

Abstract
Bioassay-guided fractionation of an ethanol extract of the roots of the endemic Malagasy plant Pongamiopsis pervilleana (Baill.) R. Vig. led to the isolation of the three new compounds 2′R,4′-hydroxyemoroidocarpan (1), pongavilleanine (3), and epipervilline (4) together with two known compounds identified as emoroidocarpan (2) and rotenolone (5). The structures of all compounds were determined by physical, chemical and spectroscopic evidence. The stereochemistry at C-2′ of the previously reported compound emoroidocarpan was determined to be R by the observation of a negative Cotton effect at 474 nm in the CD spectrum of its osmate ester derivative. Compounds 2–5 displayed moderate antiproliferative activity against the A2780 human ovarian cancer cell line, and rotenolone also showed micromolar antiproliferative activity towards the breast cancer BT-549, prostate cancer DU 145, NSCLC NCI-H460, and colon cancer HCC-2998 cell lines.
In continuation of our ongoing systematic bioassay-guided investigation aiming to discover new and strongly antiproliferative natural products from Malagasy plant extracts, we selected the EtOH extract of the roots of Pongamiopsis pervilleana (Baill.) R. Vig. (Leguminosae, subfam. Papilionoideae) on the basis of its activity against the A2780 ovarian cancer cell line (IC50 5.8 μg/mL). Plants from this family are a rich source of bioactive prenylated flavonoids, isoflavonoids, and pterocarpans.2,3 The genus Pongamiopsis consists of the three species P. amygdalina (Baill.) R. Vig., P. pervilleana, and P. viguieri Du Puy & Labat, and is a genus endemic to Madagascar.4 P. pervilleana is a deciduous shrub to small tree ca. 1.5 to 14 m tall with mauve to lilac pink flowers growing in dry deciduous woodland and xerophitic scrubland in both the southern and northern parts of Madagascar.5 No previous phytochemical investigation has been reported from any of the species of this genus.
Results and Discussion
Liquid-liquid fractionation of an EtOH extract of the roots of P. pervilleana yielded a bioactive hexane fraction, and HPLC separation of this fraction on a C-18 reversed phase column led to the isolation of the four compounds 2–5. Compound 1 was obtained by preparative TLC of an HPLC fraction.
High resolution ESIMS analysis of compound 1 gave a quasi-molecular ion peak at m/z 367.1176 [M+H]+, corresponding to the molecular formula C21H18O6. Its 1H NMR spectroscopic data (Table 1) exhibited four singlet resonances due to four aromatic protons (δ 7.24, 6.81, 6.37, and 6.29), signals for an exomethylene group (δ 5.21, br s), two oxymethylene groups (δ 4.22, 1H, dd, J = 10.7, 4.6 Hz and 3.57, 1H, t, J = 10.7 Hz, and δ 4.15, 2H, s), two oxygen-bearing methines (δ 5.47, d, J = 7.0 Hz and δ 5.31, t, J = 7.5 Hz), a methylenedioxy group (δ 5.87, d, J = 12.5 Hz) and a benzylic methylene (δ 3.07, 1H, dd, J = 14.9, 7.5 Hz and δ 3.37, 1H, m). The UV spectrum showed a major absorption at 295 nm and a smaller band at 220 nm, typical absorptions of pterocarpans.6 The 13C NMR data (Table 1) displayed 21 signals which were similar to those of emoroidocarpan (2) except for the presence of a signal for an oxymethylene carbon at δ 62.6 instead of the methyl group at δ 17.1 in 2. Two-dimensional NMR experiments including COSY, HMQC and HMBC were carried out in order to assign the functional groups present in the molecule. The location of the oxymethylene group at C-3′ was substantiated by the observation of HMBC cross-peaks from CH2-4′ to C-2′ (δ 85.4) and C-5′ (δ 110.8). The presence of long-range correlations between the methylenedioxy protons at δ 5.87 and the carbon signals at δ 143.2 (C-8) and δ 149.4 (C-9) indicated that the methylenedioxy group must be attached at C-8 and C-9. From the above evidence, compound 1 was determined to be (2′R)-4′-hydroxyemoroidocarpan.
Table 1.
1H and 13C NMR Data for Compounds 1, 2, and 4 (500 and 125 MHz)a
| position | 1 (methanol-d4) | 2 (methanol-d4) | 4 (methanol-d4) | |||
|---|---|---|---|---|---|---|
| 1H | 13C | 1H | 13C | 1H | 13C | |
| 1 | 7.24 s | 127.9 | 7.23 s | 127.7 | 7.31 s | 128.1 |
| 1a | 113.8 b | 113.7 | 113.8 | |||
| 2 | 121.8 b | 122.0 | 122.0 | |||
| 3 | 162.2 | 162.4 | 162.3 | |||
| 4 | 6.29 s | 98.7 | 6.27 s | 98.5 | 6.28 s | 98.5 |
| 4a | 157.3 | 157.4 | 157.5 | |||
| 6 | 4.22 dd (10.7, 4.6) 3.57 t (10.7) |
67.5 | 4.23 dd (10.7, 4.6) 3.56 t (10.7) |
67.5 | 4.22 dd (10.7, 3.6) (6α) 3.55 t, (10.7) (6β) |
67.6 |
| 6a | 3.49 ddd (10.7, 7.0, 4.6) | 41.5 | 3.49 ddd (10.7, 7.0, 4.6) | 41.4 | 3.51 m | 41.4 |
| 6b | 121.8 b | 120.9 | 120.5 | |||
| 7 | 6.81 s | 106.0 | 6.81 s | 105.9 | 6.74 d, (8.1) | 115.4 |
| 8 | 143.2b | 143.6 | 6.50 d (8.1) | 105.8 | ||
| 9 | 149.4 b | 149.3 | 150.2 | |||
| 10 | 6.37 s | 94.2 | 6.37 s | 94.1 | 131.7 | |
| 10a | 155.5 b | 155.4 | 156.4 | |||
| 11a | 5.47 d (7.0) | 80.3 | 5.47 d (7.0) | 80.3 | 5.49 d (6.3) | 80.6 |
| 1′ | 3.07 dd (14.9, 7.5) 3.37 m |
35.3 | 2.98 dd (14.9, 7.5) 3.33 overlapped |
34.7 | 3.31 m 2.99 dd, (15.3, 7.8) |
34.8 |
| 2′ | 5.31 t (7.5) | 85.4 | 5.20 t (7.5) | 87.8 | 5.20 t (8.75) | 87.9 |
| 3′ | 143.2 b | 145.6 | 145.8 | |||
| 4′ | 4.15 s | 62.6 | 1.75 s | 17.1 | 1.75 s | 17.2 |
| 5′ | 5.21 br s | 110.8 | 5.05 br s | 112.0 | 5.05 brs | 112.1 |
| -OCH2O- | 5.85 d (1.2) 5.88 d (1.2) |
102.5 | 5.85 br d (0.8) 5.88 br d (0.8) |
102.4 | ||
| OCH3 | 3.82 s | 56.9 | ||||
All assignment were based on the COSY, HSQC and HMBC experiments
The 13C NMR shifts of C-1a, C-2, C-6b, C-8, C-9, C-10a and C-3′ for compound 1 were observed by HMBC.
The 6a,11a cis-configuration was assigned on the basis of the coupling constants of the 6a and11a protons (d, J = 7.0 Hz), and the (6aR,11aR) absolute configuration on the negative Cotton effect observed at 240 nm in the CD spectrum of 1.7 The 2′R absolute configuration was assigned on the basis of the negative Cotton effect observed at 475 nm in the CD spectrum of the osmate ester/pyridine complex of 1. 8 Thus the structure of 1 was deduced as 2′R,4′-hydroxyemoroidocarpan.
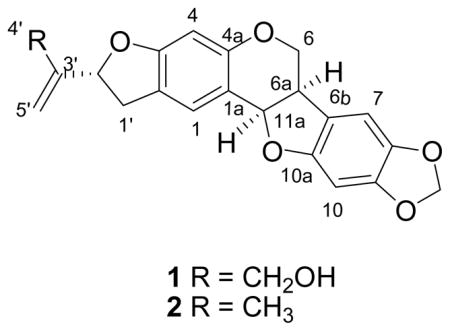
Compound 2 was identified as the known emoroidocarpan by interpretation of its physical and spectroscopic data, and by comparison with reported values.2,9 Its configuration at C-2′ was determined to be R by measuring the CD of its osmate ester/pyridine complex, as described below for compound 4.
Compound 3, named pongavilleanine, had the molecular formula C22H18O6 as determined by positive ion HRESIMS. Its IR spectrum showed absorption bands at 1713, 1615, 1591, and 1224 cm−1 ascribable to a conjugated lactone carbonyl function and an aromatic ring. The 1H NMR spectrum displayed signals for the three aromatic protons of an ABX system (δ 6.94, d, J= 1.6 Hz, H-2′, δ 6.88, d, J = 8.0 Hz, H-5′ and δ 6.90, dd, J = 8.0, 1.6 Hz, H-6′) and the two protons of an AB system (δ 7.59, d, J = 8.7 Hz, H-5, and δ 6.74, d, J = 8.7 Hz, H-6), two cis-coupled olefin protons (δ 6.92, d, J = 10.0 Hz, H-1″, δ 5.71, d, J = 10.0 Hz, H-2″), a methylenedioxy group (δ 6.00, s, -OCH2O-), a methoxy group at δ 3.58 (s) and two quaternary methyl groups (δ 1.47, s, 6H). The 13C NMR spectrum displayed signals ascribable to a lactone carbonyl (δ 164.0, C-2), an oxygen-bearing unsaturated quaternary carbon (δ 163.9, C-4), oxygenated aromatic carbons (δ 156.6, 147.8, C-7 and C-8a, respectively), together with a set of signals due to a gem-dimethyldihydropyran ring (δ 130.6, 115.6, 77.7, 28.2, 28.2). Analysis of its 1H and 13C NMR data (Table 2) revealed that compound 3 is a prenylated 3-arylcoumarin.
Table 2.
1H and 13C NMR Data for Compound 3 (500 and 125 MHz)a
| position | 1H | 13C |
|---|---|---|
| 2 | 164.0 | |
| 3 | 109.1c | |
| 4 | 163.9 | |
| 4a | 109.1c | |
| 5 | 7.59 d (8.7) | 124.0 |
| 6 | 6.74 d (8.7) | 113.4 |
| 7 | 156.6 c | |
| 8 | 111.1 c | |
| 8a | 147.8c | |
| 1′ | 126.1 | |
| 2′ | 6.94 d (1.6) | 111.7 c |
| 3′ | 147.8c | |
| 4′ | 147.8c | |
| 5′ | 6.88 d (8.0) | 108.4 |
| 6′ | 6.90 dd (8.0, 1.6) | 125.0 |
| 1″ | 6.92 d (10.0) | 115.7 |
| 2″ | 5.71 d (10.0) | 130.6 |
| 3″ | 77.7c | |
| 4″ | 1.47 s | 28.2c |
| 5″ | 1.47 s | 28.2 |
| -OCH2O- | 6.00 s | 101.4 |
| OCH3 | 3.58 s | 61.2 |
InCDCl3. All assignment were based on COSY, HSQC and HMBC experiments
The 13C NMR shifts of C-3, C-7, C-8, C-4a, C-8a, C-2′, C-3′, C-4′ and C-3″ were observed by HMBC.
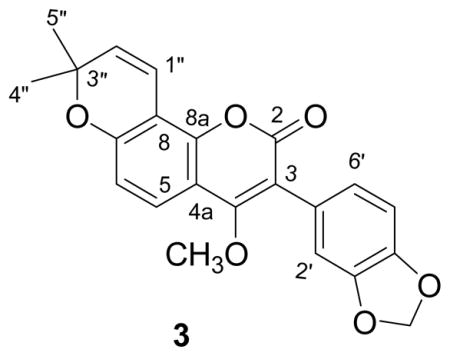
Since not all of the expected 22 carbon resonances of the molecular formula were observed in its 13C NMR spectrum, 2D-NMR experiments were carried out in order to assign all the protons and carbons of 3. An HMBC correlation was observed between the methoxy protons at δ 3.58 and the carbon signal at δ 163.9, demonstrating that the methoxy group must be attached at C-4. The second signal at δ 164.0 was assigned to the C-2 carbonyl group. The methylenedioxy protons at δ 6.00 showed only one long range correlation to a carbon or carbons at δ 147.8, indicating that C-3′ and C-4′ are magnetically equivalent. Moreover, the HMBC correlations (Figure 1) observed between the proton at δ 6.94 (H-2′) and C-3, C-3′, C-4′, and C-6′ indicated that the attachment of the methylenedioxy group must be at C-3′ and C-4′. The location of the gem-dimethyldihydropyran ring at C-7 and C-8 was substantiated by the observation of long range correlations from the cis-coupled olefin proton signal at δ 5.71 (H-2″) and the carbon signals due to the methyl groups (δ C 28.2) to the oxygen-bearing quaternary carbon at C-3″ (δ C 77.7). H-2″ also had a correlation to the C-8 carbon signal at δ C 111.7. In addition the HMBC cross peak between H-5 and the carbon signals at δ 163.9 (C-4), δ 156.6 (C-7), and δ 147.8 (C-8a) indicated the location of the remaining oxygenated aromatic carbon to be at C-7, which must be the site of cyclization of the gem-dimethyldihydropyran. A ROESY experiment confirmed the position of the methoxy group with correlations between the methoxy signal and H-5 and H-2′ (Figure 1). No HMBC correlation was observed from the lactone carbonyl at δ 164.0. These data led to the assignment of structure 3 to pongavilleanine.
Figure 1.

Important HMBC (left) and ROESY (right) correlations observed for 3
Confirmation of the structure of pongavilleanine was obtained by single-crystal X-ray diffraction, and an anisotropic displacement ellipsoid drawing is shown in Figure 2. Its structure was thus firmly established as 3.
Figure 2.
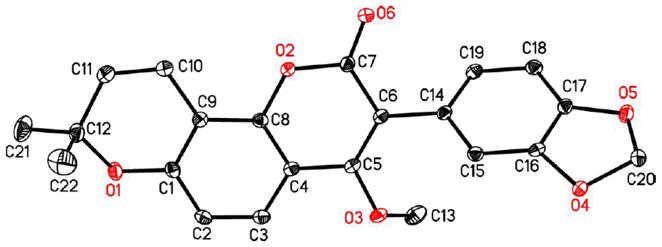
Crystal structure of 3.
Anisotropic displacement ellipsoid drawing of compound 3.
The molecular composition of compound 4 was determined to be C21H20O5 by HREIMS, and its 1H- and 13C NMR data were similar to but not identical with those of pervilline (6) (Table 1).2 The absolute configuration of pervilline has been determined as 6aR,11aR,2 but the configuration of C-2′ has not been determined. We thus prepared the osmate ester/pyridine complex of 4, since such non-planar complexes are known to be twisted, and the direction of the twist is determined by the configuration of an adjacent stereogenic center.8 The osmate ester/pyridine complex of 4 had a negative Cotton effect at 468 nm, indicating an R configuration at C-2′. Although the 1H-NMR data of compound 4 and pervilline were obtained in different solvents (CD3OD and CDCl3, respectively), it is worth noting that the 1H-NMR chemical shifts (in ppm) of H-1 and H-4 are shifted upfield in 4 (7.31, H-1; 6.28, H-4), compared with those of pervilline (7.33, H-1; 6.35, H-4), while those of H-7 and H-8 are shifted downfield in 4 (6.74, H-7; 6.50, H-8) versus 6.72 and 6.42 in pervilline. Interestingly, a positive Cotton effect was observed at 240 nm, which is the opposite of that observed for pervilline, demonstrating that both C-6a and C-11a have the S configuration. The structure and absolute configuration of 4 were thus determined as 6a,11a-epipervilline.6
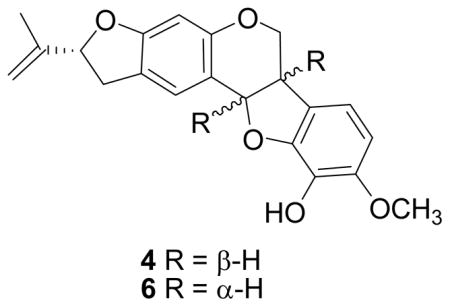
Since compounds 4 and 6 both have negative optical rotations they cannot be enantiomers, and since they differ in their configurations at C-6a and C-11a it follows that they must be diastereomers having the same configuration at C-2′. This work thus assigns the 2′R,6aR,11aR configuration to pervilline and the 2′R,6aS,11aS configuration to compound 4.
The structure of rotenolone (5) was identified by comparison of its spectroscopic data with reported values.3,10
Compounds 1–5 were evaluated for their antiproliferative activity against the A2780 human ovarian cancer cell line. Among the five compounds, compound 5 showed significant activity (IC50 0.95 μM), while compounds 2–4 displayed only weak activity (IC50 26.5, 9.5, and 23.2μM, respectively). Compound 1 (IC50>54.4 μM), which is a simple C-4′ oxidation product of 2, had even less antiproliferative activity than 2, suggesting that the presence of the 2″-isopropenyldihydrofuran unit is important for this activity. Rotenolone (5) was also evaluated against the breast cancer BT-549, prostate cancer DU 145, NSCLC NCI-H460, and colon cancer HCC-2998 cell lines, and it had IC50 values of 1.6, 2.7, 2.0, and 2.9 μM, respectively, in these assays. This work indicates that rotenolone (5) is the major antiproliferative component in P. pervilleana, just as it is the one of the major antiproliferative constituents of Mundulea chapelieri3 and of Derris trifoliate.11 It is interesting to note that rotenolone is one of the acaricidal constituents of Neorautanenia mitis,12 which also contains the pterocarpan neoduline, the structure of which was determined by X-ray crystallography.13
Experimental Section
General Experimental Procedures
Optical rotations were recorded on a JASCO P-2000 polarimeter. IR and UV spectra were measured on MIDAC M-series FTIR and Shimadzu UV-1201 spectrophotometers, respectively. CD analysis was performed on a JASCO J-810 spectropolarimeter with a 0.1 cm cell in DMSO at room temperature under the following conditions: speed 50 nm/min, time constant 1 s, bandwidth 2.0 nm. 1H and 13C NMR spectra were recorded on JEOL Eclipse 500 and Bruker 600 spectrometers in CDCl3 and methanol-d4 with TMS as internal standard. Mass spectra were obtained on JEOL JMS-HX-110, Agilent 6220 LC-TOF-MS, or Finnigan LTQ LC/MS instruments. Preparative HPLC was performed using Shimadzu LC-10AT pumps coupled with a semipreparative Varian Dynamax C-18 column (5μm, 250x10 mm), a Shimadzu SPD M10A diode array detector (DAD) and a SCL-10A system controller.
Antiproliferative Bioassays
The A2780 ovarian cancer cell line assay was performed at Virginia Polytechnic Institute and State University as previously reported.14 The A2780 cell line is a drug-sensitive ovarian cancer cell line.15 Assays against the breast cancer BT-549, prostate cancer DU 145, NSCLC NCI-H460, and colon cancer HCC-2998 cell lines were carried out at Eisai, Inc., as previously described for similar cell lines.16
Plant Material
Roots of P. pervilleana (collection: S. Randrianasolo et al. 558) were collected in the forest of South Bekaraoka, Andranotsimaty (10 km from Daraina, Antsiranana, Sava region, 13°11′13″S 049°42′40″E, Northern Madagascar). The sample collected was from a tree 15 m tall, 40 cm diameter at breast height, with violet floral buttons and yellow anthers attractive to bees. The identification of the plant was assured by M. Callmander. Duplicate voucher specimens were deposited at Centre National d’Application des Recherches Pharmaceutiques (CNARP), the Herbarium of the Parc Botanique et Zoologique de Tsimbazaza, Antananarivo, Madagascar (TAN), the Missouri Botanical Garden, St. Louis, Missouri (MO), and the Museum National d’Histoire Naturelle in Paris, France (P).
Extraction and Isolation
A ground sample of P. pervilleana roots (250 g) was extracted with EtOH at room temperature to yield 12.2 g of crude EtOH extract designated MG 3698. A total of 3.1 g of this extract was made available to Virginia Polytechnic Institute and State University. The crude EtOH extract (1.0 g) was dissolved in MeOH (300 mL) and extracted with n-hexane (3 x 200 mL) to afford 151.2 mg of residue after evaporation of the hexane soluble fraction. The MeOH layer was then evaporated, suspended in H2O (300 mL) and extracted with EtOAc (3 x 200 mL) to yield 179.3 mg of EtOAc soluble fraction. The aqueous layer was concentrated to give 603.7 mg of brown residue. The n-hexane extract was found to be cytotoxic (IC50 0.38 μg/mL) and was subjected to HPLC on a C-18 column with a solvent gradient from H2O:MeOH 30:70 to 20:80 for 10 min, to 10:90 from 10 to 15 min, to 05:95 from 15 to 20 min and to 0:100 from 20 min to 25 min, ending with 100% MeOH to 35 min. This yielded compounds: 5 (2.8 mg, tR: 14.99 min), 4 (1.5 mg, tR: 19.02 min), 3 (2.5 mg, tR: 23.27 min), and 2 (1.8 mg, tR: 24.48 min). A peak at tR 16.04 min was further purified by preparative TLC on silica gel (solvent system: hexanes: EtOAc; 1:1) to give compound 1 (2.2 mg).
2′R,4′-Hydroxyemoroidocarpan (1)
Amorphous powder, [α]D −220 (c 0.1, CHCl3); CD: [θ]240−41300 (c 0.5, MeOH); UV (MeOH) λmax nm (log ε): 295 (2.9), 220 (3.8); 1H NMR and 13C NMR spectral data, see Table 1; positive HRESIMS m/z 367.1176 [M+H]+ (calcd for C21H19O6, 367.1171).
Pongavilleanine (3)
Crystals from EtOAc/hexanes, mp 178-182 °C; UV (MeOH) λmax nm (log ε): 325 (4.22), 240 (4.22); IR (film) 1713, 1615, 1591, 1224 and 1024cm−1; 1H NMR and 13C NMR spectra, see Table 2; positive ion HRESIMS m/z 379.1176 [M+H]+ (calcd for C22H19O6, 379.1176).
X-ray Crystallography of 3
A colorless plate was cut (0.33 x 0.28 x 0.14 mm3), mounted, and centered on the goniometer of an Oxford Diffraction Gemini A Ultra diffractometer operating with MoKα radiation. The data collection routine, unit cell refinement, and data processing were carried out with the program CrysAlisPro.17 The Laue symmetry and systematic absences were consistent with the orthorhombic space group Pbca. The structure was solved by direct methods and refined using SHELXTL NT.18 The final refinement model involved anisotropic displacement parameters for all non-hydrogen atoms and a riding model for all hydrogen atoms.
Crystal data
Colorless crystals; C22H18O6, Mr =378.36, orthorhombic, P212121, a = 16.9599(3) Å, b = 9.85826(14) Å, c = 21.1449(3) Å, a = 90.00, b = 90.00, c = 90.00,V = 3535.34(9) Å3, 31228 reflections, 256 parameters. The atomic coordinates and equivalent isotropic displacement parameters, as well as a full list of bond distances and angles, and the structure factor table are deposited as supplementary material at the Cambridge Crystallographic Data Centre (Deposition No. CCDC 760992).
Epipervilline (4)
Amorphous powder, (c 0.2, CHCl3); [θ]240+47,800 (c 2, MeOH); UV (MeOH) λmax nm (log ε): 290 (4.02), 230 (3.5); 1H NMR and 13C NMR spectra, see Table 1; Positive ion HRESIMS m/z 353.1383 [M+H]+ (calcd for C21H21O5, 353.1380).
Preparation and CD Measurement of Osmate Esters
Compounds 1, 2, 4, and 5 (each 1.3 μmol) were dissolved in CH2Cl2 (62 μL) containing 25 μmol (2.3 μL) of pyridine, and the mixtures were then treated with OsO4 (1.36 μmol in 10 μL of CH2Cl2) for about 30 min at rt. Each mixture was diluted with CH2Cl2 to give a final volume of 2.8 mL. The CD spectra of the resulting osmate ester/pyridine complexes showed negative Cotton effects at 475 nm ([θ] −3298 (1), 474 nm ([θ] −2382 (2), 468 nm ([θ] −3627 (4), and 479 nm ([θ] −4957 (5).
Supplementary Material
Acknowledgments
This project was supported by the Fogarty International Center, the National Cancer Institute, the National Science Foundation, the National Heart, Lung and Blood Institute, the National Institute of Mental Health, the Office of Dietary Supplements, and the Office of the Director of NIH, under Cooperative Agreement U01 TW000313 with the International Cooperative Biodiversity Groups. This project was also supported by the National Research Initiative of the Cooperative State Research, Education and Extension Service, USDA, Grant #2008-35621-04732. These supports are gratefully acknowledged. This work was also supported by the National Science Foundation under Grant No CHE-0619382 for purchase of the Bruker Avance 600 NMR spectrometer and Grant No. CHE-0722638 for the purchase of the Agilent 6220 mass spectrometer. We thank Mr. B. Bebout for obtaining the mass spectra. Field work essential for this project was conducted under a collaborative agreement between the Missouri Botanical Garden and the Parc Botanique et Zoologique de Tsimbazaza and a multilateral agreement between the ICBG partners, including the Centre National d’Applications des Recherches Pharmaceutiques. We gratefully acknowledge courtesies extended by the Government of Madagascar (Ministère des Eaux et Forêts).
Footnotes
Supporting Information Available: 1H and 13C NMR spectra of compounds 1–5, HMBC spectra of compounds 1, 3 and 4; and 1H and 13C-NMR data of compound 5. This information is available free of charge via the Internet at http://pubs.acs.org. Crystallographic data for compound 3 reported in this paper have been deposited with the Cambridge Crystallographic Data Centre, Deposition No. CCDC 760992. Copies of the data can be obtained, free of charge, on application to the Director, CCDC, 12 Union Road, Cambridge CB2 1EZ, UK (fax: +44 1223 336 033 or deposit@ccdc.cam.ac.uk).
References and Notes
- 1.Biodiversity Conservation and Drug Discovery in Madagascar, Part 44. For Part 43, see Cao S, Hou Y, Brodie P, Miller JS, Randrianaivo R, Rakotobe E, Rasamison VE, Kingston DGI. Chem Biodiversity. 2010 doi: 10.1002/cbdv.201000061.
- 2.Pallazzino G, Rasoanaivo P, Federici E, Nicoletti M, Galeffi C. Phytochemistry. 2003:63, 471–474. doi: 10.1016/s0031-9422(02)00489-2. [DOI] [PubMed] [Google Scholar]
- 3.Cao S, Schilling JK, Miller JS, Andriantsiferana R, Rasamison V, Kingston DGI. J Nat Prod. 2004:67, 454–456. doi: 10.1021/np0303815. [DOI] [PubMed] [Google Scholar]
- 4.Du Puy DJ, Labat J-N, Rabevohitra R, Villiers J-F, Bosser J, Moat J. The Leguminosae of Madagascar. Royal Botanical Gardens; Kew: 2002. p. 720. [Google Scholar]
- 5. [Accessed June 25, 2010]; http://www.tropicos.org/SpecimenDetails.aspx?specimenid=2930626&langid=10.
- 6.Slade D, Ferreira D, Marais JPJ. Phytochemistry. 2005;63:2177–2215. doi: 10.1016/j.phytochem.2005.02.002. [DOI] [PubMed] [Google Scholar]
- 7.Baruah P, Barua NC, Sharma RP, Baruah JN, Kulanthaivel P, Herz W. Phytochemistry. 1984:23, 443–447. [Google Scholar]
- 8.Tahara S, Ingham JL, Mizutani J. Agric Biol Chem. 1987:51, 211–216. [Google Scholar]
- 9.Machocho AK, Lwande W, Jondiko JI, Moreka LVC, Hassanali A. Int J Pharmacog. 1995:33, 222–227. [Google Scholar]
- 10.Malgahaes AF, Tozzi AGMA, Sales NLHB, Malgahaes GE. Phytochemistry. 1996:42, 1459–1471. [Google Scholar]
- 11.Cheenpracha S, Karalai C, Ponglimanont C, Chantrapromma K. Can J Chem. 2007:85, 1019–1022. [Google Scholar]
- 12.Van Puyvelde L, De Kimpe N, Mudaheranwa JP, Gasiga A, Schamp N, Declercq JP, Van Meerssche M. J Nat Prod. 1987:50, 349–356. [Google Scholar]
- 13.Declercq JP, Van Puyvelde L, De Kimpe N, Schamp N. Bull Soc Chim Belg. 1986:95, 677–678. [Google Scholar]
- 14.Cao S, Brodie PJ, Randrianaivo R, Ratovoson F, Callmander M, Andriantsiferana R, Rasamison VE, Kingston DGI. J Nat Prod. 2007:70, 679–681. doi: 10.1021/np060627g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Louie KG, Behrens BC, Kinsella TJ, Hamilton TC, Grotzinger KR, McKoy WM, Winker MA, Ozols RF. Cancer Res. 1985:45, 2110–2115. [PubMed] [Google Scholar]
- 16.Murphy BT, Brodie PJ, Slebodnick C, Miller JS, Birkinshaw C, Randrianjanaka LM, Andriantsiferana R, Rasamison VE, TenDyke K, Suh EM, Kingston DGI. J Nat Prod. 2008:71, 325–329. doi: 10.1021/np070487q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.CrysAlisPro v171.33.31. Oxford Diffraction; Wroclaw, Poland: 2009. [Google Scholar]
- 18.Sheldrick GM. Acta Cryst. 2008:A64, 112–122. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.