

Abstract
We described in a previous communication (ref. 13) a variant of the popular CuI-catalyzed azide-alkyne cycloaddition (AAC) process where 5 mol% Cu(OAc)2 in the absence of any added reducing agent is sufficient to enable the reaction. 2-Picolylazide (1) and 2-azidomethylquinoline (2) were found to be by far the most reactive carbon azide substrates that convert to 1,2,3-triazoles in as short as a few minutes under the discovered conditions. We hypothesized that the abilities of 1 and 2 to chelate CuII contribute significantly to the observed high reaction rates. The current work examines the effect of auxiliary ligands near the azido group other than pyridyl for CuII on the efficiency of the Cu(OAc)2-accelerated AAC reaction. The carbon azides capable of binding to the catalytic copper center at the alkylated azido nitrogen in a chelatable fashion were indeed shown to be superior substrates under the reported conditions. The chelation between carbon azide 11 and CuII was demonstrated in an X-ray single crystal structure. In a limited set of examples, the ligand tris(benzyltriazolylmethyl)amine (TBTA), developed by Fokin et al. for assisting the original CuI-catalyzed AAC reactions (ref. 8), also dramatically enhances the Cu(OAc)2-accelerated AAC reactions involving non-chelating azides. This observation leads to the hypothesis of an additional effect of chelating azides on the efficiencies of Cu(OAc)2-accelerated AAC reactions, which is to facilitate the rapid reduction of CuII to highly catalytic CuI species. Mechanistic studies on the AAC reactions with particular emphasis on the role of carbon azide/copper interactions will be conducted based on the observations reported in this work. Finally, the immediate utility of the product 1,2,3-triazole molecules derived from chelating azides as multidentate metal coordination ligands is demonstrated. The resulting triazolyl-containing ligands are expected to bind with transition metal ions via the N(2) nitrogen of the 1,2,3-triazolyl group to form non-planar coordination rings. The CuII complexes of bidentate T1 and tetradenetate T6, and the ZnII complex of T6 were characterized by X-ray crystallography. The structure of [Cu(T1)2(H2O)2](ClO4)2 reveals the interesting synergistic effect of hydrogen bonding, π-π stacking interactions, and metal coordination in forming a one-dimensional supramolecular construct in the solid state. The tetradentate coordination mode of T6 may be incorporated into designs of new molecule sensors and organometallic catalysts.
Keywords: Azide-alkyne cycloaddition, copper(II) acetate, azide-copper complex, triazole ligands, TBTA, click reaction
Introduction
As one of the most operationally simple organic transformations, the CuI-catalyzed azide-alkyne cycloaddition (AAC) reaction has facilitated advances in many areas of science.1 The rich, yet elusive mechanistic variability of this reaction has engendered a large number of procedures developed for specific substrate classes and environmental constraints.2 A procedure for an AAC reaction typically consists of the following components – copper source (e.g. CuII salts,3 CuI salts4), reducing agent (e.g. sodium ascorbate,3 elemental copper,5 phosphorous(III) agents,6 electrochemical reduction,7 etc.), solvent (aqueous, organic, or mixed), base (presumably for facilitating copper acetylide formation2), and accelerating ligand (various polyaza ligands).8,9 One major focus in the method development of CuAAC reactions has been to find conditions that are suited to various substrate classes to rapidly generate CuI-acetylide (blue steps in Figure 1) while protecting the +1 oxidation state of the copper catalyst.10 In a simplified, generally accepted mechanistic model (Figure 1),3 CuI-acetylide subsequently forms a complex with the carbon azide substrate, or reacts with it bimolecularly, in the rate-determining step11,12 to afford the metallacycle intermediate (red step in Figure 1) en route to the 1,2,3-triazole product. In this proposed mechanism, the involvement of CuI oxidation state is required. Therefore, when a CuII salt is used as the pre-catalyst, judicious choice for a reducing agent has to be made in order to generate and sustain an adequate amount of the highly catalytic CuI species without interfering with other functional groups in the reaction system.
Figure 1.

A simplified mechanistic proposal by Sharpless, Fokin et al.3 The CuI-acetylide formation steps are marked blue; the formation of six-membered metallacycle is marked red. L: a ligand or a counter ion associated with CuI/II. Not shown is the possible bimolecular pathway between CuI-acetylide and carbon azide to afford the metallacycle.
Our group is interested in developing 1,2,3-triazolyl-containing polydentate metal coordination ligands.14,15 Motivated by the need to develop simple protocols for CuAAC reactions to ease the purification of highly polar triazolyl-containing multidentate product molecules, we discovered that Cu(OAc)2 accelerates the AAC reaction in organic solvents, particularly in alcohols without deliberate addition of a reducing agent such as sodium ascorbate (see an example in Figure 2A).13,16 We postulated that in this procedure the catalytic CuI species emerges in a short induction period via reducing Cu(OAc)2 by alcoholic solvent or terminal alkyne in an oxidative homocoupling reaction.13 The direct observation of the homocoupled products and the formation of aldehyde species under similar AAC conditions by Mizuno et al.17 and Heaney et al.18,19 substantiated our hypothesis on the catalyst-producing induction process.
Figure 2.

(A) A Cu(OAc)2-accelerated AAC “click” reaction. (B) 2-Picolylazide forms a complex with CuCl2.13 The formal charges on Nβ and Cu are noted. (C) A simplified chelation model of the intermediate prior to the metallacycle formation en route to the 1,4-substituted 1,2,3-triazole.
In addition to the absence of an added reducing agent, consistent observations suggest that 2-picolylazide (1) and 2-azidomethylquinoline (2) are exceptional substrates under the CuII-accelerated conditions, where the nature of the alkyne component carries less impact in the efficiency of the reaction (unpublished observations). Due to the ability of these two carbon azides to chelate CuII (Figure 2B, the X-ray crystal structure was reported in ref. 13), the catalytic CuI center generated via the reductive induction period is biased to bind at the alkylated nitrogen (Nα in Figure 2B) of the azido group (CuI in the absence of chelation effect may favor coordination at the terminal Nγ position of the azido group, as shown by Dias et al.20). We hypothesized that the electrophilicity of the azido group is dramatically enhanced via chelation-assisted binding between CuI and Nα, thus accelerating the nucleophilic attack by the copper acetylide and the subsequent formation of the metallacycle intermediate (see a structural model prior to the metallacycle formation in Figure 2C) in the rate-determining step (red step in Figure 1).3 In this work, to test whether the chelation-elevated reactivity of carbon azide in Cu(OAc)2-accelerated AAC reactions observed in compounds 1 and 2 is generally applicable, we examined the efficiencies of the AAC reactions involving a series of carbon azides whose Nα may bind CuII via chelation. In the following section, we report (1) the reactivities of various carbon azides designed to chelate CuII, (2) the ligand-acceleration effect on the efficiencies of the AAC reactions involving non-chelating azides in the presence of Cu(OAc)2, and (3) the utility of the triazole products derived from the chelating azides as new multidentate metal coordination ligands. The conclusion from the first two points leads to testable mechanistic hypothesis on the Cu(OAc)2-accelerated AAC reactions which is currently under investigation in our laboratory.
Results and Discussion
1. Reactivities of chelating carbon azides with N-containing auxiliary ligands
Various carbon azides containing neighboring nitrogen donor ligands (1–11) were prepared. The yields of the AAC reactions in tBuOH involving these azides in the presence of 5 mol% Cu(OAc)2 without the addition of reducing agent are shown in Table 1. The azides capable of chelating CuII between the Nα of the azido group and a nitrogen-based auxiliary ligand to afford five-membered rings (1–7) undergo rapid AAC reactions (from minutes up to 3 h) with high yields (Table 1, entries 1, 3–6, 8–10). Nitrogen donor ligands of sp2 or sp3 hybridization yield no significant difference in reactivity. The results of two control experiments emphasize the significance of chelation over the basicity of the amino/pyridyl-based auxiliary ligands. First, the AAC reactions between 1-azidooctane and p-ethynylanisole in the presence of 1 molar equivalent of pyridine (entry 2) or N-methylpyrrolidine (entry 7) proceed rather sluggishly. Second, the completion times for reactions between 2-picolylazide (1) or N-(2-azidoethyl)pyrrolidine (4) and phenylacetylene are progressively longer as pyridine or N-methylpyrrolidine of increasing amounts were added to compete in coordination to CuII (see SI). When the auxiliary ligand is a 2-fluoropyridyl group (entries 11 and 12), the reactions take a considerably longer time to reach completion. Presumably the donor strength of the pyridyl nitrogen is weakened by the presence of the electron-withdrawing fluorine substituent.
Table 1.
Cu(OAc)2-accelerated AAC reactions of chelating azides with N-containing auxiliary ligandsa
| entry | azide | product | timec | yieldd |
|---|---|---|---|---|
| 1 |  |
 |
1 h | 94% |
| 2 |  |
 |
6 h | 16% |
| 3 |  |
 |
40 min | 95% |
| 4 |  |
 |
3 h | 96% |
| 5 |  |
 |
23 min | 88% |
| 6 |  |
 |
3 h | 76% |
| 7 |  |
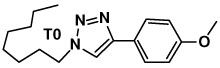 |
24 h | 5% |
| 8 |  |
 |
2 h | 84% |
| 9b |  |
 |
10 min | 88% |
| 10 |  |
 |
10 min | 68% |
| 11 |  |
 |
24 h | 64% |
| 12 |  |
 |
24 h | 75% |
| 13 |  |
 |
24 h | 91% |
| 14 |  |
24 h | 92% | |
| 15 |  |
 |
3 h | 99% |
| 16 |  |
 |
5 min | 96% |
General conditions: azide (0.2 mmol), alkyne (0.2–0.22 mmol), tBuOH (0.5 mL) and Cu(OAc)2 (25 µL, 0.4 M solution in H2O, 10 µmol).
py = 2-pyridyl.
Reaction time refers to the time it takes for the azide substrate to be undetectable by TLC.
Isolated yield after purification by column chromatography.
Azides 10 and 11 are able to chelate with CuII to form six-membered rings. Azide 11 reacts smoothly to afford the triazole products (Table 1, entries 15 and 16). More time was required for 10 to reach complete conversion in the AAC reactions (Table 1, entries 13 and 14), which may be in part attributed to the impaired binding of 10 to the copper center due to the presence of a longer alkyl chain.
The chelating ability of azide 11 was revealed by the X-ray crystal structure of its complex with CuCl2 (Figure 3). Green, air-stable [Cu2(11)2Cl4] crystallizes in P2(1)/c space group via vapor diffusion of diethyl ether to a methanolic solution of 11 and CuCl2 at equal molar ratio.21 The Nα of the azido group in conjunction with the pyridyl nitrogen binds CuII to form a six-membered chelation ring. In contrast to our previously reported [Cu2(1)2Cl4] where the square pyramidal CuII geometry was observed,13,22 the CuII centers in this dimeric complex adopt a distorted trigonal bipyramidal form where the pyridyl nitrogen (N4 in Figure 3) and one of the bridging chlorides (Cl1a) are at the apical positions. A [Cu2Cl2] rectangle core results from the ~90° angle of Cl1-Cu1-Cl1a. The six-membered chelation ring is in a boat conformation. The bond angles around the alkylated nitrogen N1 are ~120°, suggesting a sp2 hybridized N1.
Figure 3.

ORTEP (50% ellipsoids) diagrams of [Cu2(11)2Cl4]. Selected distances (Å): Cu1–Cl1 2.419, Cu1–Cl2 2.245, Cu1–N1 2.201, Cu1–N4 1.994, Cu-Cl1a 2.317, N1–N2 1.243, N2–N3 1.123.
2. Reactivities of carbon azides with O- and S-containing auxiliary ligands
Carbon azides containing potentially chelating oxygen- and sulfur-based donor ligands were evaluated. The reactions between the neutral carboxylic acids 12 and 13 and p-ethynylanisole proceed very sluggishly. In the presence of 1 molar equivalent of Hünig’s base (N,N-diisopropylethylamine), the deprotonated azidocarboxylates undergo rapid AAC reactions aided by 5 mol% Cu(OAc)2 (Table 2, entries 1 and 2). As shown in a control experiment, the reaction between 1-azidooctane and p-ethynylanisole was affected little by the addition of Hünig’s base. After 55 h, the isolated reaction yields in the presence and absence of Hünig’s base were only 25% and 19%, respectively. Evidently, in Entries 1 and 2 (Table 2), the role of Hünig’s base is restricted to deprotonation of the azidocarboxylic acids. It is at best only marginally involved in the deprotonation of the alkyne and/or CuII reduction. This observation reaffirms that chelation of CuII enabled by carboxylate formation is instrumental in the rate acceleration.
Table 2.
AAC reactions of azides with O and S auxiliary donorsa











General conditions: azide (0.2 mmol), alkyne (0.2–0.22 mmol), tBuOH (0.5 mL) and Cu(OAc)2 (25 µL, 0.4 M solution in H2O, 10 µmol).
N,N-diisopropylethylamine (0.2 mmol) was added.
Isolated yield after purification by column chromatography.
Carbon azides containing ether- and sulfide-based auxiliary donors were allowed to react over extended periods of time (15–24 h) with poor yields (Table 2, entries 3–6). The weaker Lewis basicities of the oxygen and sulfur donors in ethers and sulfides than that of amino/pyridyl groups when paired with the Lewis acidic CuII may result in weak binding, hence insufficient activation of the azido group in the AAC reactions.
3. Assisting ligand effect in the Cu(OAc)2-accelerated process
During the development of CuI-catalyzed AAC reactions, the likelihood of autocatalysis to result in rate acceleration was considered by Fokin, Finn, and others.8,9,23,24 Similar autocatalytic processes might operate in the Cu(OAc)2-accelerated AAC reactions where the bidentate triazole product serves as an accelerating ligand. Three bidentate triazole compounds derived from 2-picolylazide (T1, T18, and T19)25 were applied as additives in the AAC reactions that barely proceed in the presence of 5 mol% Cu(OAc)2 (Table 3, entries 1–6). The ligands were found to moderately accelerate the AAC reactions.26 The moderate, yet consistent accelerating effect of bidentate triazoles suggests the possibility of autocatalysis.
Table 3.
Effect of triazole ligands as additivesa
| entry | additive | product | time | yield (%)b |
|---|---|---|---|---|
| 1 |  |
 |
24 h | 12 (13) |
| 2 |  |
 |
3 h | 38 (13) |
| 3 |  |
 |
24 h | 23 (13) |
| 4 |  |
 |
4 h | 40 (17) |
| 5 |  |
 |
3 h | 46 (5) |
| 6 |  |
 |
3 h | 54 (5) |
| 7c |  |
 |
1 h | 90 |
| 8c | TBTA |  |
1 h | 98 |
| 9d | TBTA |  |
2 h | 93 |
| 10d | TBTA |  |
2 h | 90 |
| 11d | TBTA |  |
2 h | 95 |
General conditions: azide (0.2 mmol), triazole additive (0.02 mmol), alkyne (0.2–0.22 mmol), tBuOH (0.5 mL) and Cu(OAc)2 (25 µL, 0.4 M solution in H2O, 10 µmol).
Isolated yields after column chromatography; values in parentheses represent yields of isolated products in the absence of triazole additives.
10 mol% TBTA.
2 mol% TBTA.
In a study by Fokin et al., a polytriazole compound, tris(benzyltriazolylmethyl)amine (TBTA) was found to be an outstanding co-catalyst for the CuI-catalyzed AAC reactions.8 We also observed an astounding acceleration effect by TBTA in the presence of 5 mol% Cu(OAc)2 without the addition of sodium ascorbate. For the sluggish reactions involving non-chelating azides, the addition of as low as 2 mol% TBTA resulted in complete conversions in less than 2 h (entries 7–11, Table 3), much faster than what could be achieved with the assistance of bidentate triazole additives. The stabilization effect on the CuI oxidation state and delicately balanced ligand exchange properties of TBTA have been cited as the source of acceleration in the CuI-catalyzed AAC reactions.8,10,24 Under the Cu(OAc)2-accelerated conditions, the presence of TBTA may increase the thermodynamic driving force for the reduction of CuII during the induction period to rapidly produce a highly catalytic CuI species for the AAC reactions. This consideration leads to the hypothesis that chelating azides may also accelerate the CuII reduction step (blue in Figure 1) to afford highly catalytic CuI species in a similar manner as that of TBTA, in addition to aid the metallacycle formation step (red step in Figure 1). The detailed mechanistic studies on the effect of chelation-enabling auxiliary ligands on both CuII reduction and metallacycle formation steps in the AAC reactions is currently under way.
4. The metal coordination properties of polyaza 1,2,3-triazolyl-containing product molecules
As a corollary of this work, we note that an immediate utility of most of the triazole products derived from chelating carbon azides is metal ion chelation. We and Crowley et al. described earlier that 2-picolylazide-derived 1,2,3-triazoles (e.g. T19 in Table 3) chelate CuII via coordination at pyridyl and triazole N2 nitrogen atoms.13,27 Complexes of other metal ions of T19 were subsequently reported.28 In this work, the 2:1 (ligand/CuII) complex of T1 was characterized by X-ray crystallography (Figure 4A). A Jahn-Teller distorted octahedral CuII center was observed where the two bidentate T1 ligands constitute the equatorial plane. Instead of CH3CN solvent molecule, perchlorate, or chloride counter ions as shown in the previously two reported analogous structures,13,27 two water molecules assume the elongated apical positions. The unique lability of the apical coordination at a CuII center has been exploited in CuII-based Lewis acid catalytic processes for rapid binding and activation of eletrophilic substrates and turnover of product molecules.29,30
Figure 4.

ORTEP (50% ellipsoids) diagrams of (A) [Cu(T1)2(H2O)2]2+. Selected distances (Å): Cu1-N1 2.073, Cu1-N3 1.995, Cu1-O2 2.330. (B) Hydrogen bond-linked, π-π interaction-stabilized interdigitating chain structures found in the solid state (hydrogen atoms are omitted for clarity).
Complex [Cu(T1)2(H2O)2](ClO4)2 forms an extended structure in the solid state via a rich variety of supramolecular forces (Figure 4B). All the oxygen atoms are involved in hydrogen bonding interactions. The complexes align in one-dimensional chains through the hydrogen bonds between the apical water ligands (O···O = 3.030 Å). The interdigitating adjacent chains are primarily stabilized by the π-π interactions (~3.5 Å) between electron-rich methoxyphenyl and the electron-poor CuII-bound triazolyl groups (only a partial complex in the adjacent chain is shown in Figure 4B). The chains are further locked in place via the hydrogen bonds between the water ligands and the methoxyl oxygens. In the absence of the methoxy groups as in the previously reported CuII complexes of ligand T19 (Table 3), the one-dimensional polymeric chain structure was not afforded.13,27 Although the combined use of metal coordination and hydrogen bonding has been applied in crystal engineering to afford one-dimensional organometallic polymers,31,32 it is rare to observe that both hydrogen bond donors and acceptors originate from the metal centers. The observed one-dimensional chain structure was by no means obtained via deliberate design. However, the emerged synergistic effect of metal coordination, bifurcated hydrogen bonding, and π-π interaction may be considered a viable approach in crystal engineering to afford tightly stacked extended metal-containing periodic structures.
The triazole product T6 (entry 9 in Table 1) is a tetradentate metal coordination ligand. X-ray crystal structures of the ZnII and CuII complexes of T6 display coordination of N2 of the 1,2,3-triazolyl group with metal ions to form six-membered rings (Figure 5), in addition to the binding from the more conventional dipicolylamino motif. The ZnII center adopts a trigonal bipyramidal geometry. In the CuII structures Jahn-Teller distorted octahedrons were observed where the N2 of the triazolyl group and the perchlorate ion are found at the long apical positions. The unit cell of the CuII complex contains two chemically distinct species where the CuII centers bind at equatorial positions with a CH3CN molecule (Figure 5B) and a water molecule (Figure 5C), respectively. As characterized in a similar compound, the ZnII affinity of this tetradentate ligand core is in the nM (Kd) range.14 Therefore, T6 represents a new high-affinity ligand motif for transition metal ions which may be utilized in applications such as metal ion sensing14 or organometallic catalyst design.33
Figure 5.

ORTEP (50% ellipsoids) diagrams of (A) [Zn(T6)(CH3CN)]2+. Selected distances (Å): Zn1-N1 2.056, Zn1-N2 2.049, Zn1-N3 2.251, Zn1-N5 2.063, Zn1-N7 2.061. (B) [Cu(T6)(CH3CN)(ClO4)]+. Select distances (Å): Cu1-N6 1.985, Cu1-N7 2.021, Cu1-O4 2.605, Cu1-N1 2.222, Cu1-N5 1.976, Cu1-N4 2.048. (C) [Cu(T6)(H2O)(ClO4)]+: Select distances (Å): Cu31-N34 2.024; Cu31-N35 1.983; Cu31-N33 2.270; Cu31-N36 1.992; Cu31-O22 1.962; Cu1-O16 2.742.
Conclusion
In summary, we have studied the reactivities of a series of carbon azides capable of chelating with CuII. Azides with auxiliary nitrogen donor ligands show higher reactivity than those with oxygen and sulfur donors (with the exceptions of the two azidocarboxylates in Table 2, entries 1 and 2). The high reactivities of chelating carbon azides are attributed to the fact that chelation-assisted binding between the alkylated azido nitrogen and the catalytic copper center greatly enhances the electrophilicity of the azido group, which lowers the kinetic barrier to form the key metallacycle intermediate upon nucleophilic attack by the copper acetylide. The bidentate triazolyl-containing compounds were shown to moderately accelerate AAC reactions with unactivated (non-chelating) azides, suggesting the possibility of autocatalysis in AAC reactions under the CuII-accelerated conditions. The reactions involving non-chelating azides were dramatically accelerated by the polytriazole ligand TBTA developed by Fokin et al. (although the overall rates as represented by the time required for completion are still slower than those involving chelating azides such as 1, 2, 4, 6, 7, and 11). This observation leads to the hypothesis that chelating azides may have similar capacities to that of TBTA in assisting the reduction of CuII to highly catalytic CuI species, in addition to enhancing the electrophilicity of the azido group in the step of metallacycle formation. Lastly, the utility of triazolyl-containing product molecules derived from chelating carbon azides as effective polydentate ligands for transition metal ions such as CuII and ZnII were demonstrated in two cases by X-ray crystallography. These observations motivate us to explore further on (1) the mechanistic front of this operationally simple yet mechanistically complex reaction, especially the role of copper/carbon azide interactions, and (2) the functional potential of the triazole molecules derived from chelating azides as multidentate ligands for transition metal ions.
Experimental Section
Materials and general methods
Reagents and solvents were purchased from various commercial sources and used without further purification unless otherwise stated. The purity of Cu(OAc)2 is over 98+%. Analytical thin-layer chromatography (TLC) was performed using TLC plates pre-coated with silica gel 60 F254. Flash column chromatography was performed using 40–63 µm (230–400 mesh ASTM) silica gel as the stationary phase. Silica gel was carefully flame-dried under vacuum to remove absorbed moisture before use. 1H and 13C NMR spectra were recorded at 300 MHz and 75 MHz, respectively. All chemical shifts were reported in δ units relative to tetramethylsilane. The syntheses and characterizations of carbon azide substrates 1, 2, 7, 16 and triazole products T2, T7, T16, T18, T19 were reported previously.8,13,14
General procedure for the Cu(OAc)2-accelerated AAC reaction
Carbon azide (0.20 mmol), tBuOH (0.5 mL), and p-ethynylanisole (29 mg, 0.22 mmol) were added to a 2-dram sample vial followed by the addition of Cu(OAc)2 (25 µL, 0.4 M solution in H2O, 10 µmol). The vial was capped and the reaction mixture was stirred at rt until the reaction was completed as shown by TLC (CH2Cl2/EtOAc = 10/1). The reaction mixture was directly loaded onto a short plug of silica gel. After elution with EtOAc (~100 mL), the solvent was removed in vacuo and the isolated product was characterized by 1H NMR, 13C NMR, and high-resolution mass spectrometry (HRMS). If needed, the triazole product was further chromatographed using a finer solvent gradient to afford the analytically pure material.
Synthesis of azide 3
Unless otherwise stated, this is a common procedure for the preparation of all azide substrates. Warning! Low molecular weight carbon azides used in this study are potentially explosive. Appropriate protection measures should always be taken when handling these compounds. 1-Benzyl-2-(chloromethyl)-H-imidazole hydrochloride (244 mg, 1 mmol) was dissolved in CH3CN (5 mL). N,N-diisopropylethylamine (130 mg, 1 mmol), sodium azide (282 mg, 4.3 mmol), 18-crown-6 (catalytic amount) and tetrabutylammonium iodide (catalytic amount) were added to the reaction vessel sequentially. The reaction mixture was stirred at rt for 19 h before EtOAc (30 mL) was added. The white precipitate was filtered off and the solution was washed with basic brine (saturated NaCl solution that is basified using concentrated NaOH to pH 11, 2 × 40 mL). The organic portion was dried over anhydrous Na2SO4. The solvent was evaporated under vacuum to afford compound 3 (170 mg, 80%). 1H NMR (300 MHz, CDCl3): δ (ppm) 7.72 (d, J = 7.5 Hz, 3H), 7.47 (d, J = 7.9 Hz, 2H), 7.42 (s, 1H), 7.31 (s, 1H), 5.55 (s, 2H), 4.73 (s, 2H). 13C NMR (75 MHz, CDCl3): δ (ppm) 142.2, 135.6, 128.9, 128.2, 128.1, 126.6, 121.6, 49.7, 46.3.
The preparations of non-arylmethyl azides required longer reaction time and/or higher temperature.
Procedure for preparing [Cu2(11)2Cl4]
The methanolic solutions of 11 (50 mg, 0.335 mmol, 0.5 mL) and CuCl2 (anhydrous, 46 mg, 0.343 mmol, 0.5 mL) were combined and mixed well. The solvent was then removed in vacuo and the solid product was rinsed with diethyl ether three times. The product was dried under vacuum for 10 min before re-dissolved in methanol (2 mL). The solution was passed through a plug of fiberglass filter paper before being set up for vapor diffusion of diethyl ether to obtain the single crystals of the copper complex [Cu2(11)2Cl4] suitable for X-ray crystallographic analysis.
X-ray crystallography
Crystals were mounted on a nylon loop by the use of heavy oil. The samples were held at about −100 °C for data collection. After finding a crystal that indexed to give a satisfactory unit cell, a full data set was taken at low temperature on an APEX II using a detector distances of 6 cm. The number of frames taken was typically 2,400 using 0.3 degree omega scans with 20 seconds of frame collection time. Integration was performed using the program SAINT which is part of the Bruker suite of programs. Absorption corrections were made using SADABS. XPREP was used to obtain an indication of the space group and the structure was solved by direct methods and refined by SHELXTL. The nonhydrogen atoms were refined anisotropically. The hydrogens were typically constrained as a riding model, though with rare exceptions, the data were good enough that hydrogen assignment could be done directly.
The crystal for the T6/Zn(ClO4)2 complex was found to be a non-mereohedral twin. It was indexed by CELL_NOW and the data integrated by the program SAINT and the scaling done by TWINABS. The structure was solved using the hkl4 data set and refined by the hkl5 corrected data set. The main molecule resolved nicely giving chemically reasonable bond distances and ellipsoids. Not surprisingly, the perchlorate counter ions are somewhat disordered. The disorder could be refined using the part commands and a few restraints, but the data in the .cif file are as in the final refinement before attempting to model the disorder.
The T6/Cu(ClO4)2 complex was refined in P2(1)/c and contains two independent and slightly different copper complexes with four perchlorate counter ions. Solvent molecules within the structure are two CH3CN molecules and one water molecules. The copper in the complex can be thought of as being a distorted octahedron with a perchlorate ion and a solvent molecule occupying cis positions. In one molecule, CH3CN was the solvent and in the other, a water molecule occupies the sixth spot. The solvent water molecule is hydrogen bonded to the ligand water. The hydrogens in both cases were refined giving reasonable distances and angles. As is quite common with these compounds, the only significant disorder was found around a couple of the perchlorate ions. Most of the hydrogens atoms were assigned and refined as riding molecules, but the data were good enough to all the hydrogens around the water oxygen to be resolved.
Supplementary Material
ACKNOWLEDGMENT
This work was supported in part by NIGMS (R01GM-081382) and NSF (CHE-0809201).
Footnotes
Supporting Information Available. Experimental procedures, characterization of new compounds, and .cif files for [Cu2(11)2Cl4], [Cu(T1)2(H2O)2](ClO4)2, [Cu(T6)(CH3CN/H2O)(ClO4)](ClO4), and [Zn(T6)(CH3CN)](ClO4)2. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- 1.Finn MG, Fokin VV. Chem. Soc. Rev. 2010;39:1231–1232. doi: 10.1039/c003740k. [DOI] [PubMed] [Google Scholar]
- 2.Meldal M, Tornøe CW. Chem. Rev. 2008;108:2952–3015. doi: 10.1021/cr0783479. [DOI] [PubMed] [Google Scholar]
- 3.Rostovtsev VV, Green LG, Fokin VV, Sharpless KB. Angew. Chem. Int. Ed. 2002;41:2596–2599. doi: 10.1002/1521-3773(20020715)41:14<2596::AID-ANIE2596>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 4.Tornøe CW, Christensen C, Meldal M. J. Org. Chem. 2002;67:3057–3064. doi: 10.1021/jo011148j. [DOI] [PubMed] [Google Scholar]
- 5.Brik A, Muldoon J, Lin Y-C, Elder JH, Goodsell DS, Olson AJ, Fokin VV, Sharpless KB, Wong C-H. ChemBioChem. 2003;4:1246–1248. doi: 10.1002/cbic.200300724. [DOI] [PubMed] [Google Scholar]
- 6.Wang Q, Chan TR, Higraf R, Fokin VV, Sharpless KB, Finn MG. J. Am. Chem. Soc. 2003;125:3192–3193. doi: 10.1021/ja021381e. [DOI] [PubMed] [Google Scholar]
- 7.Hong V, Udit AK, Evans RA, Finn MG. ChemBioChem. 2008;9:1481–1486. doi: 10.1002/cbic.200700768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chan TR, Hilgraf R, Sharpless KB, Fokin VV. Org. Lett. 2004;6:2853–2855. doi: 10.1021/ol0493094. [DOI] [PubMed] [Google Scholar]
- 9.Rodionov NO, Presolski SI, Díaz DD, Fokin VV, Finn MG. J. Am. Chem. Soc. 2007;129:12705–12712. doi: 10.1021/ja072679d. [DOI] [PubMed] [Google Scholar]
- 10.Hein JE, Fokin VV. Chem. Soc. Rev. 2010;39:1302–1315. doi: 10.1039/b904091a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Himo F, Lovell T, Hilgraf R, Rostovtsev VV, Noodleman L, Sharpless KB, Fokin VV. J. Am. Chem. Soc. 2005;127:210–216. doi: 10.1021/ja0471525. [DOI] [PubMed] [Google Scholar]
- 12.Ahlquist M, Fokin VV. Organometallics. 2007;26:4389–4391. [Google Scholar]
- 13.Brotherton WS, Michaels HA, Simmons JT, Clark RJ, Dalal NS, Zhu L. Org. Lett. 2009;11:4954–4957. doi: 10.1021/ol9021113. [DOI] [PubMed] [Google Scholar]
- 14.Huang S, Clark RJ, Zhu L. Org. Lett. 2007;9:4999–5002. doi: 10.1021/ol702208y. [DOI] [PubMed] [Google Scholar]
- 15.Michaels HA, Murphy CS, Clark RJ, Davidson MW, Zhu L. Inorg. Chem. 2010;49:4278–4287. doi: 10.1021/ic100145e. [DOI] [PubMed] [Google Scholar]
- 16.Earlier observations that Cu(II) is capable of enabling AAC reactions appeared in the following reports. Differing from our conclusions, however, Cu(II) was postulated as the directly participating catalytic species in AAC reactions without being reduced to Cu(I) in these reports: Reddy KR, Rajgopal K, Kantam ML. Synlett. 2006:957–959. Fukuzawa S-I, Shimizu E, Kikuchi S. Synlett. 2007:2436–2438. Reddy KR, Rajgopal K, Kantam ML. Cat. Lett. 2007;114:36–40. Song Y-J, Yoo C, Hong J-T, Kim S-J, Son SU, Jang H-Y. Bull. Korean Chem. Soc. 2008;29:1561–1564. Namitharan K, Kumaaraja M, Pitchumani K. Chem. Eur. J. 2009;15:2755–2758. doi: 10.1002/chem.200802384.
- 17.Kamata K, Nakagawa Y, Yamaguchi K, Mizuno N. J. Am. Chem. Soc. 2008;130:15304–15310. doi: 10.1021/ja806249n. [DOI] [PubMed] [Google Scholar]
- 18.Buckley BR, Dann SE, Harris DP, Heaney H, Stubbs EC. Chem. Commun. 2010;46:2274–2276. doi: 10.1039/b924649e. [DOI] [PubMed] [Google Scholar]
- 19.Buckley BR, Dann SE, Heaney H. Chem. Eur. J. 2010;16:6278–6284. doi: 10.1002/chem.201000447. [DOI] [PubMed] [Google Scholar]
- 20.Dias HVR, Polach SA, Goh S-K, Archibong EF, Marynick DS. Inorg. Chem. 2000;39:3894–3901. doi: 10.1021/ic0004232. [DOI] [PubMed] [Google Scholar]
- 21.CuCl2 was shown to be a capable pre-catalyst under the crystallization conditions. Azide 11 and phenylacetylene in the presence of 5 mol% CuCl2 in CH3OH convert readily to the triazole product.
- 22.See an earlier example: Barz M, Herdtweck E, Thiel WR. Angew. Chem. Int. Ed. 1998;37:2262–2265. doi: 10.1002/(SICI)1521-3773(19980904)37:16<2262::AID-ANIE2262>3.0.CO;2-X.
- 23.Lewis WG, Magallon FG, Fokin VV, Finn MG. J. Am. Chem. Soc. 2004;126:9152–9153. doi: 10.1021/ja048425z. [DOI] [PubMed] [Google Scholar]
- 24.Donnelly PS, Zanatta SD, Zammit SC, White JM, Williams SJ. Chem. Commun. 2008:2459–2461. doi: 10.1039/b719724a. [DOI] [PubMed] [Google Scholar]
- 25.T18 and T19 were chosen because they were generated in very rapid (< 10 min) AAC reactions under Cu(OAc)2-accelerated conditions. T1 was chosen as a representative from the triazole molecules reported in this work.
- 26.There is an exception for 1-azidooctane, which showed no improvements in overall yield in the presence of T18 over 24 h (Entry 1, Table 3).
- 27.Crowley JD, Bandeen PH, Hanton LR. Polyhedron. 2010;29:70–83. [Google Scholar]
- 28.Urankar D, Pinter B, Pevec A, Rroft FD, Turel I, Kosmrlj J. Inorg. Chem. 2010;49:4820–4829. doi: 10.1021/ic902354e. [DOI] [PubMed] [Google Scholar]
- 29.Johnson JS, Evans DA. Acc. Chem. Res. 2000;33:325–335. doi: 10.1021/ar960062n. [DOI] [PubMed] [Google Scholar]
- 30.Evans DA, Seidel D, Rueping M, Lam HW, Shaw JT, Downey CW. J. Am. Chem. Soc. 2003;125:12692–12693. doi: 10.1021/ja0373871. [DOI] [PubMed] [Google Scholar]
- 31.Braga D, Grepioni F, Desiraju GR. Chem. Rev. 1998;98:1375–1405. doi: 10.1021/cr960091b. [DOI] [PubMed] [Google Scholar]
- 32.Brammer L, Rivas JCM, Atencio R, Fang S, Pigge FC. J. Chem. Soc., Dalton Trans. 2000:3855–3867. [Google Scholar]
- 33.Bergbreiter DE, Hamilton PN, Koshti NM. J. Am. Chem. Soc. 2007;129:10666–10667. doi: 10.1021/ja0741372. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.