

Abstract
Following intravascular delivery, an important route of administration for many clinical applications, the liver is the predominant site of adenovirus serotype 5 (Ad5) sequestration thereby posing a risk of toxicity. In this regard, it has recently been demonstrated that the Ad5 capsid binds to the blood coagulation factor X (FX) via the Ad5 hexon protein. This interaction mediates the majority of Ad5 liver transduction. Patient FX levels can be diminished by the administration of warfarin, a vitamin K inhibitor in the liver which decreases FX production; however, warfarin is a potent anticoagulant and can have a number of undesired side effects. Therefore, genetic modification of the virus to ablate FX binding is the preferred approach. Modifications of the hexon protein, specifically within the hypervariable 5 (HVR5) and 7 (HVR7) regions, have produced Ad5 vectors that show minimal liver sequestration. Our laboratory has pioneered adenovirus hexon modifications, including insertion of peptide ligands into the hypervariable regions and substitution of the adenovirus hexon with hexon proteins from alternate serotypes. Substitution of the adenovirus serotype 3 (Ad3) hexon protein onto the Ad5 capsid has been further characterized in regard to its interaction with FX and incorporated into an infectivity enhanced conditionally replicative adenovirus (CRAd). In vitro evaluation of these hexon modified vectors demonstrated decreased binding to FX and decreased cell transduction via FX mediated pathways. Furthermore, in vivo biodistribution studies in mice exhibited a decrease in liver sequestration. Utilizing xenograft tumor models, anti-tumor efficacy of the hexon-modified CRAds was enhanced over non-modified controls.
Keywords: Adenovirus, Gene Therapy, Oncolytic Virotherapy, Cancer
Introduction
Adenovirus (Ad)-based vectors represent the most commonly utilized vectors in gene therapy clinical trials (1), with the majority of these vectors based on serotype 5 (Ad5). Furthermore, cancer represents the disease most commonly addressed. To achieve an effective Ad-based cancer therapy, systemic administration (i.e., intravenous (IV) administration) is the ideal mode of delivery. However, effective IV administration of Ad-based vectors has been hindered by the innate liver tropism of Ad5 and concerns of hepatotoxicity. Liver tropism also results in a sequestration of the administered Ad vector away from the desired tumor targets and a rapid clearance, thereby necessitating higher doses of administered vector and resulting in poor anti-tumor efficacy (2).
Key components of this process and how to overcome the liver tropism of Ad5 have recently been elucidated and may involve multiple redundant and synergistic mechanisms (3). In this regard, the Ad5 capsid interacts with multiple vitamin-K dependent coagulation factors (4). Specifically, coagulation factor X (FX) binds to the Ad5 capsid protein hexon and mediates the majority of the Ad5 liver tropism, presumably via heparin sulfate proteoglycan mediated pathways (5–7). Through crystal structure analysis and mutagenesis studies, it has been discovered that hypervariable regions (HVRs) 5 and 7 are critical for the interaction with FX (8). Multiple vectors have been published that involve mutation of one or more of these HVRs, either by point mutation or peptide insertion. These hexon modifications have been incorporated into non-replicative vectors and some of the modifications are complex and may not be easily translated to other vectors. Our group initially pioneered Ad5 hexon modifications with substitution of the Ad serotype 3 (Subspecies B, Ad3) hexon onto the Ad5 capsid (9) and peptide insertions into the various HVRs (10).
Pre-treatment with the drug warfarin, which inhibits vitamin-K dependent coagulation factor production, can deplete circulating FX levels and decrease the liver tropism of IV administered Ad5. Pre-treatment with warfarin prior to the administration of an oncolytic Ad did increase the therapeutic window of the oncolytic Ad and resulted in an enhanced anti-tumor efficacy in two separate studies (11, 12). However, warfarin has numerous side effects, multiple drug interactions, a narrow therapeutic index, and can place the patient in a hypocoagable state that may present other problems. Therefore, incorporation of a genetic modification of the hexon protein is the preferred approach, eliminating the need for anti-coagulation therapy. One of the initial hexon modifications demonstrating FX binding has been advanced to a replicative oncolytic Ad and has shown improved anti-tumor efficacies compared to its unmodified control (13).
Ablating Ad5 liver tropism is only a portion of improving Ad tumor targeting. In this context, Ad5-based vectors selectively transduce target cells via the Ad fiber protein binding to the primary cellular receptor, the Coxsackie and Adenovirus Receptor (CAR). However, CAR is poorly expressed on many tumor targets, thereby limiting transduction (14). One means of improving tumor target transduction is by enhancing the infectivity of the vector with substitution of the knob domain of the fiber protein with that of another Ad serotype whose primary receptor has a better tumor cell expression profile. We have previously shown that substitution of the Ad3 knob domain improves tumor cell transduction for a variety of cancer types and improves anti-tumor efficacy of oncolytic Ads (15, 16).
We hypothesized that ablating Ad5 capsid binding to FX via a genetic modification could be incorporated into an infectivity enhanced oncolytic Ad to reduce liver tropism and increase the anti-tumor efficacy.
Methods and Materials
Cell lines
HEK-293, A549, HepG2, and Skov3.ip1 cells were purchased from ATCC (Manassas, VA, July 2009 – no further authentication was performed). All cells were grown in DMEM/F12 50:50 supplemented with 10% FBS, 1% penicillin/streptomycin, and 1% L-glutamine, cultured in a humidified incubator at 37° C with 5% CO2.
Animals
All animal care was performed under IACUC approved protocols. Five week old, female C57BL/6 mice and 4 week old, female athymic, nude mice were purchased from Frederick Cancer Research (Hartford, CT) and housed in UAB Animal Resources Program facilities.
Reagents, antibodies
Purified human factor X and XI, and anti-factor X monoclonal mouse antibodies were purchased from Haematologic Technologies (Essex Junction, VT). Anti-Adenovirus capsid polyclonal antibody was a gift from Dr. Joanne Douglas (UAB). Goat anti-mouse antibodies conjugated to horseradish peroxidase (HRP) and goat anti-rabbit antibodies conjugated to HRP were purchased from Dako, Inc. (Carpinteria, CA). CM5 biocore sensor chips were purchased from GE Healthcare (Piscataway, NJ).
Adenoviral vectors
Wild-type Ad48 was purchased from ATCC (Manassas, VA). Construction of non-replicative Ad5, Ad5HVR5-6H, and Ad5H3 encoding a green fluorescent protein (GFP)-luciferase (luc) expression cassette under the CMV promoter in the E1 coding region were previously published (9, 10). Non-replicative Ad5HBAP encoding a red fluorescent protein (RFP) expression cassette under the CMV promoter in the E1 coding region was provided as a gift by Dr. Michael Barry (Rochester, MN) (17). To generate the non-replicative Ad5HBAP encoding the isogenic GFP-luc cassette as the above vectors, the Ad5 hexon sequence with insertion of the BAP peptide was cloned by PCR using the Dr. Barry provided virus as a template and using the primers (F: TAACAAGTTTAGAAACCCCACG and R: CATGGGATCCACCTCAAAAGTC). The PCR product was digested with DraIII and BamHI and inserted into the previously published hexon shuttle vector pHS (9) to generate HBAP/pHS. Homologous recombination in Escherichia coli strain BJ5183 (Stratagene, La Jolla, CA) with the SwaI-digested Ad5 backbone containing the E1 CMV GFP-Luc cassette and hexon deletion was utilized to generate the final non-replicative Ad5HBAP vector encoding GFP-Luc in E1. Restriction digests and sequencing confirmed the appropriate sequences. Replicative vectors were constructed on an Ad backbone with the Δ24 E1 mutation and deletion of E3 with replacement of the E3 protein ADP and with either Ad5 fiber or with substitution of the Ad3 fiber knob domain onto the Ad5 fiber. Hexon modifications were introduced by digesting the non-replicative backbones with SfiI (unique SfiI enzyme sites flank the hexon protein coding region) and then insertion into the replicative backbones by digestion with SfiI and subsequent ligation. Adenoviral vector genomes were then digested with PacI and transfected into HEK-293 cells. Non-replicative viruses were upscaled via HEK-293 cells whereas, replicative viruses were upscaled with A549 cells after initial transfection with HEK-293 cells. Virus was purified by CsCl double ultracentrifugation and dialysis into the appropriate buffer.
Slot blot analysis
PVDF membrane (Bio-Rad, Hercules, CA) was pre-incubated with 100% methanol and then rinsed with TBS. Virus particles (vp) (1010 vp per sample) were then exposed to the PVDF membrane using a slot blot chamber. The PVDF membrane was washed 5 times with TBST and then incubated with TBS + 1% non-fat dry milk blocking reagent (Bio-Rad) for 30 minutes at room temperature. Purified human FX and all antibodies were diluted in TBS + 1% non-fat dry milk blocking reagent. The membrane was incubated with either purified human FX (8 ug/ml) or anti-Ad polyclonal rabbit antibody (1:1000 dilution) for 1 hour at room temperature. The membranes were washed 5 times with TBST between all incubations and incubated with TBS +non-fat dry milk blocking reagent for 30 minutes at room temperature prior to the next antibody incubation. The FX-incubated samples were incubated with anti-FX monoclonal mouse antibodies (Haematologic Technologies) followed by anti-mouse goat-HRP antibodies (DAKO, Inc.). Anti-Ad incubated samples were incubated with anti-rabbit goat-HRP (DAKO, Inc.). Following a final wash cycle (5 times with TBST), both PVDF membranes were developed with diaminobenzidine (DAB) (Sigma-Aldrich, St. Louis, MO).
Surface Plasmon resonance (SPR) analysis
Purified human FX and FXI proteins were covalently immobilized to CM5 biocore sensor chips by amine coupling. Viruses (1010 virus particles) were diluted in 10 mM hepes pH 7.4, 150 mM NaCl, 5mM CaCl2. Samples were processed at 37° C. Samples were analyzed using a Biocore 2000 (GE Healthcare). Virus samples were passed over the coagulation factor coupled chips at 30 μl/min. Chip surfaces were regenerated between virus samples by injection of 10 mM hepes pH 7.4, 150 mM NaCl, 5mM CaCl2, 3mM EDTA, 0.005% Tween-20. All virus samples were analyzed in duplicate.
In vitro factor X studies
A549, HepG2, Skov3.ip1 cells were seeded in 24 well plates at a density of 5 × 104 cells/well and allowed to grow 24 hours as described above. Viruses were diluted to quantities sufficient for 10 virus particles (vp)/cell and 100 vp/cell in a volume of 200 μl/well. Selected virus samples were then incubated for 30 minutes at room temperature with FX at 4 μg/ml, 8 μg/ml, or 16 μg/ml. Virus was then added to wells and infection allowed for 2 hours at 37° C. After 2 hours, virus and media were removed and fresh growth media was added to each well. Cells were allowed to grow for 48 hours and then analyzed for luciferase expression via Promega luciferase assay kit (Promega, Madison, WI). Protein concentrations were calculated with Bio-Rad direct protein assay kit (Bio-Rad). All samples were performed in triplicate.
Crystal Violet assay
Skov3.ip1 cells were seeded in 12 well plates at a density of 2.5 × 104 cells/well and allowed to grow for 24 hours ad described above. Viruses were diluted to quantities sufficient for a range of 0.01 to 1000 vp/cell in 200 μl/well of 2% infection media. Viruses were allowed to infect cells for 2 hours, after which virus and media was removed and cells were grown in 2% growth media for 10 days. After 10 days, media was removed, cells were washed with PBS and then fixed with 10% formaldehyde. Remaining cells were stained with crystal violet solution 10 minutes and rinsed with deionized water.
In Vivo biodistribution studies
For biodistribution studies, one group (three to five mice per group) of five week old C57BL/6 mice were pretreated with warfarin (5 mg/kg) dissolved in peanut oil administered subcutaneously three days and one day prior to vector injections. Mice were then injected with 1011 vp/mouse via tail vein injections. After 48 hours mice were sacrificed and organs harvested. Organs were lysed with cell culture lysis reagent (Promega) and mechanically homogenized with glass beads. Cell lysate was analyzed for luciferase expression with Promega luciferase assay kit and protein concentrations obtained with Bio-Rad direct protein assay kit. For tumor studies, skov3.ip1 cells were grown to near confluency as described above, cells were harvested and 1×106 cells/mouse were injected into the flank of 4 week old athymic nude mice. Tumors were allowed to establish over 4–6 weeks. Mice in the warfarin pre-treatment group were dosed with warfarin (5 mg/kg) at three days and one day prior to vector injections. Mice were injected with 1011 vp/mouse via tail vein injections. After 48 hours mice were sacrificed and organs and tumors harvested. DNA was isolated using Qiagen DNeasy Blood and Tissue Kit (Qiagen, Valencia, CA). Quantitative PCR was performed on DNA samples using primers specific for luciferase transgene and mouse or human β–actin.
In vivo anti-tumor efficacy studies
Skov3.ip1 cells were grown to near confluency as described above, cells were harvested and 1×106 cells/mouse were injected into the flank of 4 week old athymic nude mice. Tumors were allowed to establish over 4–6 weeks and mice placed into experimental groups with tumors ranging from 0.3–0.5 cm in diameter. Mice in the warfarin pre-treatment group were dosed with warfarin (5 mg/kg) at three days and one day prior to vector injections. Mice were injected with 1011 vp/mouse via tail vein injections. Tumors were measured with calipers at days 0, 3, 7, 14, 21, and 28 by measuring the average diameter and using the formula length × width2 × 0.5 (18). Five mice from each experimental group were sacrificed at each time point beyond day 0 and tumor and liver samples were harvested for viral dna analysis. All remaining mice were sacrificed at 28 days and liver and tumor samples were harvested. DNA was isolated from liver and tumor samples using Qiagen DNeasy Blood and Tissue Kit (Qiagen, Valencia, CA). Quantitative PCR was performed on DNA samples using primers specific for Ad E4 coding region and mouse or human β–actin.
Statistical Analysis
All data are expressed as means. In vitro experiments were performed in triplicate. In vivo experiments were performed with at least 4 animals per group. Statistical analysis was performed using Student’s T-test or for multiple comparisons, analysis of variance and Tukey’s pairwise comparison using SAS software. Statistical significance was set at p < 0.05.
Results
Modification of the Ad5 capsid hexon protein ablates adenovirus binding to human coagulation factor X
To further investigate the interaction of FX with the Ad capsid we examined several Ad mutants that either had six-histidine (6-His) peptide insertions into the Ad5 hexon protein or substitution of the Ad5 hexon protein with that of another Ad serotype, Ad3 (Ad5H3). Characteristics of the vectors used in this study are presented in table 1. Slot blot analysis (figure 1A) demonstrated binding of FX to wild-type Ad5 hexon containing virus (Ad5), as well as to an Ad5 virus with a 6-His peptide inserted into HVR5 (Ad5HVR5-6H). No detectable binding was detected between an Ad5 based capsid with the Ad3 hexon substituted capsid (Ad5H3) or with two of the previously published viruses that don’t bind FX, Ad5HBAP and Ad48. These FX-Ad interactions were further confirmed by SPR analysis (figure 1B). Purified human factor X and XI were covalently bound to biocore sensor chips and viruses were exposed to each protein with differential binding measured. Wild-type Ad5 and Ad5HVR5-6H both demonstrated specific binding to FX, whereas, no specific binding was observed for Ad5H3, Ad5HBAP, or Ad48. Additional Ad mutants were evaluated with these initial assays but failed to show any significant decrease in binding to FX compared to wild-type Ad5 (data not shown).
Table 1.
Characteristics of Ad vectors utilized.
| Virus | Hexon Modification | Hexon Amino Acid Sequence | Fiber (Knob Domain) (F) | E1 Transgene (for non-replicative) |
|---|---|---|---|---|
| Ad5 | none (wild-type Ad5) | …F 265FSTTEATAGNGDNLTPKV 284… | Ad5 or Ad3 (F5/3) | GFP-Luc |
| Ad5HVR5-6H | Ad5 capsid with 6-histidine insertion into HVR5 of hexon protein | …F 265FSTTLGSHHHHHHLGSLTPKV 284… | Ad5 | GFP-Luc |
| Ad5H3 | Ad5 capsid with substitution for Ad3 hexon | first 55 aa of Ad5 hexon, then Ad3 hexon aa sequence, then last 45 aa of Ad5 hexon | Ad5 or Ad3 (F5/3) | GFP-Luc |
| Ad5HBAP | Ad5 capsid with Biotin Acceptor Protein (BAP) inserted into HVR5 of hexon protein | …F 265FSGST … 82 aa BAP sequence … GTPKV 284… |
Ad5 or Ad3 (F5/3) | GFP-Luc |
| Ad48 | none (wild-type Ad48 capsid) | Wild-type Ad48 | Ad48 | none |
Figure 1.
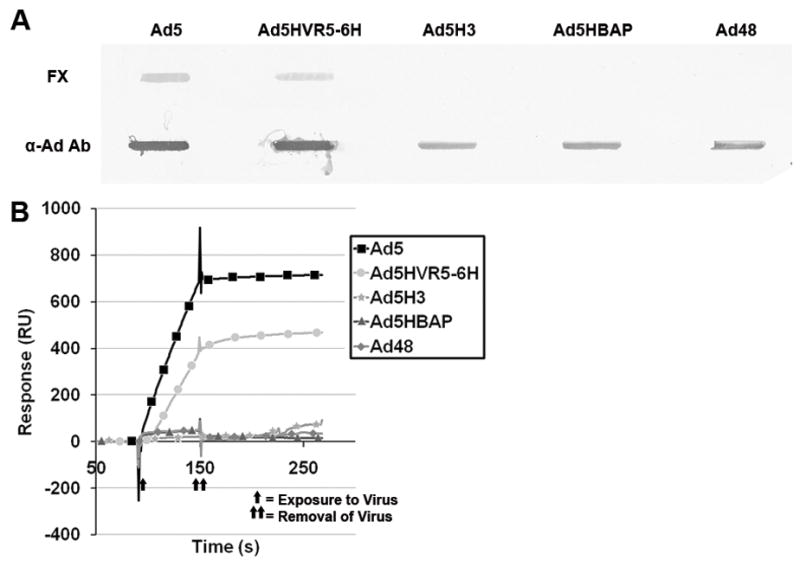
Substitution of Ad3 hexon onto Ad5 capsid ablates binding to FX. A, Slot blot assay. Viruses bound to PVDF membrane were incubated with FX or with polyclonal anti-Ad capsid antibody. B, Surface Plasmon Resonance (SPR) analysis of FX binding to hexon variants. Purified human FX was covalently coupled to a SA biosensor chip. Ad vectors were then exposed to the chip and binding interactions were analyzed. B, Overlay sensograms are presented. The sensograms presented are representative of two replicates at 37° C (additional replicates were performed at 25° C showing similar results, data not shown).
Hexon modifications decrease factor X-mediated uptake in vitro
To investigate whether the biochemical binding FX to our Ad mutants observed in the slot blot and SPR analysis translated to differences in cell transduction we examined the effect of pre-incubation of Ad vectors with FX prior to in vitro cell infection. Ad vectors were pre-incubated with increasing concentrations of purified human FX and then allowed to infect either A549 (high CAR expression), HepG2 (liver representative, moderate CAR expression), or SKOV3 (low CAR expression) cells. Luciferase transgene expression was measured and normalized for transgene expression of Ad vectors without pre-incubation with FX (figure 2). Ad5 vectors with wild-type hexon (Ad5) had an increase in transgene expression with increasing FX concentrations as would be expected and this effect was most notable in the SKOV3 cells secondary to their low CAR expression resulting in a low level of Ad5 transduction without the FX. Ad5HVR5-6H exhibited some increased transduction with FX pre-incubation but to a lesser extent than Ad5. Ad5H3 and Ad5HBAP showed little to no change in transduction with pre-incubation with FX, however Ad5H3 did exhibit some increase in transduction at the highest levels of FX (16 μg/ml), whereas, physiological levels are around 8 μg/ml.
Figure 2.

Hexon modifications decrease FX-mediated uptake in vitro. Ad vectors were pre-incubated with increasing concentrations of FX and then allowed to infect either A549 (high CAR expression), HepG2 (liver representative, moderate CAR expression), or SKOV3 (low CAR expression) cells. Luciferase transgene expression was measured at 36 hours and normalized for protein and divided over vectors without pre-incubation with FX. Ad5 vectors with wild-type hexon (Ad5) had an increase in transgene expression with increasing FX concentrations. * = p-value of < 0.05 compared to Ad5.
Modification of Ad hexon decreases liver transduction in mice
The biodistribution of our panel of vectors was investigated in C57BL/6 mice (figure 3A). The mice were injected with Ad vectors via tail vein injections and at 36 hours organs were harvested and luciferase transgene expression was assayed. Ad5H3 and Ad5HBAP exhibited approximately a hundred-fold decrease in liver transduction, comparable to Ad5 in warfarin pre-treated mice (Ad5 + warf). Ad5HVR5-6H had a slight decrease in liver transduction, but to a lesser extent than Ad5H3 and Ad5HBAP. There were no other significant variations in the biodistribution of the other vectors with the exception of an increase in the Ad5HVR5-6H vector to the spleen and a decrease in the Ad5 + warfarin to the spleen.
Figure 3.

Substitution of Ad3 hexon onto Ad5 capsid results in improved biodistribution in mice. A, Modification of Ad5 hexon decreases liver transduction in mice. C57BL/6 mice were injected with Ad vectors and at 36 hours organs were harvested and luciferase transgene expression was assayed. * = p-value of < 0.05. B, Substitution of Ad3 hexon onto Ad5 capsid results in improved tumor to liver transgene ratio. Athymic nude mice with skov3.ip1 flank tumors underwent tail vein injections with Ad vectors. Luciferase transgene transcript copy numbers in liver and tumors were measured by quantitative PCR and normalized for genomic β–actin DNA. Furthermore, comparison was made between infectivity enhanced vectors with substitution of Ad3 fiber knob domain (right grouping) compared to vectors with Ad5 fiber knob domain (left grouping). * = p < 0.05 compared to liver control, ** = p < 0.05 compared to tumor control.
Substitution of Ad3 hexon onto Ad5 capsid results in improved tumor to liver transgene ratio
Flank tumors using skov3.ip1 cells were established in athymic nude (nu/nu) mice. Following establishment of tumors Ad vectors were injected via tail vein injections. Luciferase transgene transcript copy numbers in liver and tumors were measured by quantitative PCR and normalized for genomic β–actin DNA (figure 3B). Two panels of vectors were investigated: 1) vectors with wild-type Ad5 fiber protein (left grouping of figure 3b, Ad5, Ad5 + warf, Ad5H3, Ad5HBAP) and 2) infectivity enhanced vectors with substitution of Ad3 fiber knob domain (right grouping of figure 3b, Ad5/3, Ad5/3 + warf, Ad5H3F5/3, Ad5HBAPF5/3). Vectors with wild-type Ad5 hexon proteins (Ad5 and Ad5F5/3) without warfarin showed the highest levels of transcript copy numbers in the liver with a 100-fold decrease in luciferase transcript copy numbers when pre-treated with warfarin or with hexon modification (Ad5H3 & Ad5H3F5/3, and Ad5HBAP & Ad5HBAPF5/3). The infectivity enhanced vectors (right grouping of figure 3B) that had a hexon modification or were administered to animals pre-treated with warfarin resulted in a 10-fold increase in luciferase transcript copy numbers in tumors compared to vectors with wild-type Ad5 fiber knob domains (left grouping of figure 3B).
Incorporation of hexon modification into CRAd context ablates factor X binding but does not impair cell killing
The Ad3 hexon substitution was translated to a condifionally replicative Ad (CRAd) vector. The backbone of the vector contained the delta 24 mutation (24 base pair deletion) in the E1A protein, thereby, conferring replication to pRb deficient cells only and a deletion of the E3 region with preservation of the E3 protein ADP. Two series of CRAds were constructed: 1) one set with wild type Ad5 hexon or with Ad3 hexon, and 2) one set with the wild-type Ad5 fiber or with the Ad3 fiber knob domain in place of the Ad5 fiber knob domain (table 1). Surface Plasmon Resonance (SPR) analysis of human FX binding to these CRAd vectors was performed to ensure the ablation of FX binding (figure 4A). The same procedure utilized earlier was used to evaluate the CRAd vectors and it further confirmed that substitution of Ad3 hexon ablated binding to FX. To ensure the substitution of the Ad3 hexon was compatible with cell killing, a crystal violet assay was performed to investigate the cytolytic effect of the CRAds (figure 4B). There was no impairment of cell killing by substitution of the Ad3 hexon onto the CRAd capsids (right samples in B2 and B3 panels) versus wild-type Ad5 hexon (left samples in B2 and B3 panels). Skov3 cells which are low in CAR expression were used to further illustrate the infectivity enhancement seen with substitution of the Ad3 fiber knob domain onto CRAd vectors (panel B3), which resulted in approximately 100 to 1000 fold increases in cell killing compared to CRAds with Ad5 fiber knob (panel B2).
Figure 4.

Incorporation of hexon modification into CRAd context ablates FX binding but does not impair cell killing. A, Surface Plasmon Resonance (SPR) analysis of purified human FX binding to CRAd vectors. Purified human FX was covalently coupled to a SA biosensor chip. Ad vectors were then exposed to the chip and binding interactions were analyzed. A, Overlay sensograms are presented. The sensograms presented are representative of two replicates performed at 37° C. B, Crystal violet assay demonstrating no impairment of cell killing by substitution of Ad3 hexon onto CRAd capsid. Cytocidal effect of vectors on skov3.ip1 cells was assayed using crystal violet dye assay at 10 days post-infection (vp/cell ratio in bottom left corner of sample). Images presented are representative of three replicates performed for each vector and dose.
Adenovirus hexon modification by substitution of the Ad3 hexon onto Ad5 capsid improves anti-tumor efficacy after systemic vector administration
To evaluate the efficacy of these infectivity enhanced CRAds in vivo we utilized a subcutaneous flank tumor xenograft model and administered the CRAds via tail vein injection. We established Skov3.ip1 flank xenograft tumors in athymic nude mice. The vectors (1011 vp) were administered IV by tail vein injection (day 0), with one group of animals being pre-treated with warfarin at the time of injection as a control. The mice injected with the Ad3 hexon-containing vectors (Ad5Δ24E1H3F5/3, light gray/diamonds) had significantly smaller flank tumors through 28 days post-injection compared to placebo treated animals (black/squares) (figure 5A). The Ad5 hexon containing vector (Ad5Δ24E1F5/3, dark gray/triangles) showed a mild decrease in tumor size immediately after injection of vector but had negligible size differences at each time point through 28 days compared to placebo (PBS) treated animals. Mice that were pretreated with warfarin (Ad5Δ24E1F5/3 + warf, gray/circles) at the time of injection showed a decrease in tumor size for early time points, but this effect was only temporary as the warfarin wore off and FX levels were repleted.
Figure 5.

Hexon modification results in effective systemic targeting of oncolytic Ad. Athymic nude mice with skov3.ip1 subcutaneous flank tumors were treated with the panel of infectivity enhanced CRAds. A, Tumor size was followed up to 28 days. Ad5Δ24E1F5/3 = wild-type Ad5 hexon-containing vector (dark gray/triangle), Ad5Δ24E1F5/3 + warfarin = wild-type Ad5 hexon-containing vector pre-treated with warfarin at days -3 and -1 (gray/circles), Ad5Δ24E1H3F5/3 = Ad3 hexon substituted vectors (light gray/diamonds) B, Viral replication was measured by E4 copy number in liver and tumor specimens. Closed bars = liver, open bars = tumor. * = p < 0.05 compared to placebo.
The tumor sizes correlated with viral replication as measured by E4 copy numbers in liver and tumor specimens (figure 5B). The wild-type Ad5 hexon-containing vector (Ad5Δ24E1F5/3) had the highest levels of virus present in the liver at day 3 with very low E4 copy numbers in the tumor specimens. The warfarin treated mice and Ad3 hexon substituted vectors demonstrated an improvement in replication of virus in tumor compared to liver. Viral replication in the tumors of the warfarin pre-treated mice was only significantly measurable up to day 7, whereas, CRAd replication persisted with the Ad3 hexon substituted vectors until between 14 and 21 days. It should also be noted that persistant virus in the liver wouldn’t be expected as human Ad doesn’t replicate in mice tissues. This data further demonstrates that effective IV administration of Ad vectors can be achieved by appropriate ablation of Ad binding to FX.
Discussion
Substitution of the Ad3 hexon onto the Ad5 capsid confers ablation of binding to FX and thereby decreases liver tropism and enhances the anti-tumor therapeutic window. Herein, we have demonstrated that these hexon substituted vectors do not bind FX which translates into decreased liver transduction and improved tumor transduction. Furthermore, by ablating FX binding and decreasing liver sequestration the effects of infectivity enhancement are improved as demonstrated by improved tumor transduction with the Ad3 fiber knob modified vectors in conjunction with hexon modification.
It was surprising that the Ad5HVR5-6H vector bound to FX and didn’t exhibit any decreased liver tranduction based on previously published findings of the role of HVR5 in binding to FX. However, after further investigation it is likely the lack of ablation of binding was due to a combination of factors. Some of these factors are that enough of the residues in HVR5 responsible for binding to FX were preserved (8), HVR7 remained intact, and the peptide insertion was insufficient to confer a conformational change in the hexon protein. Previously published HVR5 mutants didn’t preserve the threonine amino acid at position 269 (which was preserved in our vectors) and included insertions of at least 8 amino acids (6–8).
The treatment of animals with warfarin during the administration of Ad vectors has been previously shown to result in improved anti-tumor efficacies (11, 12). The lack of a lasting effect of the warfarin treated animals with the CRAd agent containing the wild-type Ad5 hexon is likely because the progeny virus contain the wild-type Ad5 hexon and can bind to FX, which is repleted after dosing with warfarin wears off and therefore the circulating FX would help clear the progeny virus. In our experiments therapeutic doses of warfarin were only administered immediately prior to the initial Ad injection.
In this study we substituted the Ad3 hexon onto the Ad5 capsid, therefore, it remains to be clearly determined if the ablation to binding FX of this vector results from an intrinsic lack of binding by the Ad3 hexon or if it is the conformational change from incorporation of this foreign hexon into the Ad5 capsid that ablates any binding to FX. Although previous experiences have noted a decrease in viral titer production with this modification, we were able to generate adequate viral titers (1 × 1012 vp/ml) for our experimental purposes with comparable TCID50 (data not shown).
Overcoming the obstacles to safe and effective systemic administration of Ad vectors is essential in advancing an effective anti-cancer Ad based therapy. Several key components of achieving this have been recently elucidated, notably, the liver tropism mediated by Ad5 hexon binding to coagulation FX. There are now several published Ad hexon mutants that demonstrate ablation of FX binding with resultant decreased liver tropism. Which one of these mutants is the “best” will be dependent on further comparison analysis and the result may differ for the particular application. Beyond its role in binding to FX, hexon serves as one of the major capsid proteins and modification of it may lead to vector instability. However, modifications may also serve multiple purposes beyond ablating FX binding, such as re-targeting through peptide insertions or changing the immunogenicity of the vector.
Three major approaches will likely be advanced for FX-ablating hexon modifications. Two strategies retain the majority of the Ad5 hexon: 1) minimal point mutations of the required residues in HVR5 and HVR7 to minimally affect the vector capsid yet ablate FX binding. 2) Insertion of peptides into HVR5 and/or HVR7 that ablate FX binding and allow further benefits such as re-targeting, epitope display, or avoidance of immune recognition. 3) hexon pseudotyping with Ad serotypes that don’t bind FX. The first approach has been successfully achieved by substitution of 3 residues (2 in HVR5 and 1 in HVR7) with corresponding residues from Ad26 (8). The second approach has been initiated by some groups but the full potential of such hasn’t been completely demonstrated. The third approach involves substitution of the majority of the hexon protein with that of an alternate serotype that ablates FX binding and potentially provides additional benefits such as decreased immunogenicity for repeat vector administration.
The findings presented herein, offer another strategy for Ad hexon modification for ablation of FX binding and decreasing liver tropism. Furthermore, the synergistic effect of combining infectivity enhancement strategies with decreased liver sequestration provide promise that effective targeting and therapeutic effects following IV administration can be achieved. To apply Ad vectors for several systemic disease targets, including metastatic cancer, will necessitate effective IV administration. Several obstacles will be necessary to overcome to achieve this end, these include decreasing liver toxicity and sequestration of injected vector, improving target cell transduction, and decreasing vector-immune system interactions. Adenovirus pseudotyping of the hexon and fiber proteins with alternate serotypes, as presented here, may offer the opportunity to overcome several of these obstacles.
Acknowledgments
Financial Support: American College of Surgeons Resident Research Scholarship (Short); NIH grants: 1R01HL092941 (Curiel), RO1CA094084 (Yamamoto), P20CA101955 (Yamamoto)
Footnotes
Conflicts of interest: none
References
- 1.Edelstein ML, Abedi MR, Wixon J. Gene therapy clinical trials worldwide to 2007--an update. J Gene Med. 2007;9:833–42. doi: 10.1002/jgm.1100. [DOI] [PubMed] [Google Scholar]
- 2.Imperiale MJ. Keeping adenovirus away from the liver. Cell Host Microbe. 2008;3:119–20. doi: 10.1016/j.chom.2008.02.007. [DOI] [PubMed] [Google Scholar]
- 3.Di Paolo NC, van Rooijen N, Shayakhmetov DM. Redundant and synergistic mechanisms control the sequestration of blood-born adenovirus in the liver. Mol Ther. 2009;17:675–84. doi: 10.1038/mt.2008.307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Parker AL, Waddington SN, Nicol CG, et al. Multiple vitamin K-dependent coagulation zymogens promote adenovirus-mediated gene delivery to hepatocytes. Blood. 2006;108:2554–61. doi: 10.1182/blood-2006-04-008532. [DOI] [PubMed] [Google Scholar]
- 5.Waddington SN, McVey JH, Bhella D, et al. Adenovirus serotype 5 hexon mediates liver gene transfer. Cell. 2008;132:397–409. doi: 10.1016/j.cell.2008.01.016. [DOI] [PubMed] [Google Scholar]
- 6.Vigant F, Descamps D, Jullienne B, et al. Substitution of hexon hypervariable region 5 of adenovirus serotype 5 abrogates blood factor binding and limits gene transfer to liver. Mol Ther. 2008;16:1474–80. doi: 10.1038/mt.2008.132. [DOI] [PubMed] [Google Scholar]
- 7.Kalyuzhniy O, Di Paolo NC, Silvestry M, et al. Adenovirus serotype 5 hexon is critical for virus infection of hepatocytes in vivo. Proc Natl Acad Sci U S A. 2008;105:5483–8. doi: 10.1073/pnas.0711757105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Alba R, Bradshaw AC, Parker AL, et al. Identification of coagulation factor (F)X binding sites on the adenovirus serotype 5 hexon: effect of mutagenesis on FX interactions and gene transfer. Blood. 2009;114:965–71. doi: 10.1182/blood-2009-03-208835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wu H, Dmitriev I, Kashentseva E, Seki T, Wang M, Curiel DT. Construction and characterization of adenovirus serotype 5 packaged by serotype 3 hexon. J Virol. 2002;76:12775–82. doi: 10.1128/JVI.76.24.12775-12782.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wu H, Han T, Belousova N, et al. Identification of sites in adenovirus hexon for foreign peptide incorporation. J Virol. 2005;79:3382–90. doi: 10.1128/JVI.79.6.3382-3390.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Koski A, Rajecki M, Guse K, et al. Systemic adenoviral gene delivery to orthotopic murine breast tumors with ablation of coagulation factors, thrombocytes and Kupffer cells. J Gene Med. 2009;11:966–77. doi: 10.1002/jgm.1373. [DOI] [PubMed] [Google Scholar]
- 12.Shashkova EV, Doronin K, Senac JS, Barry MA. Macrophage depletion combined with anticoagulant therapy increases therapeutic window of systemic treatment with oncolytic adenovirus. Cancer Res. 2008;68:5896–904. doi: 10.1158/0008-5472.CAN-08-0488. [DOI] [PubMed] [Google Scholar]
- 13.Shashkova EV, May SM, Doronin K, Barry MA. Expanded anticancer therapeutic window of hexon-modified oncolytic adenovirus. Mol Ther. 2009;17:2121–30. doi: 10.1038/mt.2009.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kanerva A, Mikheeva GV, Krasnykh V, et al. Targeting adenovirus to the serotype 3 receptor increases gene transfer efficiency to ovarian cancer cells. Clin Cancer Res. 2002;8:275–80. [PubMed] [Google Scholar]
- 15.Zhu ZB, Lu B, Park M, et al. Development of an optimized conditionally replicative adenoviral agent for ovarian cancer. Int J Oncol. 2008;32:1179–88. doi: 10.3892/ijo_32_6_1179. [DOI] [PubMed] [Google Scholar]
- 16.Kanerva A, Zinn KR, Chaudhuri TR, et al. Enhanced therapeutic efficacy for ovarian cancer with a serotype 3 receptor-targeted oncolytic adenovirus. Mol Ther. 2003;8:449–58. doi: 10.1016/s1525-0016(03)00200-4. [DOI] [PubMed] [Google Scholar]
- 17.Campos SK, Barry MA. Rapid construction of capsid-modified adenoviral vectors through bacteriophage lambda Red recombination. Hum Gene Ther. 2004;15:1125–30. doi: 10.1089/hum.2004.15.1125. [DOI] [PubMed] [Google Scholar]
- 18.Sun J, Blaskovich MA, Jain RK, et al. Blocking angiogenesis and tumorigenesis with GFA-116, a synthetic molecule that inhibits binding of vascular endothelial growth factor to its receptor. Cancer Res. 2004;64:3586–92. doi: 10.1158/0008-5472.CAN-03-2673. [DOI] [PubMed] [Google Scholar]