subpleural ground glass opacities (white arrow) and traction bronchiectasis suggestive of nonspecific interstitial pneumonia, and (b) honeycombing (black arrows) with ground glass opacities suggestive of usual interstitial pneumonia.
Pathogenesis of connective tissue disease-associated interstitial lung diseases
Mediators of lung fibrosis in systemic sclerosis
Interstitial lung involvement in SSc develops from an interplay of autoimmunity, inflammation, and vascular injury. Endothelial or epithelial injury is thought to precede inflammation and fibrosis, but the mechanisms that perpetuate pulmonary fibrosis are still not fully understood (Figure 2).
Figure 2.

Mechanisms perpetuating pulmonary fibrosis. Pathogenesis of pulmonary fibrosis is initiated by microvascular injury, which leads to endothelial cell damage and alveolar epithelial injury. This leads to activation of the coagulation cascade, release of various cytokines and growth factors, and ultimately activation of fibroblasts, a key event in the development of fibrosis. CTGF, connective tissue growth factor; IGF-1, insulin-like growth factor-1; LPA, lysophosphatidic acid; TGF-β, transforming growth factor beta.
A number of proinflammatory and profibrotic extra-cellular mediators have been implicated in the pathogenesis of interstitial lung diseases and IPF, and are also likely to have important roles in SSc-associated ILD. These include chemokines, cytokines, growth factors, lipids, and prostanoids. The pivotal mediator of fibrosis is the multifunctional cytokine, transforming growth factor beta (TGFβ). Substantial evidence implicates TGFβ - along with platelet-derived growth factor, endothelin-1 (ET-1), and other cytokines - in the pathogenesis of SSc. Accordingly, targeting the intracellular signaling pathways activated by TGFβ and other profibrotic mediators is a rational treatment strategy for controlling fibrosis and is an active area of current research.
Mediators of TGFβ responses
Canonical Smad signaling
The canonical TGFβ signal transduction pathway involves sequential phosphorylation of the activin-like kinase-5 type I TGFβ receptor and a group of intracellular signaling proteins called Smads [8]. When bound by active TGFβ, the cell surface TGFβ receptors transmit signals through phosphorylation of cytoplasmic Smad proteins, which translocate into the cell nucleus and trigger transcription of genes such as type I collagen, fibronectin, α-smooth muscle actin and connective tissue growth factor (CTGF), each of which plays important roles in fibrogenesis [9]. Smad3 null mice are protected against bleomycin-induced fibrosis of the skin and lungs [10,11]. In addition, pharmacologic blockade of activin-like kinase-5 activity with small molecule inhibitors such as SB431542 and SD208 results in complete abrogation of profibrotic responses induced by TGFβ, and normalization of the autonomously activated phenotype of SSc fibroblasts in vitro [12,13]. Selective blockade of Smad phosphorylation or of non-Smad signaling down-stream of TGFβ using small molecules are promising novel approaches to the treatment of fibrosis that are under investigation.
c-Abelson tyrosine kinase
In normal fibroblasts TGFβ induces activation of c-Abelson tyrosine kinase (c-Abl), a member of the Src family of nonreceptor protein tyrosine kinases [14]. Transforming mutations of c-Abl are found in 95% of patients with chronic myelogenous leukemia, and result in constitutive kinase activity that is directly responsible for myeloid cell hyperproliferation [15]. Recent studies of c-Abl function in nonmyeloid cells reveal that c-Abl is directly activated by TGFβ, and integrates serine-threonine kinase signaling with nonreceptor tyrosine kinase pathways [16].
Imatinib mesylate is a potent small molecule inhibitor of c-Abl, as well as of platelet-derived growth factor receptor activity. Inhibition of c-Abl kinase activity using imatinib was recently demonstrated to abrogate the stimulation of collagen gene expression in vitro, and to prevent the development of skin and lung fibrosis in vivo in animal models [14,17]. Preclinical studies show that, in explanted normal skin and lung fibroblasts, imatinib effectively blocked TGFβ-induced stimulation of collagen synthesis and myofibroblast transformation, which are key events in the fibrotic response [18]. Furthermore, imatinib partially reversed the abnormal phenotype of SSc fibroblasts [17]. Since one of the downstream targets of c-Abl is the profibrotic transcription factor Egr-1 (see below), blockade of c-Abl activity might prevent fibrosis by inhibiting Egr-1 activation [19].
Anecdotal reports indicate therapeutic efficacy of imatinib in SSc, graft versus host disease, nephrogenic fibrosis, and other fibrosing conditions. Ongoing clinical trials are evaluating the efficacy and safety of imatinib in SSc-associated ILD. Of note, however, a recently completed randomized controlled trial showed no benefit of imatinib compared with placebo in patients with IPF [20].
Egr-1
Egr-1 is a zinc finger DNA binding transcription factor that is rapidly and transiently induced at sites of injury. Egr-1 is implicated in cell proliferation, differentiation and survival, and plays a central role in orchestrating acute tissue responses to injury [21]. Egr-1 null mice were protected from pulmonary and skin fibrosis induced by TGFβ or by bleomycin, and Egr-1 was shown to be sufficient and necessary for the stimulation of type I collagen production in vitro [22]. Genome-wide expression profiling using microarrays has demonstrated that abnormal Egr-1 expression in the lung was strongly associated with rapid progression of lung fibrosis in patients with IPF [23]. In addition, both Egr-1 mRNA and protein were elevated in explanted SSc skin fibroblasts in vitro [24]. Egr-1 was also shown to be a key mediator of lung fibroblast activation induced by insulin-like growth factor (IGF) binding protein 5 [25].
These observations identify Egr-1 as a critical intra-cellular mediator of lung fibrosis in humans and in mouse models. Ongoing studies are investigating blocking Egr-1 expression or activity with drugs such as imatinib as potential strategies to control pathologic fibrosis.
Peroxisome proliferator-activated receptor gamma
Peroxisome proliferator-activated receptor gamma (PPARγ) is a nuclear steroid hormone receptor and a ligand-activated transcription factor. Originally described in adipocytes, it is now recognized that PPARγ is widely expressed in tissues and plays key regulatory roles not only in adipogenesis and insulin sensitivity, but also in inflammation and immunity.
An emerging novel function for PPARγ is as an endogenous anti-fibrotic defense mechanism. Ligand activation of cellular PPARγ potently inhibited the activation of TGFβ-inducible responses in normal skin and lung fibroblasts [26]. It is notable that the expression of PPARγ is markedly reduced in lung biopsies from patients with SSc-associated ILD [27]. Ligands for inducing the activity of PPARγ include endogenous natural agonists such as fatty acids or prostaglandins (PGJ2), and synthetic pharmacologic agents such as rosiglitazone and pioglitazone [28]. These drugs are in wide use for the treatment of type 2 diabetes. Rosiglitazone was recently shown to attenuate bleomycin-induced dermal fibrosis and inflammation in vivo. Furthermore, rosiglitazone prevents alveolar epithelial mesenchymal transition and also TGFβ-induced stimulation of collagen gene transcription, myofibroblast transdierentiation, and cell migration in normal fibroblasts [29].
In light of its potent anti-inflammatory and anti-fibrotic activities and relative safety in clinical practice, studies of existing PPARγ agonists - and novel selective agonists under development - are now warranted for treatment of ILD.
Endothelin-1
Endothelial injury in small and medium-sized arteries is a defining feature of SSc that leads to activation of the coagulation cascade followed by myofibroblast differentiation, activation of endothelial cells, and capillary loss. ET-1 is a potent vasoconstrictor released by endothelial cells, epithelial cells and mesenchymal cells. In lung injury, ET-1 binds to ET-1A and ET-1B receptors, recruits fibroblasts and stimulates matrix production [30]. Transgenic mice overexpressing ET-1 develop lung fibrosis [31] and ET-1 levels are elevated in mouse models of bleomycin-induced fibrosis [32]. ET-1 also has been found to stimulate TGFβ secretion in lung fibroblasts [33]. Studies with bosentan, a dual-receptor ET-1 antagonist, are underway for the treatment of IPF and SSc-associated ILD.
Growth factors and chemokines
Lysophosphatidic acid
The bioactive phospholipid lysophosphatidic acid (LPA) and its receptor LPA1 have recently been implicated in the pathogenesis of IPF [34]. LPA is produced by activated platelets, as well as by fibroblasts. The LPA1 receptor is expressed in fibroblasts, endothelial cells, and epithelial cells, and enables LPA to induce diverse biologic effects involved in tissue responses to injury.
Both LPA and its receptor are required for the development of lung fibrosis in a mouse model of IPF induced by bleomycin [34]. These studies revealed that the fibroblast chemoattractant activity present in the lungs of IPF patients is largely attributable to LPA, suggesting that LPA mediates fibroblast recruitment during the development of lung fibrosis. Preliminary results indicate that mice lacking the LPA1 receptor are protected from bleomycin-induced dermal fibrosis compared with wild-type mice (FV Castelino, AM Tager, unpublished data). The LPA-LPA1 pathway therefore appears to be a promising novel therapeutic target for SSc-associated pulmonary fibrosis.
Insulin-like growth factor
IGFs and their binding proteins have been implicated in the pathogenesis of pulmonary fibrosis and SSc. Increased levels of IGF-1 are detected in the serum as well as in the bronchoalveolar lavage of patients with SSc-associated ILD [35]. In addition, blockade of the IGF pathway leads to resolution of pulmonary fibrosis in a mouse model of pulmonary fibrosis [36]. These observations raise the possibility that targeting the IGF pathway may be a potential treatment of CTD-ILD.
Connective tissue growth factor
CTGF, also known as CCN2, is a small cysteine-rich matricellular protein with an important role in angiogenesis and the formation of connective tissue [37]. Although the specific receptors for CTGF or the precise mechanism of action are poorly understood, CTGF acts as a downstream mediator of TGFβ, and may play a role in the stimulation of extracellular matrix production and myofibroblast dierentiation.
Levels of CTGF are elevated in the skin and lungs from patients with SSc, as serum levels of CTGF reflect disease severity. Lung fibroblasts explanted from bleomycin-injected mice have a high expression of CTGF [38]. CTGF is therefore an attractive target for the treatment of pulmonary fibrosis, and clinical trials using a monoclonal anti-CTGF antibody are under preparation.
Treatment considerations
To date there is no cure or effective disease-modifying therapy for any form of CTD-ILD. D-Penicillamine and colchicine are largely ineffective [39,40]. Because evidence of inflammation is commonly present in early-stage disease, current therapies for SSc-associated ILD target the inflammatory response. The immunosuppressive agents most widely used for this purpose are corticosteroids, cyclophosphamide, azathioprine, and mycophenolate mofetil. While corticosteroids are generally ineffective, other agents have demonstrated a modest beneficial effect.
In contrast to various rheumatic diseases where immunosuppressives have been helpful, immunosuppressive therapies in CTD-ILD have not led to complete responses. There is only limited experience with newer biologicals such as anti-TNF therapies or rituximab.
Cyclophosphamide
Multiple studies and uncontrolled trials of CTD-ILD have reported beneficial effects of cyclophosphamide administered orally or intravenously [40-42]. These studies showed improvement in respiratory symptoms, lung function, radiologic findings, and bronchoalveolar lavage inflammation, as well as survival.
The Scleroderma Lung Study was the first multicenter, randomized placebo-controlled clinical trial to evaluate the effectiveness of oral cyclophosphamide in SSc-associated ILD [43]. In the study, 158 patients with earlystage SSc and symptomatic ILD with radiologic or bronchoalveolar lavage evidence of alveolar inflammation were randomized to cyclophosphamide or placebo. A 12-month course of active therapy was associated with a modest but statistically significant improvement in forced vital capacity (FVC), but no change in diffusing capacity for carbon monoxide. Furthermore, respiratory symptoms and chest radiologic abnormalities showed improvement [43,44]. The response in pulmonary function was most pronounced in those patients with the most advanced lung disease at baseline. At 24-month follow-up, the beneficial effect of cyclophosphamide on pulmonary function largely disappeared. In contrast, beneficial responses in skin score and quality of life measures persisted at 2 years.
A randomized, double-blind, placebo-controlled study from the UK compared the efficacy of intravenous cyclophosphamide combined with corticosteroids and followed by azathioprine with that of placebo. This study of 45 SSc patients with early ILD demonstrated a favorable outcome in the treatment group, but, due to the small size of the study, the results did not achieve statistical significance [45].
Mycophenolate mofetil
Mycophenolate mofetil is an immunosuppressive drug with less toxicity compared with cyclophosphamide. One study evaluated 17 patients with SSc-associated ILD treated with mycophenolate mofetil for up to 24 months [46]. At 12 months the FVC and diffusing capacity for carbon monoxide had improved by 2.6% and 1.4%, respectively, while at 24 months the increase in FVC was 2.4% [46]. Gerbino and colleagues evaluated mycophenoate mofetil in 13 patients with early SSc-associated ILD [47]. The FVC improved by a mean of 4% predicted at a median of 21 months. The ongoing Scleroderma Lung Study II will compare the efficacy and safety of mycophenolate mofetil with cyclophosphamide in patients with SSc-associated ILD.
Azathioprine
Azathioprine is an alternative agent for SSc-associated ILD. Patients with a milder form of ILD or those unable to tolerate cyclophosphamide may be potential candidates.
A retrospective analysis described 11 patients with SSc-associated ILD who received azathioprine and prednisone [48]. In this study, 8/11 patients showed an improvement in FVC and dyspnea scores at 12 months.
Data also suggest a role for azathioprine as maintenance therapy following intravenous cyclophosphamide. A retrospective series of 27 patients with SSc-associated ILD showed stabilization or improvement of lung function with a combination regimen of monthly intravenous cyclophosphamide given for 6 months followed by 18 months of azathioprine [49].
Endothelin-1 receptor antagonists
Bosentan is a dual ET-1 receptor antagonist approved for the treatment of pulmonary arterial hypertension. ET-1 is overexpressed in SSc skin and lungs, and can act as a profibrotic cytokine that promotes myofibroblast proliferation.
The Bosentan in Interstitial Lung Disease (BUILD 1) study examined the potential anti-fibrotic efficacy of bosentan in IPF. One hundred and fifty-eight patients with IPF were randomized to receive either bosentan or placebo [50]. With the 6-minute walk test as primary outcome at 12 months, bosentan was no better than placebo. Moreover, a recent placebo-controlled trial of bosentan for the treatment of SSc-associated ILD patients (BUILD 2) was terminated due to lack of efficacy [51], and BUILD 3 - evaluating the safety and efficacy of bosentan in IPF patients - did not meet the primary endpoint of a reduction in morbidity and mortality (unpublished data).
Tyrosine kinase inhibitors
The therapeutic use of small molecule kinase inhibitors for nonmalignant diseases has generated a great deal of interest, but their use is limited by toxicity. In contrast to inhibitors of ubiquitous protein kinases such as p38, imatinib mesylate (Gleevec®; Imatinib mesylate, Novartis, Basel, Switzerland) selectively blocks the activity of the c-Abl tyrosine kinase and, to a lesser degree, the platelet-derived growth factor receptor and c-kit, and appears to have a relatively favorable long-term safety profile in a large number of chronic myelogenous leukemia patients.
Individual case reports provide support for the use of imatinib in SSc. Van Daele and colleagues described a patient with SSc who had progressive pulmonary fibrosis despite treatment with intravenous cyclophosphamide [52]. After 5 months of imatinib, improvement in skin score (from 18 to 12) and pulmonary function was noted. Another report described a woman with longstanding and progressive SSc unresponsive to intravenous cyclophosphamide and mycophenolate mofetil [53]. This patient showed improved skin and stabilization in lung function after 6 months of imatinib therapy [53]. In contrast, a recent large, multicenter, randomized controlled trial of imatinib versus placebo in the treatment of IPF showed no significant benefit [20].
Pirfenidone
Pirfenidone is a pyridone with both anti-inflammatory and anti-fibrotic effects. Pirfenidone was shown to inhibit collagen synthesis and TGFβ production in vivo in animal models of IPF [54]. In clinical studies, pirfenidone slowed a decline in lung function and exercise capacity [55]. A randomized, double-blind, placebo-controlled phase III trial in IPF (CAPACITY 1 trial) demonstrated a decrease in the rate of decline of vital capacity and an increase in progression-free survival time over 52 weeks; however, the primary endpoint of change in the percentage predicted FVC at week 72 was not met [56]. The CAPACITY 2 trial, using a lower dose of pirfenidone, reached its primary endpoint [57]. Pirfenidone was administered to two patients with SSc-associated ILD [58]. These patients showed no significant radiological progression or functional deterioration. One should note, however, that the Food and Drug Administration recently rejected the use of pirfenidone for the treatment of IPF, citing the need for an additional clinical trial to prove efficacy given that the drug worked in one of the two trials and questioning whether the benefit provided by the drug was meaningful (InterMune press release).
Lung transplantation
Lung transplantation remains an option for SSc patients with ILD who fail to respond to pharmacologic therapy. A recent study comparing lung transplantation in 29 patients presenting SSc-associated ILD with 70 patients presenting IPF showed comparable cumulative survival (64%) at 2 years [59]. In another retrospective analysis, 23 of 47 SSc patients were alive at 24 months post lung transplantation [60].
Prognosis of systemic sclerosis-associated interstitial lung disease
ILD is a leading cause of morbidity and mortality in SSc. The prognosis of SSc-associated ILD depends on the underlying pathology. The NSIP pattern has a more favorable outcome compared with the UIP pattern characteristically associated with IPF [61]. A recent retrospective review of 80 patients with SSc-associated ILD showed that 76% had an NSIP pattern and 11% had a UIP pattern [61,62]. In this study, the 5-year survival rates were similar for patients with the NSIP pattern and the UIP pattern (82% and 91%, respectively). It is difficult to predict whether IPF patients with a UIP pattern would have similar survival as SSc patients with a UIP pattern.
Other connective tissue disease-associated interstitial lung diseases
Rheumatoid arthritis
Lung disease is a leading cause of death in RA, second only to infection. Evidence of ILD is seen in 20 to 30% of patients, but the reported prevalence varies depending on the criteria used for diagnosis [63]. Most RA patients show pulmonary parenchymal abnormalities on HRCT, including bronchial wall thickening, bronchial dilation, micronodules, and opacities, along with pleural effusions.
The main finding in patients with ILD is bibasilar symmetrical reticular infiltrates followed by honeycombing. The histologic NSIP pattern was previously thought to be most frequent in RA, but recent studies of surgical lung biopsies reveal that the UIP pattern may be more common [64]. The 5-year survival in RA patients with UIP is less than 50%. In one study, RA patients had a greater number of CD4-positive T cells in the bronchoalveolar lavage fluid than IPF patients [65].
Little is known regarding the optimal therapy of RA-associated lung disease, and randomized trials are lacking. Corticosteroids and immunosuppressive agents are widely used, but corticosteroids by themselves are of limited benefit in RA UIP [66]. In IPF, N-acetylcysteine added to a regimen of prednisone and azathioprine slowed deterioration of the FVC and diffusing capacity for carbon monoxide at 12 months [67]. Mycophenolate mofetil is another therapeutic consideration in patients with RA-associated ILD. One report of two RA patients with ILD showed benefit of mycophenolate mofetil on pulmonary function and radiologic abnormalities [68]. While the course of RA-associated ILD varies from a slow progression to a fulminant course, the prognosis is generally better than that of IPF.
Polymyositis/dermatomyositis
The presence of ILD markedly influences the disease course in inflammatory myositis. The reported incidence of ILD varies from 20 to 54% depending on the criteria used for diagnosis [69,70]. The strongest predictive factor is the presence of autoantibodies to aminoacyl tRNA synthetase, most commonly anti-Jo-1 [71]. Another serum marker of increased risk for ILD is antibody to KL-6, a glycoprotein expressed on type II alveolar and bronchiolar epithelial cells [72]. Amyopathic dermatomyositis is also associated with ILD and can have a poor prognosis [73]. In addition, anti-clinically amyopathic dermatomyositis antibodies associated with this subset suggest rapidly progressive ILD [74].
The histopathology of ILD in PM/DM includes cryptogenic organizing pneumonia, diffuse alveolar damage, and NSIP and UIP patterns [75]. One study suggested that patients with a cryptogenic organizing pneumonia pattern respond to corticosteroids, while those with diffuse alveolar damage and UIP patterns do not [75].
There are no controlled trials evaluating the treatment of PM/DM-associated ILD. The most common initial therapy uses corticosteroids, generally at a dose of 1 mg/kg/day prednisone for 6 to 8 weeks, followed by a gradual taper. Steroid-sparing immunosuppressive agents such as cyclophosphamide, azathioprine, and metho-trexate are frequently used. For some patients whose disease is rapidly progressive, either oral steroids or pulse methylprednisolone combined with monthly intravenous cyclophosphamide has been reported to show a favorable response [76].
One report described the use of tacrolimus in two myositis patients with progressive ILD who had failed cyclophosphamide and high-dose corticosteroid treatment [77]. These patients showed significant improvement in symptoms and radiologic abnormalities. In another report, 12 out of 15 PM/DM patients treated with tacrolimus for up to 36 months showed significant improvement in all pulmonary parameters [78].
Rituximab has also been used in the treatment of myositis and anti-synthetase syndromes. In a retrospective case series, rituximab appeared to stabilize ILD in seven out of 11 patients during the first 6 months after treatment [79]. In addition, in a study of 49 patients with DM/PM, 75% showed a good response in myositis features after treatment with rituximab [80].
Sjögren's syndrome
ILD develops in approximately 25% of patients with Sjögren's syndrome [81]. In these patients, ILD characteristically presents with cough, dyspnea, and bilateral pulmonary infiltrates on chest radiographs.
The lymphocytic interstitial pneumonia pattern was previously suggested to be the most characteristic histopathology in Sjögren's syndrome, but recent studies show that the NSIP pattern is more prevalent [81,82]. Lymphocytic interstitial pneumonia represents a benign polyclonal proliferation of mature B cells or T cells that can involve the lung either diffusely or focally. Lymphocytic interstitial pneumonia is also considered relatively responsive to steroid therapy [81].
The optimal treatment for patients with Sjögren's syndrome-associated ILD is not known. Anecdotal reports and small case series suggest the disease is steroid responsive. While the majority of patients experienced rapid subjective improvement, pulmonary function tests and radiological abnormalities showed a slower response over several months [82]. Some patients require additional immunosuppressive agents such as azathioprine or cyclophosphamide.
Systemic lupus erythematosus
Pulmonary involvement is frequent in SLE, and can affect the pleura, pulmonary vasculature, and parenchyma. The prevalence and severity of ILD appears to be lower in SLE than in the other CTDs. Acute lupus pneumonitis is an uncommon manifestation of SLE [83]. The disease typically presents with acute dyspnea, cough, fever, and pleuritic pain, and occasionally with pulmonary hemorrhage. Diffuse ILD or chronic pneumonitis in SLE occurs in 3 to 8% of patients [83].
The treatment for SLE-associated ILD is to some extent dictated by the predominant lung pathology. In patients with acute lupus pneumonitis, the mainstay of treatment is oral prednisone (1 mg/kg/day). If there is no prompt improvement, then intravenous methylprednisolone with an immunosuppressive agent such as cyclophosphamide is commonly used. One report described a patient with acute lupus pneumonitis who responded to weekly rituximab with a rapid improvement in subjective symptoms and pulmonary function test abnormalities [84].
Undifferentiated connective tissue disease
Patients with undierentiated CTD often have some features of a rheumatic disease but do not have sufficient findings for a discrete rheumatic diagnosis [85]. These patients may have a concomitant ILD that either precedes or occurs concomitantly with their rheumatic symptoms. In a case-control study evaluating 28 patients with idiopathic interstitial pneumonia, 88% of patients classified with an initial histologic pattern of idiopathic NSIP had features of an undierentiated CTD [86]. In addition, patients with undifferentiated CTD had a substantial improvement in FVC during a follow-up period of 8 months compared with IPF patients [87]. Treatment of undierentiated CTD-ILD is similar to other CTD-ILDs with an NSIP pattern.
Mixed connective tissue disease
Pulmonary involvement is a common complication of mixed CTD. Up to two-thirds of patients have a reduced diffusing capacity for carbon monoxide, and approximately one-half have evidence of restrictive abnormalities on pulmonary function tests [88]. The predominant radiologic abnormality in the chest is ground glass opacities associated with septal thickening with a lower lobe predominance [89]. These findings are similar to those seen in SSc-associated ILD. Treatment of ILD in mixed CTD is similar to that of other CTD-ILDs. In one study, 47% of patients with mixed CTD-ILD responded to corticosteroids at a dose of 2 mg/kg/day [89].
Conclusion
ILD is now increasingly recognized as a frequent and serious complication of rheumatic diseases and CTDs. Effective disease-modifying therapies are still lacking, and many of the currently used treatments are largely ineffective. Stem cell therapies and novel agents including rituximab, angiotensin II inhibitors, tyrosine kinase inhibitors, PPARγ agonists, intravenous immunoglobulin, and biologicals targeting chemokines, cytokines, and growth factors are in preclinical or clinical studies. There is progress towards better understanding the pathogenesis of CTD-ILD, and the role of growth factors, chemokines, and lipid mediators. Serum biomarkers as either indicators of pulmonary fibrosis or indicators of disease progression are under active investigation. Despite these impressive recent advances, the management of patients with CTD-ILD remains unsatisfactory. Further study into the cell types, mediators, and pathways involved in lung fibrosis is urgently needed. These further studies may lead to a better understanding of lung fibrosis, and to the development of safer and more effective rational therapies.
Abbreviations
BUILD: Bosentan in Interstitial Lung Disease study; C-ABL: c-Abelson tyrosine kinase; CTD: connective tissue disease; CTD-ILD: connective tissue disease-associated interstitial lung disease; CTGF: connective tissue growth factor; EGR-1: early growth response-1; ET-1: endothelin-1; FVC: forced vital capacity; HRCT: high-resolution computed tomography; IGF: insulin-like growth factor; ILD: interstitial lung disease; IPF: idiopathic pulmonary fibrosis; KL-6: Krebs von den lungen-6; LPA: lysophosphatidic acid; NSIP: nonspecific interstitial pneumonia; PM/DM: polymyositis/dermatomyositis; PPARγ: peroxisome proliferator-activated receptor gamma; RA: rheumatoid arthritis; SLE: systemic lupus erythematosus; SP-A/D: surfactant protein A/D; SSC: systemic sclerosis; TGFβ: transforming growth factor beta; TNF: tumor necrosis factor; UIP: usual interstitial pneumonia.
Competing interests
The authors declare that they have no competing interests.
Contributor Information
Flavia V Castelino, Email: FCASTELINO@PARTNERS.ORG.
John Varga, Email: j-varga@northwestern.edu.
References
- Castelino FV, Goldberg H, Dellaripa PF. American College of Rheumatology Annual Meeting. San Francisco, CA; 2008. The impact of rheumatologic evaluation on the management of patients with interstitial lung disease (ILD) [abstract 637] [Google Scholar]
- McNearney TA, Reveille JD, Fischbach M, Friedman AW, Lisse JR, Goel N, Tan FK, Zhou X, Ahn C, Feghali-Bostwick CA, Fritzler M, Arnett FC, Mayes MD. Pulmonary involvement in systemic sclerosis: associations with genetic, serologic, sociodemographic, and behavioral factors. Arthritis Rheum. 2007;57:318–326. doi: 10.1002/art.22532. [DOI] [PubMed] [Google Scholar]
- Kane GC, Varga J, Conant EF, Spirn PW, Jimenez S, Fish JE. Lung involvement in systemic sclerosis (scleroderma): relation to classification based on extent of skin involvement or autoantibody status. Respir Med. 1996;90:223–230. doi: 10.1016/S0954-6111(96)90291-7. [DOI] [PubMed] [Google Scholar]
- Walker UA, Tyndall A, Czirjak L, Denton C, Farge-Bancel D, Kowal-Bielecka O, Muller-Ladner U, Bocelli-Tyndall C, Matucci-Cerinic M. Clinical risk assessment of organ manifestations in systemic sclerosis: a report from the EULAR Scleroderma Trials And Research group database. Ann Rheum Dis. 2007;66:754–763. doi: 10.1136/ard.2006.062901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanaba K, Hasegawa M, Takehara K, Sato S. Comparative study of serum surfactant protein-D and KL-6 concentrations in patients with systemic sclerosis as markers for monitoring the activity of pulmonary fibrosis. J Rheumatol. 2004;31:1112–1120. [PubMed] [Google Scholar]
- Ober C, Chupp GL. The chitinase and chitinase-like proteins: a review of genetic and functional studies in asthma and immune-mediated diseases. Curr Opin Allergy Clin Immunol. 2009;9:401–408. doi: 10.1097/ACI.0b013e3283306533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gotway MB, Freemer MM, King TE Jr. Challenges in pulmonary fibrosis. 1: use of high resolution CT scanning of the lung for the evaluation of patients with idiopathic interstitial pneumonias. Thorax. 2007;62:546–553. doi: 10.1136/thx.2004.040022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mauviel A. Transforming growth factor-beta: a key mediator of fibrosis. Methods Mol Med. 2005;117:69–80. doi: 10.1385/1-59259-940-0:069. [DOI] [PubMed] [Google Scholar]
- Varga J, Abraham D. Systemic sclerosis: a prototypic multisystem fibrotic disorder. J Clin Invest. 2007;117:557–567. doi: 10.1172/JCI31139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakos G, Takagawa S, Chen SJ, Ferreira AM, Han G, Masuda K, Wang XJ, DiPietro LA, Varga J. Targeted disruption of TGF-β/Smad3 signaling modulates skin fibrosis in a mouse model of scleroderma. Am J Pathol. 2004;165:203–217. doi: 10.1016/s0002-9440(10)63289-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Shi W, Wang YL, Chen H, Bringas P Jr, Datto MB, Frederick JP, Wang XF, Warburton D. Smad3 deficiency attenuates bleomycin-induced pulmonary fibrosis in mice. Am J Physiol Lung Cell Mol Physiol. 2002;282:L585–L593. doi: 10.1152/ajplung.00151.2001. [DOI] [PubMed] [Google Scholar]
- Ishida W, Mori Y, Lakos G, Sun L, Shan F, Bowes S, Josiah S, Lee WC, Singh J, LE Ling, Varga J. Intracellular TGF-β receptor blockade abrogates Smad-dependent fibroblast activation in vitro and in vivo. J Invest Dermatol. 2006;126:1733–1744. doi: 10.1038/sj.jid.5700303. [DOI] [PubMed] [Google Scholar]
- Chen Y, Shi-wen X, Eastwood M, Black CM, Denton CP, Leask A, Abraham DJ. Contribution of activin receptor-like kinase 5 (transforming growth factor beta receptor type I) signaling to the fibrotic phenotype of scleroderma fibroblasts. Arthritis Rheum. 2006;54:1309–1316. doi: 10.1002/art.21725. [DOI] [PubMed] [Google Scholar]
- Daniels CE, Wilkes MC, Edens M, Kottom TJ, Murphy SJ, Limper AH, Leof EB. Imatinib mesylate inhibits the profibrogenic activity of TGF-β and prevents bleomycin-mediated lung fibrosis. J Clin Invest. 2004;114:1308–1316. doi: 10.1172/JCI19603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman JM, Melo JV. BCR-ABL in chronic myelogenous leukemia - how does it work? Acta Haematol. 2008;119:212–217. doi: 10.1159/000140633. [DOI] [PubMed] [Google Scholar]
- Wilkes MC, Leof EB. Transforming growth factor beta activation of c-Abl is independent of receptor internalization and regulated by phosphatidylinositol 3-kinase and PAK2 in mesenchymal cultures. J Biol Chem. 2006;281:27846–27854. doi: 10.1074/jbc.M603721200. [DOI] [PubMed] [Google Scholar]
- Distler JH, Jungel A, Huber LC, Schulze-Horsel U, Zwerina J, Gay RE, Michel BA, Hauser T, Schett G, Gay S, Distler O. Imatinib mesylate reduces production of extracellular matrix and prevents development of experimental dermal fibrosis. Arthritis Rheum. 2007;56:311–322. doi: 10.1002/art.22314. [DOI] [PubMed] [Google Scholar]
- Soria A, Cario-Andre M, Lepreux S, Rezvani HR, Pasquet JM, Pain C, Schaeverbeke T, Mahon FX, Taieb A. The effect of imatinib (Glivec) on scleroderma and normal dermal fibroblasts: a preclinical study. Dermatology. 2008;216:109–117. doi: 10.1159/000111507. [DOI] [PubMed] [Google Scholar]
- Bhattacharyya S, Ishida W, Wu M, Wilkes M, Mori Y, Hinchcliff M, Leof E, Varga J. A non-Smad mechanism of fibroblast activation by transforming growth factor-beta via c-Abl and Egr-1: selective modulation by imatinib mesylate. Oncogene. 2009;28:1285–1297. doi: 10.1038/onc.2008.479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniels CE, Lasky JA, Limper AH, Mieras K, Gabor E, Schroeder DR. Imatinib treatment for IPF: randomized placebo controlled trial results. Am J Respir Crit Care Med. 2009;181:604–610. doi: 10.1164/rccm.200906-0964OC. [DOI] [PubMed] [Google Scholar]
- Thiel G, Cibelli G. Regulation of life and death by the zinc finger transcription factor Egr-1. J Cell Physiol. 2002;193:287–292. doi: 10.1002/jcp.10178. [DOI] [PubMed] [Google Scholar]
- Wu M, Melichian DS, de la Garza M, Gruner K, Bhattacharyya S, Barr L, Nair A, Shahrara S, Sporn PH, Mustoe TA, Tourtellotte WG, Varga J. Essential roles for early growth response transcription factor Egr-1 in tissue fibrosis and wound healing. Am J Pathol. 2009;175:1041–1055. doi: 10.2353/ajpath.2009.090241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boon K, Bailey NW, Yang J, Steel MP, Groshong S, Kervitsky D, Brown KK, Schwarz MI, Schwartz DA. Molecular phenotypes distinguish patients with relatively stable from progressive idiopathic pulmonary fibrosis (IPF) PLoS One. 2009;4:e5134. doi: 10.1371/journal.pone.0005134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharyya S, Chen SJ, Wu M, Warner-Blankenship M, Ning H, Lakos G, Mori Y, Chang E, Nihijima C, Takehara K, Feghali-Bostwick C, Varga J. Smad-independent transforming growth factor-beta regulation of early growth response-1 and sustained expression in fibrosis: implications for scleroderma. Am J Pathol. 2008;173:1085–1099. doi: 10.2353/ajpath.2008.080382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuoka H, Hsu E, Ruiz XD, Steinman RA, Choi AM, Feghali-Bostwick CA. The fibrotic phenotype induced by IGFBP-5 is regulated by MAPK activation and egr-1-dependent and -independent mechanisms. Am J Pathol. 2009;175:605–615. doi: 10.2353/ajpath.2009.080991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh AK, Bhattacharyya S, Lakos G, Chen SJ, Mori Y, Varga J. Disruption of transforming growth factor beta signaling and profibrotic responses in normal skin fibroblasts by peroxisome proliferator-activated receptor gamma. Arthritis Rheum. 2004;50:1305–1318. doi: 10.1002/art.20104. [DOI] [PubMed] [Google Scholar]
- Wei J, Ghosh A, Komura K, Qi-Qunag Hunag, Sargent J, Jain M, Whitfield M, Feghali-Bostwick C, Varga J. Smad-dependent inhibition of peroxisome proliferator activated receptor-gamma expression and defective expression and function in systemic sclerosis: a novel mechanism for persistent fibrogenesis. PLoS One. 2010. in press . [DOI] [PMC free article] [PubMed]
- Sime PJ. The antifibrogenic potential of PPARgamma ligands in pulmonary fibrosis. J Investig Med. 2008;56:534–538. doi: 10.2310/JIM.0b013e31816464e9. [DOI] [PubMed] [Google Scholar]
- Milam JE, Keshamouni VG, Phan SH, Hu B, Gangireddy SR, Hogaboam CM, Standiford TJ, Thannickal VJ, Reddy RC. PPAR-γ agonists inhibit profibrotic phenotypes in human lung fibroblasts and bleomycin-induced pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol. 2008;294:L891–L901. doi: 10.1152/ajplung.00333.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clozel M, Salloukh H. Role of endothelin in fibrosis and anti-fibrotic potential of bosentan. Ann Med. 2005;37:2–12. doi: 10.1080/07853890410018925. [DOI] [PubMed] [Google Scholar]
- Hocher B, Schwarz A, Fagan KA, Thone-Reineke C, El-Hag K, Kusserow H, Elitok S, Bauer C, Neumayer HH, Rodman DM, Theuring F. Pulmonary fibrosis and chronic lung inflammation in ET-1 transgenic mice. Am J Respir Cell Mol Biol. 2000;23:19–26. doi: 10.1165/ajrcmb.23.1.4030. [DOI] [PubMed] [Google Scholar]
- Wendel M, Petzold A, Koslowski R, Kasper M, Augstein A, Knels L, Bleyl JU, Koch T. Localization of endothelin receptors in bleomycin-induced pulmonary fibrosis in the rat. Histochem Cell Biol. 2004;122:507–517. doi: 10.1007/s00418-004-0708-7. [DOI] [PubMed] [Google Scholar]
- Shi-Wen X, Renzoni EA, Kennedy L, Howat S, Chen Y, Pearson JD, Bou-Gharios G, Dashwood MR, du Bois RM, Black CM, Denton CP, Abraham DJ, Leask A. Endogenous endothelin-1 signaling contributes to type I collagen and CCN2 overexpression in fibrotic fibroblasts. Matrix Biol. 2007;26:625–632. doi: 10.1016/j.matbio.2007.06.003. [DOI] [PubMed] [Google Scholar]
- Tager AM, LaCamera P, Shea BS, Campanella GS, Selman M, Zhao Z, Polosukhin V, Wain J, Karimi-Shah BA, Kim ND, Hart WK, Pardo A, Blackwell TS, Xu Y, Chun J, Luster AD. The lysophosphatidic acid receptor LPA1 links pulmonary fibrosis to lung injury by mediating fibroblast recruitment and vascular leak. Nat Med. 2008;14:45–54. doi: 10.1038/nm1685. [DOI] [PubMed] [Google Scholar]
- Hamaguchi Y, Fujimoto M, Matsushita T, Hasegawa M, Takehara K, Sato S. Elevated serum insulin-like growth factor (IGF-1) and IGF binding protein-3 levels in patients with systemic sclerosis: possible role in development of fibrosis. J Rheumatol. 2008;35:2363–2371. doi: 10.3899/jrheum.080340. [DOI] [PubMed] [Google Scholar]
- Choi JE, Lee SS, Sunde DA, Huizar I, Haugk KL, Thannickal VJ, Vittal R, Plymate SR, Schnapp LM. Insulin-like growth factor-I receptor blockade improves outcome in mouse model of lung injury. Am J Respir Crit Care Med. 2009;179:212–219. doi: 10.1164/rccm.200802-228OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perbal B. CCN proteins: multifunctional signalling regulators. Lancet. 2004;363:62–64. doi: 10.1016/S0140-6736(03)15172-0. [DOI] [PubMed] [Google Scholar]
- Ponticos M, Holmes AM, Shi-wen X, Leoni P, Khan K, Rajkumar VS, Hoyles RK, Bou-Gharios G, Black CM, Denton CP, Abraham DJ, Leask A, Lindahl GE. Pivotal role of connective tissue growth factor in lung fibrosis: MAPK-dependent transcriptional activation of type I collagen. Arthritis Rheum. 2009;60:2142–2155. doi: 10.1002/art.24620. [DOI] [PubMed] [Google Scholar]
- Steen VD, Owens GR, Redmond C, Rodnan GP, Medsger TA Jr. The effect of D-penicillamine on pulmonary findings in systemic sclerosis. Arthritis Rheum. 1985;28:882–888. doi: 10.1002/art.1780280807. [DOI] [PubMed] [Google Scholar]
- Steen VD, Lanz JK Jr, Conte C, Owens GR, Medsger TA Jr. Therapy for severe interstitial lung disease in systemic sclerosis. A retrospective study. Arthritis Rheum. 1994;37:1290–1296. doi: 10.1002/art.1780370904. [DOI] [PubMed] [Google Scholar]
- Akesson A, Scheja A, Lundin A, Wollheim FA. Improved pulmonary function in systemic sclerosis after treatment with cyclophosphamide. Arthritis Rheum. 1994;37:729–735. doi: 10.1002/art.1780370518. [DOI] [PubMed] [Google Scholar]
- White B, Moore WC, Wigley FM, Xiao HQ, Wise RA. Cyclophosphamide is associated with pulmonary function and survival benefit in patients with scleroderma and alveolitis. Ann Intern Med. 2000;132:947–954. doi: 10.7326/0003-4819-132-12-200006200-00004. [DOI] [PubMed] [Google Scholar]
- Tashkin DP, Elashoff R, Clements PJ, Roth MD, Furst DE, Silver RM, Goldin J, Arriola E, Strange C, Bolster MB, Seibold JR, Riley DJ, Hsu VM, Varga J, Schraufnagel D, Theodore A, Simms R, Wise R, Wigley F, White B, Steen V, Read C, Mayes M, Parsley E, Mubarak K, Connolly MK, Golden J, Olman M, Fessler B, Rothfi eld N. et al. Effects of 1-year treatment with cyclophosphamide on outcomes at 2 years in scleroderma lung disease. Am J Respir Crit Care Med. 2007;176:1026–1034. doi: 10.1164/rccm.200702-326OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldin JG, Lynch DA, Strollo DC, Suh RD, Schraufnagel DE, Clements PJ, Elashoff RM, Furst DE, Vasunilashorn S, McNitt-Gray MF, Brown MS, Roth MD, Tashkin DP. High-resolution CT scan findings in patients with symptomatic scleroderma-related interstitial lung disease. Chest. 2008;134:358–367. doi: 10.1378/chest.07-2444. [DOI] [PubMed] [Google Scholar]
- Hoyles RK, Ellis RW, Wellsbury J, Lees B, Newlands P, Goh NS, Roberts C, Desai S, Herrick AL, McHugh NJ, Foley NM, Pearson SB, Emery P, Veale DJ, Denton CP, Wells AU, Black CM, du Bois RM. A multicenter, prospective, randomized, double-blind, placebo-controlled trial of corticosteroids and intravenous cyclophosphamide followed by oral azathioprine for the treatment of pulmonary fibrosis in scleroderma. Arthritis Rheum. 2006;54:3962–3970. doi: 10.1002/art.22204. [DOI] [PubMed] [Google Scholar]
- Zamora AC, Wolters PJ, Collard HR, Connolly MK, Elicker BM, . Webb WR, King TE Jr, Golden JA. Use of mycophenolate mofetil to treat scleroderma-associated interstitial lung disease. Respir Med. 2008;102:150–155. doi: 10.1016/j.rmed.2007.07.021. [DOI] [PubMed] [Google Scholar]
- Gerbino AJ, Goss CH, Molitor JA. Effect of mycophenolate mofetil on pulmonary function in scleroderma-associated interstitial lung disease. Chest. 2008;133:455–460. doi: 10.1378/chest.06-2861. [DOI] [PubMed] [Google Scholar]
- Dheda K, Lalloo UG, Cassim B, Mody GM. Experience with azathioprine in systemic sclerosis associated with interstitial lung disease. Clin Rheumatol. 2004;23:306–309. doi: 10.1007/s10067-004-0906-7. [DOI] [PubMed] [Google Scholar]
- Berezne A, Ranque B, Valeyre D, Brauner M, Allanore Y, Launay D, Le Guern V, Kahn JE, Couderc LJ, Constans J, Cohen P, Mahr A, Pagnoux C, Hachulla E, Kahan A, Cabane J, Guillevin L, Mouthon L. Therapeutic strategy combining intravenous cyclophosphamide followed by oral azathioprine to treat worsening interstitial lung disease associated with systemic sclerosis: a retrospective multicenter open-label study. J Rheumatol. 2008;35:1064–1072. [PubMed] [Google Scholar]
- King TE Jr, Behr J, Brown KK, du Bois RM, Lancaster L, de Andrade JA, Stahler G, Leconte I, Roux S, Raghu G. BUILD-1: a randomized placebo-controlled trial of bosentan in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2008;177:75–81. doi: 10.1164/rccm.200705-732OC. [DOI] [PubMed] [Google Scholar]
- Silver RM. Endothelin and scleroderma lung disease. Rheumatology (Oxford) 2008;47(Suppl 5):v25–v26. doi: 10.1093/rheumatology/ken283. [DOI] [PubMed] [Google Scholar]
- van Daele PL, Dik WA, Thio HB, van Hal PT, van Laar JA, Hooijkaas H, van Hagen PM. Is imatinib mesylate a promising drug in systemic sclerosis? Arthritis Rheum. 2008;58:2549–2552. doi: 10.1002/art.23648. [DOI] [PubMed] [Google Scholar]
- Sfikakis PP, Gorgoulis VG, Katsiari CG, Evangelou K, Kostopoulos C, Black CM. Imatinib for the treatment of refractory, diffuse systemic sclerosis. Rheumatology (Oxford) 2008;47:735–737. doi: 10.1093/rheumatology/ken104. [DOI] [PubMed] [Google Scholar]
- Oku H, Shimizu T, Kawabata T, Nagira M, Hikita I, Ueyama A, Matsushima S, Torii M, Arimura A. Antifibrotic action of pirfenidone and prednisolone: different effects on pulmonary cytokines and growth factors in bleomycin-induced murine pulmonary fibrosis. Eur J Pharmacol. 2008;590:400–408. doi: 10.1016/j.ejphar.2008.06.046. [DOI] [PubMed] [Google Scholar]
- Azuma A, Nukiwa T, Tsuboi E, Suga M, Abe S, Nakata K, Taguchi Y, Nagai S, Itoh H, Ohi M, Sato A, Kudoh S. Double-blind, placebo-controlled trial of pirfenidone in patients with idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2005;171:1040–1047. doi: 10.1164/rccm.200404-571OC. [DOI] [PubMed] [Google Scholar]
- Taniguchi H, Ebina M, Kondoh Y, Ogura T, Azuma A, Suga M, Taguchi Y, Takahashi H, Nakata K, Sato A, Takeuchi M, Raghu G, Kudoh S, Nukiwa T. Pirfenidone in idiopathic pulmonary fibrosis. Eur Respir J. 2009;35:821–829. doi: 10.1183/09031936.00005209. [DOI] [PubMed] [Google Scholar]
- Noble PW, Albera C, Bradford W, Costabel U, Kardatzke D, King T, Jr, Sahn S, Szwarcberg J, Valeyre D, du Bois R. American Thoracic Society International Conference. San Diego, CA; 2009. The CAPACITY (CAP) trials: randomized, double-blind, placebo-controlled, phase III trials of pirfenidone (PFD) in patients with idiopathic pulmonary fibrosis (IPF) [abstract 216] [Google Scholar]
- Nagai S, Hamada K, Shigematsu M, Taniyama M, Yamauchi S, Izumi T. Open-label compassionate use one year-treatment with pirfenidone to patients with chronic pulmonary fibrosis. Intern Med. 2002;41:1118–1123. doi: 10.2169/internalmedicine.41.1118. [DOI] [PubMed] [Google Scholar]
- Schachna L, Medsger TA Jr, Dauber JH, Wigley FM, Braunstein NA, White B, Steen VD, Conte JV, Yang SC, McCurry KR, Borja MC, Plaskon DE, Orens JB, Gelber AC. Lung transplantation in scleroderma compared with idiopathic pulmonary fibrosis and idiopathic pulmonary arterial hypertension. Arthritis Rheum. 2006;54:3954–3961. doi: 10.1002/art.22264. [DOI] [PubMed] [Google Scholar]
- Massad MG, Powell CR, Kpodonu J, Tshibaka C, Hanhan Z, Snow NJ, Geha AS. Outcomes of lung transplantation in patients with scleroderma. World J Surg. 2005;29:1510–1515. doi: 10.1007/s00268-005-0017-x. [DOI] [PubMed] [Google Scholar]
- Bouros D, Wells AU, Nicholson AG, Colby TV, Polychronopoulos V, Pantelidis P, Haslam PL, Vassilakis DA, Black CM, du Bois RM. Histopathologic subsets of fibrosing alveolitis in patients with systemic sclerosis and their relationship to outcome. Am J Respir Crit Care Med. 2002;165:1581–1586. doi: 10.1164/rccm.2106012. [DOI] [PubMed] [Google Scholar]
- Wells AU, Cullinan P, Hansell DM, Rubens BM, Black CM, Newman-Taylor AJ, Du Bois RM. Fibrosing alveolitis associated with systemic sclerosis has a better prognosis than lone cryptogenic fibrosing alveolitis. Am J Respir Crit Care Med. 1994;149:1583–1590. doi: 10.1164/ajrccm.149.6.8004317. [DOI] [PubMed] [Google Scholar]
- McDonagh J, Greaves M, Wright AR, Heycock C, Owen JP, Kelly C. High resolution computed tomography of the lungs in patients with rheumatoid arthritis and interstitial lung disease. Br J Rheumatol. 1994;33:118–122. doi: 10.1093/rheumatology/33.2.118. [DOI] [PubMed] [Google Scholar]
- Lee HK, Kim DS, Yoo B, Seo JB, Rho JY, Colby TV, Kitaichi M. Histopathologic pattern and clinical features of rheumatoid arthritis-associated interstitial lung disease. Chest. 2005;127:2019–2027. doi: 10.1378/chest.127.6.2019. [DOI] [PubMed] [Google Scholar]
- Turesson C, Matteson EL, Colby TV, Vuk-Pavlovic Z, Vassallo R, Weyand CM, Tazelaar HD, Limper AH. Increased CD4+ T cell infiltrates in rheumatoid arthritis-associated interstitial pneumonitis compared with idiopathic interstitial pneumonitis. Arthritis Rheum. 2005;52:73–79. doi: 10.1002/art.20765. [DOI] [PubMed] [Google Scholar]
- Lynch JP, Saggar R, Weigt SS, Zisman DA, White ES. Usual interstitial pneumonia. Semin Respir Crit Care Med. 2006;27:634–651. doi: 10.1055/s-2006-957335. [DOI] [PubMed] [Google Scholar]
- Demedts M, Behr J, Buhl R, Costabel U, Dekhuijzen R, Jansen HM, MacNee W, Thomeer M, Wallaert B, Laurent F, Nicholson AG, Verbeken EK, Verschakelen J, Flower CD, Capron F, Petruzzelli S, De Vuyst P, van den Bosch JM, Rodriguez-Becerra E, Corvasce G, Lankhorst I, Sardina M, Montanari M. High-dose acetylcysteine in idiopathic pulmonary fibrosis. N Engl J Med. 2005;353:2229–2242. doi: 10.1056/NEJMoa042976. [DOI] [PubMed] [Google Scholar]
- Saketkoo LA, Espinoza LR. Rheumatoid arthritis interstitial lung disease: mycophenolate mofetil as an antifibrotic and disease-modifying antirheumatic drug. Arch Intern Med. 2008;168:1718–1719. doi: 10.1001/archinte.168.15.1718. [DOI] [PubMed] [Google Scholar]
- Chen IJ, Jan Wu YJ, Lin CW, Fan KW, Luo SF, Ho HH, Liou LB, Tsai WP, Chen JY, Yang CH, Kuo CF, Yu KH. Interstitial lung disease in polymyositis and dermatomyositis. Clin Rheumatol. 2009;28:639–646. doi: 10.1007/s10067-009-1110-6. [DOI] [PubMed] [Google Scholar]
- Hayashi S, Tanaka M, Kobayashi H, Nakazono T, Satoh T, Fukuno Y, Aragane N, Tada Y, Koarada S, Ohta A, Nagasawa K. High-resolution computed tomography characterization of interstitial lung diseases in polymyositis/dermatomyositis. J Rheumatol. 2008;35:260–269. [PubMed] [Google Scholar]
- Tillie-Leblond I, Wislez M, Valeyre D, Crestani B, Rabbat A, Israel-Biet D, Humbert M, Couderc LJ, Wallaert B, Cadranel J. Interstitial lung disease and anti-Jo-1 antibodies: difference between acute and gradual onset. Thorax. 2008;63:53–59. doi: 10.1136/thx.2006.069237. [DOI] [PubMed] [Google Scholar]
- Kubo M, Ihn H, Yamane K, Kikuchi K, Yazawa N, Soma Y, Tamaki K. Serum KL-6 in adult patients with polymyositis and dermatomyositis. Rheumatology (Oxford) 2000;39:632–636. doi: 10.1093/rheumatology/39.6.632. [DOI] [PubMed] [Google Scholar]
- Mukae H, Ishimoto H, Sakamoto N, Hara S, Kakugawa T, Nakayama S, Ishimatsu Y, Kawakami A, Eguchi K, Kohno S. Clinical differences between interstitial lung disease associated with clinically amyopathic dermatomyositis and classic dermatomyositis. Chest. 2009;136:1341–1347. doi: 10.1378/chest.08-2740. [DOI] [PubMed] [Google Scholar]
- Sato S, Hoshino K, Satoh T, Fujita T, Kawakami Y, Kuwana M. RNA helicase encoded by melanoma differentiation-associated gene 5 is a major autoantigen in patients with clinically amyopathic dermatomyositis: association with rapidly progressive interstitial lung disease. Arthritis Rheum. 2009;60:2193–2200. doi: 10.1002/art.24621. [DOI] [PubMed] [Google Scholar]
- Fujisawa T, Suda T, Nakamura Y, Enomoto N, Ide K, Toyoshima M, Uchiyama H, Tamura R, Ida M, Yagi T, Yasuda K, Genma H, Hayakawa H, Chida K, Nakamura H. Differences in clinical features and prognosis of interstitial lung diseases between polymyositis and dermatomyositis. J Rheumatol. 2005;32:58–64. [PubMed] [Google Scholar]
- Yamasaki Y, Yamada H, Yamasaki M, Ohkubo M, Azuma K, Matsuoka S, Kurihara Y, Osada H, Satoh M, Ozaki S. Intravenous cyclophosphamide therapy for progressive interstitial pneumonia in patients with polymyositis/dermatomyositis. Rheumatology (Oxford) 2007;46:124–130. doi: 10.1093/rheumatology/kel112. [DOI] [PubMed] [Google Scholar]
- Ochi S, Nanki T, Takada K, Suzuki F, Komano Y, Kubota T, Miyasaka N. Favorable outcomes with tacrolimus in two patients with refractory interstitial lung disease associated with polymyositis/dermatomyositis. Clin Exp Rheumatol. 2005;23:707–710. [PubMed] [Google Scholar]
- Wilkes MR, Sereika SM, Fertig N, Lucas MR, Oddis CV. Treatment of antisynthetase-associated interstitial lung disease with tacrolimus. Arthritis Rheum. 2005;52:2439–2446. doi: 10.1002/art.21240. [DOI] [PubMed] [Google Scholar]
- Sem M, Molberg O, Lund MB, Gran JT. Rituximab treatment of the anti-synthetase syndrome: a retrospective case series. Rheumatology (Oxford) 2009;48:968–971. doi: 10.1093/rheumatology/kep157. [DOI] [PubMed] [Google Scholar]
- Rios Fernandez R, Callejas Rubio JL, Sanchez Cano D, Saez Moreno JA, Ortego Centeno N. Rituximab in the treatment of dermatomyositis and other inflammatory myopathies. A report of 4 cases and review of the literature. Clin Exp Rheumatol. 2009;27:1009–1016. [PubMed] [Google Scholar]
- Ito I, Nagai S, Kitaichi M, Nicholson AG, Johkoh T, Noma S, Kim DS, Handa T, Izumi T, Mishima M. Pulmonary manifestations of primary Sjogren's syndrome: a clinical, radiologic, and pathologic study. Am J Respir Crit Care Med. 2005;171:632–638. doi: 10.1164/rccm.200403-417OC. [DOI] [PubMed] [Google Scholar]
- Parambil JG, Myers JL, Lindell RM, Matteson EL, Ryu JH. Interstitial lung disease in primary Sjogren syndrome. Chest. 2006;130:1489–1495. doi: 10.1378/chest.130.5.1489. [DOI] [PubMed] [Google Scholar]
- Pego-Reigosa JM, Medeiros DA, Isenberg DA. Respiratory manifestations of systemic lupus erythematosus: old and new concepts. Best Pract Res Clin Rheumatol. 2009;23:469–480. doi: 10.1016/j.berh.2009.01.002. [DOI] [PubMed] [Google Scholar]
- Lim SW, Gillis D, Smith W, Hissaria P, Greville H, Peh CA. Rituximab use in systemic lupus erythematosus pneumonitis and a review of current reports. Intern Med J. 2006;36:260–262. doi: 10.1111/j.1445-5994.2006.01055.x. [DOI] [PubMed] [Google Scholar]
- Mosca M, Tani C, Bombardieri S. Undifferentiated connective tissue diseases (UCTD): a new frontier for rheumatology. Best Pract Res Clin Rheumatol. 2007;21:1011–1023. doi: 10.1016/j.berh.2007.09.004. [DOI] [PubMed] [Google Scholar]
- Kinder BW, Collard HR, Koth L, Daikh DI, Wolters PJ, Elicker B, Jones KD, King TE Jr. Idiopathic nonspecific interstitial pneumonia: lung manifestation of undifferentiated connective tissue disease? Am J Respir Crit Care Med. 2007;176:691–697. doi: 10.1164/rccm.200702-220OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinder BW, Shariat C, Collard HR, Koth LL, Wolters PJ, Golden JA, Panos RJ, King TE Jr. Undifferentiated connective tissue disease-associated interstitial lung disease: changes in lung function. Lung. 2010;188:143–149. doi: 10.1007/s00408-009-9226-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prakash UB. Respiratory complications in mixed connective tissue disease. Clin Chest Med. 1998;19:733–746. doi: 10.1016/S0272-5231(05)70113-1. ix. [DOI] [PubMed] [Google Scholar]
- Bodolay E, Szekanecz Z, Devenyi K, Galuska L, Csipo I, Vegh J, Garai I, Szegedi G. Evaluation of interstitial lung disease in mixed connective tissue disease (MCTD) Rheumatology (Oxford) 2005;44:656–661. doi: 10.1093/rheumatology/keh575. [DOI] [PubMed] [Google Scholar]