

Abstract
Hydrogels are commonly used as extracellular matrix mimetics for applications in tissue engineering and increasingly as cell culture platforms with which to study the influence of biophysical and biochemical cues on cell function in 3D. In recent years, a significant number of studies have focused on linking substrate mechanical properties to cell function using standard methodologies to characterize the bulk mechanical properties of the hydrogel substrates. However, current understanding of the correlations between the microstructural mechanical properties of hydrogels and cell function in 3D is poor, in part because of a lack of appropriate techniques. Here we have utilized a laser tracking system, based on passive optical microrheology instrumentation, to characterize the microstructure of viscoelastic fibrin clots. Trajectories and mean square displacements were observed as bioinert PEGylated (PEG: polyethylene glycol) microspheres (1, 2 or 4.7 μm in diameter) diffused within confined pores created by the protein phase of fibrin hydrogels. Complementary confocal reflection imaging revealed microstructures comprised of a highly heterogeneous fibrin network with a wide range of pore sizes. As the protein concentration of fibrin gels was increased, our quantitative laser tracking measurements showed a corresponding decrease in particle mean square displacements with greater resolution and sensitivity than conventional imaging techniques. This platform-independent method will enable a more complete understanding of how changes in substrate mechanical properties simultaneously influence other microenvironmental parameters in 3D cultures.
1. Introduction
Biomaterials that are natural, synthetic, or hybrids of both, hold promise as scaffolding materials for engineered tissues, and are proving useful as model extracellular matrices (ECMs) to study cell–cell, cell–ECM interactions, and their effects on cell response. Naturally occurring biomacromolecules, such as type-I collagen, glycosaminoglycans, or fibrinogen, self-assemble to form viscoelastic hydrogels that can serve as 3D models for the in vivo cellular microenvironment [1]. Proteins can also be modified based on relatively simple biochemistries to contain reactive groups that can be cross-linked to form polymer gels [2–4]. Hydrogels that mimic the cellular microenvironment require consideration of a multitude of factors, including mechanical properties [5–8], biocompatibility, reproducibility, ligand density [9], as well as biotransport and pore size [10–14]. The interplay between these factors will contribute to transduction of cellular signals, which, in turn, determines cell survival, proliferation, and phenotype. In many cases, the stochastic nature of the polymerization processes by which many commonly used hydrogels are constructed can lead to a heterogeneous microstructure. As a result, cells distributed within an apparently uniform gel will experience different microenvironments. This may translate into significant variation in cell–ECM interactions and consequently downstream signaling and ultimately behavior, all within a single gel. ECM fibers can be forced to align in a preferred orientation using magnetic fields, stretch, or other techniques [15, 16]. However, starting with a random orientation and studying the processes by which cells impose structure on their surroundings, may in fact model morphogenesis more closely than an entirely homogeneous structure. Regardless, it is evident that better characterization of the physical properties of the hydrogels on a length scale relevant for individual cells and cell processes is needed.
It is well established that substrate mechanical properties influence cell fate in two-dimensional (2D) cultures [17–22]. Multipotent stem cells [23, 24], tumor cells [25], and more committed cells [26] have been shown to change their phenotype based on the stiffness or compliance of their underlying substrate. Cells are thought to sense ECM mechanical properties in part by generating actin mediated traction forces [27–29]. There is also evidence suggesting that cells may be affected by substrate compliance when embedded within three-dimensional (3D) ECMs [30–33]. However, the influence of mechanics on cells in the 3D setting is ill-defined, since the compliance of the surrounding matrix depends not only upon the material properties of individual protein fibers, but also the complex organization of macromolecules that are cross-linked into an asymmetrical 3D network. In contrast to cells cultured on a 2D surface, cells seeded into a 3D scaffold are living in a porous protein network with mechanical and chemical stimuli on all sides. Cell morphology, migration speed [34, 35], and cytoskeletal organization are affected not only by matrix compliance, but also the density of ligands in the microenvironment [36]. Furthermore, geometric confinement of cells by the ECM has been shown to be directly linked to morphology [37–40], cell death [41], and stem cell differentiation [42]. As a result, the distribution of stresses about the cell will depend on the local geometry of fibers and on stresses originating both from the cell's cytoskeleton and the surrounding matrix.
In an effort to better understand the mechanics of cell–ECM interactions in a 3D biomaterial, a wide variety of methodologies have been implemented to characterize substrates. Tensile/compressive testing and shear rheology are routinely used to examine bulk mechanical properties [26, 43–45]. Such measurements are essential in biomaterial design; however, these methods typically do not reveal any information about microstructure. Confocal imaging relying on fluorescently tagged proteins [46, 47] or back scattered light [48, 49], multiphoton and second-harmonic imaging [14, 50], and even electron microscopy [51] have all given insight into the geometric microstructure of proteins in 3D. Traction force microscopy, in which immobilized microspheres are tracked in response to a cell-generated force, has been implemented in 2D [52, 53], with some recent success in 3D [54, 55]. If mechanical properties of a substrate are well characterized, microsphere displacements can also be used to back calculate the local forces transmitted by a cell to the substrate.
Microrheology is a field dedicated to characterizing the viscoelastic nature of materials on the micron scale, typically through tracking the movement of microspheres. Unlike `macro' rheology, which measures ensemble average properties, microrheology uses micron-sized beads that are on a similar length scale to subcellular structures involved in mechanosensing and force generation. Passive tracking methods of microrheology have been used to measure low strain, high frequency, high sensitivity information about viscoelastic biopolymer solutions [56], as probed under the influence of weak Brownian forces. In a physiologically relevant hydrogel, passive measurements of mechanical properties only probe small-deformation mechanics, which can be significantly different from those sensed by cells as they induce considerably larger local deformations. However, passive measurements can report the fluid properties within pores and the scale of the pore. A wide variety of technologies have been used to enable microrheology, including diffuse wave spectroscopy/dynamic light scattering [57, 58], video particle tracking [59–61], atomic force microscopy [62], and magnetic [63], and laser tweezers [64, 65]. Microrheology has also been used to look at the mechanical properties of the viscoelastic cytoplasm inside a cell [66–69], cell membranes [70], as well as ECM polymers like hyaluronan [71] and collagen [72].
In this study, we have implemented passive back focal plane laser tracking methods, based on passive optical microrheology, to investigate the microstructure of natural fibrin hydrogels. Fibrin gels are biocompatible, modifiable, easy to make, and serve as a good model substrate for studying common biological processes such as angiogenesis, wound healing, and cell–ECM interactions [51]. Plasma concentrations of fibrinogen (the precursor of fibrin) for a normal individual range from 2.0 to 4.0 mg ml−1 [73]. In vivo, fibrin clot formation, structure, and stability depend not only on the concentration of circulating fibrinogen, but also on the concentration of thrombin, which is dynamic [74]. To simulate microstructural variability in fibrin clots and to investigate the influence of matrix density on microstructure, here we characterize fibrin hydrogels composed of 3 different fibrinogen concentrations. These concentrations both bracket the physiological range of fibrinogen concentration in vivo, and have been shown to elicit variable phenotypic response in vitro [28].
2. Methods
2.1. Bead functionalization
Carboxylated silica beads (1, 2, and 4.74 μm diameter, Bangs Labs, Fishers, IN) were PEGylated with aminefunctionalized polyethylene glycol (amine-PEG, Creative PEGWorks, Winston Salem, NC) via carbodiimide reactive chemistry. Beads were suspended in 1 ml MES Buffer (Fischer Scientific, Pittsburg, PA), pH 4.7, at 2% w/v and were sonicated until there was no visible precipitate. Carboxyl groups on beads were activated at this low pH by addition of 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride (EDC) to form O-acylisourea intermediate which was further stabilized by addition of N-hydroxysuccinimide (NHS). Both EDC and NHS were added at 10× molar excess of bead surface groups at room temperature for 30 min under rotation. Excess EDC and NHS were removed by repeated centrifugation at 6000 rpm and washing with DI water (at least 3 times). Two milliliters of 5 kD amine-PEG dissolved in 200 mM PBS, pH 7, was added to resuspend the beads in solution at 10× molar excess. Raising the pH from the initial activation step helps drive the PEGylation reaction. The reaction between the amine-PEG and the beads was allowed to occur for 4 h or overnight at room temperature, under rotation and shielded from light with metal foil. Unreacted amine-PEG was removed by repeated washing with DI water. If further coating with bovine serum albumin (BSA, Sigma) was desired, PEGylated beads were incubated in 5% w/v BSA and washed once again. BSA may adsorb to unreacted functional groups on the bead surface to help reduce non-specific binding between beads and the protein phase of the fibrin hydrogels. The final concentration of the 1 ml bead solution, approximated from dilution of initial concentration, was 20 μg ml−1.
2.2. Fibrin hydrogel construction
The day of the experiment, bovine fibrinogen (Sigma) solutions of the desired concentrations (2.5, 5, or 10 mg ml−1) were prepared in 1× PBS under sterile conditions. After addition of 20 μl of 20 μg ml−1 PEGylated 1 μm silica beads and 5% fetal bovine serum (FBS), polymerization of 1 ml of the solution was initiated by adding 20 μl of thrombin (Sigma, 50 U ml−1) in a 35 mm optical dish (MatTek, Ashland, MA). FBS contains factor XIII, a zymogen that contributes to the cross-linking of the fibrin gel when activated to Factor XIIIa. The solutions were left undisturbed for 30 min at room temperature until gelation was complete. Gels were immersed in PBS to maintain hydration throughout the experiment.
2.3. Confocal reflection microscopy
Unlabeled 3D fibrin matrices were imaged by confocal reflection microscopy, using a laser scanning confocal microscope at the Laboratory of Fluorescence Dynamics (LFD). Briefly, confocal reflection microscopy was performed using a fluoView 1000 (Olympus) microscope equipped with a 60×, 1.2 NA UPLSAPO water immersion objective (Olympus). Samples were illuminated with 488 nm Argon laser (Melles Griot) through an 80/20 filter, and scanned at a rate of 2 μs/pixel.
2.4. Laser tweezers particle tracking
2.4.1. System setup and data acquisition
We have constructed a custom back focal plane detection system around an IX81 Olympus microscope on a vibration dampening SMART table (Newport, Irvine, Ca) as shown in figure 1. Laser light from a 1064 nm ytterbium fiber laser (IPG) is expanded by lenses L1 (f = 400 mm) and L2 (f = 500 mm), and is steeply focused onto the sample using a 1.45 NA oil immersion PlanApo objective (Olympus). Both a stepper motor sample stage (MS 2000, Applied Scientific Instruments (ASI), Eugene, OR) as well as a piezoelectric sample stage (XY PZT, PI), objectives, filter cubes, camera (Orca, Hamamatsu), and data acquisition are controlled through custom LabVIEW software. The XY PZT nanostage has 0.1 nm precision and is mounted by a custom adapter into the ASI stage. Forward scattered light is refocused by lenses L3 (f = 50 mm) and L4 (f = 35 mm) onto a quadrant photodiode (QPD, New Focus, San Jose, CA, 900–1700 nm operating range) positioned conjugate to the back focal plane of our objective. Fluctuating microspheres steer the laser across the QPD, which outputs thee analog signals: diff(X), diff(Y), and sum. Signals are digitized through a NIPXI M-series DAQ board (National Instruments). The X and Y signals were normalized by the sum signal to compensate for small changes in average laser intensity. In addition, an IR band pass filter is mounted in front of the QPD to suppress broadband ambient light. Signals were sampled at 10 kHz for 10 s. Sufficiently low laser power (~0.1 mW) was used during gel experiments to avoid the contribution of significant laser trap forces, as validated experimentally (data not shown).
Figure 1.
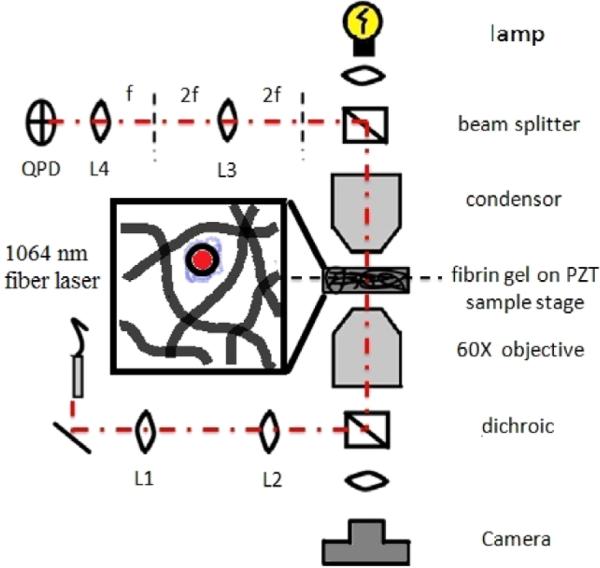
Schematic of laser tweezers microscope. A magnified portion of the sample is shown with a microsphere diffusing in the pore space created by fibrin ECM proteins.
2.4.2. Calibration and system characterization
To calibrate QPD signals, a 1 μm bead was scanned through the laser focus using the XY PZT in a stepwise manner (1 nm per step), and QPD signals were recorded at each position. System sensitivity of approximately 1 nm and a conversion factor of 750 ± 6.7 nm V−1 were determined. We used the mean-squared-displacement (MSD) 〈ΔR2(τ)〉 = 〈(r(t+τ)−r(t))2〉 of the particle's trajectory, r(t) calculated over various lag-times, τ, to quantify the amplitude of trajectories over different time and length scales [56, 75]. Unlike the MSD of purely diffusive particles, the MSDs of particles bound by a pore will exhibit a plateau as τ increases indicative of the mean maximum path the particle can diffuse along until it encounters a physical boundary. The value of τ at which the plateau is first encountered in indicative of the mean time to traverse the pore. By analogy, and to validate our system, we first observe the MSD of optically trapped beads in water at room temperature under various trap stiffness. To calibrate trap stiffness, power spectral densities were computed from (X, Y) displacement signals, and corner frequencies were used to calculate trap stiffness (k(pN nm−1)) as previously described [76, 77]. An optically trapped bead represents a viscoelastic system driven by Brownian forces and subject to viscous dissipation and spring-like `elastic' laser trap forces tending to return the bead to the trap center. As the trap stiffness increases with increasing laser power, the particle becomes more constrained and the MSD plateau decreases in magnitude. Likewise, as the trap stiffness increases, the MSD plateau will be reached at smaller values of τ since the average maximum path length and the time to traverse that path will have a monotonic relationship. MSD curves were obtained by trapping 1, 2 and 4.74 μm beads in water at increasing laser powers (figure 2(a)). As laser power and trap stiffness increases, both the magnitude of the MSD plateau and the value of τ at which the curves plateau decreases, consistent with optical tweezers theory and representative of increased confinement. The dashed line shows the theoretical MSD for beads diffusing in a purely viscous medium with slope equivalent to the diffusion coefficient in water; a flat slope would indicate a purely elastic system with no viscous dissipation [75]. Furthermore, as bead size is increased, the value of τ at which the curves plateau decreases (data not shown). This is expected since larger beads will experience larger viscous resistance and therefore, will take longer to achieve their maximum displacement. To further characterize the system, 1, 2, and 4.7 μm diameter beads were trapped at one of three laser powers and the MSD values were quantified. A linear relationship between maximum MSD and trap stiffness was observed for all three bead sizes (figure 2(b)), with trap stiffness highest for the 1 μm beads and lowest for the 4.7 μm beads. These data are consistent with theory and published observations for particles transitioning from the Mie regime (particle diameter near the trapping wavelength) to the ray optics regime (particle diameter larger than the trapping wavelength) [78].
Figure 2.

(a) MSD calibration curves for a 1 μm bead trapped in water. Corresponding laser trap stiffness for each MSD curve is shown. (b) The relationship between trap stiffness, k, and maximum MSD was generated for three different bead sizes.
2.4.3. Fibrin hydrogel
Microbeads were dispersed within a given fibrin gel and were randomly selected and positioned into the laser focus so that the beam and bead centers were co-aligned. We applied the following criteria before measuring the bead: (i) no other beads within a few diameters distance; (ii) bead must not be near the glass surface of the dish; (iii) bead must be sufficiently confined within a single pore so that it does not diffuse completely out of focus, transversely out of the beam. After a short period to allow for any vibrations excited through the sample stage movement to dissipate, custom LabVIEW software directed the acquisition of five sequential recordings of bead movement at 10 kHz with 10 s duration. A total of 25 beads were selected per bead size, resulting in 75 measurements for each gel concentration. All recordings were made in a dark, quiet room to reduce the possibility for externally excited vibrations.
2.4.4. Analysis
Raw signals acquired in LabVIEW were imported into Matlab for further analysis. Diff(X) and diff(Y) spectra were normalized by dividing by the mean of the sum signal, and scaled using the previously determined calibration factor for conversion of volts to nanometers. X versus Y trajectories were plotted to visualize the geometry of the bead's movement. MSDs were calculated from (X, Y) displacement trajectories (figures 4(a)–(c)). Probability density functions were estimated using a normal kernel function encapsulated by the `ksdensity' function in Matlab (figure 4(d)).
Figure 4.

MSDs for 1 (left), 2 (center), and 4.7 μm beads (right) in (a) 2.5 mg ml−1, (b) 5 mg ml−1, and (c) 10 mg ml−1 fibrin gels. (d) Probability density functions of plateau MSD values for each gel concentration show the range of displacements measured for each bead size. MSD values for 4.7 μm beads in both 5 mg ml−1 and 10 mg ml−1 gels are indistinguishable from those of beads adherent to glass (not shown).
3. Results and discussion
In this study, we have characterized the microstructure of fibrin-based ECM mimetics through the use of laser tweezers-based passive microrheology. Our initial experiments involved dispersing plain hydrophobic 1 μm polystyrene beads (Bangs Labs, Fishers, IN) inside the hydrogels. However, in these experiments, no diffusive movement was ever observed with unmodified beads; instead, the beads appeared physically adsorbed to the protein phase of the gels (data not shown). Reflection confocal images (figure 6) confirmed beads were immobilized due to physical adsorption on the fibers; beads were never observed floating in the pores of the matrices. To further establish that microspheres movement was limited as a result of adsorption to the protein phase of the gels rather than mechanical confinement by the matrix, a high concentration solution of non-functionalized 1 μm microspheres was injected into a 2.5 mg ml−1 fibrin gel using a 22 gauge needle (figure 3). The injection process created a physical insult that left a void within the gel. Microspheres were immobilized and physically adsorbed to filaments at the interface between the void and the gel. Penetration into the gel was limited to tens of microns, but microspheres were not freely diffusing.
Figure 6.

Reflection confocal images of (a) 2.5 mg ml−1, (b) 5 mg ml−1, and (c) 10 mg ml−1 fibrin gels shown at different magnifications (bar = 20 μm). Phase contrast images showing multiple beads of different sizes are on the far right. Arrows show the location of beads.
Figure 3.

Reflection confocal images of non-functionalized 1 μm beads adsorbed to the protein phase of the gel. The dark region in the upper part of each image was the site of bead injection (bar = 20 μm). Arrows show the location of microspheres.
These data are consistent with observations made by Valentine et al, who previously reported on the importance of bead surface chemistry for particle tracking measurements in different materials [79]. To eliminate non-specific binding between probe particles and ECM proteins, carboxylated silica microspheres were functionalized with amine polyethylene glycol (amine-PEG) using a PEGylation procedure adapted from Ehrenberg and McGrath [80], similar to that described by Valentine, et al [79]. Beads were PEGylated with 5 kDa amine-PEG, and further incubation with BSA in some cases. There were no significant differences in thermal fluctuations between PEGylated beads and PEGylated beads incubated in a BSA solution (data not shown). Complete functionalization of the microspheres should prevent adsorption of any protein, including BSA; thus our observations suggest functionalization of the beads was successful and complete. All formulations dramatically improved results, effectively shielding the beads from non-specific interaction with the protein phase of fibrin gels. The diffusion of functionalized microspheres within the pores of the ECM proteins was easily observed at 60× magnification.
While some particles may have an inherent `biological inertness', most are synthetic objects with a highly energetic attraction to protein. Protein adsorption occurs immediately upon implantation of many biomaterials into tissue [81], similar to the way in which untreated microspheres adhere to a fibrin matrix. This issue was previously addressed for very low concentration (0.44 mg ml−1) fibrin gels [79], but the strong association between unmodified beads and fibrin gels shown here (figures 3 and 6) suggests it may be even more critical in physiologically relevant gels used for 3D cell cultures. Adsorption of protein to the probe particles may give a misleading decrease in the measured MSD, resulting in an overestimate of ensemble material stiffness or an underestimate in pore size. The apparent displacement of beads adsorbed to relatively stiff, cross-linked, viscoelastic ECM hydrogels, such as those composed of fibrin or type-I collagen, may move in a correlated manner with bulk gel movement caused by external vibrations, which may also lead to misinterpretations of data. Furthermore, non-functionalized synthetic beads will have an increasingly highly energetic attraction to proteins when the concentration of proteins is very high, such as within a small pore. Blocking nonspecific interactions between the beads and the solid phase of ECM mimetics will also be particularly important in fully synthetic (e.g. PEG) or hybrid gels (e.g. PEGylated fibrinogen) whose structure cannot be easily imaged. Thus, when using passive microrheology to probe hydrogel microstructure, it is imperative to prevent undesirable protein adsorption. On the other hand, specific protein–bead interactions will undoubtedly be critical to assess the mechanical properties of individual filaments via active microrheology.
Using PEGylated beads of 3 different sizes, MSDs were then measured in fibrin gel matrices synthesized from 2.5, 5, or 10 mg ml−1 fibrinogen. Analyses of 25 beads per gel revealed maximum MSD values listed in table 1. MSD curves were characteristic of subdiffusion or diffusion of constrained particles with a sub-viscous linear slope followed by an elastic plateau [82], as can be seen in figure 4. The qualitative shape of these MSD curves are in agreement with those for microbeads diffusing within confined volumes within entangled F-actin networks for values of tau = 0.05 s and above [83]. The similarities in MSD are well explained by figure 6, in which beads appear similarly confined within the 2.5 and 5 mgml−1 gels. As expected, the 2.5 mg ml−1 gels exhibit the largest pores of the three gel concentrations tested. In those gels, both 1 and 2 μm diameter spheres exhibit a linear viscous response throughout most values of tau, with the 2 μm spheres `flattening out' at tau = 0.5 s (figure 4(a)). This is supported by the images in figure 6, which show the 2.5 mg ml−1 gel contains large protein-free volumes with respect to the smaller beads. The 4.7 μm spheres exhibit an anomalous response with respect to viscous subdiffusion within a confined volume and may contain information regarding elasticity at the pore's boundary. The 5 mg ml−1 gels are more `crowded' as compared to the 2.5 mg ml−1 gels, with more pores of qualitatively smaller dimension. In these gels, one would expect to observe greater confinement represented by a lower plateau value in the MSD curves occurring at lower values of tau. Both 1 and 2 μm beads exhibit such a plateau (figure 4(b)), indicating both beads are found in pores larger than their diameters. The 4.7 μm beads were fully confined, with MSD curves similar to our background measurements of a microbeads adherent to the glass coverslip. In these cases, the MSD represents system noise and vibration. In the 10 mg ml−1 gels 1 and 2 μm beads exhibited subdiffusion with reduced plateau values occurring at lower tau, while the motion of all 4.7 μm beads were indistinguishable from background (figure 4(c)). The distribution of maximum MSD values for each bead size in all three gel concentrations are shown in figure 4(d). The gray box in the 4.7 μm bead distribution represents values indistinguishable from background. The distributions indicate that 1 μm beads exhibit similar distributions of maximum MSDs in both 2.5 and 5 mg ml−1 gels, whereas the 2 μm beads become more confined in the 5 mg ml−1 gels. This suggests that 5 mg ml−1 gels contain pores both larger than and on the scale of 2 μm beads, as suggested by figure 6(b). In the 10 mg ml−1 gels, only the 1 and 2 μm beads show detectable movement, as would be expected from the mesh geometry (figure 6(c)).
Table 1.
Range of MSD Plateau values (in nm2).
| Gel Concentration (mg ml−1) | Range of MSD Plateau values (nm2) |
||
|---|---|---|---|
| 1μm (×103) | 2μm (×103) | 4.7μm (×103) | |
| 2.5 | (0.350–190) | (1.6–170) | (0.24–1.2) |
| 5 | (0.520–44) | (0.50–55) | (0.18–0.36) |
| 10 | (0.420–9.7) | (0.57–7.6) | (0.18–0.36) |
In addition to looking at MSD values, x versus y displacements were studied qualitatively to observe the anisotropy of microsphere movement. A wide variety of trajectories were observed with different magnitudes and geometries (figure 5), especially in the 2.5 and 5 mg ml−1 gels. No recurring trajectory geometry was consistently observed during measurements in any of the gels. Some trajectories reflected the triangular fractal structure of the ECM seen by reflection confocal (figure 6) in the shape of their distribution. Our system is currently limited in its ability to definitively extract pore geometries from trajectories since they are a 2D representation of a 3D event. Beads are diffusing in a volume of water encased by protein but recorded signals are from laser deflection on a single plane. Our system does not account for movement in 3D, nor any scaling that may occur as a result of the defocusing of the laser as a result of this movement. This can be overcome using a beam splitter and a second QPD. If the QPD is overfilled, then changes in the z-direction can be calibrated to changes in focus on the QPD with resolution equivalent to that of the optical axis. Despite these limitations, our results clearly show a range of MSD values, suggesting a variety of pore geometries and the random nature of the polymerized hydrogel structure as perceived in 2D. As a rule of thumb, any laser tweezer-based passive microrheology method cannot measure bead movements larger than the bead diameter. This is due to the fact that the laser power used is not strong enough to trap (it is intended to only detect), and any bead that is not caged in protein will become undetectable as it escapes the path of the laser beam. Video-based particle tracking methods allow for larger displacements to be measured, but at the expense of spatial and temporal resolution. Specifically, when compared to such methods, our laser tweezer-based microrheology method enables MSD values to be obtained at significantly lower values of tau (two additional decades) [79]. In a protein gel, this increased resolution allows the intrapore viscous properties of physiologically relevant ECM hydrogels to be identified and characterized, features that would be missed by most current video-based methods.
Figure 5.

Sample XY trajectories of bead movement inside the pores of a 2.5 mg ml−1 fibrin gel. Trajectories depict a wide array of ECM pore geometry.
To complement the laser tracking MSD measurements, we also visualized the geometric properties of fibrin matrices using confocal laser scanning microscopy in reflection mode (figure 6). Clear images of hydrophobic fibrin fibers were obtained by collecting backscattered light from scanning a 488 nm laser at minimal power. Reflection confocal images are readily obtainable for nearly any biopolymer given a large enough difference in index of refraction in comparison to water. This technique revealed that the 2.5 mg ml−1 fibrin gels had a wide range of pore sizes ranging from hundreds of nm to as big as 5–10 μm, consistent with the wider range of MSD measurements (figure 4(a)). These images also demonstrated a non-repeating pattern made up of polygonal geometries. The range of visible pore sizes decreased as the protein concentration of gels was increased, resulting in a higher density of protein as well as an increase in cross-link frequency. It becomes harder to distinguish any geometric regularity in the matrix as the pore sizes decrease beyond the resolution of the laser, and the fibrin density converges on the pixel resolution of the image. Some fibril-like proteins may not be visible since they are beyond the diffraction limit of the light, but this would be reflected in particle tracking experiments.
Reflection confocal is limited by its inability to visualize proteins and other molecules that do not strongly scatter light or whose size is smaller than the diffraction limit of the laser being used. This may be problematic when trying to use reflection confocal to image many synthetic polymer hydrogels or hybrid hydrogels [3] (i.e. those with both native and synthetic components) that commonly use highly hydrophilic molecules, such as PEG. PEG attracts water molecules, and thus, does not scatter light very well in a hydrogel. However, the microstructure of such gels can still be interrogated (in effect, imaged) by quantifying the MSDs of laser-tracked microspheres as we have done here. Our laser tweezer microrheology system is successful in mapping out pore geometries by tracking microbeads confined by the protein phase of ECMs, as demonstrated here in fibrin hydrogels. Additionally our method has sufficient temporal resolution to observe a linear viscous response at low tau to determine if a bead can freely diffuse within a pore. This method is limited, however, in that once the bead leaves the beam, it is undetectable. By this limitation a bead can only probe a pore of approximately twice its diameter. In naive material for which pore structure is unknown and cannot be imaged, a hybrid video particle tracking—passive laser detection system may be most appropriate. By mixing in a cocktail of beads spanning a range of diameters, the video methods can quickly determine the range of pore size. At that point, laser tracking can be implemented to determine the local pore viscosity as well as a higher resolution measurement of the distribution of pore size and shape. Furthermore, an additional advantage of using laser tweezer-based methods is the ability to drive bead displacement for active microrheology measurements (a topic we and other investigators are pursuing, but beyond the scope of this paper).
The structural properties of complex soft materials, such as ECM hydrogels, are difficult to characterize due to the small resolution of their mesh and the fact that they are predominantly composed of water. High resolution electron microscopy is capable of visualizing hydrogels with profound detail; however, not only is real-time bioimaging impossible, a vacuumed dry environment is typically needed, which may impose artifacts that make the reported structure questionable. The confocal reflection imaging method utilized here is an excellent means for imaging highly scattering static ECM hydrogels such as fibrin and type-I collagen, and provides for consistent time-lapse imaging capabilities. Here, we have proposed the novel application of using passive optical tweezers microrheology to quantify MSD values for multiple particles sizes in ECM gels of different protein concentrations. The structural information provided by confocal reflection imaging, in combination with high frequency, high sensitivity, laser particle tracking provides a powerful and robust tool for investigating the microarchitecture of the ECM. Both of these tools are platform independent, and can be used for virtually any ECM, including collagen, elastin, and hyaluronan.
Via passive laser tracking and reflection confocal, we have characterized the structure of fibrin-based ECMs on the micron and sub-micron scales. Subtleties in pore size distributions and material heterogeneity that are not attainable using typical bulk mechanical testing were directly addressed via our methodology, thereby enabling characterization of biomaterial structure on the scale of cellular and subcellular mechanosensing. We have shown that increased protein concentration both increases the density of protein in a given volume and further limits the range of pore sizes as shown by MSDs of functionalized microspheres. This has many implications in studying cell fate processes in 3D ECM hydrogels and their design for tissue engineering. In particular, increasing protein concentration is often used to achieve a desirable increase in material stiffness, with other effects including increased ligand concentration, decreased pore size, and greater confinement ignored or dismissed. Previous studies have shown that both protein ligand concentration and adhesion area are linked to a cell's ability to migrate [35, 38]. Cells tend to remain firmly anchored and unable to escape from highly adhesive sites, and may be unable to penetrate, or squeeze through regions where protein concentrations are too high in the absence of protease [84]. Furthermore, a cell's ability to spread is directly linked to cell survival [41]; thus limited pore size, due to an increasingly dense matrix, may contribute to apoptosis and necrosis. Ideally, new biomaterials designed to address the influence of substrate mechanical properties on cell fate in 3D should possess the flexibility to manipulate mechanical properties independently of these other features. Realistically, this is much simpler in theory than in practice. Importantly, our method here provides a new way to characterize how changing protein concentration, as a way to manipulate substrate mechanical properties, simultaneously alters microstructure and pore size, and may enable us to better understand the ECMs complex influence on cell function in 3D.
Acknowledgments
We gratefully acknowledge Dr Ken Shea and his lab members, Yu-Hsiang Hsu and Zhiyang Zeng, for their assistance and guidance regarding microsphere surface modification, Samir Shreim for his assistance in machining a stage adapter for the XY PZT stage, Bhupinder Shergill and Ekaterina Kniazeva for their technical advice and helpful discussion, and Enrico Gratton and the Laboratory of Fluorescence Dynamics for graciously permitting use of their reflection confocal system.
Support for this research was provided by the National Science Foundation (DMR-0805164) and National Institutes of Health (LAMMP: P41RR001192).
References
- [1].Mano JF, Silva GA, Azevedo HS, Malafaya PB, Sousa RA, Silva SS, Boesel LF, Oliveira JM, Santos TC, Marques AP, Neves NM, Reis RL. J. R. Soc. Interface. 2007;4:999–1030. doi: 10.1098/rsif.2007.0220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Frisman I, Seliktar D, Bianco-Peled H. Acta Biomater. 2009 doi: 10.1016/j.actbio.2009.07.015. at press (doi:10.1016/j.actbio.2009.07.015) [DOI] [PubMed] [Google Scholar]
- [3].Kim PD, Peyton SR, VanStrien AJ, Putnam AJ. Biomaterials. 2009;30:3854–64. doi: 10.1016/j.biomaterials.2009.04.013. [DOI] [PubMed] [Google Scholar]
- [4].Almany L, Seliktar D. Biomaterials. 2005;26:2467–77. doi: 10.1016/j.biomaterials.2004.06.047. [DOI] [PubMed] [Google Scholar]
- [5].Winer JP, Oake S, Janmey PA. PLoS One. 2009;4:e6382. doi: 10.1371/journal.pone.0006382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Plant AL, Bhadriraju K, Spurlin TA, Elliott JT. Biochim. Biophys. Acta. 2009;1793:893–902. doi: 10.1016/j.bbamcr.2008.10.012. [DOI] [PubMed] [Google Scholar]
- [7].Peyton SR, Ghajar CM, Khatiwala CB, Putnam AJ. Cell Biochem. Biophys. 2007;47:300–20. doi: 10.1007/s12013-007-0004-y. [DOI] [PubMed] [Google Scholar]
- [8].Khatiwala CB, Peyton SR, Putnam AJ. Am. J. Physiol. Cell Physiol. 2006;290:C1640–50. doi: 10.1152/ajpcell.00455.2005. [DOI] [PubMed] [Google Scholar]
- [9].Behravesh E, Sikavitsas VI, Mikos AG. Biomaterials. 2003;24:4365–74. doi: 10.1016/s0142-9612(03)00338-7. [DOI] [PubMed] [Google Scholar]
- [10].Liao H, Munoz-Pinto D, Qu X, Hou Y, Grunlan MA, Hahn MS. Acta Biomater. 2008;4:1161–71. doi: 10.1016/j.actbio.2008.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Munoz-Pinto DJ, Bulick AS, Hahn MS. J. Biomed. Mater. Res. A. 2009;90:303–16. doi: 10.1002/jbm.a.32492. [DOI] [PubMed] [Google Scholar]
- [12].Snyders R, Shingel KI, Zabeida O, Roberge C, Faure MP, Martinu L, Klemberg-Sapieha JE. J. Biomed. Mater. Res. A. 2007;83:88–97. doi: 10.1002/jbm.a.31217. [DOI] [PubMed] [Google Scholar]
- [13].Dikovsky D, Bianco-Peled H, Seliktar D. Biomaterials. 2006;27:1496–506. doi: 10.1016/j.biomaterials.2005.09.038. [DOI] [PubMed] [Google Scholar]
- [14].Raub CB, Suresh V, Krasieva T, Lyubovitsky J, Mih JD, Putnam AJ, Tromberg BJ, George SC. Biophys. J. 2007;92:2212–22. doi: 10.1529/biophysj.106.097998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Brown AE, Litvinov RI, Discher DE, Purohit PK, Weisel JW. Science. 2009;325:741–4. doi: 10.1126/science.1172484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Erikson A, Andersen HN, Naess SN, Sikorski P, Davies Cde L. Biopolymers. 2008;89:135–43. doi: 10.1002/bip.20874. [DOI] [PubMed] [Google Scholar]
- [17].Janmey PA, McCulloch CA. Annu. Rev. Biomed. Eng. 2007;9:1–34. doi: 10.1146/annurev.bioeng.9.060906.151927. [DOI] [PubMed] [Google Scholar]
- [18].Ingber DE. Ann. Med. 2003;35:564–77. doi: 10.1080/07853890310016333. [DOI] [PubMed] [Google Scholar]
- [19].Kumar S, Weaver VM. Cancer Metastasis Rev. 2009;28:113–27. doi: 10.1007/s10555-008-9173-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Wang Y, Botvinick EL, Zhao Y, Berns MW, Usami S, Tsien RY, Chien S. Nature. 2005;434:1040–5. doi: 10.1038/nature03469. [DOI] [PubMed] [Google Scholar]
- [21].Botvinick EL, Wang Y. Methods Cell Biol. 2007;82:497–523. doi: 10.1016/S0091-679X(06)82018-2. [DOI] [PubMed] [Google Scholar]
- [22].Discher DE, Janmey P, Wang YL. Science. 2005;310:1139–43. doi: 10.1126/science.1116995. [DOI] [PubMed] [Google Scholar]
- [23].Saha K, Keung AJ, Irwin EF, Li Y, Little L, Schaffer DV, Healy KE. Biophys. J. 2008;95:4426–38. doi: 10.1529/biophysj.108.132217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Engler AJ, Sen S, Sweeney HL, Discher DE. Cell. 2006;126:677–89. doi: 10.1016/j.cell.2006.06.044. [DOI] [PubMed] [Google Scholar]
- [25].Paszek MJ, Zahir N, Johnson KR, Lakins JN, Rozenberg GI, Gefen A, Reinhart-King CA, Margulies SS, Dembo M, Boettiger D, Hammer DA, Weaver VM. Cancer Cell. 2005;8:241–54. doi: 10.1016/j.ccr.2005.08.010. [DOI] [PubMed] [Google Scholar]
- [26].Peyton SR, Putnam AJ. J. Cell Physiol. 2005;204:198–209. doi: 10.1002/jcp.20274. [DOI] [PubMed] [Google Scholar]
- [27].Wood W, Martin P. Int. J. Biochem. Cell Biol. 2002;34:726–30. doi: 10.1016/s1357-2725(01)00172-8. [DOI] [PubMed] [Google Scholar]
- [28].Kniazeva E, Putnam AJ. Am. J. Physiol. Cell Physiol. 2009;297:C179–87. doi: 10.1152/ajpcell.00018.2009. [DOI] [PubMed] [Google Scholar]
- [29].Berrier AL, Yamada KM. J. Cell Physiol. 2007;213:565–73. doi: 10.1002/jcp.21237. [DOI] [PubMed] [Google Scholar]
- [30].Hadjipanayi E, Mudera V, Brown RA. Cell Motil. Cytoskeleton. 2009;66:121–8. doi: 10.1002/cm.20331. [DOI] [PubMed] [Google Scholar]
- [31].Freyman TM, Yannas IV, Pek YS, Yokoo R, Gibson LJ. Exp. Cell Res. 2001;269:140–53. doi: 10.1006/excr.2001.5302. [DOI] [PubMed] [Google Scholar]
- [32].Pizzo AM, Kokini K, Vaughn LC, Waisner BZ, Voytik-Harbin SL. J. Appl. Phys. 2005;98:1909–21. doi: 10.1152/japplphysiol.01137.2004. [DOI] [PubMed] [Google Scholar]
- [33].Even-Ram S, Yamada KM. Curr. Opin. Cell Biol. 2005;17:524–32. doi: 10.1016/j.ceb.2005.08.015. [DOI] [PubMed] [Google Scholar]
- [34].Zaman MH, Trapani LM, Sieminski AL, Mackellar D, Gong H, Kamm RD, Wells A, Lauffenburger DA, Matsudaira P. Proc. Natl Acad. Sci. USA. 2006;103:10889–94. doi: 10.1073/pnas.0604460103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Harley BA, Kim HD, Zaman MH, Yannas IV, Lauffenburger DA, Gibson LJ. Biophys. J. 2008;95:4013–24. doi: 10.1529/biophysj.107.122598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Engler A, Bacakova L, Newman C, Hategan A, Griffin M, Discher D. Biophys. J. 2004;86:617–28. doi: 10.1016/S0006-3495(04)74140-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Thery M, Racine V, Piel M, Pepin A, Dimitrov A, Chen Y, Sibarita JB, Bornens M. Proc. Natl Acad. Sci. USA. 2006;103:19771–6. doi: 10.1073/pnas.0609267103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Vogel V, Sheetz M. Nat. Rev. Mol. Cell Biol. 2006;7:265–75. doi: 10.1038/nrm1890. [DOI] [PubMed] [Google Scholar]
- [39].Nelson CM, Vanduijn MM, Inman JL, Fletcher DA, Bissell MJ. Science. 2006;314:298–300. doi: 10.1126/science.1131000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Moon JJ, Hahn MS, Kim I, Nsiah BA, West JL. Tissue Eng. A. 2009;15:579–85. doi: 10.1089/ten.tea.2008.0196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Chen CS, Mrksich M, Huang S, Whitesides GM, Ingber DE. Science. 1997;276:1425–8. doi: 10.1126/science.276.5317.1425. [DOI] [PubMed] [Google Scholar]
- [42].McBeath R, Pirone DM, Nelson CM, Bhadriraju K, Chen CS. Dev. Cell. 2004;6:483–95. doi: 10.1016/s1534-5807(04)00075-9. [DOI] [PubMed] [Google Scholar]
- [43].Vanderhooft JL, Alcoutlabi M, Magda JJ, Prestwich GD. Macromol. Biosci. 2009;9:20–8. doi: 10.1002/mabi.200800141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Janmey PA, Georges PC, Hvidt S. Methods Cell Biol. 2007;83:3–27. doi: 10.1016/S0091-679X(07)83001-9. [DOI] [PubMed] [Google Scholar]
- [45].Janmey PA, Schliwa M. Curr. Biol. 2008;18:R639–41. doi: 10.1016/j.cub.2008.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Ghajar CM, Blevins KS, Hughes CC, George SC, Putnam AJ. Tissue Eng. 2006;12:2875–88. doi: 10.1089/ten.2006.12.2875. [DOI] [PubMed] [Google Scholar]
- [47].Hall H, Baechi T, Hubbell JA. Microvasc. Res. 2001;62:315–26. doi: 10.1006/mvre.2001.2348. [DOI] [PubMed] [Google Scholar]
- [48].Roeder BA, Kokini K, Sturgis JE, Robinson JP, Voytik-Harbin SL. J. Biomech. Eng. 2002;124:214–22. doi: 10.1115/1.1449904. [DOI] [PubMed] [Google Scholar]
- [49].Hartmann A, Boukamp P, Friedl P. Blood Cells Mol. Dis. 2006;36:191–3. doi: 10.1016/j.bcmd.2005.12.033. [DOI] [PubMed] [Google Scholar]
- [50].Raub CB, Unruh J, Suresh V, Krasieva T, Lindmo T, Gratton E, Tromberg BJ, George SC. Biophys. J. 2008;94:2361–73. doi: 10.1529/biophysj.107.120006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Janmey PA, Winer JP, Weisel JW. J. R. Soc. Interface. 2009;6:1–10. doi: 10.1098/rsif.2008.0327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Gardel ML, Sabass B, Ji L, Danuser G, Schwarz US, Waterman CM. J. Cell. Biol. 2008;183:999–1005. doi: 10.1083/jcb.200810060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Sabass B, Gardel ML, Waterman CM, Schwarz US. Biophys. J. 2008;94:207–20. doi: 10.1529/biophysj.107.113670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Bloom RJ, George JP, Celedon A, Sun SX, Wirtz D. Biophys. J. 2008;95:4077–88. doi: 10.1529/biophysj.108.132738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Hur SS, Botvinick EL, Li Y, Chien S. Cell. Mol. Bioeng. 2009;2:425–36. doi: 10.1007/s12195-009-0082-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Yamada S, Wirtz D, Kuo SC. Biophys. J. 2000;78:1736–47. doi: 10.1016/S0006-3495(00)76725-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Oppong FK, Rubatat L, Frisken BJ, Bailey AE, de Bruyn JR. Phys. Rev. E. 2006;73:041405. doi: 10.1103/PhysRevE.73.041405. [DOI] [PubMed] [Google Scholar]
- [58].Dasgupta BR, Weitz DA. Phys. Rev. E. 2005;71:021504. doi: 10.1103/PhysRevE.71.021504. [DOI] [PubMed] [Google Scholar]
- [59].Panorchan P, Lee JS, Kole TP, Tseng Y, Wirtz D. Biophys. J. 2006;91:3499–507. doi: 10.1529/biophysj.106.084988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Daniels BR, Masi BC, Wirtz D. Biophys. J. 2006;90:4712–9. doi: 10.1529/biophysj.105.080606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Wirtz D. Annu. Rev. Biophys. 2009;38:301–26. doi: 10.1146/annurev.biophys.050708.133724. [DOI] [PubMed] [Google Scholar]
- [62].Alcaraz J, Buscemi L, Grabulosa M, Trepat X, Fabry B, Farre R, Navajas D. Biophys. J. 2003;84:2071–9. doi: 10.1016/S0006-3495(03)75014-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Puig-De-Morales M, Grabulosa M, Alcaraz J, Mullol J, Maksym GN, Fredberg JJ, Navajas D. J. Appl. Phys. 2001;91:1152–9. doi: 10.1152/jappl.2001.91.3.1152. [DOI] [PubMed] [Google Scholar]
- [64].Brau RR, Ferrer JM, Lee H, Castro CE, Tam BK, Tarsa PB, Matsudaira P, Boyce MC, Kamm RD, Lang MJ. J. Opt. A: Pure Appl. Opt. 2007;9:S103–12. [Google Scholar]
- [65].Addas KM, Schmidt CF, Tang JX. Phys. Rev. E. 2004;70:021503. doi: 10.1103/PhysRevE.70.021503. [DOI] [PubMed] [Google Scholar]
- [66].Thompson MS, Wirtz D. Methods Cell Biol. 2008;89:467–86. doi: 10.1016/S0091-679X(08)00618-3. [DOI] [PubMed] [Google Scholar]
- [67].Rogers SS, Waigh TA, Lu JR. Biophys. J. 2008;94:3313–22. doi: 10.1529/biophysj.107.123851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Kole TP, Tseng Y, Wirtz D. Methods Cell Biol. 2004;78:45–64. doi: 10.1016/s0091-679x(04)78003-6. [DOI] [PubMed] [Google Scholar]
- [69].Baker EL, Bonnecaze RT, Zaman MH. Biophys. J. 2009;97:1013–21. doi: 10.1016/j.bpj.2009.05.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Jonas M, Huang H, Kamm RD, So PT. Biophys. J. 2008;95:895–909. doi: 10.1529/biophysj.107.120303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Nijenhuis N, Mizuno D, Schmidt CF, Vink H, Spaan JA. Biomacromolecules. 2008;9:2390–8. doi: 10.1021/bm800381z. [DOI] [PubMed] [Google Scholar]
- [72].Velegol D, Lanni F. Biophys. J. 2001;81:1786–92. doi: 10.1016/S0006-3495(01)75829-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Weisel JW. Adv. Protein Chem. 2005;70:247–99. doi: 10.1016/S0065-3233(05)70008-5. [DOI] [PubMed] [Google Scholar]
- [74].Wolberg AS. Blood Rev. 2007;21:131–42. doi: 10.1016/j.blre.2006.11.001. [DOI] [PubMed] [Google Scholar]
- [75].Jonas M, Huang H, Kamm RD, So PT. Biophys. J. 2008;94:1459–69. doi: 10.1529/biophysj.106.098111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Tolic-Norrelykke SF, Schaffer E, Howard J, Pavone FS, Julicher F, Flyvbjerg H. Rev. Sci. Instrum. 2006;77:103101–11. [Google Scholar]
- [77].Berg-Sorensen K, Flyvbjerg H. Rev. Sci. Instrum. 2004;75:594–612. [Google Scholar]
- [78].Simmons RM, Finer JT, Chu S, Spudich JA. Biophys. J. 1996;70:1813–22. doi: 10.1016/S0006-3495(96)79746-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Valentine MT, Perlman ZE, Gardel ML, Shin JH, Matsudaira P, Mitchison TJ, Weitz DA. Biophys. J. 2004;86:4004–14. doi: 10.1529/biophysj.103.037812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Ehrenberg M, McGrath JL. Acta Biomater. 2005;1:305–15. doi: 10.1016/j.actbio.2005.02.002. [DOI] [PubMed] [Google Scholar]
- [81].Anderson JM, Rodriguez A, Chang DT. Semin. Immunol. 2008;20:86–100. doi: 10.1016/j.smim.2007.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Condamin S, Tejedor V, Voituriez R, Benichou O, Klafter J. Proc. Natl Acad. Sci. USA. 2008;105:5675–80. doi: 10.1073/pnas.0712158105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Wong IY, Gardel ML, Reichman DR, Weeks ER, Valentine MT, Bausch AR, Weitz DA. Phys. Rev. Lett. 2004;92:178101. doi: 10.1103/PhysRevLett.92.178101. [DOI] [PubMed] [Google Scholar]
- [84].Sabeh F, Shimizu-Hirota R, Weiss SJ. J. Cell. Biol. 2009;185:11–9. doi: 10.1083/jcb.200807195. [DOI] [PMC free article] [PubMed] [Google Scholar]