

Abstract
Since the 1920s, it has been known that the repetitive brain trauma associated with boxing may produce a progressive neurological deterioration, originally termed “dementia pugilistica” and more recently, chronic traumatic encephalopathy (CTE). We review the 47 cases of neuropathologically verified CTE recorded in the literature and document the detailed findings of CTE in 3 professional athletes: one football player and 2 boxers. Clinically, CTE is associated with memory disturbances, behavioral and personality changes, Parkinsonism, and speech and gait abnormalities. Neuropathologically, CTE is characterized by atrophy of the cerebral hemispheres, medial temporal lobe, thalamus, mammillary bodies, and brainstem, with ventricular dilatation and a fenestrated cavum septum pellucidum. Microscopically, there are extensive tau-immunoreactive neurofibrillary tangles, astrocytic tangles, and spindle-shaped and threadlike neurites throughout the brain. The neurofibrillary degeneration of CTE is distinguished from other tauopathies by preferential involvement of the superficial cortical layers, irregular, patchy distribution in the frontal and temporal cortices, propensity for sulcal depths, prominent perivascular, periventricular and subpial distribution, and marked accumulation of tau-immunoreactive astrocytes. Deposition of beta amyloid, most commonly as diffuse plaques, occurs in fewer than half the cases. CTE is a neuropathologically distinct, slowly progressive tauopathy with a clear environmental etiology.
Keywords: Athletes, Concussion, Dementia, Encephalopathy, Neurodegeneration, Tau protein, Traumatic brain injury
INTRODUCTION
Over recent years there has been increasing attention focused on the neurological sequelae of sports-related traumatic brain injury, particularly concussion. Concussion is a frequent occurrence in contact sports: 1.6 to 3.8 million sports-related concussions occur annually in the United States (1–3). Most sport-related head injury is minor and although the majority of athletes who suffer a concussion recover within a few days or weeks a small number of individuals develop long-lasting or progressive symptoms. This is especially true in cases of repetitive concussion or mild traumatic brain injury in which at least 17% of individuals develop chronic traumatic encephalopathy (CTE) (4). The precise incidence of CTE after repetitive head injury is unknown, however, and it is likely much higher. It is also unclear what severity or recurrence of head injury is required to initiate CTE; no well-designed prospective studies have addressed these important public health issues (5–10).
Repetitive closed head injury occurs in a wide variety of contact sports, including football, boxing, wrestling, rugby, hockey, lacrosse, soccer, and skiing. Furthermore, in collision sports such as football and boxing, players may experience thousands of subconcussive hits over the course of a single season (11, 12). Although the long-term neurological and neuropathological sequelae associated with repetitive brain injury are best known in boxing, pathologically verified CTE has been reported in professional football players, a professional wrestler and a soccer player, as well as in epileptics, head bangers and domestic abuse victims (13–21). Other sports associated with a post-concussive syndrome include hockey, rugby, karate, horse riding, and parachuting (22–25), although the list is almost certainly more inclusive. Furthermore, additional large groups of individuals prone to repetitive head trauma, such as military veterans, may be at risk for CTE.
In this review, we present a summary of the 47 cases of neuropathologically verified CTE in the literature. We also report the clinical and immunocytochemical findings of CTE in 3 retired professional athletes, i.e. 1 football player and 2 boxers, ranging in age from 45 to 80 years. Although the cases previously reported in the literature detailed some of the characteristic gross and histological features of CTE, the spectrum of unique, regionally specific immunocytochemical abnormalities of phosphorylated tau that occur in this disorder have not been previously described. We demonstrate that although CTE shares many features of other neurodegenerative disorders, including Alzheimer disease (AD), progressive supranuclear palsy, post-encephalitic Parkinsonism, and the amyotrophic lateral sclerosis/Parkinson’s-dementia complex of Guam (ALS/PDC), CTE is a neuropathologically distinct, progressive tauopathy with a clear environmental etiology.
Clinical and Demographic Features of CTE
The concept of CTE was first introduced by Martland in 1928 who introduced the term ‘punch-drunk’ to a symptom complex that appeared to be the result of repeated sublethal blows to the head (26). This syndrome, long recognized in professional boxers, was termed “dementia pugilistica” by Millspaugh (27) and “the psychopathic deterioration of pugilists” by Courville (28). The symptoms of CTE are insidious, first manifest by deteriorations in attention, concentration, and memory, as well as disorientation and confusion, and occasionally accompanied by dizziness and headaches. With progressive deterioration, additional symptoms, such as lack of insight, poor judgment, and overt dementia, become manifest. Severe cases are accompanied by a progressive slowing of muscular movements, a staggered, propulsive gait, masked facies, impeded speech, tremors, vertigo, and deafness (27). Corsellis, Bruton, and Freeman-Browne described 3 stages of clinical deterioration as follows: The first stage is characterized by affective disturbances and psychotic symptoms. Social instability, erratic behavior, memory loss, and initial symptoms of Parkinson disease appear during the second stage. The third stage consists of general cognitive dysfunction progressing to dementia and is often accompanied by full-blown Parkinsonism, as well as speech and gait abnormalities. Other symptoms include dysarthria, dysphagia, and ocular abnormalities, such as ptosis (29). The severity of the disorder appears to correlate with the length of time engaged in the sport and the number of traumatic injuries, although whether a single traumatic brain injury can trigger the onset of CTE remains a matter of speculation.
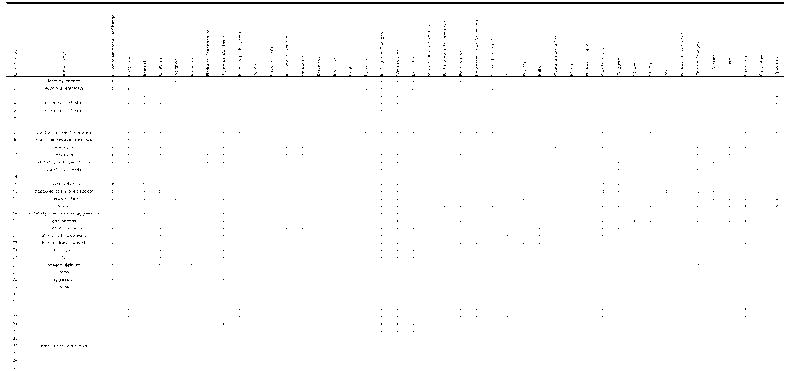
Of the 51 neuropathologically confirmed cases of CTE, 46 (90%) occurred in athletes. The athletes included 39 boxers (85%) who fought as amateurs and as professionals for varying lengths of time (range: 4 to 25 years; mean: 14.4 years), 5 football players (11%), whose playing time ranged between 14 and 23 years (M = 18.4 years, SD = 3.9); 1 professional wrestler, and 1 soccer player. The athletes began their respective sports at young ages, i.e. between 11 and 19 years (M = 15.4 years, SD = 2.2) (Tables 1, 2). The first symptoms of CTE were noticed at ages ranging from 25 to 76 years (M = 42.8 years, SD = 12.7). One third were symptomatic at the time of their retirement from the sport and half were symptomatic within 4 years of stopping play. Common presenting symptoms included memory loss, irritability, outbursts of aggressive or violent behavior, confusion, speech abnormalities, cognitive decline, gait abnormalities, unsteadiness, headaches, slurred speech and Parkinsonism. In 14 cases (30%) there was a prominent mood disturbance, usually depression (28%); 1 boxer was described as having a “euphoric dementia” (31); another boxer was described as manic-depressive (35); and a football player was considered “bipolar” (40). In most of the reported cases, the disease progressed slowly over several decades (range: 2–46 years; M = 18.6 years, SD = 12.6) with increasing abnormalities in behavior and personality, memory loss, cognitive decline, and visuospatial difficulties. Movement abnormalities were eventually found in 42% subjects, consisting of Parkinsonism, staggered, slowed or shuffled gait, slowed, slurred or dysarthric speech, ataxia, ocular abnormalities and dysphagia. As Critchley noted in 1957, “Once established it not only does not permit reversibility, but ordinarily advances steadily, even though the boxer has retired from the ring”(42).
Table 1.
Demographic Information
| Case Number | Reference | Gender | Sport/actiivity | Age Sport Begun (years) | Years of Play | Age at Onset Symptoms (years) | Interval between retirement and symptoms (years) |
Interval between symptom onset and death (years) |
Age at Death (years) | ApoE Genotype |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 30 | M | Boxing | 17 | 11 | 38 | 10 | 13 | 51 | |
| 2 | 31 | M | Boxing | 15 | 14 | 36 | 6 | 12 | 48 | |
| 3 | 32 | M | Boxing | 14 | 10 | 46 | 10 | 7 | 53 | |
| 4 | 32 | M | Boxing | 18 | 6 | 48 | 24 | 10 | 58 | |
| 5 | 28 | M | Boxing | 4 | 49 | |||||
| 6 | 33 | M | Boxing | |||||||
| 7 | 33 | M | Boxing | |||||||
| 8 | 34 | M | Boxing | 16 | 7 | 25 | 1 | 33 | 58 | |
| 9 | 35 | M | Boxing | 12 | 12 | 30 | 6 | |||
| 10 | 35 | M | Boxing | 15 | 20 | 36 | 0 | 10 | 46 | |
| 11 | 35 | M | Boxing | 19 | 12 | 31 | 0 | 15 | 46 | |
| 12 | 35 | M | Boxing | 16 | 16 | 40 | 8 | 5 | 45 | |
| 13 | 35 | M | Boxing | 15 | 13 | 28 | 0 | 16 | 44 | |
| 14 | 35 | M | Boxing | 12 | 12 | 4 | 28 | |||
| 15 | 29 | M | Boxing | 11 | 14 | 25 | 0 | 38 | 63 | |
| 16 | 29 | M | Boxing | 20 | 50 | 20 | 27 | 77 | ||
| 17 | 29 | M | Boxing | 16 | 14 | 30 | 0 | 33 | 63 | |
| 18 | 29 | M | Boxing | 15 | 25 | 35 | 0 | 34 | 69 | |
| 19 | 29 | M | Boxing | 18 | 18 | 36 | 0 | 25 | 61 | |
| 20 | 29 | M | Boxing | 13 | 25 | 37 | 0 | 46 | 83 | |
| 21 | 29 | M | Boxing | 16 | 20 | 54 | 18 | 8 | 62 | |
| 22 | 29 | M | Boxing | 17 | 23 | 60 | 20 | 11 | 71 | |
| 23 | 29 | M | Boxing | >10 | 31 | 0 | 41 | 72 | ||
| 24 | 29 | M | Boxing | 40 | 27 | 67 | ||||
| 25 | 29 | M | Boxing | 48 | 19 | 67 | ||||
| 26 | 29 | M | Boxing | 14 | 16 | 43 | 4 | 14 | 57 | |
| 27 | 29 | M | Boxing | 18 | 10 | 61 | ||||
| 28 | 29 | M | Boxing | 91 | ||||||
| 29 | 29 | M | Boxing | 58 | ||||||
| 30 | 18 | F | Physical Abuse | 76 | ||||||
| 31 | 14 | F | Autistic Head banging | 24 | ||||||
| 32 | 36 | M | Boxing | >25 | 63 | |||||
| 33 | 36 | M | Boxing | >25 | 69 | |||||
| 34 | 19 | M | Circus Clown | 15 | 33 | |||||
| 35 | 15 | M | Boxing | >11 | 61 | 37 | 10 | 71 | ||
| 36 | 13,37 | M | Boxing | 11 | 12 | 23 | ε3/ε4 | |||
| 37 | 13 | M | Boxing | 16 | 5 | 0 | 28 | |||
| 38 | 13 | M | Head banging | 28 | ||||||
| 39 | 13 | M | Epilepsy | 27 | ||||||
| 40 | 13 | M | Soccer | 23 | ε3/ε3 | |||||
| 41 | 38 | M | Boxing | 10 | 64 | 67 | ε3/ε4 | |||
| 42 | 39 | M | Boxing | 76 | 78 | |||||
| 43 | 17 | M | Football | 16 | 22 | 50 | ε3/ε3 | |||
| 44 | 16 | M | Football | 18 | 14 | 35 | 2 | 10 | 45 | ε3/ε3 |
| 45 | 21 | M | Football | 15 | 23 | 38 | 0 | 6 | 44 | ε3/ε4 |
| 46 | 20 | M | Wrestling | 18 | 22 | 38 | 0 | 2 | 40 | ε3/ε3 |
| 47 | 40 | M | Football | 16 | 17 | 36 | 3 | 3 | 36 | |
| 48 | 41 | M | Boxing | 16 | 17 | 58 | 13 | 61 | ε3/ε3 | |
| 49 | Case 1 | M | Football | 16 | 16 | 40 | 9 | 5 | 45 | ε4/ε4 |
| 50 | Case 2 | M | Boxing | 17 | 5 | 63 | 33 | 17 | 80 | ε3/ε4 |
| 51 | Case 3 | M | Boxing | 11 | 22 | 58 | 25 | 15 | 73 | ε3/ε3 |
M =48 male; F = female
Table 2.
Clinical Manifestations
 |
 |
New cases 1–3 of this series
x = Clinical feature was noted as present; blank = clinical feature was not mentioned
CTE in Football Players
Five football players, including our Case 1, had neuropathologically verified CTE at autopsy. All died suddenly in middle age (age at death, range 36–50 years, M = 44.0 years, SD = 5.0) and were younger at the time of death compared to boxers with CTE (boxers age at death, range: 23–91 years, M = 60.0 years, SD = 15.2). The duration of symptomatic illness was also shorter in the football players (range 3–10 years, M = 6.0 years, SD = 2.9) compared to the boxers (range 5–46 years, M = 20.6 years, SD = 12.3). All 5 football players played similar positions: 3 were offensive lineman, one was a defensive lineman and the other was a linebacker. In the football players, the most common symptoms were mood disorder (mainly depression), memory loss, paranoia, and poor insight or judgment (each found in 80%), outbursts of anger or aggression, irritability, and apathy (each found in 60%), confusion, reduced concentration, agitation, or hyperreligiosity (each found in 40%). Furthermore, 4 of the 5 experienced tragic deaths, i.e. 2 from suicide (16, 17), 1 during a high-speed police chase (40), and another from an accidental gunshot while cleaning his gun (Case 1). Case 1 exemplifies these clinical features.
Case 1
A 45-year-old right-handed Caucasian man died unexpectedly as a result of an accidental gunshot wound to the chest while he was cleaning a gun. He was a retired professional football player who played football in high school, 3 years of college and 10 years in the NFL as a linebacker. According to his wife, he was concussed 3 times during his college football years and at least 8 times during his NFL career; however, only 1 concussion was medically confirmed. He was never formally diagnosed with post-concussive syndrome and never sought medical attention for residual cognitive or behavioral difficulties. There was no history of ever losing consciousness for more than a few seconds and he never required being carried off the field or hospitalization.
At age 40, his family began to notice minor impairments in his short-term memory, attention, concentration, organization, planning, problem solving, judgment, and ability to juggle more than one task at one time. His spatial abilities were mildly impaired and his language was unaffected. He repeatedly asked the same questions over and over, he did not recall why he went to the store unless he had a list, and he would ask to rent a movie that he had already seen. These symptoms gradually increased and became pronounced by the end of his life 5 years later. Using a modification of the Family Version of the Cognitive Difficulties Scale (43, 44) he had a moderate amount of cognitive difficulties. On a modified AD8 informant interview for dementia, he received a total score of 4, which indicated “cognitive impairment is likely to be present.” (45). By contrast, the Functional Activities Questionnaire (FAQ) (46), an informant-based measure of Instrumental Activities of Daily Living, did not indicate significant functional dependence despite his difficulty assembling tax records, shopping alone, and understanding television (total FAQ = 3). Moreover, he continued to perform his job as a hunting and fishing guide in a satisfactory manner.
Towards the end of his life, he tended to become angry and verbally aggressive over insignificant issues and was more emotionally labile. He also began to consume more alcohol but did not show other signs or symptoms of depression. He had no significant psychiatric history and he had never taken performance-enhancing or illicit drugs. His family history was negative for dementia and psychiatric illness.
CTE in Boxers
Boxing is the most frequent sport associated with CTE and disease duration is the longest in boxers, with case reports of individuals living for 33, 34, 38, 41 and 46 years with smoldering, yet symptomatic disease (29). Boxers with long-standing CTE are frequently demented (46%) and may be misdiagnosed clinically as AD (47), as occurred in Cases 2 and 3.
Case 2
An 80-year-old African-American/American Indian man was first noted to have difficulty remembering things in his mid-twenties. He began boxing when he was 17, quickly rose to professional ranks and fought professionally for 5 years until he retired at age 22. He suffered a mild head injury in his early teenage years while moving farm equipment although he did not lose consciousness or suffer any permanent disability. By his mid-thirties he had brief, occasional episodes of confusion and a tendency to fall. His wife attributed his occasional forgetfulness, falls and confusion to being mildly “punch-drunk.” His symptoms remained more or less stable over the following 4 decades except for an increased tendency to become disoriented when traveling to unfamiliar places. By age 70, he got lost driving on familiar roads; he became increasingly confused and disoriented and did not recognize his daughter. By age 78, he was paranoid, his memory loss had increased, his gait was unsteady, his speech slowed and he fell frequently. He was easily agitated and required multiple hospitalizations for aggressive behaviors. He died at age 80 from complications of septic shock.
He had a period of alcohol abuse as a young adult but was abstinent for the last 40 years of his life. He smoked cigarettes for 20 years. He was employed as a roofer for most of his life and was in excellent physical condition, running miles and doing daily calisthenics. He had no history of depression or anxiety and was generally pleasant and even-tempered. His family history was positive for a paternal grandfather with a history of cognitive decline and a brother with AD. Cerebral computerized axial tomography performed 2 and 3 years before death revealed progressive cerebral and cerebellar atrophy and mild ventricular enlargement.
Case 3
A 73-year-old Caucasian male began boxing at the age of 11 and fought as an amateur boxer for 9 years and as a professional boxer for 13 years. He fought a total of 48 professional bouts, accumulating 2 world championships before retiring at the age of 33. In his late 50s, he became forgetful with mood swings and restlessness. He changed from his normally happy, easy-going self to become apathetic, socially withdrawn, paranoid, irritable and sometimes violently agitated. Over the next 2 years, he began to confuse close relatives and developed increasing anxiety, aggression and agitation; on occasion, he was verbally abusive towards his wife and tried to strike her. He required neuroleptics for control of his behavior. The following year he had episodes of dizziness, which was suspected to be vertigo and resulted in a hospital admission. Neurological examination found him to be disoriented, inattentive, with very poor immediate and remote memory, and impaired visuospatial skills. Neuropsychological testing showed deficits in all cognitive domains, including executive functioning, attention, language, visuospatial abilities, and profound deficits in learning and memory. CT scan and MRI showed generalized cortical atrophy, enlargement of the cerebral ventricles, cavum septum pellucidum and a right globus pallidus lacune. An EEG, magnetic resonance angiogram and carotid ultrasound were normal. He smoked and drank alcohol occasionally until his early fifties. A first cousin developed dementia in her early 50s and 3 uncles and 1 aunt (of 11 children) were demented.
Over the following 2 years he continued to decline in all cognitive domains. He fell frequently and developed a tremor of his left hand. Repeat neuropsychological testing at age 67 revealed further global deficits, again with prominent impairments in memory. By age 70 he had severe swallowing difficulties, diminished upgaze, masked facies, garbled speech and a slow, shuffling gait. Mini-mental state examination several months prior to death was 7 out of 30. He died at age 73 from complications of pneumonia.
CTE in Other Sports and Activities
Other sports associated with neuropathologically verified CTE are professional wrestling (20), and soccer (13). The first known case of CTE in a professional wrestler involved a 40-year-old Caucasian man who began professional wrestling at age 18 and wrestled for the next 22 years (20) He was known for his rough, aggressive style and had suffered numerous concussions and a cervical fracture during his career. At age 36 he began to experience problems in his marriage with periods of depression and lapses of memory. During his 40th year he had episodes of violent behavior; he ultimately killed his wife and son and committed suicide. He was believed to have used anabolic steroids and prescription narcotics. His past medical history included a motor vehicle accident at age 6 requiring 3 days of hospitalization for mild traumatic brain injury without known neurological sequelae.
Geddes and colleagues reported finding mild changes of CTE in a 23-year-old amateur soccer player who regularly “headed” the ball while playing and had a history of a single severe head injury (13). Williams and Tannenberg reported the findings in a 33-year-old achondroplastic dwarf, with a long history of alcohol abuse, who worked for 15 years as a clown in a circus (19). He had been knocked unconscious “a dozen times” and participated in dwarf-throwing events.
Pathological Features of CTE
Gross Pathology
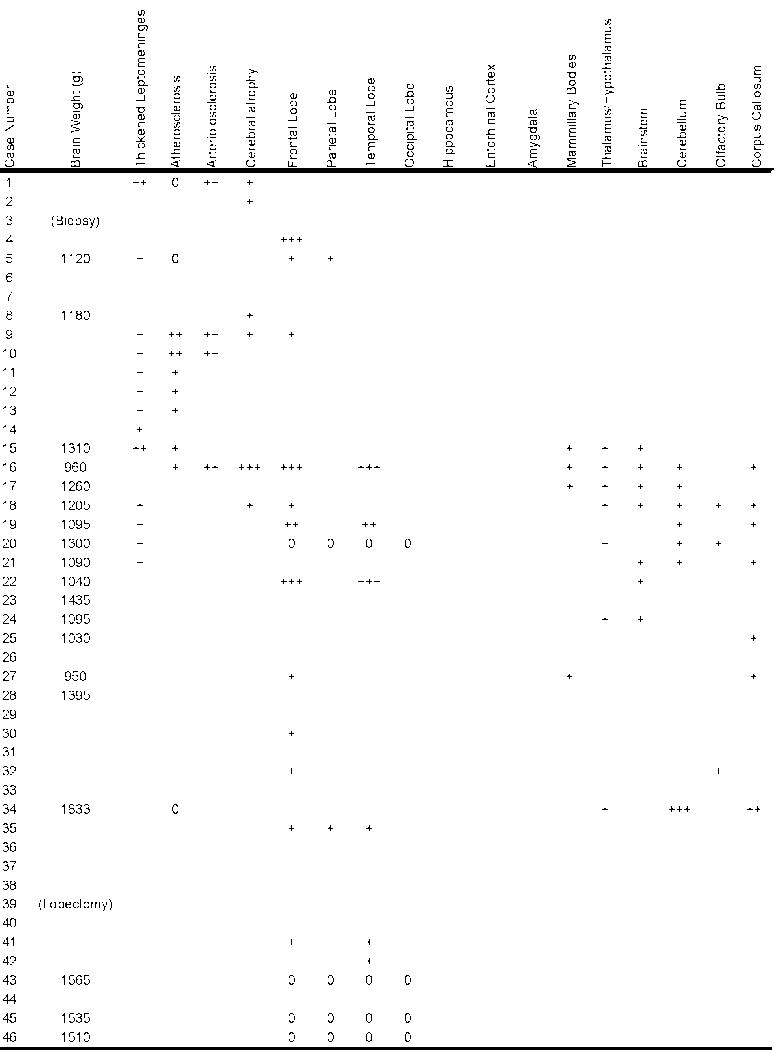
In their comprehensive description of the pathology, Corsellis and colleagues summarized the most common gross neuropathological findings, including 1) a reduction in brain weight, 2) enlargement of the lateral and third ventricles, 3) thinning of the corpus callosum, 4) cavum septum pellucidum with fenestrations, 5) scarring and neuronal loss of the cerebellar tonsils (29). The reduction in brain weight is generally mild (mean 1261 grams, range 950–1833) and associated with atrophy of the frontal lobe (36%), temporal lobe (31%), parietal lobe (22%), and less frequently, occipital lobe (3%) (Tables 3–6). With increasing severity of the disease, atrophy of the hippocampus, entorhinal cortex and amygdala may become marked. The lateral ventricles (53%) and III ventricles (29%) are frequently dilated; rarely there is dilation of the IV ventricle (4%). Cavum septum pellucidum is often present (69%), usually with fenestrations (49%). Other common gross features include pallor of the substantia nigra and locus ceruleus, atrophy of the olfactory bulbs, thalamus, mammillary bodies, brainstem and cerebellum, and thinning of the corpus callosum. Many of these gross pathological features were found in our Cases 2 and 3.
Table 3.
Gross Pathological Features: Atrophy
 |
 |
New Cases 1–3 of this series
0 = feature not present; + = mild; ++ = moderate; +++ = severe; blank = feature was not mentioned
Table 6.
Microscopic Pathological Features: Other
 |
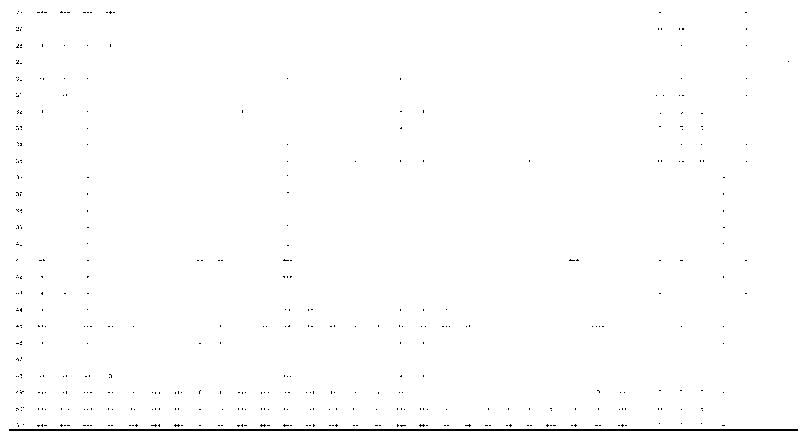 |
New cases 1–3 of this series; 0 = feature not present; + = mild; ++ = moderate; +++ = severe; blank = feature was not mentioned; NFT = neurofibrillary tangles; sp = senile plaques; SN = substantia nigra; LC = locus ceruleus
Case 2
The brain weighed 1,360 grams. There was a mild yellow-brown discoloration in the leptomeninges over the temporal poles. There was mild atrophy of the frontal, parietal and temporal lobes, most pronounced in the temporal pole. The floor of the hypothalamus was thinned and translucent and the mammillary bodies were atrophic. The medial thalamus was atrophic and concave. The frontal, temporal and occipital horns of the lateral and third ventricles were enlarged with a 0.5-cm cavum septum pellucidum. The corpus callosum was thinned in its mid-portion. The anterior hippocampus, amygdala and entorhinal cortex were severely atrophic. By contrast, the posterior hippocampus was only mildly atrophic. The substantia nigra and locus ceruleus were markedly pale.
Case 3
The brain weighed 1,220 grams. There was moderate atrophy of the frontal, parietal and temporal lobes, most pronounced in the temporal pole. The floor of the hypothalamus was markedly thinned and the mammillary bodies were atrophic. The corpus callosum was thinned, most prominently in its anterior portion. There was a large cavum septum pellucidum (0.8 cm) with fenestrations. The frontal and temporal horns of the lateral ventricles and the third ventricle were moderately enlarged. The entorhinal cortex, hippocampus, and amygdala were markedly atrophic throughout their entire extent. The medial thalamus was atrophic and concave. The perivascular spaces of the temporal and frontal white matter were prominent. A 1.0-cm lacune was present in the internal segment of the right globus pallidus. There was severe pallor of the substantia nigra and locus ceruleus with discoloration and atrophy of the frontopontine fibers in the cerebral peduncle.
Microscopic Pathology*
Neuronal Loss
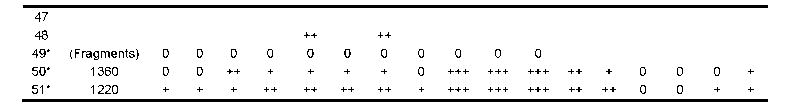
A few reports in the literature (cases 3, 4, 10, 12, 14, 29; Table 5) described neuronal loss and gliosis in the hippocampus, substantia nigra and cerebral cortex without appreciable neurofibrillary pathology. Neuronal loss and gliosis most commonly accompany neurofibrillary degeneration, however, and are pronounced in the hippocampus, particularly the CA1 and subiculum, the entorhinal cortex and amygdala. If the disease is advanced, neuronal loss is also found in the subcallosal and insular cortex and to a lesser degree in the frontal and temporal cortex. Other areas of neuronal loss and gliosis include the mammillary bodies, medial thalamus, substantia nigra, locus ceruleus and nucleus accumbens. In Cases 2 and 3, the cerebral cortex showed mild neuronal loss in the insular and septal cortices and moderate neuronal loss in the entorhinal cortex, amygdala, medial thalamus, mammillary bodies, substantia nigra pars compacta and pars reticulata and to a lesser extent, locus ceruleus. In Case 2, CA1 of the hippocampus showed moderate loss of neurons, and in Case 3, CA1 and the subiculum of the hippocampus showed severe neuronal loss and gliosis.
Table 5.
Microscopic Pathological Features: Neuronal Loss
| Case Number | Frontal Cortex | Parietal Cortex | Temporal Cortex | Occipital Cortex | Hippocampus | Entorhinal Cortex | Amygdala | Cerebellum |
|---|---|---|---|---|---|---|---|---|
| 1 | ||||||||
| 2 | ||||||||
| 3 | +++ | +++ | +++ | |||||
| 4 | +++ | +++ | +++ | |||||
| 5 | ||||||||
| 6 | ||||||||
| 7 | ||||||||
| 8 | ||||||||
| 9 | ||||||||
| 10 | ||||||||
| 11 | ||||||||
| 12 | ||||||||
| 13 | ||||||||
| 14 | ||||||||
| 15 | + | |||||||
| 16 | +++ | +++ | +++ | |||||
| 17 | ||||||||
| 18 | ||||||||
| 19 | ||||||||
| 20 | + | + | + | + | ||||
| 21 | ||||||||
| 22 | + | + | + | + | ||||
| 23 | + | + | + | + | ||||
| 24 | ||||||||
| 25 | ||||||||
| 26 | +++ | +++ | +++ | +++ | ||||
| 27 | ||||||||
| 28 | ||||||||
| 29 | ||||||||
| 30 | ||||||||
| 31 | ||||||||
| 32 | ||||||||
| 33 | ||||||||
| 34 | ||||||||
| 35 | ||||||||
| 36 | ||||||||
| 37 | ||||||||
| 38 | ||||||||
| 39 | ||||||||
| 40 | ||||||||
| 41 | ||||||||
| 42 | ||||||||
| 43 | ||||||||
| 44 | ||||||||
| 45 | ||||||||
| 46 | ||||||||
| 47 | ||||||||
| 48 | ++ | ++ | ++ | ++ | ++ | |||
| 49* | 0 | 0 | 0 | 0 | + | + | + | |
| 50* | + | + | + | + | ++ | ++ | ++ | |
| 51* | ++ | ++ | ++ | ++ | +++ | +++ | +++ |
New cases 1–3 of this series; 0 = feature not present; + = mild; ++ = moderate; +++ = severe; blank = feature was not mentioned
Tau Deposition
Neurofibrillary tangles (NFTs), astrocytic tangles, and dot-like and spindle-shaped neuropil neurites (NNs) are common in the dorsolateral frontal, subcallosal, insular, temporal, dorsolateral parietal, and inferior occipital cortices. The tau-immunoreactive neurofibrillary pathology is characteristically irregular in distribution with multifocal patches of dense NFTs in the superficial cortical layers, often in a perivascular arrangement. This superficial distribution of neocortical NFTs was originally described by Hof and colleagues who noted that the NFTs in CTE were preferentially distributed in layer II and the upper third of layer III in neocortical areas and generally more dense than in AD (47).
Geddes and colleagues drew attention to the perivascular distribution of NFTs in their description of the neuropathological alterations in the brain and frontal lobectomy specimens of 5 young men, ranging in age from 23 to 28 years (13, 37). The 5 cases included 2boxers, a soccer player, a person described as “mentally subnormal” with a long history of head banging, and an epileptic patient who frequently hit his head during seizures. Microscopically, all the brains showed argyrophilic, tau-positive neocortical NFTs, strikingly arranged in groups around small intracortical blood vessels, associated with neuropil threads and granular tau-positive neurons. There were also NFTs along the basal surfaces of the brain, usually at the depths of sulci. The hippocampi of the 4 autopsy cases were normal.
In CTE, tau-immunoreactive protoplasmic astrocytes are interspersed throughout the superficial cortical layers appearing as plaque-like accumulations composed of primarily globular neurites. The corpus callosum and subcortical white matter of the cortex show NNs and fibrillar astrocytic tangles. The U-fibers are prominently involved. Subcortical white matter structures such as the extreme and external capsule, anterior and posterior commissures, thalamic fasciculus, and fornix also show NNs and astrocytic tangles.
Dense NFTs, ghost tangles, and astrocytic tangles are found in the olfactory bulbs, hippocampus, entorhinal cortex, and amygdala, often in greater density than is found in AD. Abundant NFTs and astrocytic tangles are also found in the thalamus, hypothalamus, mammillary bodies, nucleus basalis of Meynert, medial geniculate, substantia nigra (pars compacta more than the pars reticulata), locus ceruleus, superior colliculus, periaqueductal gray, medial lemniscus, oculomotor nucleus, trochlear nucleus, ventral tegmental area, dorsal and median raphe, trigeminal motor nucleus, pontine nuclei, hypoglossal nucleus, dorsal motor nucleus of the vagus, inferior olives and reticular formation. The nucleus accumbens is usually moderately affected; the globus pallidus, caudate, and putamen are less involved. In the brainstem and spinal cord, midline white matter tracts show dense astrocytic tangles especially around small capillaries. Fibrillar astrocytic tangles are also common in the subpial and periventricular zones. Neurons in the spinal cord gray matter contain NFTs, and astrocytic tangles are frequent in the ventral gray matter. This unique pattern of tau-immunoreactive pathology was found in all 3 of our cases, with increasing severity from Case 1 to Case 3.
Case 1
NFTs immunopositive for tau epitopes (Appendix) were prominent in the inferior frontal, superior frontal, subcallosal, insular, temporal, and inferior parieto-temporal cortices (Fig. 1). Primary visual cortex showed no NFTs; anterior and posterior cingulate cortex showed only scant NFTs. NFTs occurred in irregular patches, often greatest at the sulcal depths (Fig. 2). Tau-positive fibrillar astrocytes (“astrocytic tangles”) were prominent in foci, especially in subpial regions and around small blood vessels (Figs. 2, 3). NFTs were especially numerous in cortical laminae II and III, where a prominent perivascular distribution of neuronal NFTs and fibrillar astrocytic tangles was evident (Fig. 3). Although some neuronal NFTs showed multiple tau-positive perisomatic processes, most neuronal NFTs were morphologically similar to those found in AD. In the cortex there were many tau-positive astrocytes bearing a corona of tau-positive processes. These tau-positive protoplasmic astrocytes were similar in appearance to the astrocytic plaques of corticobasal degeneration except that the perikaryon was often tau-positive (Fig. 3).
Figure 1.

(A–D) Case 1: Whole mount 50-μm coronal sections immunostained for tau with monoclonal antibody AT8 and counterstained with cresyl violet showing irregular, patchy deposition of phosphorylated tau protein in frontal, subcallosal, insular, temporal, and parietal cortices and the medial temporal lobe.
Figure 2.

(A–C) Whole mount 50-μm coronal sections of superior frontal cortex from case 1 (A), case 2 (B), case 3 (C) immunostained for tau with monoclonal antibody CP-13 showing extensive immunoreactivity that is greatest at sulcal depths (asterisks) and is associated with contraction of the cortical ribbon. (D–F) Microscopically there are dense tau-immunoreactive neurofibrillary tangles (NFTs) and neuropil neurites throughout the cortex, case 1 (D), case 2 (E) and case 3 (F). There are focal nests of NFTs and astrocytic tangles around small blood vessels (E, arrow) and plaque-like clusters of tau-immunoreactive astrocytic processes distributed throughout the cortical layers (F, arrows).
Figure 3.

Whole mount 50-μm sections from cases 1 and 2 immunostained with anti-tau monoclonal antibody AT85. (A) Case 2. There is a prominent perivascular collection of neurofibrillary tangles (NFTs) and astrocytic tangles evident in the superficial cortical layers with lesser involvement of the deep laminae. Prominent neuropil neurites (NNs) are found in the subcortical U-fibers (arrow), original magnification x150. (B) Case 1. There is a preferential distribution of NFTs in layer II and NNs extending into the subcortical white matter even in mildly affected cortex, original magnification x150. (C) Case 1. Focal subpial collections of astrocytic tangles and NFTs are characteristic of chronic traumatic encephalopathy (CTE), original magnification x150. (D) Case 1. The shape of most NFTs and NNs in CTE is similar to those found in Alzheimer disease, original magnification x150. Some NFTs have multiple perisomatic processes (E) and spindle-shaped and dot-like neurites are found in addition to thread-like forms (E, Case 1, original magnification x350. (F) Case 1. Astrocytic tangles are interspersed with NFTs in the cortex (arrows, original magnification x350). (G) Case 1. Tau-immunoreactive astrocytes are common in periventricular regions, original magnification x150. (H, I) Case 1. Tau-immunoreactive astrocytes take various forms; some appear to be protoplasmic astrocytes with short rounded processes (H, I, double immunostained section with AT8 (brown) and anti-glial fibrillary acidic protein (red), (H) original magnification x350, (I) original magnification x945). (J) Case 2: Dot-like or spindle-shaped neurites predominate in the white matter although there are also some threadlike forms, original magnification x150.
Neuropil neurites (NNs) and astrocytic tangles were abundant in the frontal and temporal white matter (Fig. 3). NNs were often dot-like and spindle-shaped in addition to threadlike forms similar to those found in AD. The hippocampus, entorhinal, and transentorhinal cortex contained dense NFTs, ghost tangles and NNs, including many ghost tangles in CA1 and subiculum; NFTs were denser in the anterior hippocampus compared to the posterior hippocampus. The amygdala showed dense tau immunoreactivity, including NFTs, astrocytic tangles and NNs (Fig. 4). NFTs were most frequent in the lateral nuclear group of the amygdala.
Figure 4.

(A–C) Whole mount 50-μm-thick coronal sections immunostained for tau (AT8) from case 1 (A), case 2 (B), case 3 (C) (counterstained with cresyl violet) showing extremely dense deposition of tau protein in the amygdala with increasing severity from left to right. (D–F) Microscopically, there is a moderate density of NFTs and astrocytic tangles in case 1 (D), the density is increased in case 2 (E), and extremely marked in case 3 (F), original magnification x350.
The nucleus basalis of Meynert, hypothalamic nuclei, septal nuclei, fornix, and lateral mammillary bodies showed dense NFTs and astrocytic tangles. NFTs and astrocytic tangles were also found in the olfactory bulb, thalamus, caudate, and putamen. The globus pallidus and subthalamic nucleus were relatively spared. The lateral substantia nigra pars compacta showed mild neuronal loss, extraneuronal pigment deposition, and moderate numbers of NFTs and NNs. The pars reticulata was unremarkable. The cerebellar peduncle showed mild perivascular hemosiderin deposition. NFTs were numerous in the dorsal and median raphe nuclei. The internal, external and extreme capsules, fornix and mammillothalamic tract showed moderate NNs, although, in general, the white matter was less affected than adjacent gray matter.
Case 2
There were abundant tau–positive NFTs, glial tangles, and dot-like and spindle-shaped NNs in the superficial layers of cerebral cortex (I–III) (Fig. 3). Cortical tau pathology was most prominent in patchy areas of the superior frontal and temporal lobes, especially the medial temporal lobe, often in a vasocentric pattern. The olfactory bulb, hippocampus, entorhinal cortex and amygdala showed extremely dense NFTs with many ghost tangles (Figs. 4–6). Tau-positive glia and NNs were also found in the subcortical white matter and corpus callosum. The olfactory bulb, thalamus, hypothalamus, nucleus basalis, striatum, globus pallidus, substantia nigra, raphe, periventricular gray, locus ceruleus, oculomotor nucleus, red nucleus, pontine base, tegmentum, reticular nuclei, inferior olives, and dentate nucleus showed dense NFTs and glial tangles. Spindle-shaped NNs and tau-positive glia were pronounced in the midline white matter tracts of the brainstem.
Figure 6.

Tau-immunoreactive (AT8) NFTs, astrocytic tangles and neuropil neurites are found in many subcortical nuclei including the substantia nigra (A, case 3, original magnification x350) and nucleus basalis of Meynert (B, case 3, original magnification x350). NFTs are also abundant in the olfactory bulb (C, case 2, Bielschowsky silver method, original magnification x150) and thalamus (case 1, original magnification x350, AT8 immunostain counterstained with cresyl violet).
Case 3
Microscopic examination showed dense accumulations of tau-immunoreactive NFTs, astrocytic tangles, and NNs in irregular patches of the dorsolateral frontal, insular, subcallosal, inferior frontal, superior parietal and posterior temporo-occipital cortices, and most severely in the medial temporal lobe. The hippocampus, entorhinal cortex and amygdala contained extremely dense NFTs with ghost tangles and severe neuronal loss (Figs. 4, 6). Tau-positive glia and NNs were also found in the subcortical white matter, particularly in the subcortical U-fibers. The olfactory bulb, thalamus, hypothalamus, nucleus basalis, striatum, globus pallidus, substantia nigra, raphe, periventricular gray, locus ceruleus, oculomotor nucleus, red nucleus, pontine base, pontine tegmentum, hypoglossal nuclei, reticular nuclei, inferior olives, midline tracts of the medulla, and dentate nucleus contained dense NFTs and astrocytic tangles (Figs. 5, 7). Subcortical white matter tracts including the anterior and posterior commissure, thalamic fasciculus, external and extreme capsule also showed astrocytic tangles and NNs.
Figure 5.

Whole mount 50-μm coronal sections of case 2 (A) and case 3 (B), immunostained for tau (AT8) and counter-stained with cresyl violet. There is extremely dense deposition of tau protein in the hippocampus and medial temporal lobe structures. There is also prominent tau deposition in the medial thalamus.
Figure 7.

Whole mount tau (AT8)-immunostained 50-μm coronal sections of the brainstem from case 3 showing severe involvement of the locus ceruleus, pontine tegmentum, pontine base, midline medulla, and hypoglossal nuclei.
The abnormal tau proteins that are found in the glial and neuronal tangles in CTE are indistinguishable from NFTs in AD and are composed of all 6 brain tau isoforms (39). Neuropathologically, CTE resembles several other neurodegenerative diseases characterized by accumulations of hyperphosphorylated tau protein in neurons or glial cells, including ALS/PDC of Guam, post-encephalitic parkinsonism, progressive supranuclear palsy (PSP), corticobasal degeneration, and frontotemporal dementia with parkinsonism linked to chromosome 17 (FTDP-17) (36, 48–50). Like ALS/PDC of Guam, neurofibrillary tau pathology in CTE is found in the medial temporal lobe structures, cerebral cortex and spinal cord with only a subset of cases showing evidence of diffuse plaques (51). Like ALS/PDC and PSP, CTE preferentially involves the superficial cortical layers and involves the accumulation of tau-immunoreactive astrocytes (36). However, CTE differs from ALS/PDC of Guam and PSP in that the cortical involvement is irregular and patchy, greatest at sulcal depths, and distributed in a prominent perivascular, periventricular and subpial pattern. Furthermore, there is a unique regional involvement of subcortical and brainstem structures in CTE (Tables 5, 6).
β-Amyloid Deposition
β-Amyloid (Aβ) deposition is an inconstant feature in CTE. Fourteen of the 15 brains originally described by Corsellis (29) and 6 additional boxers were re-examined by Roberts and colleagues using Aβ immunocytochemistry with formic acid pretreatment; 19 of the 20 cases showed widespread diffuse Aβ deposits (18). The only case that did not contain diffuse Aβ was that of a 22-year-old boxer who died during a fight. Similarly, Tokuda and colleagues who found abundant diffuse Aβ deposits in 8 cases of CTE and cerebrovascular Aβ deposits in 3 cases (52). In our series, only Case 2 showed moderate numbers of diffuse Aβ plaques in the frontal, parietal and temporal cortex, and sparse neuritic plaques; there was no vascular amyloid. Of the 51 neuropathologically verified cases of CTE, diffuse plaques were found in 22 (44%), neuritic plaques in 13 (27%), and amyloid angiopathy in 3 (6%). There was also one report of a fatal cerebral hemorrhage from amyloid angiopathy associated with CTE (15).
White Matter Changes and Other Abnormalities
Tau-positive fibrillar astrocytic tangles are found in the white matter, but the major abnormality is that of dot-like or spindle-shaped tau-positive neurites. The shape of the tau-immunoreactive neurites is distinct from the predominantly threadlike forms found in AD and suggests an axonal origin. Tokuda and colleagues characterized the neuropil neurites in CTE as shorter and less prominent than the neuropil threads found in AD and not spatially related to senile plaques (52). Generally, tau abnormalities in the white matter are not as severe as in adjacent gray matter. Other abnormalities found frequently in the cerebral and cerebellar white matter include small arterioles with thickened fibrohyalinized walls with perivascular hemosiderin-laden macrophages, widened perivascular spaces, and white matter rarefaction. In our cases 1 3, mild to moderate myelin and axonal loss was found in the corpus callosum and subcortical white matter of the frontal and temporal lobes and cerebellum with mild perivascular hemosiderin deposition.
α-Synuclein Staining
Extensive accumulation of α-synuclein has been found in axons following acute traumatic brain injury (TBI) (53) but α-synuclein immunostaining was not a feature of any of the 51 cases of CTE, including our 3 cases.
Clinicopathological Considerations
The distribution of the tau abnormalities in CTE suggests distinctive core pathology within the amygdalo-hippocampal-septo-hypothalamic-mesencephalic continuum, i.e. the Papez circuit (54, 55). The early involvement of these anatomical regions, sometimes referred to as “emotional” or “visceral” brain, may underlie many of the early behavioral symptoms, including the tendency toward emotional lability, aggression and violent outbursts. The early involvement of the hippocampus, entorhinal cortex and medial thalamus may explain episodic memory disturbance as a frequent presenting symptom (56). Neurofibrillary degeneration of the frontal cortex and underlying white matter most likely contributes to the dysexecutive symptoms. Although less common and generally less severe, neurofibrillary degeneration in the dorsolateral parietal, posterior temporal, and occipital cortices likely accounts for the visuospatial difficulties. The parkinsonian features found in 42% of cases are likely due to degeneration of the substantia nigra pars compacta. The gait disorder, variously described as staggered, slowed, shuffled, or frankly ataxic, may result from a combination of cortical and subcortical frontal damage, degeneration of cerebellar tracts in the brainstem, direct cerebellar injury, as well as Parkinsonism from substantia nigra pathology. Similarly, speech abnormalities, most often described as slowed and slurred, likely reflect multiregional degeneration. Symptoms of dysarthria, dysphagia, and ocular abnormalities probably result from degeneration of brainstem nuclei, e.g. the hypoglossal and oculomotor nuclei.
Possible Mechanisms of Cerebral Injury
Acceleration and deceleration forces are thought to be important events in concussion, particularly rotational acceleration and deceleration (57–59). Sagittal (front-to-back) injuries result in relatively good recovery whereas lateral (side-to-side) injuries produce the most injury, with injury directed related to the severity of the generating force (58). Conceivably, a concussive impact imparts a fluid wave in the lateral ventricles that produces a shearing force on the septum pellucidum; this may explain the development of an enlarged cavum septum pellucidum and, if severe or repeated, fenestrations.
The patchy, irregular location of the cortical NFTs and astrocytic tangles suggests that the distribution is related to direct mechanical injury from blows to the side or top of the head, given their multifocal dorsolateral frontal and parietal, inferior frontal and occipital, and lateral temporal distribution. The possibility that ischemia may contribute to the development of the tau pathology is suggested by the concentration of tau-immunoreactive pathology at the depths of sulci. Damage to the blood-brain barrier and release of local neurotoxins might explain some of the tendency toward perivascular nests of tau-immunoreactive NFT, tau-positive glia, and NNs (13). Buee and Hof studied the microvasculature of several cases with dementia pugilistica and found decreased microvascular density and tortuosity with a strong correlation between the laminar distribution of NFTs and pathological microvasculature. Hof and colleagues suggested that the shear forces of repetitive head trauma might lead to vascular damage followed by perivascular NFT and NN formation (60). Further supporting a possible vascular connection to the pathological changes in CTE, Bouras reported laser microprobe mass analysis of NFTs and nuclei of NFT-free neurons in CTE contained substantially higher amounts of iron and aluminum than NFTs in AD (61).
Acute Traumatic Brain Injury
Axonal Injury
Acute concussion produces diffuse axonal injury (62). The “diffuse degeneration of the cerebral white matter” was first described by Strich as the shearing or mechanical tearing of axons at the time of injury (63). It is now appreciated that axons are not sheared at the time of injury except in the most severe instances of diffuse axonal injury, but instead undergo a series of changes that may result in a secondary axotomy within 24 hours (64). The axolemma is one of the initial sites of injury; the increased permeability, uncontrolled influx of Ca++, swelling of mitochondria, disruption of microtubules, and alterations in axonal transport that follow produce axonal swelling and secondary axotomy (64–66). Rapid axonal swelling, perisomatic axotomy and Wallerian degeneration may also occur without changes in axolemmal permeability, suggesting that trauma may have diverse effects on axons. McKenzie showed that 80% of patients who died from acute head injury showed immunocytochemical evidence of axonal injury within 2 hours of injury; after 3 hours of injury, axonal bulbs were identified, and as the survival time increased, the amount of axonal damage and axonal bulb formation increased. Axonal injury was found most frequently in the brainstem, followed by the internal capsule, thalamus, corpus callosum and parasagittal white matter (67). Axonal damage may continue for weeks after the acute TBI (68).
Deposition of Abnormal Proteins
In individuals undergoing surgical brain tissue resection for acute TBI, tau-immunoreactive dystrophic axons were found in the white matter and diffuse tau immunoreactivity was found in some neuronal cell bodies, dendrites, and glial cells within 2–3 hours post injury (67). Studies of acute TBI in experimental animal models and postmortem human brain also demonstrate that Aβ deposition, amyloid precursor protein (APP) processing, production and accumulation are increased after injury (69–78). Increased APP production in experimental TBI has also been associated with heightened neuronal loss in the hippocampus (73, 79). In acute TBI, diffuse cortical Aβ plaques have been found in 30% to 38% of cases as early as 2 hours after injury (73, 76, 80). In addition, individuals with cortical Aβ plaques showed increased levels of soluble Aβ42 and half were Apolipoprotein E (ApoE) ε4 allele carriers (81). In acute TBI, Aβ deposition is widely distributed throughout the neocortex without apparent association with the injury sites (82). The predominant form of Aβ in acute TBI is Aβ42 whereas the Aβ40 form predominates in serum and CSF, a situation similar to that in AD (83). A recent report also showed that interstitial soluble Aβ concentrations in the brain appear to correlate directly with neurological outcome following TBI (84).
Neuronal Death in Acute TBI and Relationship to CTE
There are multiple reasons for neuronal loss in acute traumatic injury including neuronal death from direct physical damage, necrosis from the immediate release of excitatory transmitters such as glutamate, and diffuse, delayed cell death involving both necrotic and apoptotic death cascades (85, 86). Other contributing factors include focal ischemia, breakdown of the blood-brain barrier, inflammation and the release of cytokines. Experimental lateral percussive injury in the rat produces apoptotic and necrotic neuronal death that progresses for up to one year after injury with degeneration of the cortex, hippocampi, thalami and septum, ventriculomegaly, and impaired memory performance (62, 85, 87–89). The thalamic degeneration typically follows the cortical degeneration by weeks, suggesting that a secondary process such as deafferentation may play a role in the thalamic neuronal death. Neuronal loss in the hippocampus and thalamus has also been reported following blunt head injury in humans using stereological techniques (90, 91). One of the key features of CTE is that the disease continues to progress decades after the activity that produced traumatic injury has stopped. It is most likely that multiple pathological cascades continue to exert their effects throughout the individual’s lifetime once they are triggered by repetitive trauma; the longer the survival after the initial events and the more severe the original injuries, the greater the severity of the neurodegeneration. It is clear that neuronal loss, cerebral atrophy, and ventricular enlargement all increase with longer survival and greater exposure to repetitive trauma.
Diagnosis of CTE
Presently there are no available biomarkers for the diagnosis of CTE. Although significant decreases in CSF ApoE and Aβ concentrations have been reported that correlated with severity of the injury after TBI, there have been no similar studies in CTE (92). Nonetheless, advances in neuroimaging offer the promise of detecting subtle changes in axonal integrity in acute TBI and CTE. Standard T1- or T2-weighted structural MR imagining is helpful for quantitating pathology in acute TBI, but diffusion tensor MRI (DTI) is a more sensitive method to assess axonal integrity in vivo (93, 94). In chronic moderate to severe TBI, abnormalities on DTI have been reported in the absence of observable lesions on standard structural MRI (83). More severe white matter abnormalities on DTI have been associated with greater cognitive deficits by neuropsychological testing (94, 95) and increases in whole-brain apparent diffusion coefficient and decreases in fractional anisotropy using DTI have been found in boxers compared to controls (96, 97).
Genetic Risk and the Role of ApoE4
ApoE genotyping has been reported in 10 cases of CTE, including our most recent cases. Five of the 10 cases of CTE carried at least one ApoE ε4 allele (50%), and 1 was homozygous for ApoE ε4 (our case 1). The percentage of ApoE ε4 carriers in the general population is 15%; this suggests that the inheritance of an ApoE ε4 allele might be a risk factor for the development of CTE.
In acute TBI there is accumulating evidence that the deleterious effects of head trauma are more severe in ApoE ε4-positive individuals (98–100). Acute TBI induces Aβ deposition in 30% of people (75, 76) and a significant proportion of these individuals are heterozygous for ApoE ε4 (101, 102). ApoE4 transgenic mice suffer greater mortality from TBI than ApoE ε3 mice (102). Furthermore, transgenic mice that express ApoE ε4 and overexpress APP show greater Aβ deposition after experimental TBI (103).
Guidelines for Prevention and Treatment
Clearly, the easiest way to decrease the incidence of CTE is to decrease the number of concussions or mild traumatic brain injuries. In athletes this is accomplished by limiting exposure to trauma, for example, by penalizing intentional hits to the head (as is happening in football and hockey) and adhering to strict “return to play” guidelines. Proper care and management of mild traumatic brain injury in general and particularly in sports will also reduce CTE. No reliable or specific measures of neurological dysfunction after concussion currently exist, and most recommendations are centered on the resolution of acute symptoms such as headache, confusion, sensitivity to light, etc. (104). Asymptomatic individuals have been shown, however, to have persistent decreases in P300 amplitudes in response to an auditory stimulus at least 5 weeks after a concussion, thereby casting doubt on the validity of the absence of symptoms as a guidepost (105, 106). Neuropsychological tests have also helped provide estimates of the appropriate time for athletes to return to practice and play. Studies using event-related potentials, transcranial magnetic stimulation, balance testing, multitask effects on gait stability, PET, and DTI MRI have all shown abnormalities in concussed athletes or nonathletes with TBI lasting for 2 to 4 weeks (105, 107–109). These studies indicate that safe return to play guidelines might require at least 4 to 6 weeks to facilitate more complete recovery and to protect from reinjury, as a second concussion occurs much more frequently in the immediate period after a concussion (106, 110). In addition, experimental evidence in animals suggests that there is expansion of brain injury and inhibition of functional recovery if the animal is subjected to overactivity within the first week (111).
Conclusion
CTE is a progressive neurodegeneration clinically associated with memory disturbances, behavioral and personality change, Parkinsonism, and speech and gait abnormalities. Pathologically, CTE is characterized by cerebral and medial temporal lobe atrophy, ventriculomegaly, enlarged cavum septum pellucidum, and extensive tau-immunoreactive pathology throughout the neocortex, medial temporal lobe, diencephalon, brainstem, and spinal cord. There is overwhelming evidence that the condition is the result of repeated sublethal brain trauma that often occurs well before the development of clinical manifestations. Repetitive closed head injury occurs in a wide variety of contact sports as well as a result of accidents or in the setting of military service. Pathologically, CTE shares some features of AD, notably tau-immunoreactive NFTs, NNs and, in approximately 40% of cases, diffuse senile plaques. Furthermore, the Aβ and NFTs found in CTE are immunocytochemically identical to those found in AD, suggesting a possible common pathogenesis. Multiple epidemiological studies have shown that head injury is a risk factor for AD and there have been several case reports citing an association between a single head injury and the development of subsequent AD (112, 113). Just as acquired vascular injury may interact additively or synergistically with AD, traumatic injury may interact additively with AD to produce a mixed pathology with greater clinical impact or synergistically by promoting pathological cascades that result in either AD or CTE. In athletes, by instituting and following proper guidelines for return to play after a concussion or mild traumatic brain injury, it is possible that the frequency of sports-related CTE could be dramatically reduced or perhaps, entirely prevented.
Table 4.
Gross Pathological Features: Other
| Case Number | II Ventricle Enlarged | III Ventricle Enlarged | IV Ventricle Enlarged | Cavum Septum | Fenestrations | SN Pallor | LC Pallor |
|---|---|---|---|---|---|---|---|
| 1 | + | + | |||||
| 2 | + | + | |||||
| 3 | |||||||
| 4 | +++ | ||||||
| 5 | + | + | |||||
| 6 | + | ||||||
| 7 | + | ||||||
| 8 | + | +++ | |||||
| 9 | + | + | + | ||||
| 10 | + | + | + | ||||
| 11 | + | + | + | ||||
| 12 | + | + | |||||
| 13 | + | + | + | ||||
| 14 | + | + | |||||
| 15 | ++ | ++ | +++ | + | ++ | ||
| 16 | +++ | +++ | +++ | +++ | + | ++ | |
| 17 | ++ | ++ | ++ | + | +++ | ||
| 18 | ++ | ++ | + | + | +++ | ||
| 19 | ++ | ++ | + | +++ | + | + | |
| 20 | ++ | ++ | ++ | + | +++ | ||
| 21 | ++ | ++ | ++ | + | |||
| 22 | ++ | ++ | + | ++ | + | ||
| 23 | ++ | ++ | + | +++ | |||
| 24 | +++ | +++ | +++ | + | |||
| 25 | ++ | +++ | + | ||||
| 26 | + | + | |||||
| 27 | ++ | ++ | ++ | + | |||
| 28 | ++ | ++ | ++ | ||||
| 29 | + | + | |||||
| 30 | +++ | + | |||||
| 31 | +++ | ||||||
| 32 | + | + | |||||
| 33 | |||||||
| 34 | +++ | +++ | + | ||||
| 35 | + | ||||||
| 36 | 0 | 0 | 0 | ||||
| 37 | 0 | 0 | 0 | ||||
| 38 | 0 | 0 | 0 | ||||
| 39 | |||||||
| 40 | 0 | 0 | 0 | ||||
| 41 | + | ++ | ++ | ||||
| 42 | + | ||||||
| 43 | + | ||||||
| 44 | |||||||
| 45 | + | ||||||
| 46 | |||||||
| 47 | |||||||
| 48 | ++ | + | + | ||||
| 49* | + | 0 | 0 | ||||
| 50* | + | + | + | + | +++ | +++ | |
| 51* | ++ | ++ | ++ | + | +++ | +++ |
New cases 1–3 of this series; 0 = feature not present; + = mild; ++ = moderate; +++ = severe; blank = feature was not mentioned; SN = substantia nigra; LC = locus ceruleus
Acknowledgments
Supported by the Boston University Alzheimer’s Disease Center NIA P30 AG13846, supplement 0572063345-5 and the Department of Veterans’ Affairs.
The authors wish to thank Rafael Romero, MD, for his review of the clinical features of case 3.
Appendix. Methods for Analysis of Cases 1–3
The following anatomic regions were evaluated microscopically in paraffin sections in Cases 1 to 3: olfactory bulb, midbrain at level of red nucleus, right motor cortex, right inferior parietal cortex (Brodmann Area [BA] 39, 40), right anterior cingulate (BA 24), right superior frontal (BA 8, 9), left Inferior frontal cortex (BA 10, 11, 12), left lateral frontal (BA 45, 46), caudate, putamen, and accumbens (CAP), anterior temporal (BA 38), superior temporal (BA 20, 21, 22), middle temporal cortex, inferior temporal cortex, amygdala, entorhinal cortex (BA 28), globus pallidus, insula, substantia innominata, right hippocampal formation at the level of the lateral geniculate, hippocampus, thalamus with mamillary body, thalamus, posterior cingulate (BA 23, 31), calcarine cortex (BA 17,18), superior parietal cortex (BA 7B), cerebellar vermis, cerebellum with dentate nucleus, parastriate cortex (BA 19) pons, medulla and spinal cord.
The sections were stained with Luxol fast blue and hematoxylin and eosin, Bielschowsky silver impregnation and by immunohistochemistry with antibodies to phosphoserine 202 and phosphothreonine 205 of PHF-tau (mouse monoclonal AT8, Pierce Endogen, Rockford IL, 1:2,000), α-synuclein (rabbit polyclonal, Chemicon, Temecula, CA, 1:15,000), β-amyloid (mouse monoclonal, Dako North America Inc, Carpinteria, CA, 1:2,000) (following formic acid pretreatment), and Aβ 42 (rabbit polyclonal, Invitrogen (Biosource), Carpinteria, CA, 1:2,000). In addition, multiple large coronal fragments were cut at 50 μm on a sledge microtome and stained as free-floating sections using a mouse monoclonal antibody directed against phosphoserine 202 of tau (CP-13, courtesy of Peter Davies, 1:200); this is considered to be the initial site of tau phosphorylation in NFT formation (114–118). Other monoclonal antibodies used for immunostaining were AT8, phosphoserine 396 and phosphoserine 404 of hyperphosphorylated tau(PHF-1, courtesy of Peter Davies, 1:1000) (114–118), glial fibrillary acidic protein (GFAP) (Chemicon, 1:2,000), and HLA-DR-Class II major histocompatibility complex (LN3, Zymed, San Francisco, CA, 1:2,000); some of these sections were counterstained with cresyl violet.
References
- 1.Thurman DJ, Branche CM, Sniezek JE. The epidemiology of sports-related traumatic brain injuries in the United States: recent developments. J Head Trauma Rehabil. 1998;13:1–8. doi: 10.1097/00001199-199804000-00003. [DOI] [PubMed] [Google Scholar]
- 2.Langlois JA, Rutland-Brown W, Wald MM. The epidemiology and impact of traumatic brain injury: A brief overview. J Head Trauma Rehabil. 2006;21:375–78. doi: 10.1097/00001199-200609000-00001. [DOI] [PubMed] [Google Scholar]
- 3.Nowinski C. Head Games: Football’s Concussion Crisis from the NFL to Youth Leagues. East Bridgewater, MA: Drummond Publishing Group; 2006. [Google Scholar]
- 4.Roberts GW, Allsop D, Bruton C. The occult aftermath of boxing. J Neurol Neurosurg Psychiatry. 1990;53:373–78. doi: 10.1136/jnnp.53.5.373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Webbe FM, Barth JT. Short-term and long-term outcome of athletic closed head injuries. Clin Sports Med. 2003;22:577–92. doi: 10.1016/s0278-5919(02)00103-5. [DOI] [PubMed] [Google Scholar]
- 6.Macciocchi SN, Barth JT, Alves W, et al. Neuropsychological functioning and recovery after mild head injury in collegiate athletes. Neurosurgery. 1996;39:510–14. [PubMed] [Google Scholar]
- 7.Collins MW, Lovell MR, Iverson GL, et al. Cumulative effects of concussion in high school athletes. Neurosurgery. 2002;51:1175–9. doi: 10.1097/00006123-200211000-00011. discussion 80–81. [DOI] [PubMed] [Google Scholar]
- 8.Gaetz M, Goodman D, Weinberg H. Electrophysiological evidence for the cumulative effects of concussion. Brain Inj. 2000;14:1077–88. doi: 10.1080/02699050050203577. [DOI] [PubMed] [Google Scholar]
- 9.Gaetz M, Weinberg H. Electrophysiological indices of persistent post-concussion symptoms. Brain Inj. 2000;14:815–32. doi: 10.1080/026990500421921. [DOI] [PubMed] [Google Scholar]
- 10.Bailes JE, Cantu RC. Head injury in athletes. Neurosurgery. 2001;48:26–45. doi: 10.1097/00006123-200101000-00005. [DOI] [PubMed] [Google Scholar]
- 11.Beckwith JG, Chu JJ, Greenwald RM. Validation of a noninvasive system for measuring head acceleration for use during boxing competition. J Appl Biomech. 2007;23:238–44. doi: 10.1123/jab.23.3.238. [DOI] [PubMed] [Google Scholar]
- 12.Greenwald RM, Gwin JT, Chu JJ, et al. Head impact severity measures for evaluating mild traumatic brain injury risk exposure. Neurosurgery. 2008;62:789–98. doi: 10.1227/01.neu.0000318162.67472.ad. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Geddes JF, Vowles GH, Nicoll JA, et al. Neuronal cytoskeletal changes are an early consequence of repetitive head injury. Acta Neuropathol. 1999;98:171–78. doi: 10.1007/s004010051066. [DOI] [PubMed] [Google Scholar]
- 14.Hof PR, Knabe R, Bovier P, et al. Neuropathological observations in a case of autism presenting with self-injury behavior. Acta Neuropathol. 1991;82:321–26. doi: 10.1007/BF00308819. [DOI] [PubMed] [Google Scholar]
- 15.Jordan BD, Kanik AB, Horwich MS, et al. Apolipoprotein E epsilon 4 and fatal cerebral amyloid angiopathy associated with dementia pugilistica. Ann Neurol. 1995;38:698–99. doi: 10.1002/ana.410380429. [DOI] [PubMed] [Google Scholar]
- 16.Omalu BI, DeKosky ST, Hamilton RL, et al. Chronic traumatic encephalopathy in a national football league player: part II. Neurosurgery. 2006;59:1086–92. doi: 10.1227/01.NEU.0000245601.69451.27. [DOI] [PubMed] [Google Scholar]
- 17.Omalu BI, DeKosky ST, Minster RL, et al. Chronic traumatic encephalopathy in a National Football League player. Neurosurgery. 2005;57:128–34. doi: 10.1227/01.neu.0000163407.92769.ed. [DOI] [PubMed] [Google Scholar]
- 18.Roberts GW, Whitwell HL, Acland PR, et al. Dementia in a punch-drunk wife. Lancet. 1990;335:918–19. doi: 10.1016/0140-6736(90)90520-f. [DOI] [PubMed] [Google Scholar]
- 19.Williams DJ, Tannenberg AE. Dementia pugilistica in an alcoholic achondroplastic dwarf. Pathology. 1996;28:102–4. doi: 10.1080/00313029600169653. [DOI] [PubMed] [Google Scholar]
- 20.Cajigal S. Brain damage may have contributed to former wrestler’s violent demise. Neurology Today. 2007;7:1–16. [Google Scholar]
- 21.Schwarz A. [Accessed January 26, 2008];Expert ties ex-player’s suicide to brain damage [ New York Times Web site] 2007 January 18; Available at: http://www.nytimes.com/2007/01/18/sports/football/18waters.html.
- 22.Aotsuka A, Kojima S, Furumoto H, et al. Punch drunk syndrome due to repeated karate kicks and punches. Rinsho Shinkeigaku. 1990;30:1243–46. [PubMed] [Google Scholar]
- 23.Matser JT, Kessels AG, Jordan BD, et al. Chronic traumatic brain injury in professional soccer players. Neurology. 1998;51:791–96. doi: 10.1212/wnl.51.3.791. [DOI] [PubMed] [Google Scholar]
- 24.McCrory P, Turner M, Murray J. A punch drunk jockey? Br J Sports Med. 2004;38:e3. doi: 10.1136/bjsm.2003.006338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tysvaer AT, Storli OV, Bachen NI. Soccer injuries to the brain. A neurologic and electroencephalographic study of former players. Acta Neurol Scand. 1989;80:151–56. doi: 10.1111/j.1600-0404.1989.tb03858.x. [DOI] [PubMed] [Google Scholar]
- 26.Martland HS. Punch drunk. JAMA. 1928;91:1103–7. [Google Scholar]
- 27.Millspaugh JA. Dementia pugilistica. US Naval Medicine Bulletin. 1937;35:297–303. [Google Scholar]
- 28.Courville CB. Punch drunk. Its pathogenesis and pathology on the basis of a verified case. Bull Los Angel Neuro Soc. 1962;27:160–68. [PubMed] [Google Scholar]
- 29.Corsellis JA, Bruton CJ, Freeman-Browne D. The aftermath of boxing. Psychol Med. 1973;3:270–303. doi: 10.1017/s0033291700049588. [DOI] [PubMed] [Google Scholar]
- 30.Brandenburg W, Hallervorden J. Dementia pugilistica with anatomical findings. Virchows Arch. 1954;325:680–709. doi: 10.1007/BF00955101. [DOI] [PubMed] [Google Scholar]
- 31.Grahmann H, Ule G. Diagnosis of chronic cerebral symptoms in boxers (dementia pugilistica & traumatic encephalopathy of boxers) Psychiatr Neurol (Basel) 1957;134:261–83. [PubMed] [Google Scholar]
- 32.Neubuerger KT, Sinton DW, Denst J. Cerebral atrophy associated with boxing. AMA Arch Neurol Psychiatry. 1959;81:403–8. doi: 10.1001/archneurpsyc.1959.02340160001001. [DOI] [PubMed] [Google Scholar]
- 33.Mawdsley C, Ferguson FR. Neurological Disease in Boxers. Lancet. 1963;2:799–801. doi: 10.1016/s0140-6736(63)90498-7. [DOI] [PubMed] [Google Scholar]
- 34.Constantinidis J, Tissot R. Generalized Alzheimer’s neurofibrillary lesions without senile plaques. (Presentation of one anatomo-clinical case) Schweiz Arch Neurol Neurochir Psychiatr. 1967;100:117–30. [PubMed] [Google Scholar]
- 35.Payne EE. Brains of boxers. Neurochirurgia (Stuttg) 1968;11:173–88. doi: 10.1055/s-0028-1095326. [DOI] [PubMed] [Google Scholar]
- 36.Hof PR, Delacourte A, Bouras C. Distribution of cortical neurofibrillary tangles in progressive supranuclear palsy: a quantitative analysis of six cases. Acta Neuropathol. 1992;84:45–51. doi: 10.1007/BF00427214. [DOI] [PubMed] [Google Scholar]
- 37.Geddes JF, Vowles GH, Robinson SF, et al. Neurofibrillary tangles, but not Alzheimer-type pathology, in a young boxer. Neuropathol Appl Neurobiol. 1996;22:12–16. [PubMed] [Google Scholar]
- 38.Newell KL, Drachman DA. Case records of the Massachusetts General Hospital. Weekly clinicopathological exercises. Case 12–1999. A 67-year-old man with three years of dementia. N Engl J Med. 1999;340:1269–77. doi: 10.1056/NEJM199904223401609. [DOI] [PubMed] [Google Scholar]
- 39.Schmidt ML, Zhukareva V, Newell KL, et al. Tau isoform profile and phosphorylation state in dementia pugilistica recapitulate Alzheimer’s disease. Acta Neuropathol. 2001;101:518–24. doi: 10.1007/s004010000330. [DOI] [PubMed] [Google Scholar]
- 40.Schwarz A. [Accessed March 11, 2009];Lineman, dead at 36, exposes brain injuries [New York Times Web site] 2007 June 15; Available at: http://www.nytimes.com/2007/06/15/sports/football/15brain.html.
- 41.Areza-Fegyveres R, Rosemberg S, Castro RM, et al. Dementia pugilistica with clinical features of Alzheimer’s disease. Arq Neuropsiquiatr. 2007;65(3B):830–33. doi: 10.1590/s0004-282x2007000500019. [DOI] [PubMed] [Google Scholar]
- 42.Critchley M. Medical aspects of boxing, particularly from a neurological standpoint. Br Med J. 1957;1:357–62. doi: 10.1136/bmj.1.5015.357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.McNair DM, Kahn RJ. Self-assessment of cognitive deficits. In: Crook T, Ferris S, Bartus R, editors. Assessment in geriatric psychopharmacology. New Canaan, CT: Mark Powley; 1984. [Google Scholar]
- 44.Spitznagel MB, Tremont G. Cognitive reserve and anosognosia in questionable and mild dementia. Arch Clin Neuropsychol. 2005;20:505–15. doi: 10.1016/j.acn.2004.11.003. [DOI] [PubMed] [Google Scholar]
- 45.Galvin JE, Roe CM, Powlishta KK, et al. The AD8: A brief informant interview to detect dementia. Neurology. 2005;65:559–64. doi: 10.1212/01.wnl.0000172958.95282.2a. [DOI] [PubMed] [Google Scholar]
- 46.Pfeffer RI, Kurosaki TT, Harrah CH, Jr, et al. Measurement of functional activities in older adults in the community. J Gerontol. 1982;37:323–29. doi: 10.1093/geronj/37.3.323. [DOI] [PubMed] [Google Scholar]
- 47.Hof PR, Bouras C, Buee L, et al. Differential distribution of neurofibrillary tangles in the cerebral cortex of dementia pugilistica and Alzheimer’s disease cases. Acta Neuropathol. 1992;85:23–30. doi: 10.1007/BF00304630. [DOI] [PubMed] [Google Scholar]
- 48.Feany MB, Mattiace LA, Dickson DW. Neuropathologic overlap of progressive supranuclear palsy, Pick’s disease and corticobasal degeneration. J Neuropathol Exp Neurol. 1996;55:53–67. doi: 10.1097/00005072-199601000-00006. [DOI] [PubMed] [Google Scholar]
- 49.Litvan I, Hauw JJ, Bartko JJ, et al. Validity and reliability of the preliminary NINDS neuropathologic criteria for progressive supranuclear palsy and related disorders. J Neuropathol Exp Neurol. 1996;55:97–105. doi: 10.1097/00005072-199601000-00010. [DOI] [PubMed] [Google Scholar]
- 50.Perl DP, Hof PR, Purohit DP, et al. Hippocampal and entorhinal cortex neurofibrillary tangle formation in Guamanian Chamorros free of overt neurologic dysfunction. J Neuropathol Exp Neurol. 2003;62:381–88. doi: 10.1093/jnen/62.4.381. [DOI] [PubMed] [Google Scholar]
- 51.Hirano A. Amyotrophic lateral sclerosis and parkinsonism-dementia complex on Guam: Immunohistochemical studies. Keio J Med. 1992;41:6–9. doi: 10.2302/kjm.41.6. [DOI] [PubMed] [Google Scholar]
- 52.Tokuda T, Ikeda S, Yanagisawa N, et al. Re-examination of ex-boxers’ brains using immunohistochemistry with antibodies to amyloid beta-protein and tau protein. Acta Neuropathol. 1991;82:280–85. doi: 10.1007/BF00308813. [DOI] [PubMed] [Google Scholar]
- 53.Uryu K, Chen XH, Martinez D, et al. Multiple proteins implicated in neurodegenerative diseases accumulate in axons after brain trauma in humans. Exp Neurol. 2007;208:185–92. doi: 10.1016/j.expneurol.2007.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Eggers AE. Redrawing Papez’ circuit: A theory about how acute stress becomes chronic and causes disease. Med Hypotheses. 2007;69:852–57. doi: 10.1016/j.mehy.2007.01.074. [DOI] [PubMed] [Google Scholar]
- 55.Papez JW. A proposed mechanism of emotion. 1937. J Neuropsychiatry Clin Neurosci. 1995;7:103–12. doi: 10.1176/jnp.7.1.103. [DOI] [PubMed] [Google Scholar]
- 56.Bird CM, Burgess N. The hippocampus and memory: Insights from spatial processing. Nat Rev Neurosci. 2008;9:182–94. doi: 10.1038/nrn2335. [DOI] [PubMed] [Google Scholar]
- 57.Gaetz M. The neurophysiology of brain injury. Clin Neurophysiol. 2004;115:4–18. doi: 10.1016/s1388-2457(03)00258-x. [DOI] [PubMed] [Google Scholar]
- 58.Holbourn AHS. Mechanics of head injury. Lancet. 1943;2:438–41. [Google Scholar]
- 59.Ommaya AK, Gennarelli TA. Cerebral concussion and traumatic unconsciousness. Correlation of experimental and clinical observations of blunt head injuries. Brain. 1974;97:633–54. doi: 10.1093/brain/97.1.633. [DOI] [PubMed] [Google Scholar]
- 60.Buee L, Hof PR, Bouras C, et al. Pathological alterations of the cerebral microvasculature in Alzheimer’s disease and related dementing disorders. Acta Neuropathol. 1994;87:469–80. doi: 10.1007/BF00294173. [DOI] [PubMed] [Google Scholar]
- 61.Bouras C, Giannakopoulos P, Good PF, et al. A laser microprobe mass analysis of brain aluminum and iron in dementia pugilistica: Comparison with Alzheimer’s disease. Eur Neurol. 1997;38:53–58. doi: 10.1159/000112903. [DOI] [PubMed] [Google Scholar]
- 62.Graham DI, McIntosh TK, Maxwell WL, et al. Recent advances in neurotrauma. J Neuropathol Exp Neurol. 2000;59:641–51. doi: 10.1093/jnen/59.8.641. [DOI] [PubMed] [Google Scholar]
- 63.Strich SJ. Diffuse degeneration of the cerebral white matter in severe dementia following head injury. J Neurol Neurosurg Psychiatry. 1956;19:163–85. doi: 10.1136/jnnp.19.3.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Maxwell WL, McCreath BJ, Graham DI, et al. Cytochemical evidence for redistribution of membrane pump calcium-ATPase and ecto-Ca-ATPase activity, and calcium influx in myelinated nerve fibres of the optic nerve after stretch injury. J Neurocytol. 1995;24:925–42. doi: 10.1007/BF01215643. [DOI] [PubMed] [Google Scholar]
- 65.Giza CC, Hovda DA. The neurometabolic cascade of concussion. J Athl Train. 2001;36:228–35. [PMC free article] [PubMed] [Google Scholar]
- 66.Hovda DA, Lee SM, Smith ML, et al. The neurochemical and metabolic cascade following brain injury: moving from animal models to man. J Neurotrauma. 1995;12:903–6. doi: 10.1089/neu.1995.12.903. [DOI] [PubMed] [Google Scholar]
- 67.McKenzie KJ, McLellan DR, Gentleman SM, et al. Is beta-APP a marker of axonal damage in short-surviving head injury? Acta Neuropathol. 1996;92:608–13. doi: 10.1007/s004010050568. [DOI] [PubMed] [Google Scholar]
- 68.Blumbergs PC, Scott G, Manavis J, et al. Staining of amyloid precursor protein to study axonal damage9 in mild head injury. Lancet. 1994;344:1055–56. doi: 10.1016/s0140-6736(94)91712-4. [DOI] [PubMed] [Google Scholar]
- 69.Gentleman SM, Nash MJ, Sweeting CJ, et al. Beta-amyloid precursor protein (beta APP) as a marker for axonal injury after head injury. Neurosci Lett. 1993;160:139–44. doi: 10.1016/0304-3940(93)90398-5. [DOI] [PubMed] [Google Scholar]
- 70.Graham DI, Gentleman SM, Lynch A, et al. Distribution of beta-amyloid protein in the brain following severe head injury. Neuropathol Appl Neurobiol. 1995;21:27–34. doi: 10.1111/j.1365-2990.1995.tb01025.x. [DOI] [PubMed] [Google Scholar]
- 71.Masumura M, Hata R, Uramoto H, et al. Altered expression of amyloid precursors proteins after traumatic brain injury in rats: in situ hybridization and immunohistochemical study. J Neurotrauma. 2000;17:123–34. doi: 10.1089/neu.2000.17.123. [DOI] [PubMed] [Google Scholar]
- 72.McKenzie JE, Gentleman SM, Roberts GW, et al. Increased numbers of beta APP-immunoreactive neurones in the entorhinal cortex after head injury. Neuroreport. 1994;6:161–64. doi: 10.1097/00001756-199412300-00041. [DOI] [PubMed] [Google Scholar]
- 73.Murakami N, Yamaki T, Iwamoto Y, et al. Experimental brain injury induces expression of amyloid precursor protein, which may be related to neuronal loss in the hippocampus. J Neurotrauma. 1998;15:993–1003. doi: 10.1089/neu.1998.15.993. [DOI] [PubMed] [Google Scholar]
- 74.Pierce JE, Trojanowski JQ, Graham DI, et al. Immunohistochemical characterization of alterations in the distribution of amyloid precursor proteins and beta-amyloid peptide after experimental brain injury in the rat. J Neurosci. 1996;16:1083–90. doi: 10.1523/JNEUROSCI.16-03-01083.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Roberts GW, Gentleman SM, Lynch A, et al. βA4 amyloid protein deposition in brain after head trauma. Lancet. 1991;338:1422–23. doi: 10.1016/0140-6736(91)92724-g. [DOI] [PubMed] [Google Scholar]
- 76.Roberts GW, Gentleman SM, Lynch A, et al. Beta amyloid protein deposition in the brain after severe head injury: Implications for the pathogenesis of Alzheimer’s disease. J Neurol Neurosurg Psychiatry. 1994;57:419–25. doi: 10.1136/jnnp.57.4.419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Smith DH, Chen XH, Nonaka M, et al. Accumulation of amyloid beta and tau and the formation of neurofilament inclusions following diffuse brain injury in the pig. J Neuropathol Exp Neurol. 1999;58:982–92. doi: 10.1097/00005072-199909000-00008. [DOI] [PubMed] [Google Scholar]
- 78.Uryu K, Laurer H, McIntosh T, et al. Repetitive mild brain trauma accelerates Abeta deposition, lipid peroxidation, and cognitive impairment in a transgenic mouse model of Alzheimer amyloidosis. J Neurosci. 2002;22:446–54. doi: 10.1523/JNEUROSCI.22-02-00446.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Smith DH, Nakamura M, McIntosh TK, et al. Brain trauma induces massive hippocampal neuron death linked to a surge in beta-amyloid levels in mice overexpressing mutant amyloid precursor protein. Am J Pathol. 1998;153:1005–10. doi: 10.1016/s0002-9440(10)65643-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ikonomovic MD, Uryu K, Abrahamson EE, et al. Alzheimer’s pathology in human temporal cortex surgically excised after severe brain injury. Exp Neurol. 2004;190:192–203. doi: 10.1016/j.expneurol.2004.06.011. [DOI] [PubMed] [Google Scholar]
- 81.DeKosky ST, Abrahamson EE, Ciallella JR, et al. Association of increased cortical soluble abeta42 levels with diffuse plaques after severe brain injury in humans. Arch Neurol. 2007;64:541–44. doi: 10.1001/archneur.64.4.541. [DOI] [PubMed] [Google Scholar]
- 82.Graham DI, Gentleman SM, Nicoll JA, et al. Altered beta-APP metabolism after head injury and its relationship to the aetiology of Alzheimer’s disease. Acta Neurochir Suppl. 1996;66:96–102. doi: 10.1007/978-3-7091-9465-2_17. [DOI] [PubMed] [Google Scholar]
- 83.Gentleman SM, Greenberg BD, Savage MJ, et al. A beta 42 is the predominant form of amyloid beta-protein in the brains of short-term survivors of head injury. Neuroreport. 1997;8:1519–22. doi: 10.1097/00001756-199704140-00039. [DOI] [PubMed] [Google Scholar]
- 84.Brody DL, Magnoni S, Schwetye KE, et al. Amyloid-beta dynamics correlate with neurological status in the injured human brain. Science. 2008;321:1221–24. doi: 10.1126/science.1161591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Geddes DM, LaPlaca MC, Cargill RS., 2nd Susceptibility of hippocampal neurons to mechanically induced injury. Exp Neurol. 2003;184:420–27. doi: 10.1016/s0014-4886(03)00254-1. [DOI] [PubMed] [Google Scholar]
- 86.Colicos MA, Dixon CE, Dash PK. Delayed, selective neuronal death following experimental cortical impact injury in rats: Possible role in memory deficits. Brain Res. 1996;739:111–19. doi: 10.1016/s0006-8993(96)00819-0. [DOI] [PubMed] [Google Scholar]
- 87.Bramlett HM, Kraydieh S, Green EJ, et al. Temporal and regional patterns of axonal damage following traumatic brain injury: A beta-amyloid precursor protein immunocytochemical study in rats. J Neuropathol Exp Neurol. 1997;56:1132–41. doi: 10.1097/00005072-199710000-00007. [DOI] [PubMed] [Google Scholar]
- 88.Dixon CE, Kochanek PM, Yan HQ, et al. One-year study of spatial memory performance, brain morphology, and cholinergic markers after moderate controlled cortical impact in rats. J Neurotrauma. 1999;16:109–22. doi: 10.1089/neu.1999.16.109. [DOI] [PubMed] [Google Scholar]
- 89.Smith DH, Chen XH, Pierce JE, et al. Progressive atrophy and neuron death for one year following brain trauma in the rat. J Neurotrauma. 1997;14:715–27. doi: 10.1089/neu.1997.14.715. [DOI] [PubMed] [Google Scholar]
- 90.Maxwell WL, Domleo A, McColl G, et al. Post-acute alterations in the axonal cytoskeleton after traumatic axonal injury. J Neurotrauma. 2003;20:151–68. doi: 10.1089/08977150360547071. [DOI] [PubMed] [Google Scholar]
- 91.Maxwell WL, MacKinnon MA, Smith DH, et al. Thalamic nuclei after human blunt head injury. J Neuropathol Exp Neurol. 2006;65:478–88. doi: 10.1097/01.jnen.0000229241.28619.75. [DOI] [PubMed] [Google Scholar]
- 92.Kay AD, Day SP, Kerr M, et al. Remodeling of cerebrospinal fluid lipoprotein particles after human traumatic brain injury. J Neurotrauma. 2003;20:717–23. doi: 10.1089/089771503767869953. [DOI] [PubMed] [Google Scholar]
- 93.Hughes DG, Jackson A, Mason DL, et al. Abnormalities on magnetic resonance imaging seen acutely following mild traumatic brain injury: Correlation with neuropsychological tests and delayed recovery. Neuroradiology. 2004;46:550–58. doi: 10.1007/s00234-004-1227-x. [DOI] [PubMed] [Google Scholar]
- 94.Kraus MF, Susmaras T, Caughlin BP, et al. White matter integrity and cognition in chronic traumatic brain injury: a diffusion tensor imaging study. Brain. 2007;130:2508–19. doi: 10.1093/brain/awm216. [DOI] [PubMed] [Google Scholar]
- 95.Salmond CH, Menon DK, Chatfield DA, et al. Diffusion tensor imaging in chronic head injury survivors: correlations with learning and memory indices. Neuroimage. 2006;29:117–24. doi: 10.1016/j.neuroimage.2005.07.012. [DOI] [PubMed] [Google Scholar]
- 96.Chappell MH, Ulug AM, Zhang L, et al. Distribution of microstructural damage in the brains of professional boxers: A diffusion MRI study. J Magn Reson Imaging. 2006;24:537–42. doi: 10.1002/jmri.20656. [DOI] [PubMed] [Google Scholar]
- 97.Zhang L, Ravdin LD, Relkin N, et al. Increased diffusion in the brain of professional boxers: a preclinical sign of traumatic brain injury? AJNR Am J Neuroradiol. 2003;24:52–57. [PMC free article] [PubMed] [Google Scholar]
- 98.Ariza M, Pueyo R, Matarin Mdel M, et al. Influence of APOE polymorphism on cognitive and behavioural outcome in moderate and severe traumatic brain injury. J Neurol Neurosurg Psychiatry. 2006;77:1191–93. doi: 10.1136/jnnp.2005.085167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Chiang MF, Chang JG, Hu CJ. Association between apolipoprotein E genotype and outcome of traumatic brain injury. Acta Neurochir (Wien) 2003;145:649–53. doi: 10.1007/s00701-003-0069-3. [DOI] [PubMed] [Google Scholar]
- 100.Friedman G, Froom P, Sazbon L, et al. Apolipoprotein E-epsilon4 genotype predicts a poor outcome in survivors of traumatic brain injury. Neurology. 1999;52:244–48. doi: 10.1212/wnl.52.2.244. [DOI] [PubMed] [Google Scholar]
- 101.Nicoll JA, Roberts GW, Graham DI. Apolipoprotein E epsilon 4 allele is associated with deposition of amyloid beta-protein following head injury. Nat Med. 1995;1:135–37. doi: 10.1038/nm0295-135. [DOI] [PubMed] [Google Scholar]
- 102.Nicoll JA, Roberts GW, Graham DI. Amyloid beta-protein, APOE genotype and head injury. Ann N Y Acad Sci. 1996;777:271–75. doi: 10.1111/j.1749-6632.1996.tb34431.x. [DOI] [PubMed] [Google Scholar]
- 103.Hartman RE, Laurer H, Longhi L, et al. Apolipoprotein E4 influences amyloid deposition but not cell loss after traumatic brain injury in a mouse model of Alzheimer’s disease. J Neurosci. 2002;22:10083–87. doi: 10.1523/JNEUROSCI.22-23-10083.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Cantu RC. Recurrent athletic head injury: Risks and when to retire. Clin Sports Med. 2003;22:593–603. doi: 10.1016/s0278-5919(02)00095-9. [DOI] [PubMed] [Google Scholar]
- 105.Gosselin N, Theriault M, Leclerc S, et al. Neurophysiological anomalies in symptomatic and asymptomatic concussed athletes. Neurosurgery. 2006;58:1151–61. doi: 10.1227/01.NEU.0000215953.44097.FA. Discussion 1151–61. [DOI] [PubMed] [Google Scholar]
- 106.Mayers L. Return-to-play criteria after athletic concussion: A need for revision. Arch Neurol. 2008;65:1158–61. doi: 10.1001/archneur.65.9.1158. [DOI] [PubMed] [Google Scholar]
- 107.Arfanakis K, Haughton VM, Carew JD, et al. Diffusion tensor MR imaging in diffuse axonal injury. AJNR Am J Neuroradiol. 2002;23:794–802. [PMC free article] [PubMed] [Google Scholar]
- 108.Bergsneider M, Hovda DA, Lee SM, et al. Dissociation of cerebral glucose metabolism and level of consciousness during the period of metabolic depression following human traumatic brain injury. J Neurotrauma. 2000;17:389–401. doi: 10.1089/neu.2000.17.389. [DOI] [PubMed] [Google Scholar]
- 109.De Beaumont L, Brisson B, Lassonde M, et al. Long-term electrophysiological changes in athletes with a history of multiple concussions. Brain Inj. 2007;21:631–44. doi: 10.1080/02699050701426931. [DOI] [PubMed] [Google Scholar]
- 110.Guskiewicz KM, McCrea M, Marshall SW, et al. Cumulative effects associated with recurrent concussion in collegiate football players: The NCAA Concussion Study. JAMA. 2003;290:2549–55. doi: 10.1001/jama.290.19.2549. [DOI] [PubMed] [Google Scholar]
- 111.Kozlowski DA, James DC, Schallert T. Use-dependent exaggeration of neuronal injury after unilateral sensorimotor cortex lesions. J Neurosci. 1996;16:4776–86. doi: 10.1523/JNEUROSCI.16-15-04776.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Corsellis JA, Brierley JB. Observations on the pathology of insidious dementia following head injury. J Ment Sci. 1959;105:714–20. doi: 10.1192/bjp.105.440.714. [DOI] [PubMed] [Google Scholar]
- 113.Rudelli R, Strom JO, Welch PT, et al. Posttraumatic premature Alzheimer’s disease. Neuropathologic findings and pathogenetic considerations. Arch Neurol. 1982;39:570–75. doi: 10.1001/archneur.1982.00510210040009. [DOI] [PubMed] [Google Scholar]
- 114.Klein RL, Lin WL, Dickson DW, et al. Rapid neurofibrillary tangle formation after localized gene transfer of mutated tau. Am J Pathol. 2004;164:347–53. doi: 10.1016/S0002-9440(10)63124-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Weaver CL, Espinoza M, Kress Y, et al. Conformational change as one of the earliest alterations of tau in Alzheimer’s disease. Neurobiol Aging. 2000;21:719–27. doi: 10.1016/s0197-4580(00)00157-3. [DOI] [PubMed] [Google Scholar]
- 116.Su JH, Cummings BJ, Cotman CW. Early phosphorylation of tau in Alzheimer’s disease occurs at Ser-202 and is preferentially located within neurites. Neuroreport. 1994;5:2358–62. doi: 10.1097/00001756-199411000-00037. [DOI] [PubMed] [Google Scholar]
- 117.Lewis J, McGowan E, Rockwood J, et al. Neurofibrillary tangles, amyotrophy and progressive motor disturbance in mice expressing mutant (P301L) tau protein. Nat Genet. 2000;25:402–5. doi: 10.1038/78078. [DOI] [PubMed] [Google Scholar]
- 118.Lewis J, Dickson DW, Lin WL, et al. Enhanced neurofibrillary degeneration in transgenic mice expressing mutant tau and APP. Science. 2001;293:1487–91. doi: 10.1126/science.1058189. [DOI] [PubMed] [Google Scholar]