

Abstract

Enedione-diazoesters formed from 3-TBSO-2-diazo-3-butenoates undergo base catalyzed pericyclization that with dinitrogen extrusion and methyl migration provide a novel and efficient route to 2-carboalkoxyresorcinols. Intercepting the intermediate enolate anion with methyl vinyl ketone leads to the corresponding 4-substituted-2-carboalkoxyresorcinol and suggests generalization of this methodology.
Resorcinol and its derivatives are important ingredients for the total synthesis of a number of natural products and phenolic compounds of pharmaceutical interest.1,2 However, with few exceptions3,4 general methods for their synthesis are difficult to achieve except through traditional methodologies that originate with resorcinol.1 We wish to report a new methodology for the synthesis of 2-carboalkoxyresorcinols, based in part on serendipity, that relies on a convenient procedure that we recently reported for the synthesis of diverse α-diazo-β-keto esters.5 This procedure uses the readily accessible 3-TBSO-substituted vinyldiazoacetate 1 for zinc triflate-catalyzed Mukaiyama-Michael reactions with α,β-unsaturated ketones resulting in functionalized 3-keto-2-diazoalkanoates in high yield and selectivity (Scheme 1). The methodology for resorcinol synthesis employs a functionalized α,β-unsaturated ketone that undergoes elimination to an enedione-diazo ester which is susceptible to an unprecedented pericyclic reaction and rearrangement.
Scheme 1.
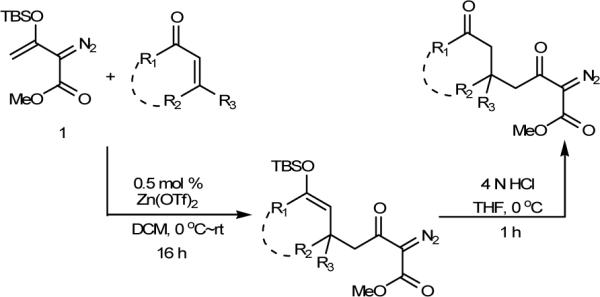
Mukaiyama-Michael Reactions of TBSO-Substituted Vinyldiazoacetate 1
In examining the breadth of the Mukaiyama-Michael transformation of 1 and its applications, we employed trans-4-methoxy-3-buten-2-one (2) as the substrate with the expectation that an enolizable enedione6 would be formed whose chemistry could lead us to cycloaddition products that contained the diazoester functionality. As anticipated, the direct Mukaiyama-Michael product (3) was unstable, undergoing elimination under the reaction conditions to form α,β-unsaturated ketone 4 exclusively (Scheme 2),7 but the resulting enedione underwent an unexpected transformation resulting in the formation of a substituted resorcinol. Examination of this process showed a diverse chemistry that we now report.
Scheme 2.
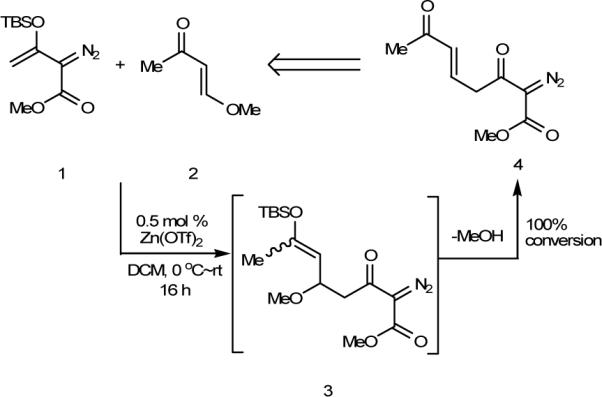
Mukaiyama-Michael Reaction with 4-Methoxy-3(E)-buten-2-one
Attempted chromatographic purification of 4 on silica gel resulted in the loss of 4, but the highly substituted resorcinol 5 was isolated in low yield (13%) (Scheme 3). Various conditions were employed to optimize formation of this unexpected reaction product by first treating 1 and 2 with zinc triflate and then, after concentrating the mixture but without isolating 4, adding a suitable promoter to catalyze the formation of the resorcinol product. Since compound 4 decomposes upon contact with silica gel, silica gel was added to the reaction system; however, only migration of the enone double bond occurred (36% conversion to 6). Performing the reaction at elevated temperature with silica gel or by adding acetic acid to the system produced the same results. When 2.0 molar equiv of 4 N aqueous HCl was used, 5 was produced in 21% isolated yield. However, assuming that enolization of 4 is one of the key steps for this transformation, we anticipated that the addition of base would facilitate this reaction. With 5.0 equivalents of triethylamine, 5 was obtained in 45% isolated yield, but the yield of 5 was further improved when only a catalytic amount of aqueous sodium hydroxide (0.10 mol/L NaOH, 10 mol %) was employed; and under these catalytic conditions 5 was isolated in 83% yield.
Scheme 3.
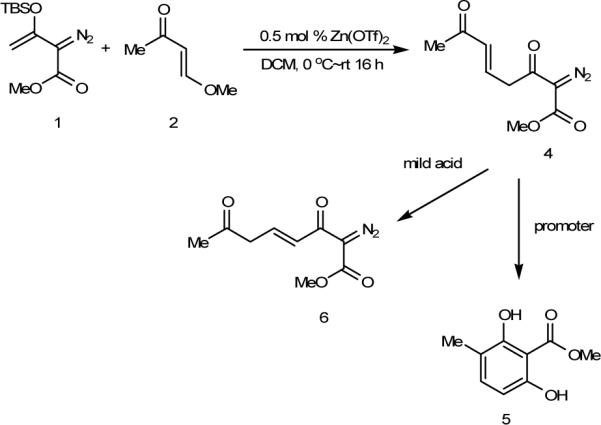
Reactions of Methyl 2-Diazo-3,7-dioxo-5(E)-octenoate
A plausible mechanistic pathway for resorcinol formation is presented in Scheme 4. Removal of the most acidic proton from compound 4 produces the conjugated enolate anions that are depicted by intermediates 7. Isomerization of 7a in which the 5,6-positions have the E-geometry to 7b in which the 5,6-positions have the Z-geometry is critical to the second isomerization in which the two ends are wrapped together (7c) through a conjugated triene that is appropriately arranged for pericyclization. Similar 6-π electrocyclizations involving enolate derivatives have been reported,8 although none have involved a diazo compound.
Scheme 4.

Base-catalyzed Pericyclization-rearrangement of 4
The diazonium ion intermediate 8 resulting from pericyclization is suitably positioned to undergo loss of dinitrogen in concert with methyl migration to form intermediate 9 that is the tautomer of the observed resorcinol product. The conversion of 8 to 9 is, to our knowledge, unprecedented; also, instead of undergoing methyl migration to the carbon bearing the diazonium ion that would be a semi-pinacol rearrangement9 or, alternatively, forming an epoxide10 with loss of dinitrogen, the methyl group migrates in the reverse direction to that of dinitrogen extrusion. The formation of the phenolate anion that can continue proton removal from reactant 4 is consistent with the need for only a substoichiometric amount of base to complete the reaction; after the reaction is initiated, the transformation is self sustained.
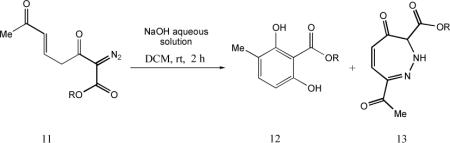
Ester derivatives of 4 were prepared and subjected to the same conditions as those used for the synthesis of 5. The coresponding resorcinol products were formed (Table 1), but their isolated yields were only moderate, and an additional product (13) accompanied the derivative resorcinol (eq 1). This compound, which was not observed from reactions with 4, resisted interpretation until an x-ray crystal structure of compound 13b revealed the 1,2-diazepine structure (Figure 1).
Table 1.
Influence of Alkyl Ester on Resorcinol Formation from Alkyl 2-Diazo-3,7-dioxo-5(E)-octenoate.
 |
(1) |
| R | equivalents of NaOH | yielda 12+13 (%) | ratiob (12:13) | |
|---|---|---|---|---|
| a | i-Pr | 0.1 equiv | 73 | 81:19 |
| i-Pr | 0.2 equiv | 67 | 79:21 | |
| b | t-Bu | 0.1 equiv | 68 | 79:22 |
| t-Bu | 0.2 equiv | 72 | 73:27 | |
| c | Bn | 0.1 equiv | 66 | 71:29 |
| Bn | 0.2 equiv | 73 | 67:33 |
Yield of isolated products after column chromatography.
Ratio based on isolated yields of compounds 5 and 14.
Figure 1.
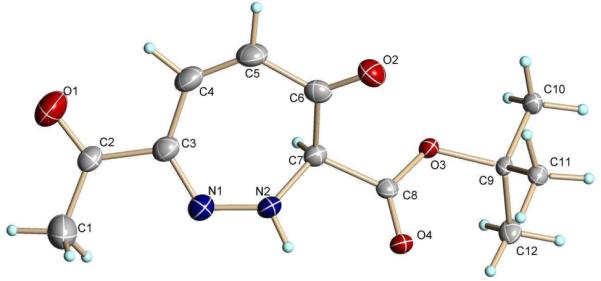
X-ray structure of 1,2-Diazepine 13b.
The formation of 13 can be understood as arising from an 8π-electrocyclization for which we are aware of few previous examples.11 The origin of the 1,2-diazepine precursors in prior studies has been cyclobutene-substituted diazoacetates or TMS-diazomethyl compounds that undergo electrocyclic ring opening to the requisite cisdienyldiazo intermediate that is structurally situated to undergo 8π-electrocyclization. The production of 13, and its absence in the reactions of 4 that produce 5, may be due to steric resistance to the coiling of 7a to 7c that preceeds electrocyclization in the formation of resorcinol derivatives (Scheme 4) and, indeed, the yield of 13 varies with the steric bulk of the ester. Accordingly, the pathway to 13 can be understood as arising from deprotonation of 4 to form enolate 7a that isomerizes to 7d before undergoing electrocyclization (Scheme 5).
Scheme 5.
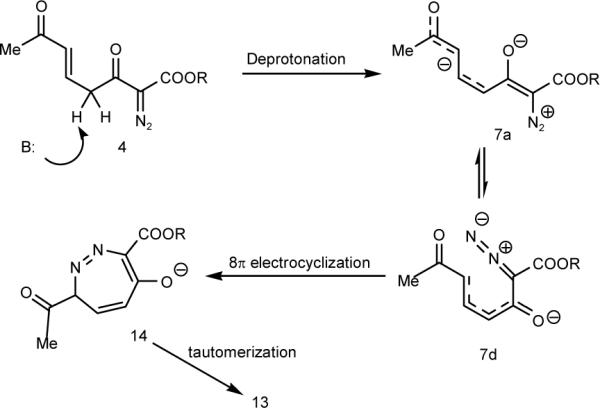
Pathway to 1,2-Diazepines 13
In an effort to examine the generality of this novel cyclization process, substituted vinyl ketone 15 was employed, and the resultant resorcinol 16 was formed in moderate yield (Scheme 6). Reactant 15 was prepared from methyl 2-diazo-3-ketopenanoate. Compound 17 has been used as an atraric acid derivative for treatment of benign prostate hyperplasia, prostate carcinoma and spinobulbar muscular atrophy.12
Scheme 6.
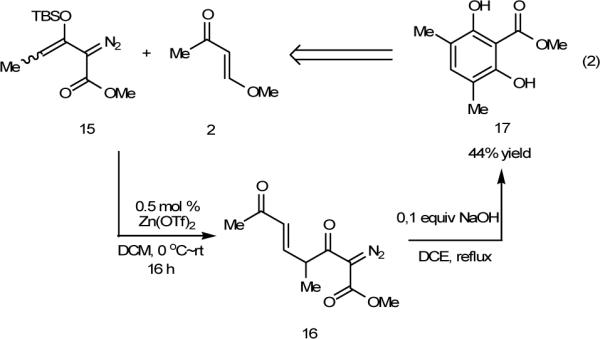
Synthesis of Atraric Acid Derivative
The pathway to substituted resorcinols that is described in Scheme 4 involves initial formation of enolate 7 that was expected to be suitable for trapping by Michael acceptors.13 If isomerization from the original E-isomer to the Z-isomer (7a to 7b in Scheme 4) is the limiting factor, then reaction of 7 with a Michael acceptor should not only be possible, yielding product with the same E olefin geometry, but also make possible further reaction that results in additional functionalization of the resorcinol core. Accordingly, treatment of 4 with a catalytic amount of sodium hydroxide in the presence of 4.0 molar equiv. of methyl vinyl ketone at 0°C resulted in the formation of Michael addition product 18 in 65% isolated yield (Scheme 7); 18 was the sole addition product, and resorcinol 19 was not formed under these conditions. The Michael addition product 18 was sufficiently robust to survive silica gel purification, which was not the case for 4. Subsequent treatment of 18 with triethylamine at room temperature resulted in the formation of resorcinol 19 in 45% isolated yield, thus further demonstrating the versatility of the methodology.
Scheme 7.
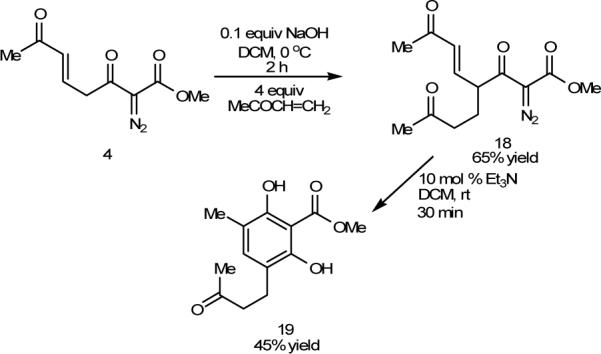
Michael Addition/Pericyclization/Rearrangement
In summary, enedione-diazoesters undergo a novel base-catalyzed pericyclization that, with subsequent dinitrogen extrusion and alkyl migration, forms 2-carboalkoxyresorcinols in good yield. Interception of the enolate intermediate on the pathway to pericyclization by a Michael acceptor, and its subsequent conversion to a 4-substituted-2-carboalkoxyresorcinol, suggests the synthetic potential of this methodology. Efforts are underway to further develop this novel synthetic process.
Supplementary Material
Acknowledgment
We are grateful to the National Institutes of Health for their financial support of this research (GM46503). We wish to thank Arnold R. Romero Bohórquez of the Laboratorio de Química Orgánica y Biomolecular, Escuela de Química, Universidad Industrial de Santander in Bucaramanga, Colombia for early efforts in this research.
Footnotes
Supporting Information Available. General experimental procedures, structure of 13b, and spectroscopic data for all new compounds. This material is available free of charge via the internet at http://pubs.org.edu.
References
- (1).(a) Durairaj RB. Resorcinol:Chemistry, Technology, and Applications. Springer; New York: 2005. [Google Scholar]; (b) Dressler H. Resorcinol: Its Uses and Derivatives. Plenum Press; New York: 1994. [Google Scholar]
- (2).(a) Khatib S, Nerya O, Musa R, Tamir S, Peter T, Vaya J. J. Med. Chem. 2007;50:2676. doi: 10.1021/jm061361d. [DOI] [PubMed] [Google Scholar]; (b) Brizzi A, Cascio MG, Brizzi V, Bisogno T, Dinatolo MT, Martinelli A, Tuccinardi T, Di Marzo V. Biorg. Med. Chem. 2007;15:5406. doi: 10.1016/j.bmc.2007.05.060. [DOI] [PubMed] [Google Scholar]; (c) Marsini MA, Huang Y, Van De Water R, Pettus TRR. Org. Lett. 2007;9:3229. doi: 10.1021/ol0710257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Kim A, Powers JD, Toczko JF. J. Org. Chem. 2006;71:2170. doi: 10.1021/jo0523868. [DOI] [PubMed] [Google Scholar]
- (4).Ceglia SS, Kress MH, Nelson TD, McNamara JM. Tetrahedron Lett. 2005;46:1731. [Google Scholar]
- (5).Liu Y, Zhang Y, Jee N, Doyle MP. Org. Lett. 2008;10:1605. doi: 10.1021/ol800298n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Schuler M, Duvvuru D, Retailleau P, Betzer J-F, Marinetti A. Org. Lett. 2009;11:4406. doi: 10.1021/ol901758k. [DOI] [PubMed] [Google Scholar]
- (7).Compound 3 was produced when the zinc triflate catalyzed reaction was performes in the presence of molecular sieves 4A; A mixture of cis and trans isomer in about 2/1 ratio was observed by NMR.
- (8).(a) Magomedov NA, Ruggiero PL, Tang Y. J. Am. Chem. Soc. 2004;126:1624. doi: 10.1021/ja0399066. [DOI] [PubMed] [Google Scholar]; (b) Austin WF, Zhang Y, Danheiser RL. Org. Lett. 2005;7:3905. doi: 10.1021/ol051307b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).For a recent review of the semi-pinacol rearrangement, see: Snape TJ. Chem. Soc. Revs. 2007;36:1823. doi: 10.1039/b709634h.; For a relevant example, see: Kirmse W, Hellwig G, Van Chiem P. Chem. Ber. 1986;119:1511..
- (10).Padwa A, Cimiluca P, Eastman D. J. Org. Chem. 1972;37(6):805. [Google Scholar]
- (11).(a) Sharp JT. In: Comprehensive Heterocyclic Chemistry. 7. Katritzky AR, Rees CW, editors. V.1. Pergamon Press; Oxford: 1984. pp. 595–604. [Google Scholar]; (b) Ohno M, Noda M, Yamamoto Y, Eguchi S. J. Org. Chem. 1999;64:707. doi: 10.1021/jo980523d. [DOI] [PubMed] [Google Scholar]; (c) Matsuya Y, Ohsawa N, Nemoto H. J. Am. Chem. Soc. 2006;128:13072. doi: 10.1021/ja065277z. [DOI] [PubMed] [Google Scholar]
- (12).Hoffmann H, Matusch R, Baniahmad A. PCT Int. Appl. 2006081997. [Google Scholar]
- (13).Permutter P. Conjugate Addition Reactions in Organic Synthesis. V.9. Oxford, New York: 1992. (Tetrahedron Organic Chemistry Series). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.