

Abstract
Synaptic efficacy requires that presynaptic and postsynaptic specializations align precisely and mature coordinately. The underlying mechanisms are poorly understood, however. We propose that adenomatous polyposis coli protein (APC) is a key coordinator of presynaptic and postsynaptic maturation. APC organizes a multiprotein complex that directs nicotinic acetylcholine receptor (nAChR) localization at postsynaptic sites in avian ciliary ganglion neurons in vivo. We hypothesize that the APC complex also provides retrograde signals that direct presynaptic active zones to develop in register with postsynaptic nAChR clusters. In our model, the APC complex provides retrograde signals via postsynaptic neuroligin that interacts extracellularly with presynaptic neurexin. S-SCAM (synaptic cell adhesion molecule) and PSD-93 (postsynaptic density-93) are scaffold proteins that bind to neuroligin. We identify S-SCAM as a novel component of neuronal nicotinic synapses. We show that S-SCAM, PSD-93, neuroligin and neurexin are enriched at α3*-nAChR synapses. PSD-93 and S-SCAM bind to APC and its binding partner β-catenin, respectively. Blockade of selected APC and β-catenin interactions, in vivo, leads to decreased postsynaptic accumulation of S-SCAM, but not PSD-93. Importantly, neuroligin synaptic clusters are also decreased. On the presynaptic side, there are decreases in neurexin and active zone proteins. Further, presynaptic terminals are less mature structurally and functionally. We define a novel neural role for APC by showing that the postsynaptic APC multiprotein complex is required for anchoring neuroligin and neurexin at neuronal synapses in vivo. APC human gene mutations correlate with autism spectrum disorders, providing strong support for the importance of the association, demonstrated here, between APC, neuroligin and neurexin.
Introduction
Nicotinic synapses are essential for normal cognitive and autonomic functions. However, little is known about mechanisms that direct their assembly. Further, proper synaptic function requires that presynaptic and postsynaptic specializations align precisely and mature coordinately, but the underlying mechanisms are poorly understood. Our studies are defining these molecular mechanisms in vivo.
We have identified the only protein known to function in localizing nicotinic acetylcholine receptors (nAChRs) at neuronal synapses. The key synapse organizer is adenomatous polyposis coli protein (APC). APC organizes a multimolecular complex that stabilizes the local cytoskeleton and links α3*-nAChRs to APC at postsynaptic sites (Rosenberg et al., 2008).
We predict that APC coordinates presynaptic and postsynaptic maturation and thereby ensures synaptic efficacy. We speculate that the APC multiprotein complex provides retrograde signals that direct presynaptic active zones to form opposite postsynaptic nAChR clusters. In our model, the APC complex contains scaffold proteins that bind to the transmembrane adhesion molecule neuroligin (NL). Postsynaptic NL signals retrogradely via extracellular interactions with presynaptic neurexin (Nrx) that, in turn, directs functional active zone assembly (Dean et al., 2003; Missler et al., 2003).
NL and Nrx are sufficient to induce functional presynaptic and postsynaptic specializations in cultured neurons (Scheiffele et al., 2000; Craig and Kang, 2007; Ko et al., 2009). NL and Nrx are essential, in vivo, for functional maturation of synapses (Missler et al., 2003; Varoqueaux et al., 2006). The identity of proteins that position NL at synapses is controversial. The scaffold proteins PSD-95 (postsynaptic density-95), PSD-93, and synaptic cell adhesion molecule (S-SCAM) directly bind to NL (Kim and Sheng, 2004). Reports suggest that these scaffold proteins have unique, redundant or no role in positioning NL at glutamatergic synapses in vitro (Dresbach et al., 2004; Iida et al., 2004; Levinson et al., 2005).
We predict that scaffold proteins of the APC complex are required, in vivo, for localizing NL at synapses and coordinating presynaptic and postsynaptic maturation. To test our hypothesis, we employ experimentally amenable avian ciliary ganglion (CG) neurons. APC and its binding partners are enriched at CG nicotinic synapses (Temburni et al., 2004). APC binds to PSD-93 and β-catenin (β-cat). β-Catenin binds to and recruits S-SCAM to glutamatergic synapses in vitro (Nishimura et al., 2002). Here, we identify S-SCAM as a novel nicotinic synaptic component. We show that dominant-negative blockade of selected APC and β-catenin interactions leads to decreases in postsynaptic clusters of S-SCAM, but not PSD-93 or PSD-95. Importantly, we also find decreases in clusters of postsynaptic NL, presynaptic Nrx and active zone proteins, and in structural and functional maturation of presynaptic terminals. Our results demonstrate that the APC multiprotein complex is essential for anchoring NL and Nrx at synapses in vivo. The association of NL and Nrx mutations with autism (Südhof, 2008; Blundell et al., 2010) highlights the importance of defining molecules that direct NL and Nrx synaptic localization. Similarly, APC mutations correlate with autism and mental retardation (Raedle et al., 2001; Zhou et al., 2007), underscoring the importance of our uncovering an association between APC, NL and Nrx.
Materials and Methods
Antibodies.
Primary antibodies used were as follows: anti-MAGI-2/S-SCAM (Sigma); anti-NL2 polyclonal antibody (sc-14087, Santa Cruz Biotechnology); anti-NL1 (clone 4F9), anti-piccolo and anti-RIM1,2 (Synaptic Systems); pan-Nrx antibody (generous gift from Dr. Peter Scheiffele, Biozentrum, University of Basel, Basel, Switzerland) (Dean et al., 2003), anti-PSD95 (clone K28/86.2, Millipore); anti-β-catenin (Invitrogen); mAb35 (Developmental Studies Hybridoma Bank, Iowa City, IA) to detect α3*-nAChRs; anti-APC (clone Ali12-28, Abcam); anti-SV2 for synaptic vesicles (Developmental Studies Hybridoma Bank); anti-dephospho S37-T41 β-catenin (clone 8E7, Millipore); and anti-HA (clone 3F10, Roche Diagnostics). Secondary reagents used were Alexa-Fluor-488, -555 and -594-conjugated secondary antibodies raised in rabbit, rat, mouse, chicken, goat and guinea pig (Invitrogen).
Chicken embryos.
White Leghorn embryonated chicken eggs, obtained from Charles River Spafas or University of Connecticut Poultry Farm (Storrs, CT), were maintained at 37°C in a forced air-draft humidified incubator until use. Embryos were staged according to the Hamburger and Hamilton classification scheme (Hamburger and Hamilton, 1951), the embryonic days of development refer to the stage (st) rather than the actual days of incubation. The developmental stages used include embryonic day (E) 11–14 (st 37–40).
Immunolabeling.
CGs from β-cat::S-SCAM-dn-infected or APC::EB1-dn-infected and age-matched uninfected control embryos were processed in parallel for frozen sectioning and immunolabeling as previously described (Olsen et al., 2007). For confocal microscopy, CGs were acutely dissociated and processed for immunolabeling as previously described (Williams et al., 1998; Rosenberg et al., 2008). Controls for specific binding in double-labeling studies included omitting the first or second primary antibody in separate tests; only background labeling was detected (data not shown).
NL2 polyclonal antibody (sc-14087, Santa Cruz Biotechnology) was used for immunolabeling NL1 in chicken CG neurons, based on the NL2 antibody being raised against an epitope that is completely identical between chicken NL1 and rat NL2 (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). Further, NL1 is expressed in the chicken CG, whereas the chicken NL2 sequence has not yet been identified based on chicken genome database searches (this study; Ross and Conroy, 2008). To verify specific binding of the NL2 antibody to NL1 in CG neurons, immunolabeling was used in combination with knockdown of endogenous NL1 using shRNA that targets nucleotides 417-437 of chicken NL1 RNA (NCBI accession number NP_001074971.1; kind gift from Dr. Darwin Berg, University of California, San Diego, San Diego, CA) (Conroy et al., 2007). Dissociated CG neurons in culture (see below) were transfected with the NL1 shRNA or control scrambled RNA (similar length and composition but different sequence) in pRNAT-U6.1/Neo (Genescript) that carries the GFP marker (Conroy et al., 2007). CG neurons were transfected at the time of plating using Effectene (Qiagen), as previously described (Conroy et al., 2007). The neurons were immunostained at 5 d in vitro.
Quantitative light microscopy.
Immunolabeled sections were examined with a Zeiss Axioscope microscope and photographed either with a SPOT color CCD camera and software (Diagnostic Instruments) or with a Q-Imaging Retiga 200R Fast 1394 black and white CCD camera and Nikon Instruments NIS Elements software. For each primary antibody, images were acquired using identical gain and exposure settings such that pixel intensities were below saturation levels.
To quantify changes in immunolabeling levels in β-cat::S-SCAM-dn or APC::EB1-dn-expressing neurons compared with uninfected control neurons, we assessed pixel intensity profiles along ∼3- to 5-μm-length segments of the labeled surface regions per neuron using either ImageJ (http://rsb.info.nih.gov/ij/) or NIS elements software. Individual neurons were selected for analysis based on the criterion that they showed brightest staining for each protein analyzed for each experimental and control condition. For each immunolabel, we analyzed pixel intensities for at least three different line segments per neuron (15–20 μm total length) and a total of 10–49 dominant negative-infected neurons and uninfected control neurons from 6 to 12 separate embryos. Pixel intensity distribution analyses: the pixel intensities of sampled surface segments were binned into incremental groups of 10 pixel intensity steps (e.g., 0–9, 10–19 etc., up to 255, saturation) for images acquired with SPOT camera (dynamic range 0–255) or 100 pixel intensity steps for images acquired with Q-imaging camera (dynamic range 0–4095). We calculated the percentage of pixels that belonged to each pixel intensity category and plotted the relative frequency distributions. For each protein, we also calculated the mean pixel intensity for each neuron analyzed and then determined the SEMs and Student's t test values with Microsoft Excel.
To quantify changes in the length of presynaptic terminals that contact dominant negative (dn)-expressing ciliary neurons versus age-matched control ciliary neurons, we measured the length of continuous SV2 immunolabeled segments on HA-APC::EB1-dn-positive and control neurons. We then calculated the mean lengths of continuous SV2 labeling, SEM and Student's t test values with Microsoft Excel.
Quantitative confocal microscopy.
Immunolabeled CG neurons were examined by confocal microscopy with a Leica TCS SP2 microscope using Kr (568 nm) and Ar (488 nm) laser sources, and a 63× 1.32 numerical aperture lens. Optical sections were acquired from the top to the bottom of each neuron at 0.8–1 μm step intervals. Laser intensity and photomultiplier tube gain were kept constant across experiments. Settings were chosen such that pixel intensities fell below saturation levels. In addition, the wavelengths of light collected in each detection channel were set such that no detectable bleed-through occurred between the different channels. Pixel intensity profiles along ∼3 μm segments of labeled neuronal surface regions were assessed as described above using Leica imaging software.
Retroviral vector-mediated gene transfer.
APC::EB1-dn cDNA (amino acids 1943-2184; corresponding to the EB1-binding domain of chicken APC sequence NCBI accession number XP_001233411) was generated and coupled to the hemagglutinin (HA) tag (YPYDVPDYA) at its N terminus by PCR (Rosenberg et al., 2008). APC::EB1-dn specificity and efficacy, in vitro and in vivo, was previously verified (Rosenberg et al., 2008). β-cat::S-SCAM-dn cDNA corresponded to the C terminus PDZ binding motif of β-catenin that binds to S-SCAM (amino acids 664-781 in chicken β-catenin; NCBI accession number NP_990412.1). β-cat::S-SCAM-dn was generated and HA-tagged by PCR. β-cat::S-SCAM-dn was previously shown to selectively block β-catenin interactions with S-SCAM (Nishimura et al., 2002). The dominant-negative cDNA constructs were subcloned separately into the avian-specific retroviral vector RCASBP (B envelope subgroup type; (Homburger and Fekete, 1996). RCASBP containing GFP cDNA was a gift from Dr. Constance Cepko (Harvard Medical School, Boston, MA). Viral stocks were prepared in DF1 chicken fibroblast cells (American Type Culture Collection). CGs were infected in ovo at 36 h of development (st 8–9) and sampled 1–2 weeks later as previously described (Williams et al., 1998; Temburni et al., 2004).
Western analyses.
Standard immunoblot analyses and coimmunoprecipitations were performed using CG lysates as previously described (Temburni et al., 2004; Rosenberg et al., 2008).
FM1-43FX labeling of actively recycling synaptic vesicles.
For this assay, live CG neurons were freshly isolated with presynaptic terminals attached. The CGs were freshly dissected from E13.5 APC::EB1-dn-injected embryos versus age-matched uninjected control embryos and the CGs were partially dissociated by incubation in 1.0 mg/ml collagenase A (Roche Biochemicals) in dissociation media (DM, 150 mm NaCl, 3 mm KCl, 2 mm CaCl2, 1 mm MgCl2, 10 mm glucose, 10 mm HEPES, pH 7.4) for 10 min at 37°C. CGs were rinsed twice with DM supplemented with 10% horse serum (Invitrogen), switched into MEM (Invitrogen) supplemented with 10% horse serum and 3% embryonic chicken eye extract, and gently triturated using fire-polished Pasteur pipettes. Isolated cells were allowed to adhere to Silane-coated glass slides (Electron Microscopy Sciences) for 15 min at 37°C in a 5% CO2 incubator. The live CG neurons were then rinsed twice with DM and incubated with 1 μg/ml FM1-43FX (Invitrogen) in DM for 1 min. Vesicle recycling was stimulated by incubation in DM containing 90 mm KCl and 1 μg/ml FM1-43FX for 1 min. The neurons were washed extensively with DM to remove unbound FM1-43FX dye and then fixed with 2% paraformaldehyde in PBS for 15 min before imaging. FM1-43X dye labeling of synaptic vesicles was measured by quantifying the fluorescence pixel intensity along the neuronal surface.
LiCl treatment of CG neuron cultures.
Embryonic day 9 CGs were freshly dissected and the neurons were dissociated by gentle trituration in dissociation media (see above). The dissociated neurons were plated onto poly-l-lysine laminin-coated 35 mm dishes or glass coverslips (Fisher Scientific) in MEM supplemented with 10% Horse Serum, 3% eye extract, and penicillin/streptomycin in 5% CO2 humidified 37°C incubator as previously described (Temburni et al., 2004, Rosenberg et al., 2008). Half of the culture volume was replaced with fresh media every 2 d. After 3 d in culture, LiCl or NaCl (as control) were added to a final concentration of 20 mm and the neurons were allowed to grow for an additional 2 d before harvesting for immunoprecipitation or immunostaining. Treatment with 20 mm LiCl for 2 d has been shown to effectively inhibit GSK3β and GSK3β-mediated phosphorylation of β-catenin (Klein and Melton, 1996; Lucas et al., 1998; Hall et al., 2000).
Results
S-SCAM is a novel component of neuronal nicotinic synapses
To test our prediction that the postsynaptic APC complex provides retrograde signals required for presynaptic terminal maturation, we first determined whether the scaffold proteins that bind to NL: PSD-93, PSD-95 and S-SCAM, localize at nicotinic synapses on CG neurons in vivo. We previously identified PSD-93 and PSD-95 as nicotinic postsynaptic components in CG neurons (Temburni et al., 2004; Rosenberg et al., 2008). APC interacts predominantly with PSD-93a, the short isoform that lacks the SH3 and guanylate kinase domains (Conroy et al., 2003; Temburni et al., 2004). APC also binds to and colocalizes with β-catenin at CG nicotinic synapses, and β-catenin recruits S-SCAM to glutamatergic synapses in vitro (Nishimura et al., 2002; Temburni et al., 2004). We therefore tested for S-SCAM expression at CG nicotinic postsynaptic sites.
First, immunoblotting demonstrated that the S-SCAM antibody recognizes protein bands of the expected size in embryonic chicken CG lysates (Fig. 1a). Next, double-label immunofluorescence of CG frozen sections showed that S-SCAM colocalized (overlapped) with α3*-nAChR (Fig. 1b) and APC (Fig. 1c) surface clusters, as evidenced by the predominance of yellow fluorescence patches and covarying fluorescence intensity profiles (Fig. 1b,c, bottom). Similarly, S-SCAM immunostaining was juxtaposed to and partially overlapped with SV2 labeled presynaptic terminals on the neurons (Fig. 1d). Strong colocalization was further indicated by the Pearson's correlation coefficients (r) of 0.64 ± 0.04 for colocalization of S-SCAM with α3*-nAChRs, 0.80 ± 0.03 for S-SCAM with APC, and 0.74 ± 0.03 for S-SCAM with SV2 (r can vary from +1 to −1; measurements were taken along 15–20 μm lengths of labeled surface area per neuron; n = 12–20 different neurons). This is the first report, to our knowledge, that S-SCAM localizes at nicotinic synapses.
Figure 1.
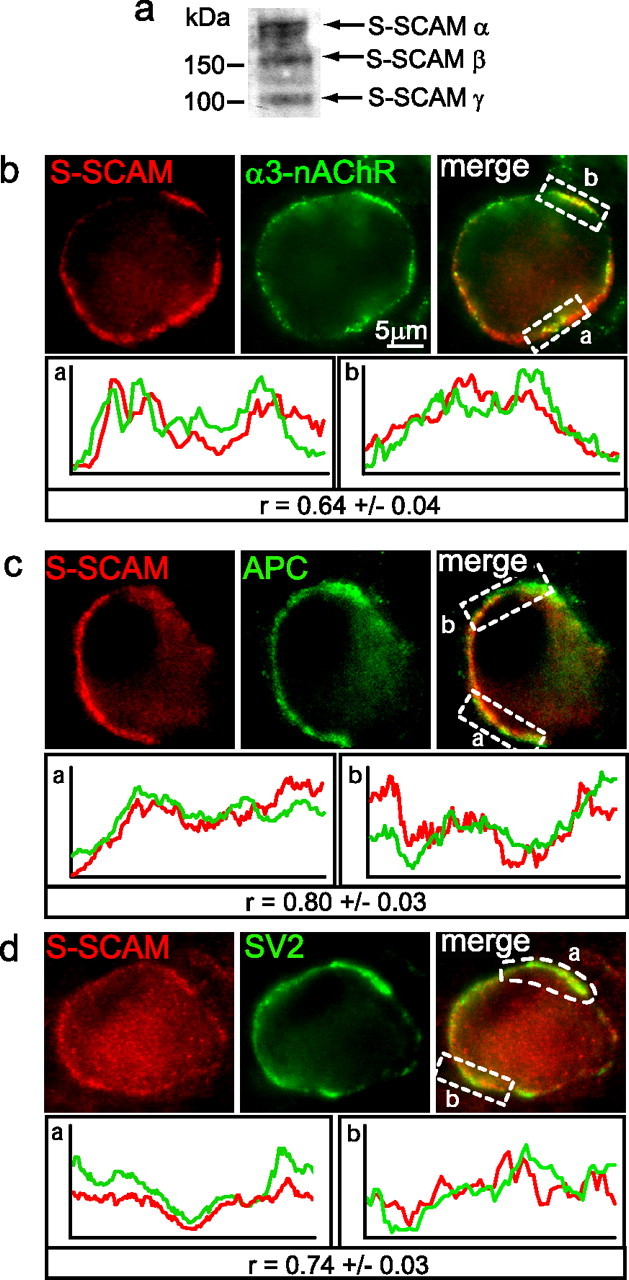
S-SCAM is enriched at nicotinic synapses on CG neurons in vivo. a, Total CG lysate proteins were separated by SDS-PAGE and immunoblotted for S-SCAM. Protein bands corresponding to expected molecular weights of S-SCAM isoforms were detected, including α (full-length), β, and γ (180, 160, and 105 kDa). b–d, Micrographs of immunofluorescence double-labeled E14 CG frozen sections. S-SCAM surface-associated clusters (red; b, c) are predominantly colocalized with α3*-nAChR (green; b) and APC (green; c) clusters (overlap, yellow). Additionally, S-SCAM clusters (red; d) are juxtaposed to and partially overlap with the presynaptic terminal stained for synaptic vesicle protein SV2 (green; d). Note that CG neurons lack dendrites and organize synapses on both somatic spines and the smooth region of the soma (see diagram of the synapse Fig. 8e). Bottom, Graphs show red and green fluorescence intensity profiles for the boxed regions. Staining intensities covaried and strongly correlated with each other for colocalization of S-SCAM with α3*-nAChRs, APC and SV2 [r, Pearson's correlation coefficient (mean ± SEM), can range from −1.0 to +1.0; n = 2–3 labeled surface areas per neuron, total of 15–20 μm in length, 12–20 neurons].
Neuroligin and neurexin are enriched at nicotinic synapses in vivo
The S-SCAM, PSD-93 and PSD-95 interacting protein, NL is also enriched at CG nicotinic synapses. In mammalian central neurons, NL1 is selectively enriched at excitatory synapses, whereas NL2 accumulates at inhibitory synapses (Chih et al., 2005; Chubykin et al., 2007; Kang et al., 2008; Patrizi et al., 2008; Dahlhaus et al., 2010). Immunoblotting and RT-PCR analyses suggested that NL1 is expressed in the avian CG (Fig. 2a) (Conroy et al., 2007; Ross and Conroy, 2008). NL2 was not analyzed; the chicken NL2 sequence has not yet been identified based on chicken genome database searches. Albeit useful for immunoblotting, the available NL1-specific antibodies were not effective for immunolabeling chicken CG neurons (data not shown; Conroy et al., 2007). Interestingly, amino acid sequence analysis of chicken NL1 indicated an identical region shared with mammalian NL2 within an alternatively spliced domain of the extracellular region (supplemental Fig. 1a, available at www.jneurosci.org as supplemental material). Based on the completely identical region in chicken NL1 and rat NL2, we used an NL2 polyclonal antibody raised against the shared epitope (Santa Cruz Biotechnology personal communication) for immunolocalization of NL1 in chicken CG neurons. While the NL2 antibody was not effective for immunoblotting (chicken CG, chicken brain or rodent brain lysates; data not shown and Santa Cruz Biotechnology personal communication), it did selectively immunolabel CG nicotinic synapses. NL1 clusters colocalized with α3*-nAChR surface clusters (yellow= overlap; Fig. 2b) and partially overlapped with SV2-stained presynaptic terminals (Fig. 2c). The fluorescence intensity profiles covaried (Fig. 2b,c, bottom) and were strongly correlated [r = 0.78 ± 0.03 (n = 16 neurons) and 0.62 ± 0.03 (n = 18 neurons) for NL1 with α3*-nAChRs and SV2, respectively]. To verify that the immunostaining represented specific binding of the NL2 antibody to chicken NL1, endogenous NL1 levels were knocked down in cultured CG neurons using shRNA that targets nucleotides 417-437 of chicken NL1 RNA (Conroy et al., 2007). Bicistronic GFP was used as a marker of transfected cells. NL2 antibody staining was obviously decreased in CG neurons transfected with NL1 shRNA, compared with the staining in nontransfected neurons within the dish, as an internal control, and neurons transfected with scrambled RNA, as a negative control (supplemental Fig. 1b–d, available at www.jneurosci.org as supplemental material). All together, the data show that NL1 is enriched at embryonic chicken nicotinic synapses in vivo.
Figure 2.

Neuroligin and neurexin are enriched at CG nicotinic synapses in vivo. a, d, Immunoblot analyses of lysates from chicken CG and brain, for comparison, show protein bands of the expected sizes for neuroligin (NL) (∼110 kDa) and neurexin (Nrx) (∼160 kDa for α-Nrx and ∼75 kDa for β-Nrx). b, c, e, f, Micrographs of immunofluorescence double-labeled E12–E14 CG frozen sections show that NL surface clusters (red; b, c) colocalize with α3*-nAChRs clusters (green; b) and partially overlap with the SV2-positive presynaptic terminal (green; c). Similarly, Nrx surface clusters (red; e, f) partially overlap with postsynaptic α3*-nAChR clusters (green; e) and the presynaptic terminal stained for SV2 (green; f). Bottom, Red and green fluorescence intensity profiles for boxed regions; r, correlation coefficients for colocalization (n = 15–18 neurons).
Nrx, the presynaptic binding partner of postsynaptic NL1, is also concentrated at this synapse. Immunoblotting showed that the pan-Nrx antibody recognizes protein bands of the expected sizes for α- and β-Nrx in chicken CG lysates (Fig. 2d). Nrx immunolabeling partially overlapped with SV2-stained presynaptic terminals (Fig. 2f) and postsynaptic α3*nAChR surface clusters (Fig. 2e). The intensity profiles covaried and correlated with each other [Fig. 2e,f, bottom; r = 0.77 ± 0.05 (n = 15 neurons) and 0.62 ± 0.03 (n = 18 neurons) for Nrx with SV2 and α3*-nAChRs, respectively].
β-Catenin interactions are required for targeting S-SCAM to nicotinic postsynaptic sites
To test for a role of the APC multiprotein complex in localizing NL at nicotinic synapses, we developed dominant-negative peptides that block selected interactions of endogenous APC complex components. Our goal was to decrease synaptic levels of the NL binding scaffold proteins within the APC complex. The APC binding partner β-cat interacts with S-SCAM, and this interaction is essential for S-SCAM localizing at glutamatergic synapses in vitro (Nishimura et al., 2002). We tested whether β-cat's role in recruiting S-SCAM is shared at nicotinic synapses in vivo. First, we established that S-SCAM coprecipitates with β-cat and NL from CG lysates, suggesting that these proteins function in a complex at nicotinic synapses in vivo (Fig. 3). Next, we used the dn peptide previously shown to selectively and effectively block β-cat interactions with S-SCAM (Nishimura et al., 2002; Iida et al., 2004). This dn caused specific decreases in S-SCAM levels at glutamatergic synapses in cultured hippocampal neurons (Nishimura et al., 2002). We overexpressed the dn in CG neurons in vivo to block endogenous β-cat and S-SCAM interactions during nicotinic synapse formation. The blocking peptide, referred to here as β-cat::S-SCAM-dn, consisted of the β-cat C-terminal domain (chicken β-catenin amino acids 696-781, homologous to the mouse dn sequence) and included the PDZ binding motif which interacts with S-SCAM PDZ5; it should be noted that distinct domains of S-SCAM bind to β-cat (PDZ5) and NL1 (PDZ1 and ww domains) (Hirao et al., 1998; Nishimura et al., 2002; Iida et al., 2004). We used the avian-specific retroviral vector RCASBP to overexpress the dn construct and the optimized conditions that we developed for in ovo retroviral infection of neural crest precursor cells that give rise to CG neurons (Williams et al., 1998; Temburni et al., 2004; Rosenberg et al., 2008). We used hemagglutinin (HA) epitope-tagged β-cat::S-SCAM-dn to distinguish infected CG neurons.
Figure 3.

S-SCAM links to β-catenin and neuroligin in CG neurons. β-cat (lane 1) and NL (lane 3) coimmunoprecipitated with S-SCAM from CG lysates. In contrast, IQGAP (lane 8) did not. S-SCAM itself precipitated with the S-SCAM antibody (lane 5), but not with the hemagglutinin (HA) antibody, as a negative control (lane 6). CG homogenates were immunoprecipitated (IP) with S-SCAM or HA antibody. The precipitate and 1–2% of total input were separated by SDS-PAGE and immunoblotted (IB) with β-cat antibody (lanes 1 and 2), NL antibody (lanes 3 and 4), S-SCAM antibody (lanes 5–7), or IQGAP antibody (lanes 8 and 9). β-cat ∼92 kDa; NL ∼110 kDa; S-SCAM isoforms: α (full-length), β, and γ, 180, 160, and 105 kDa; IQGAP ∼190 kDa. n = 3 separate experiments.
We found obvious decreases in S-SCAM surface-associated immunofluorescence staining in CG neurons expressing the HA-tagged β-cat::S-SCAM-dn compared with uninfected control neurons (Fig. 4a–c). Controls included uninfected neurons within the same CG (internal controls) and neurons in separate uninfected, age-matched CGs that were processed in parallel. Pixel intensity distribution curves showed that S-SCAM labeling was shifted toward lower intensity levels (Fig. 4b). The mean pixel intensity was 29.6% lower in β-cat::S-SCAM-dn-expressing neurons relative to uninfected control neuron levels (Fig. 4c). Note that this semiquantitative approach may underestimate the changes because of steric hindrance limiting antibody access at control neuron synapses, based on the previous comparison of changes in α3*-nAChR surface levels on dn-expressing versus control CG neurons using quantitative immunofluorescence versus electrophysiological recording (31% lower mean intensity levels compared with 61.5% decreases in α3*-nAChR mean current densities; Rosenberg et al., 2008). Clearly, the results show that S-SCAM interaction with β-cat is essential for S-SCAM localizing at nicotinic postsynaptic sites in vivo.
Figure 4.

Expression of β-catenin::S-SCAM or APC::EB1 dominant-negative blocking peptides, in vivo, led to decreased accumulation of S-SCAM near the CG neuron surface. a, d, Micrographs of immunofluorescence double-labeled E11–E13 CG frozen sections showing S-SCAM surface-associated clusters (red) were decreased in neurons expressing the HA-tagged β-cat::S-SCAM-dn (DN, green; a) or HA-tagged APC::EB1-dn (DN, green; d) compared with control neurons (Ctl; a, d) [uninfected neurons from the same CG (internal control) or uninfected CGs age-matched and processed in parallel]. HA staining (green) shows that retroviral infection was restricted to CG neurons and occasionally a few glial cells (small HA+ cells; not seen here) that surround the neuronal somata. Insets, twofold magnification views of boxed regions. b, e, Frequency distribution graphs show reductions in the fluorescence pixel intensities of S-SCAM labeling near the surface in β-cat::S-SCAM-dn and APC::EB1-dn-infected neurons (red boxes) versus age-matched Ctl neurons (blue triangles). Dashed vertical lines indicate the median intensity values. c, f, Bar graphs showing 29.6% and 30.7% decreases in mean pixel intensity levels for S-SCAM surface-associated clusters in β-cat:::S-SCAM-dn (c) and APC::EB1-dn (f) neurons relative to Ctl neuron levels (β-cat::S-SCAM-dn: *p < 4.1 × 10−15, Student's t test, n = 21 DN and 21 Ctl neurons; APC::EB1-dn: *p < 5.8 × 10−18, Student's t test, n = 24 DN and 10 Ctl neurons). Bars represent the mean ± SEM. For the quantitative assessments, the fluorescence pixel intensities were measured along 3- to 5-μm-length segments of the brightest labeled surface regions (n = 3 different line segments per neuron, 10–49 DN and Ctl neurons, and 6–12 embryos for each immunolabeling experiment). The values were binned into incremental groups of 10 pixel intensity steps (e.g., 0–9, 10–19, etc., up to saturation). The percentage of pixels that belonged to each pixel intensity category was calculated and the data plotted as a relative frequency distribution (b, e).
In contrast to the decreases in S-SCAM, β-cat surface-associated clusters were not altered in β-cat::S-SCAM-dn-expressing neurons compared with controls (supplemental Fig. 2a–c, available at www.jneurosci.org as supplemental material), suggesting that β-cat localizing at nicotinic synapses does not require its interaction with S-SCAM. Specificity was further indicated by the lack of change in S-SCAM surface-associated clusters in neurons with retroviral-mediated overexpression of GFP, as a negative control (supplemental Fig. 4a,b, available at www.jneurosci.org as supplemental material).
The APC multiprotein complex regulates S-SCAM accumulation at nicotinic synapses
S-SCAM surface-associated accumulation was also decreased by expression of a different dn peptide, referred to as APC::EB1-dn, that blocks endogenous APC interactions with microtubule plus-end binding protein EB1 (Fig. 4d–f). APC::EB1-dn corresponds to APC's binding domain for EB1 (amino acids 1943-2184 of chicken APC sequence). We previously demonstrated the specificity and efficacy of APC::EB1-dn in vitro and in vivo (Rosenberg et al., 2008).
Blockade of APC::EB1 interactions led to reduced immunofluorescence intensities for S-SCAM surface-associated clusters and 30.7% decreases in mean pixel intensity levels relative to control neuron levels (Fig. 4d–f). These decreases in S-SCAM were similar to those caused by β-cat::S-SCAM-dn expression (Fig. 4a–c). In contrast, total surface-associated levels of β-cat were not altered in APC::EB1-dn neurons (Rosenberg et al., 2008), as also shown for β-cat::S-SCAM-dn neurons (supplemental Fig. 2a–c, available at www.jneurosci.org as supplemental material).
We propose that APC::EB1-dn expression causes changes in APC and β-cat interactions that, in turn, lead to reductions in β-cat binding to S-SCAM. Decreasing APC binding to EB1 leads to increases in APC interaction with β-cat (Wang et al., 2005). APC binding to β-cat brings it into close physical proximity with GSK3β that, in turn, phosphorylates β-cat. β-cat and APC shift binding partners and functional complexes, depending upon their phosphorylation and interacting protein levels (Xing et al., 2004; Choi et al., 2006; Daugherty and Gottardi, 2007; Xu and Kimelman, 2007). Some of the multimolecular complexes prevent β-cat binding to S-SCAM (Subauste et al., 2005).
We tested for changes in β-cat phosphorylation at GSK3β sites caused by APC::EB1-dn expression and for phosphorylation-dependent changes in β-cat interactions with S-SCAM in CG neurons. GSK3β kinase phosphorylates β-cat at serine (S)-37 and threonine (T)-41, and phospho-S37-T41 β-cat shows greater affinity for binding to APC compared with β-cat dephosphorylated at these sites (Liu et al., 2006). We found decreases in dephospho-S37-T41 β-cat surface-associated clusters in APC::EB1-dn-expressing CG neurons, and a 31.8% reduction in mean intensity levels compared with control neurons (supplemental Fig. 2d–f, available at www.jneurosci.org as supplemental material). As total surface-associated levels of β-cat were not altered, blockade of APC::EB1 interactions caused a greater proportion of the β-cat to be phosphorylated by GSK3β, increasing its affinity for APC. Further, β-cat phosphorylated at GSK3β sites may have reduced affinity for S-SCAM, based on the decreases in S-SCAM surface associated clusters in APC::EB1-dn-expressing CG neurons (Fig. 4d–f). Consistent with this model, LiCl inhibition of GSK3β led to increases in the size and overlap of β-cat and S-SCAM surface associated clusters on cultured CG neurons compared with the clusters on neurons in sister wells treated with NaCl, as a negative control (supplemental Fig. 3a,b, available at www.jneurosci.org as supplemental material). Similarly, greater amounts of β-cat coprecipitated with S-SCAM from LiCl versus NaCl-treated CG neuron cultures (supplemental Fig. 3c,d, available at www.jneurosci.org as supplemental material). All together, our findings suggest that dynamic shifts in APC, EB1 and β-cat interactions regulate S-SCAM accumulation at neuronal nicotinic postsynaptic sites.
Specific decreases in S-SCAM, but not PSD-93 or PSD-95, surface-associated clusters in dn-expressing CG neurons
In contrast to decreases in S-SCAM, PSD-93 surface-associated clusters were not altered in APC::EB1-dn-expressing neurons relative to controls (Rosenberg et al., 2008). Similarly, PSD-95 surface-associated clusters were not changed (Fig. 5). The specific decrease in S-SCAM, but not PSD-93 or PSD-95, provided the valuable opportunity to test for a role of S-SCAM in localizing NL at nicotinic synapses in vivo. Controversial reports suggest that S-SCAM, PSD-93 and PSD-95 have unique, redundant, or no role in positioning NL at glutamatergic synapses in vitro (Dresbach et al., 2004; Iida et al., 2004; Levinson et al., 2005).
Figure 5.

Expression of APC::EB1-dn does not alter PSD-95 clusters near the neuron surface. a, Micrographs of immunofluorescence double-labeled E12 CG frozen sections showing PSD-95 (red) clusters near the surface are not altered by expression of HA-tagged APC::EB1-dn (DN, green) compared with Ctl neurons from age-matched uninfected CGs processed in parallel. Insets, Twofold magnification views of boxed regions. b, Frequency distribution graph and mean intensity levels (data not shown) indicated no significant difference in pixel intensities of PSD-95 labeling in APC::EB1-dn neurons versus Ctl neurons (p = 0.22, Student's t test, n = 21 DN and 19 Ctl neurons). Dashed vertical lines indicate the median intensity values (b).
S-SCAM interactions play a role in NL localizing at neuronal nicotinic synapses in vivo
We tested for changes in synaptic accumulation of NL, the direct binding partner of S-SCAM, PSD-93 and PSD-95, in CG neurons expressing the two different blocking peptides. We found obvious decreases in NL surface clusters (Fig. 6a,d). Distribution plots showed that NL surface staining was shifted toward lower intensities (Fig. 6b,e). Mean pixel intensity levels of NL surface clusters were reduced by 29.5% and 21.5% in β-cat::S-SCAM-dn and APC::EB1-dn-expressing neurons, respectively, compared with control neurons (Fig. 6c,f). To establish specificity, we showed no change in NL surface clusters in neurons with retroviral-mediated GFP overexpression, as a negative control (supplemental Fig. 4c,d, available at www.jneurosci.org as supplemental material). Further, LiCl treatment lead to increased coprecipitation of NL with S-SCAM, in addition to increased β-cat and S-SCAM coprecipitation and surface clusters, relative to NaCl-treated sister wells (supplemental Fig. 3c,d, available at www.jneurosci.org as supplemental material).
Figure 6.

a–f, Neuroligin surface clusters are decreased in dn-expressing CG neurons. a, f, Micrographs of immunofluorescence double-labeled E11–E13 CG frozen sections. a, d, NL surface clusters (red; a, d) are decreased in neurons expressing HA-tagged β-cat::S-SCAM-dn (DN, green; a) or HA-tagged APC::EB1-dn (DN, green; d) compared with Ctl neurons (Ctl; a, d). Insets, Twofold magnification views of boxed regions. b, c, e, f, NL surface clusters show shifts to lower pixel intensity levels (b, e) as well as 29.5% and 21.5% reductions in mean intensity levels (c, f) in β-cat::S-SCAM-dn and APC::EB1-dn-expressing neurons, respectively, relative to Ctl neurons (β-cat::S-SCAM-dn: *p < 1.8 × 10−7, Student's t test, n = 16 DN and 16 Ctl neurons; APC::EB1-dn: *p < 6.9 × 10−6, Student's t test, n = 32 DN and 11 Ctl neurons). Dashed vertical lines indicate the median intensity values (b, e). Bars represent the mean ± SEM (c, f).
S-SCAM interactions play a role in NL localization and/or stable retention at nicotinic postsynaptic sites in vivo. The findings do not rule out a similar role for PSD-93 and PSD-95 in positioning NL at nicotinic synapses, but show that they are not sufficient. We propose that S-SCAM, β-cat, APC and EB1 act in concert to regulate NL synaptic localization, based on the similar reductions in S-SCAM and NL synaptic accumulation caused by the two different dns.
The postsynaptic APC multimolecular complex provides retrograde signals that promote presynaptic terminal maturation
Neurexin
The postsynaptic transmembrane protein NL signals retrogradely via its extracellular interactions with α- and β-Nrx transmembrane proteins in the presynaptic terminal. α-Nxn functionally couples Ca2+ channels to the exocytotic machinery, and β-Nxn directs active zone assembly (Dean et al., 2003; Missler et al., 2003). NL::Nrx interactions stabilize synapses by linking postsynaptic scaffold proteins to presynaptic terminal proteins, and direct functional active zones to develop in register with postsynaptic receptor clusters (Südhof, 2008).
Based on the decreases in NL clusters on the surface of dn-expressing CG neurons (Fig. 6), we tested for changes in Nrx clusters on presynaptic terminals that contact the dn-positive neurons. It is important to note that retroviral-mediated expression of the constructs (GFP, β-cat::S-SCAM-dn and APC::EB1-dn) was limited to postsynaptic CG neurons and occasional surrounding glial cells, whereas expression was never detected in the presynaptic inputs (data not shown; Williams et al., 1998; Temburni et al., 2004; Rosenberg et al., 2008). The time and site of retroviral injection in ovo targets neural crest precursor cells that give rise to the CG, but not the neural tube precursor cells that develop into the midbrain accessory oculomotor nucleus which innervates CG neurons.
Nrx surface clusters were shifted to lower pixel intensity levels and showed decreases of 31.5% and 22.6% in mean staining intensities in APC::EB1-dn and β-cat::S-SCAM-dn-expressing neurons relative to control neurons (Fig. 7). The postsynaptic APC multiprotein complex provides retrograde signals via NL that direct the localization of presynaptic Nrx at neuronal nicotinic synapses in vivo.
Figure 7.

a–f, Neurexin surface clusters are decreased on presynaptic terminals that contact postsynaptic neurons expressing APC::EB1-dn or β-cat::S-SCAM-dn. a, d, Micrographs of immunofluorescence double-labeled E13 CG frozen sections showing that Nrx surface clusters (red; a, d) are decreased on presynaptic terminals that contact APC::EB1-dn (a) and β-cat::S-SCAM-dn (d) neurons compared with Ctl neurons. Insets, twofold magnification views of boxed regions. b, c, e, f, Nrx staining shows shifts to lower pixel intensity levels (b, e) as well as 31.5% and 22.6% reductions in mean intensity levels (c, f) at synapses on APC::EB1-dn (b, c)- and β-cat::S-SCAM-dn (e, f)-expressing neurons, respectively, relative to Ctl neurons. (APC::EB1-dn: *p < 6.9 × 10−9, Student's t test, n = 21 DN and 14 Ctl neurons; β-cat::S-SCAM-dn: *p < 5.2 × 10−22 Student's t test, n = 22 DN and 21 Ctl neurons). Dashed vertical lines indicate the median intensity values (b, e). Bars represent the mean ± SEM (c, f).
Synaptic vesicles
Next, we looked for changes in presynaptic terminal maturation. First, we analyzed synaptic vesicle clusters in terminals contacting postsynaptic CG neurons expressing APC::EB1-dn. No alterations were detected in the levels of SV2 cluster immunostaining (Fig. 8a,b). The mean pixel intensity of SV2 clusters was 102.5 ± 2.9% versus 100 ± 2.9% in terminals on APC::EB1-dn-expressing neurons versus terminals on uninfected control neurons, respectively.
Figure 8.

Altered architecture of the calyx-type presynaptic terminal on postsynaptic dn-expressing ciliary neurons. a, Micrographs of immunofluorescence double-labeled E13 CG frozen sections show no decrease in SV2-stained synaptic vesicle clusters (red) in presynaptic terminals that contact postsynaptic neurons expressing APC::EB1-dn (DN) compared with terminals on Ctl neurons. b, Presynaptic SV2 labeling shows no significant difference in pixel intensity levels in APC::EB1-dn neurons versus Ctl neurons (p = 0.15, Student's t test; n = 33 DN and 83 Ctl neurons). In contrast, the architecture of the presynaptic calyx-type terminal is altered. c, Threefold magnification views of the boxed regions in a show greater discontinuity in the length of SV2-stained terminals on APC::EB1-dn-positive ciliary neurons versus Ctl ciliary neurons (Ctl). White lines mark the continuous SV2-stained segments. d, Bar graph shows a 24.9% decrease in the mean length of continuous SV2 labeling for calyx terminals on APC::EB1-dn neurons versus age-matched Ctl neurons (*p < 0.04, Student's t test, n = 67 DN and 157 Ctl neurons, 9 or more embryos, 3 independent experiments). e, Schematic representation of maturational changes in architecture of the presynaptic terminal on normal developing embryonic ciliary neurons. The red dots represent synaptic vesicles. During immature stages, multiple boutons fuse to form the calyx. Maturation is complete by E14, all ciliary neurons are contacted by a single large calyx-type terminal. The decrease in mean length of continuous SV2 labeling on E13 ciliary neurons expressing APC::EB1-dn suggests that the presynaptic terminals are less mature.
However, we did observe changes in presynaptic terminal architecture. The CG contains two neuron types, ciliary and choroid cells, which are readily distinguished on the basis of size, location in the ganglion, and type of presynaptic input (Landmesser and Pilar, 1974; Pilar et al., 1980). We analyzed the ciliary neurons because they receive a single large calyx-type presynaptic terminal that is readily detected by SV2 labeling. During development, multiple boutons fuse to form the one large calyx. This presynaptic terminal maturation is normally completed by embryonic day (E) 14 (Landmesser and Pilar, 1974).
We found greater discontinuity in the length of SV2-stained terminals on APC::EB1-dn-positive ciliary neurons versus control ciliary neurons at E12–E14 (Fig. 8c–e). The average length of continuous SV2 labeling was 24.9% smaller relative to control neuron values (Fig. 8d). This architectural change suggests that presynaptic terminals were less mature on postsynaptic neurons expressing APC::EB1-dn. Similar to our results, expression of an NL1-dn in postsynaptic CG neurons led to decreases in presynaptic SV2 cluster size and number, but not pixel intensity levels (Triana-Baltzer et al., 2008).
Active zone proteins
The presynaptic active zone proteins RIM and piccolo modulate regulated exocytosis of synaptic vesicles (Khanna et al., 2007; Kiyonaka et al., 2007; Kaeser et al., 2008; Leal-Ortiz et al., 2008). RIM and piccolo cluster intensity levels were decreased by 18.8% and 48.2%, respectively, in presynaptic terminals on APC::EB1-dn-expressing postsynaptic CG neurons (Fig. 9a–f). Similarly, β-cat::S-SCAM-dn caused 28% reductions in piccolo cluster intensity levels (Fig. 9g–i). The decreases in three different presynaptic proteins, Nrx, piccolo, and RIM, and the terminal architectural changes suggest that retrograde signals from the postsynaptic APC complex are essential for presynaptic terminal maturation.
Figure 9.

a–i, Presynaptic active zone protein clusters are decreased in terminals that contact postsynaptic neurons expressing APC::EB1-dn or β-cat::S-SCAM-dn. a, d, g, Micrographs of immunofluorescence double-labeled E11–E13 CG frozen sections showing that RIM (red; a) and piccolo (red; d, g) clusters are decreased in presynaptic terminals that contact postsynaptic neurons expressing APC::EB1-dn (a, d) or β-cat::S-SCAM-dn (g) compared with terminals on Ctl neurons. Insets, Twofold magnification views of boxed regions. b, c, e, f, RIM (b, c) and piccolo (e, f,) clusters show shifts to lower pixel intensity levels (b, e) and reductions in mean intensity levels of 18.8% for RIM (c) and 48.2% for piccolo (f) in presynaptic terminals on APC::EB1-dn neurons relative to terminals on Ctl neurons. (RIM, c; *p < 7.6 × 10−4, Student's t test; n = 28 DN and 49 Ctl neurons; piccolo, d; *p < 8.9 × 10−24, Student's t test; n = 27 DN and 10 Ctl neurons). Similarly, piccolo staining shows 28% reductions in mean intensity levels (h, i) in presynaptic terminals on β-cat::S-SCAM-dn-expressing neurons (i, *p < 6.5 × 10−13, Student's t test; n = 24 DN and 24 Ctl neurons). Dashed vertical lines indicate the median intensity values (b, e, h). Bars represent the mean ± SEM (c, f, i).
Reduced functional maturation of presynaptic terminals on postsynaptic neurons expressing APC::EB1-dn
Based on the decreases in active zone proteins, we tested for reduced presynaptic functional maturation. We used FM1-43FX styrl dye labeling to assess the levels of active synaptic vesicle recycling induced by high K+ depolarization, a well established method for measuring presynaptic function and maturation (Gaffield and Betz, 2006; Wittenmayer et al., 2009). Live neurons with attached intact presynaptic terminals were isolated from APC::EB1-dn-expressing CGs and control age-matched CGs using mild dissociation conditions. We found a 30.1% reduction in mean intensity levels of FM1-43FX labeling of presynaptic terminals on APC::EB1-dn neurons compared with terminals on control neurons (Fig. 10). The reduced active synaptic vesicle recycling during high K+ depolarization is consistent with the decreased levels of piccolo and RIM. Unfortunately, technical limitations prevented assessment of changes in synaptic transmission (spontaneous and evoked synaptic responses) because dissociated CG neurons expressing the APC::EB1-dn failed to re-form processes in vitro, preventing the formation of synaptic connections. As strong support for our model of altered functional maturation of nicotinic synapses in vivo, a new study of glutamatergic synapses in vitro showed that changes in NL1 levels (up or down) altered the levels of synaptic vesicle recycling induced by high K+ depolarization, as well as vesicle release probability and frequency of miniature EPSCs (Wittenmayer et al., 2009). Overall, our findings suggest a role for the postsynaptic APC multiprotein complex in promoting presynaptic terminal maturation in vivo.
Figure 10.

Blockade of postsynaptic APC::EB1 interactions leads to decreased presynaptic terminal functional maturation. Live neurons with attached intact presynaptic terminals were isolated from APC::EB1-dn-infected CGs and control uninfected CGs at E13.5 using mild dissociation conditions. a, Micrographs show substantial decreases in FM1-43FX labeling of recycling synaptic vesicles during high K+ depolarization of the freshly isolated APC::EB1-dn neurons versus Ctl neurons. b, Cumulative frequency distribution graph showing that FM1-43FX labeled puncta shift to lower peak intensity levels in terminals on APC::EB1-dn neurons relative to terminals on Ctl neurons (n = 13 DN and 23 Ctl neurons, two independent experiments). c, Bar graphs show 30.1% reduction in mean peak intensity levels of FM1-43X labeled clusters in presynaptic terminals on APC::EB1-dn versus Ctl neurons (*p < 0.0048, Student's t test). Bars represent the mean ± SEM.
Discussion
We have uncovered a new role for APC as a key coordinator of presynaptic and postsynaptic maturation in cholinoceptive neurons. Synaptic efficacy requires coordinated maturation of presynaptic and postsynaptic specializations. Previously, we have shown that APC is essential for high density accumulation of α3*-nAChRs at postsynaptic sites (Temburni et al., 2004; Rosenberg et al., 2008). We now show that the APC multiprotein complex also provides retrograde signals that promote presynaptic active zone assembly. Postsynaptic APC orchestrates presynaptic and postsynaptic maturation to ensure that functional active zones form in register with neurotransmitter receptor clusters. We show a schematic representation of our model of the synapse organizing function of APC (supplemental Fig. 5, available at www.jneurosci.org as supplemental material). We identify S-SCAM and NL as novel components of the APC multiprotein complex at neuronal nicotinic synapses. We show that β-cat interactions are required to recruit S-SCAM to nicotinic postsynaptic sites. Most important, we show that APC, EB1, β-cat, and S-SCAM act in concert to localize and/or retain NL at nicotinic synapses in vivo. Postsynaptic blockade of selected APC and β-cat interactions leads to decreases in S-SCAM and NL, on the postsynaptic side, and in Nrx and active zone proteins RIM and piccolo, on the presynaptic side. Presynaptic terminal functional maturation is also reduced, based on decreases in actively recycling synaptic vesicles during high K+ depolarization. Our findings provide novel insights into molecular mechanisms that coordinate presynaptic and postsynaptic maturation at neuronal nicotinic synapses.
Our work is the first report, to our knowledge, that identifies molecular interactions required for localizing NL at synapses in vivo. Our results suggest a role for S-SCAM in anchoring NL1 at nicotinic postsynaptic sites. Most studies of NL1 synaptic targeting have used cultured neurons and implicate PSD-95 interactions with NL1 at glutamatergic synapses (Levinson et al., 2005; Futai et al., 2007). In contrast, we find that PSD-95 and PSD-93 are not sufficient for normal levels of NL1 at nicotinic synapses that have reduced levels of S-SCAM. The three scaffold proteins, S-SCAM, PSD-95 and PSD-93, are enriched at α3*-nAChR synapses and directly bind to NL1 via their PDZ domains. Deletion analyses suggest, however, that the PDZ binding domain of NL1 is dispensable for its localization at synapses in vitro (Dresbach et al., 2004; Conroy et al., 2007). Interestingly, S-SCAM has a second binding site for NL1. The ww domain of S-SCAM binds to the NL1 middle cytoplasmic region (Iida et al., 2004). The NL1 cytoplasmic region is critical for synaptic targeting and some synaptic functions. The cytoplasmic region proximal to the transmembrane domain is sufficient to target exogenous NL1 to the preformed postsynaptic complex and promotes alignment of presynaptic and postsynaptic specializations (Dresbach et al., 2004; Conroy et al., 2007). The middle cytoplasmic region of NL1 is essential for presynaptic terminal functional maturation (Wittenmayer et al., 2009). Albeit the protein that binds to the NL1 proximal cytoplasmic region is undefined, Brose and colleagues (Dresbach et al., 2004) propose that protein 4.1 may bind to this region and link NL1 to the actin cytoskeleton at the synapse. The NL1 middle cytoplasmic region contains the S-SCAM binding domain (Iida et al., 2004). We propose that S-SCAM links NL1 to the postsynaptic cytoskeleton via β-cat, APC and EB1. These three proteins bind to key cytoskeletal regulators that cross-link and stabilize the local submembranous actin and microtubule cytoskeleton at postsynaptic sites (supplemental Fig. 5, available at www.jneurosci.org as supplemental material) (Rosenberg et al., 2008). Further, Dresbach and colleagues (Wittenmayer et al., 2009) speculate that S-SCAM plays a key role in presynaptic maturation by linking NL1::Nrx and N-cad::β-cat signaling functions at the synapse. NL and N-cad (N-cadherin) have separate roles in enhancing synchronous versus asynchronous release of transmitter from cholinergic presynaptic terminals (Neff et al., 2009). The results reported here provide support for a role of S-SCAM in promoting presynaptic maturation. Consistent with our findings at neuronal nicotinic synapses in vivo, S-SCAM is required for NL1 localizing at glutamatergic synapses in vitro (Iida et al., 2004). At a distinct nicotinic synapse, the neuromuscular junction, the β-cat complex provides retrograde signals that promote presynaptic differentiation and function (Li et al., 2008). Accumulating evidence supports our model of postsynaptic S-SCAM, β-cat and APC functioning in concert to anchor NL1 at nicotinic synapses and thereby promote functional maturation of presynaptic terminals.
Although our results show that PSD-95 and PSD-93 are not sufficient for normal levels of NL1 at nicotinic synapses, we cannot rule out a role for these two scaffold proteins in promoting NL1 localization and/or stable retention at postsynaptic sites. At nicotinic and glutamatergic synapses in vitro, the postsynaptic complex of PSD-95 and NL1 signals retrogradely to regulate synchronous release of neurotransmitter from presynaptic terminals and/or structural alignment of presynaptic and postsynaptic elements (Futai et al., 2007; Neff et al., 2009). However, a new study shows that PSD-95 accumulates at nascent glutamatergic synapses after NL1 is positioned there (Barrow et al., 2009). It will be important to test directly whether PSD-95/PSD-93 play a role in anchoring NL at neuronal synapses in vivo.
Trans-synaptic interactions between NL and Nrx function to align the postsynaptic density scaffold with the presynaptic neurotransmitter release machinery (Dean and Dresbach, 2006; Li et al., 2007; Südhof, 2008). Within the postsynaptic neuron, NL binds to multidomain scaffold proteins that link (directly or indirectly) to neurotransmitter receptors, signaling molecules and cytoskeletal elements (Deng et al., 2006; Keith and El-Husseini, 2008). Trans-synaptically, NL binds to α- and β-Nrx (Boucard et al., 2005; Chih et al., 2006). Within the presynaptic terminal, α-Nrx functionally couples Ca2+ channels to the exocytotic machinery (Missler et al., 2003; Zhang et al., 2005; Dudanova et al., 2006). β-Nrx recruits synaptic vesicles via direct binding to CASK and synaptotagmin (Dean and Dresbach, 2006). Coculture studies of transfected heterologous cells and neurons indicate that Nrx is sufficient to induce postsynaptic specializations and NL is sufficient to induce presynaptic specializations (Scheiffele et al., 2000; Craig and Kang, 2007). However, knock-out transgenic mouse studies show that, in vivo, NL and Nrx are dispensable for synapse assembly, whereas they are essential for functional maturation of glutamatergic and GABAergic synapses (Missler et al., 2003; Varoqueaux et al., 2006; Chubykin et al., 2007). We extend these studies by showing that: (1) NL and Nrx enhance structural and functional maturation of presynaptic terminals at neuronal nicotinic synapses in vivo and (2) the APC multiprotein complex anchors NL at these synapses.
Synaptic maturation is also regulated by Wnt signaling (Speese and Budnik, 2007; Ataman et al., 2008; Salinas and Zou, 2008). APC and β-cat are key players in the Wnt signaling pathway. Wnt signaling regulates GSK3β phosphorylation of β-cat and APC, and thereby leads to dynamic shifts in their protein interactions and functions, including intercellular adhesion complexes, cytoskeleton regulation, and Wnt-responsive gene transcription (Zumbrunn et al., 2001; Daugherty and Gottardi, 2007; Barth et al., 2008). The decreased levels of stable, dephosphorylated β-cat in dn-expressing CG neurons (supplemental Fig. 2c–e, available at www.jneurosci.org as supplemental material) suggest that Wnt signaling may be reduced. Direct modulation of Wnt is necessary, in future studies, to define its specific role at neuronal nicotinic synapses. We predict that the present findings identify molecules, both presynaptic and postsynaptic, that likely mediate the role of Wnt signaling in promoting neuronal synaptic maturation.
Our findings identify novel molecular interactions that coordinate presynaptic and postsynaptic maturation. We show that APC organizes a multiprotein postsynaptic complex that anchors NL1 and Nrx at nicotinic synapses and promotes functional active zones to develop in register with α3*-nAChR clusters. The key molecular players, APC, β-cat, S-SCAM, EB1, NL1 and Nrx, are shared components between excitatory nicotinic and glutamatergic synapses. These shared synaptic components suggest that conserved mechanisms direct presynaptic and postsynaptic maturation in the distinct neuron types. Our newly generated transgenic mouse model with targeted deletion of APC in central neurons will provide a critical test of this model.
Autism and mental retardation associate with NL and Nrx loss-of-function human gene mutations (Südhof, 2008; Glessner et al., 2009; Blundell et al., 2010), underscoring the importance of defining molecular interactions which localize NL and Nrx at vertebrate synapses. As further support for our model, APC loss-of-function human gene mutations also correlate with autism spectrum disorders and mental retardation (Raedle et al., 2001; Zhou et al., 2007), highlighting the significance of the association, demonstrated here, between APC, NL and Nrx. Our data are defining novel roles for APC as a key coordinator of excitatory synapse maturation and function in neurons in vivo.
Footnotes
This research was funded by National Institutes of Health Grants NINDS (National Institute of Neurological Disorders and Stroke) NS21725 (to M.H.J.) and the Tufts Center for Neuroscience Research NINDS P30 NS047243 (to Dr. F. Rob Jackson). We thank Dr. Rachel Blitzblau and Dr. Murali Krishna Temburni for help in developing the dominant-negative constructs, Dr. Peter Scheiffele for the gift of pan-neurexin antibody, and Dr. Darwin Berg for providing the neuroligin-1 shRNA construct.
References
- Ataman B, Ashley J, Gorczyca M, Ramachandran P, Fouquet W, Sigrist SJ, Budnik V. Rapid activity-dependent modifications in synaptic structure and function require bidirectional Wnt signaling. Neuron. 2008;57:705–718. doi: 10.1016/j.neuron.2008.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrow SL, Constable JR, Clark E, El-Sabeawy F, McAllister AK, Washbourne P. Neuroligin1: a cell adhesion molecule that recruits PSD-95 and NMDA receptors by distinct mechanisms during synaptogenesis. Neural Dev. 2009;4:17. doi: 10.1186/1749-8104-4-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barth AI, Caro-Gonzalez HY, Nelson WJ. Role of adenomatous polyposis coli (APC) and microtubules in directional cell migration and neuronal polarization. Semin Cell Dev Biol. 2008;19:245–251. doi: 10.1016/j.semcdb.2008.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blundell J, Blaiss CA, Etherton MR, Espinosa F, Tabuchi K, Walz C, Bolliger MF, Sudhof TC, Powell CM. Neuroligin-1 deletion results in impaired spatial memory and increased repetitive behavior. J Neurosci. 2010;30:2115–2129. doi: 10.1523/JNEUROSCI.4517-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boucard AA, Chubykin AA, Comoletti D, Taylor P, Südhof TC. A splice code for trans-synaptic cell adhesion mediated by binding of neuroligin 1 to alpha- and beta-neurexins. Neuron. 2005;48:229–236. doi: 10.1016/j.neuron.2005.08.026. [DOI] [PubMed] [Google Scholar]
- Chih B, Engelman H, Scheiffele P. Control of excitatory and inhibitory synapse formation by neuroligins. Science. 2005;307:1324–1328. doi: 10.1126/science.1107470. [DOI] [PubMed] [Google Scholar]
- Chih B, Gollan L, Scheiffele P. Alternative splicing controls selective trans-synaptic interactions of the neuroligin-neurexin complex. Neuron. 2006;51:171–178. doi: 10.1016/j.neuron.2006.06.005. [DOI] [PubMed] [Google Scholar]
- Choi HJ, Huber AH, Weis WI. Thermodynamics of beta-catenin-ligand interactions: the roles of the N- and C-terminal tails in modulating binding affinity. J Biol Chem. 2006;281:1027–1038. doi: 10.1074/jbc.M511338200. [DOI] [PubMed] [Google Scholar]
- Chubykin AA, Atasoy D, Etherton MR, Brose N, Kavalali ET, Gibson JR, Südhof TC. Activity-dependent validation of excitatory versus inhibitory synapses by neuroligin-1 versus neuroligin-2. Neuron. 2007;54:919–931. doi: 10.1016/j.neuron.2007.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conroy WG, Liu Z, Nai Q, Coggan JS, Berg DK. PDZ-containing proteins provide a functional postsynaptic scaffold for nicotinic receptors in neurons. Neuron. 2003;38:759–771. doi: 10.1016/s0896-6273(03)00324-6. [DOI] [PubMed] [Google Scholar]
- Conroy WG, Nai Q, Ross B, Naughton G, Berg DK. Postsynaptic neuroligin enhances presynaptic inputs at neuronal nicotinic synapses. Dev Biol. 2007;307:79–91. doi: 10.1016/j.ydbio.2007.04.017. [DOI] [PubMed] [Google Scholar]
- Craig AM, Kang Y. Neurexin-neuroligin signaling in synapse development. Curr Opin Neurobiol. 2007;17:43–52. doi: 10.1016/j.conb.2007.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahlhaus R, Hines RM, Eadie BD, Kannangara TS, Hines DJ, Brown CE, Christie BR, El-Husseini A. Overexpression of the cell adhesion protein neuroligin-1 induces learning deficits and impairs synaptic plasticity by altering the ratio of excitation to inhibition in the hippocampus. Hippocampus. 2010;20:305–322. doi: 10.1002/hipo.20630. [DOI] [PubMed] [Google Scholar]
- Daugherty RL, Gottardi CJ. Phospho-regulation of Beta-catenin adhesion and signaling functions. Physiology (Bethesda) 2007;22:303–309. doi: 10.1152/physiol.00020.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dean C, Dresbach T. Neuroligins and neurexins: linking cell adhesion, synapse formation and cognitive function. Trends Neurosci. 2006;29:21–29. doi: 10.1016/j.tins.2005.11.003. [DOI] [PubMed] [Google Scholar]
- Dean C, Scholl FG, Choih J, DeMaria S, Berger J, Isacoff E, Scheiffele P. Neurexin mediates the assembly of presynaptic terminals. Nat Neurosci. 2003;6:708–716. doi: 10.1038/nn1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng F, Price MG, Davis CF, Mori M, Burgess DL. Stargazin and other transmembrane AMPA receptor regulating proteins interact with synaptic scaffolding protein MAGI-2 in brain. J Neurosci. 2006;26:7875–7884. doi: 10.1523/JNEUROSCI.1851-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dresbach T, Neeb A, Meyer G, Gundelfinger ED, Brose N. Synaptic targeting of neuroligin is independent of neurexin and SAP90/PSD95 binding. Mol Cell Neurosci. 2004;27:227–235. doi: 10.1016/j.mcn.2004.06.013. [DOI] [PubMed] [Google Scholar]
- Dudanova I, Sedej S, Ahmad M, Masius H, Sargsyan V, Zhang W, Riedel D, Angenstein F, Schild D, Rupnik M, Missler M. Important contribution of alpha-neurexins to Ca2+-triggered exocytosis of secretory granules. J Neurosci. 2006;26:10599–10613. doi: 10.1523/JNEUROSCI.1913-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Futai K, Kim MJ, Hashikawa T, Scheiffele P, Sheng M, Hayashi Y. Retrograde modulation of presynaptic release probability through signaling mediated by PSD-95-neuroligin. Nat Neurosci. 2007;10:186–195. doi: 10.1038/nn1837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaffield MA, Betz WJ. Imaging synaptic vesicle exocytosis and endocytosis with FM dyes. Nat Protoc. 2006;1:2916–2921. doi: 10.1038/nprot.2006.476. [DOI] [PubMed] [Google Scholar]
- Glessner JT, Wang K, Cai G, Korvatska O, Kim CE, Wood S, Zhang H, Estes A, Brune CW, Bradfield JP, Imielinski M, Frackelton EC, Reichert J, Crawford EL, Munson J, Sleiman PM, Chiavacci R, Annaiah K, Thomas K, Hou C, et al. Autism genome-wide copy number variation reveals ubiquitin and neuronal genes. Nature. 2009;459:569–573. doi: 10.1038/nature07953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall AC, Lucas FR, Salinas PC. Axonal remodeling and synaptic differentiation in the cerebellum is regulated by WNT-7a signaling. Cell. 2000;100:525–535. doi: 10.1016/s0092-8674(00)80689-3. [DOI] [PubMed] [Google Scholar]
- Hamburger V, Hamilton H. A series of normal stages in the development of the chick embryo. J Morphology. 1951;88:49–82. [PubMed] [Google Scholar]
- Hirao K, Hata Y, Ide N, Takeuchi M, Irie M, Yao I, Deguchi M, Toyoda A, Sudhof TC, Takai Y. A novel multiple PDZ domain-containing molecule interacting with N-methyl-d-aspartate receptors and neuronal cell adhesion proteins. J Biol Chem. 1998;273:21105–21110. doi: 10.1074/jbc.273.33.21105. [DOI] [PubMed] [Google Scholar]
- Homburger SA, Fekete DM. High efficiency gene transfer into the embryonic chicken CNS using B-subgroup retroviruses. Dev Dyn. 1996;206:112–120. doi: 10.1002/(SICI)1097-0177(199605)206:1<112::AID-AJA10>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- Iida J, Hirabayashi S, Sato Y, Hata Y. Synaptic scaffolding molecule is involved in the synaptic clustering of neuroligin. Mol Cell Neurosci. 2004;27:497–508. doi: 10.1016/j.mcn.2004.08.006. [DOI] [PubMed] [Google Scholar]
- Kaeser PS, Kwon HB, Chiu CQ, Deng L, Castillo PE, Südhof TC. RIM1alpha and RIM1beta are synthesized from distinct promoters of the RIM1 gene to mediate differential but overlapping synaptic functions. J Neurosci. 2008;28:13435–13447. doi: 10.1523/JNEUROSCI.3235-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang Y, Zhang X, Dobie F, Wu H, Craig AM. Induction of GABAergic postsynaptic differentiation by alpha-neurexins. J Biol Chem. 2008;283:2323–2334. doi: 10.1074/jbc.M703957200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keith D, El-Husseini A. Excitation Control: Balancing PSD-95 Function at the Synapse. Front Mol Neurosci. 2008;1:4. doi: 10.3389/neuro.02.004.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khanna R, Li Q, Bewersdorf J, Stanley EF. The presynaptic CaV2.2 channel-transmitter release site core complex. Eur J Neurosci. 2007;26:547–559. doi: 10.1111/j.1460-9568.2007.05680.x. [DOI] [PubMed] [Google Scholar]
- Kim E, Sheng M. PDZ domain proteins of synapses. Nat Rev Neurosci. 2004;5:771–781. doi: 10.1038/nrn1517. [DOI] [PubMed] [Google Scholar]
- Kiyonaka S, Wakamori M, Miki T, Uriu Y, Nonaka M, Bito H, Beedle AM, Mori E, Hara Y, De Waard M, Kanagawa M, Itakura M, Takahashi M, Campbell KP, Mori Y. RIM1 confers sustained activity and neurotransmitter vesicle anchoring to presynaptic Ca2+ channels. Nat Neurosci. 2007;10:691–701. doi: 10.1038/nn1904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein PS, Melton DA. A molecular mechanism for the effect of lithium on development. Proc Natl Acad Sci U S A. 1996;93:8455–8459. doi: 10.1073/pnas.93.16.8455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko J, Zhang C, Arac D, Boucard AA, Brunger AT, Sudhof TC. Neuroligin-1 performs neurexin-dependent and neurexin-independent functions in synapse validation. EMBO J. 2009;28:3244–3255. doi: 10.1038/emboj.2009.249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landmesser L, Pilar G. Synapse formation during embryogenesis on ganglion cells lacking a periphery. J Physiol. 1974;241:715–736. doi: 10.1113/jphysiol.1974.sp010680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leal-Ortiz S, Waites CL, Terry-Lorenzo R, Zamorano P, Gundelfinger ED, Garner CC. Piccolo modulation of Synapsin1a dynamics regulates synaptic vesicle exocytosis. J Cell Biol. 2008;181:831–846. doi: 10.1083/jcb.200711167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levinson JN, Chéry N, Huang K, Wong TP, Gerrow K, Kang R, Prange O, Wang YT, El-Husseini A. Neuroligins mediate excitatory and inhibitory synapse formation: involvement of PSD-95 and neurexin-1beta in neuroligin-induced synaptic specificity. J Biol Chem. 2005;280:17312–17319. doi: 10.1074/jbc.M413812200. [DOI] [PubMed] [Google Scholar]
- Li J, Ashley J, Budnik V, Bhat MA. Crucial role of Drosophila neurexin in proper active zone apposition to postsynaptic densities, synaptic growth, and synaptic transmission. Neuron. 2007;55:741–755. doi: 10.1016/j.neuron.2007.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li XM, Dong XP, Luo SW, Zhang B, Lee DH, Ting AK, Neiswender H, Kim CH, Carpenter-Hyland E, Gao TM, Xiong WC, Mei L. Retrograde regulation of motoneuron differentiation by muscle beta-catenin. Nat Neurosci. 2008;11:262–268. doi: 10.1038/nn2053. [DOI] [PubMed] [Google Scholar]
- Liu J, Xing Y, Hinds TR, Zheng J, Xu W. The third 20 amino acid repeat is the tightest binding site of APC for beta-catenin. J Mol Biol. 2006;360:133–144. doi: 10.1016/j.jmb.2006.04.064. [DOI] [PubMed] [Google Scholar]
- Lucas FR, Goold RG, Gordon-Weeks PR, Salinas PC. Inhibition of GSK-3β leading to the loss of phosphorylated MAP-1B is an early event in axon remodeling induced by WNT-7a or lithium. J Cell Sci. 1998;111:1351–1361. doi: 10.1242/jcs.111.10.1351. [DOI] [PubMed] [Google Scholar]
- Missler M, Zhang W, Rohlmann A, Kattenstroth G, Hammer RE, Gottmann K, Südhof TC. Alpha-neurexins couple Ca2+ channels to synaptic vesicle exocytosis. Nature. 2003;423:939–948. doi: 10.1038/nature01755. [DOI] [PubMed] [Google Scholar]
- Neff RA, 3rd, Conroy WG, Schoellerman JD, Berg DK. Synchronous and asynchronous transmitter release at nicotinic synapses are differentially regulated by postsynaptic PSD-95 proteins. J Neurosci. 2009;29:15770–15779. doi: 10.1523/JNEUROSCI.4951-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimura W, Yao I, Iida J, Tanaka N, Hata Y. Interaction of synaptic scaffolding molecule and Beta-catenin. J Neurosci. 2002;22:757–765. doi: 10.1523/JNEUROSCI.22-03-00757.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen DP, Dunlap K, Jacob MH. Kainate receptors and RNA editing in cholinergic neurons. J Neurochem. 2007;101:327–341. doi: 10.1111/j.1471-4159.2006.04359.x. [DOI] [PubMed] [Google Scholar]
- Patrizi A, Scelfo B, Viltono L, Briatore F, Fukaya M, Watanabe M, Strata P, Varoqueaux F, Brose N, Fritschy JM, Sassoè-Pognetto M. Synapse formation and clustering of neuroligin-2 in the absence of GABAA receptors. Proc Natl Acad Sci U S A. 2008;105:13151–13156. doi: 10.1073/pnas.0802390105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilar G, Landmesser L, Burstein L. Competition for survival among developing ciliary ganglion cells. J Neurophysiol. 1980;43:233–254. doi: 10.1152/jn.1980.43.1.233. [DOI] [PubMed] [Google Scholar]
- Raedle J, Friedl W, Engels H, Koenig R, Trojan J, Zeuzem S. A de novo deletion of chromosome 5q causing familial adenomatous polyposis, dysmorphic features, and mild mental retardation. Am J Gastroenterol. 2001;96:3016–3020. doi: 10.1111/j.1572-0241.2001.04674.x. [DOI] [PubMed] [Google Scholar]
- Rosenberg MM, Yang F, Giovanni M, Mohn JL, Temburni MK, Jacob MH. Adenomatous polyposis coli plays a key role, in vivo, in coordinating assembly of the neuronal nicotinic postsynaptic complex. Mol Cell Neurosci. 2008;38:138–152. doi: 10.1016/j.mcn.2008.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross BS, Conroy WG. Capabilities of neurexins in the chick ciliary ganglion. Dev Neurobiol. 2008;68:409–419. doi: 10.1002/dneu.20598. [DOI] [PubMed] [Google Scholar]
- Salinas PC, Zou Y. Wnt signaling in neural circuit assembly. Annu Rev Neurosci. 2008;31:339–358. doi: 10.1146/annurev.neuro.31.060407.125649. [DOI] [PubMed] [Google Scholar]
- Scheiffele P, Fan J, Choih J, Fetter R, Serafini T. Neuroligin expressed in nonneuronal cells triggers presynaptic development in contacting axons. Cell. 2000;101:657–669. doi: 10.1016/s0092-8674(00)80877-6. [DOI] [PubMed] [Google Scholar]
- Speese SD, Budnik V. Wnts: up-and-coming at the synapse. Trends Neurosci. 2007;30:268–275. doi: 10.1016/j.tins.2007.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subauste MC, Nalbant P, Adamson ED, Hahn KM. Vinculin controls PTEN protein level by maintaining the interaction of the adherens junction protein beta-catenin with the scaffolding protein MAGI-2. J Biol Chem. 2005;280:5676–5681. doi: 10.1074/jbc.M405561200. [DOI] [PubMed] [Google Scholar]
- Südhof TC. Neuroligins and neurexins link synaptic function to cognitive disease. Nature. 2008;455:903–911. doi: 10.1038/nature07456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Temburni MK, Rosenberg MM, Pathak N, McConnell R, Jacob MH. Neuronal nicotinic synapse assembly requires the adenomatous polyposis coli tumor suppressor protein. J Neurosci. 2004;24:6776–6784. doi: 10.1523/JNEUROSCI.1826-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Triana-Baltzer GB, Liu Z, Gounko NV, Berg DK. Multiple cell adhesion molecules shaping a complex nicotinic synapse on neurons. Mol Cell Neurosci. 2008;39:74–82. doi: 10.1016/j.mcn.2008.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varoqueaux F, Aramuni G, Rawson RL, Mohrmann R, Missler M, Gottmann K, Zhang W, Südhof TC, Brose N. Neuroligins determine synapse maturation and function. Neuron. 2006;51:741–754. doi: 10.1016/j.neuron.2006.09.003. [DOI] [PubMed] [Google Scholar]
- Wang Y, Zhou X, Zhu H, Liu S, Zhou C, Zhang G, Xue L, Lu N, Quan L, Bai J, Zhan Q, Xu N. Overexpression of EB1 in human esophageal squamous cell carcinoma (ESCC) may promote cellular growth by activating beta-catenin/TCF pathway. Oncogene. 2005;24:6637–6645. doi: 10.1038/sj.onc.1208819. [DOI] [PubMed] [Google Scholar]
- Williams BM, Temburni MK, Levey MS, Bertrand S, Bertrand D, Jacob MH. The long internal loop of the alpha 3 subunit targets nAChRs to subdomains within individual synapses on neurons in vivo. Nat Neurosci. 1998;1:557–562. doi: 10.1038/2792. [DOI] [PubMed] [Google Scholar]
- Wittenmayer N, Körber C, Liu H, Kremer T, Varoqueaux F, Chapman ER, Brose N, Kuner T, Dresbach T. Postsynaptic Neuroligin1 regulates presynaptic maturation. Proc Natl Acad Sci U S A. 2009;106:13564–13569. doi: 10.1073/pnas.0905819106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing Y, Clements WK, Le Trong I, Hinds TR, Stenkamp R, Kimelman D, Xu W. Crystal structure of a beta-catenin/APC complex reveals a critical role for APC phosphorylation in APC function. Mol Cell. 2004;15:523–533. doi: 10.1016/j.molcel.2004.08.001. [DOI] [PubMed] [Google Scholar]
- Xu W, Kimelman D. Mechanistic insights from structural studies of beta-catenin and its binding partners. J Cell Sci. 2007;120:3337–3344. doi: 10.1242/jcs.013771. [DOI] [PubMed] [Google Scholar]
- Zhang W, Rohlmann A, Sargsyan V, Aramuni G, Hammer RE, Südhof TC, Missler M. Extracellular domains of alpha-neurexins participate in regulating synaptic transmission by selectively affecting N- and P/Q-type Ca2+ channels. J Neurosci. 2005;25:4330–4342. doi: 10.1523/JNEUROSCI.0497-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou XL, Giacobini M, Anderlid BM, Anckarsater H, Omrani D, Gillberg C, Nordenskjold M, Lindblom A. Association of adenomatous polyposis coli (APC) gene polymorphisms with autism spectrum disorder (ASD) Am J Med Genet B Neuropsychiatr Genet. 2007;144B:351–354. doi: 10.1002/ajmg.b.30415. [DOI] [PubMed] [Google Scholar]
- Zumbrunn J, Kinoshita K, Hyman AA, Näthke IS. Binding of the adenomatous polyposis coli protein to microtubules increases microtubule stability and is regulated by GSK3 beta phosphorylation. Curr Biol. 2001;11:44–49. doi: 10.1016/s0960-9822(01)00002-1. [DOI] [PubMed] [Google Scholar]