

Abstract
Recent evidence has demonstrated that the complement cascade is involved in a variety of physiologic and pathophysiologic processes in addition to its role as an immune effector. Research in a variety of organ systems has shown that complement proteins are direct participants in maintenance of cellular turnover, healing, proliferation and regeneration. As a physiologic housekeeper, complement proteins maintain tissue integrity in the absence of inflammation by disposing of cellular debris and waste, a process critical to the prevention of autoimmune disease. Developmentally, complement proteins influence pathways including hematopoietic stem cell engraftment, bone growth, and angiogenesis. They also provide a potent stimulus for cellular proliferation including regeneration of the limb and eye in animal models, and liver proliferation following injury. Here, we describe the complement cascade as a mediator of tissue growth and regeneration.
Keywords: Complement, Growth, Regeneration, Proliferation, Healing
Introduction
Since its discovery over 100 years ago, the complement cascade has been recognized as an effector arm of innate immunity, a set of serum proteins known to “complement” antibody-mediated pathogen clearance [1–3]. The complement cascade includes 12 soluble proteins capable of initiating a cytolytic response in the absence of other humoral or cellular elements. Complement activation and amplification occur on the surface of the target cell, and are initiated by the binding of one or more molecules of the classical, alternative, or mannose-binding lectin pathways [4]. The classical pathway destroys antibody-coated targets, apoptotic cells, gram-negative bacteria, and some viruses [3]. The mannose-binding lectin pathway destroys mannose-bearing pathogens [3]. Lastly, the alternative pathway destroys a variety of infectious agents including bacteria, viruses, and fungi, in addition to playing a role in the immune surveillance of tumors [3]. Their convergence into a final common pathway results in creation of the membrane attack complex (MAC), an amphipathic pore-forming structure made up of C5b, C6, C7, C8, and multiple C9 molecules [2].
The traditional view of the complement cascade maintains that its role is to facilitate cellular killing and inflammation. However, increasing evidence has challenged this view in diverse ways. New sites of complement protein synthesis and new locations for complement protein receptors have been uncovered [3, 5]. Immune “imbalance” via complement protein deficiency or dysregulation has been implicated in the pathogenesis of numerous autoimmune and non-immune diseases [3, 6]. Finally, complement protein regulation of cellular proliferation has been demonstrated in a broad range of tissues [7]. This exponential increase in our knowledge has established new roles for the complement cascade in both tissue health and disease that expand upon its former designation as a mere immune effector of host defense [3, 6, 8].
Evidence demonstrating the novel importance of complement proteins has opened up especially exciting avenues of translational research. Complement proteins play important roles in developmental, proliferative, and regenerative pathways in both injured and uninjured tissue (Fig. 1). The dichotomy of “defender” and “healer” in a single system represents a major departure from previous considerations of complement cascade-mediated effects, and demands a reappraisal of traditional views. The following review offers insight into the emerging body of research implicating complement proteins as mediators of tissue health via their regulatory effects on cell turnover, proliferation, maturation, and regeneration.
Fig. 1.
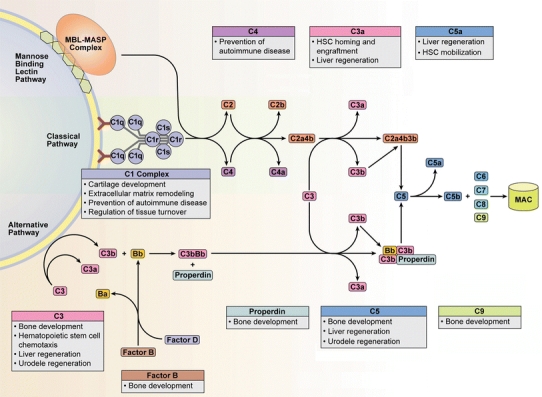
The complement cascade is comprised of the Classical, Alternative, and Mannose Binding Lectin pathways. Although the activation and amplification of each pathway is unique, they all converge with cleavage of C3 into its main split products, C3a and C3b, ultimately resulting in formation of the membrane attack complex (MAC). Historically considered a critical arm of innate immunity, emerging research has implicated various complement proteins in proliferative and regenerative processes in organs including bone, marrow, liver, and connective tissues. C1q maintains appropriate turnover and clearance of cellular debris, while C4 maintains B cell tolerance, processes critical in the prevention of autoimmune disease. Hematopoietic stem cell homing, chemotaxis, engraftment, and mobilization are heavily influenced by the anaphylatoxins C3a and C5a. Additionally, they help facilitate hepatocyte protection and regeneration in mouse models of toxic and surgical injury. Limb and lens regeneration in urodele amphibians are mediated by C3 and C5, while complement proteins such as C1s, C5, C9, factor B and properdin are distributed throughout immature, developing bone and appear to be important in osteogenesis. MBL mannose-binding lectin, MASP mannose-binding lectin associated serine proteases, HSC hematopoietic stem cell
The complement cascade as a physiologic housekeeper
Complement proteins are constitutively active, facilitate cellular turnover, and prevent autoimmune disease
Whether caused by endogenous or exogenous stimuli, cellular injury is an inciting event for activation of the classical, alternative, and mannose-binding lectin pathways. Complement proteins participate in multiple phases of acute and chronic inflammation, including local hemodynamic change, increased vascular permeability, recruitment and chemotaxis of leukocytes, lymphocyte activation and upregulation, opsonization of pathogens, and clearance of necrotic and apoptotic tissues [9]. The potentially devastating effects of uncontrolled complement activation and subsequent promotion of inflammation are attenuated by a widespread network of inhibitory membrane-bound complement receptors and soluble proteins. These endogenous receptors and proteins prevent indiscriminate complement mediated cellular destruction in healthy host tissues by inhibiting excessive amplification of the complement cascade, thereby precluding formation of the lytic MAC. Necrotic or apoptotic cells are susceptible to complement mediated destruction due to their loss of membrane-bound inhibitory molecules [6]. When these checks and balances are lost, dysregulation of the complement cascade facilitates a wide variety of autoimmune diseases in which chronic inflammation is seen, including glomerulonephritis [10–12], systemic lupus erythematosus (SLE) [13], rheumatoid arthritis [14], and multiple sclerosis [15].
Research linking incomplete complement mediated clearance of dead cells with subsequent autoimmune disease [16–20] argues that complement proteins play a vital homeostatic mechanism. Termed the “waste-disposal” hypothesis, it has been proposed that complement protein dysfunction or deficiency can lead to inadequate clearance of cellular debris, consequently facilitating exposure to potentially antigenic intracellular molecules [6]. Immature dendritic cells then take up these autoantigens, mature, and present them to T cells, leading to autoreactive B cell stimulation and subsequent production of autoantibodies [6]. Homozygous C1q deficiency provides a striking example of complement related autoimmunity. Loss of this initiator protein of the classical pathway almost invariably leads to severe SLE [13].
Complement activation due to cell death can occur secondary to inflammation or entirely independent of it. Constitutive activation of the alternative pathway via spontaneous C3 cleavage has been recognized for many years [21]. Newer evidence suggests the classical cascade is also constitutively active in humans and mice in an antigen-independent manner, with blood stasis posited as a possible inciting stimulus for its basal low-level activation [22]. Based on the observation that ex vivo blood samples from humans and mice exhibit spontaneous C3 fragment formation, the authors investigated the complement activation pathway responsible for this effect by studying plasma from mice and humans with specific complement protein deficiencies. Spontaneous generation of C3 fragments was abrogated in both C1q-deficient humans and complement knockout mice including C1qa−/−, C4−/−, and H2-Bf, C2−/− mice [22]. While plasma from mice deficient in immunoglobulins (RAG−/−) and factor B (Bf−/−) demonstrated spontaneous C3 fragment formation, it was significantly reduced compared to control mice. The authors concluded that the alternative pathway likely amplifies the production of C3 fragments initiated by the classical pathway [22]. Such continuous activation and priming of the complement cascade might provide an important physiologic mechanism that enhances clearance of apoptotic and necrotic cells, better allowing for regulation of tissue turnover.
However, complement protein dysfunction is not only thought to contribute to autoimmune disease via insufficient clearance of debris; emerging evidence points to a critical role for complement proteins in maintaining B cell tolerance, thereby precluding production of autoantibodies. In one elegant study, researchers investigated the role of complement in the prevention or production of autoreactive B cells in a mouse model of autoimmunity resembling SLE [23]. They first created a transgenic mouse model of B cell tolerance in which the foreign protein hen egg lysozyme (HEL), an antigen capable of inducing production of autoreactive B cells, remained sequestered in the endoplasmic reticulum (ER) of all MHC-1 cells [24]. This ER-restricted mHEL (mHEL-KK) transgenic mouse closely recapitulates the pathogenesis of SLE by sequestering the HEL autoantigen intracellularly. They then crossed C1q deficient mice (C1q−/−) with MD4 IgHEL mice, a transgenic model encoding IgMa and IgDa specific to HEL. Finally, they crossed these single transgenic mice (C1q−/− IgHEL) with mHEL-KK mice to create double-transgenic C1q−/− IgHEL/mHEL-KK mice. Armed with these mouse models of autoimmunity resembling SLE, the authors found that C1q facilitates potent downregulation of numerous autoimmune phenomena. These C1q deficient mice demonstrated increased positive selection of B1b B cells responsive to intracellular HEL, decreased negative selection of conventional B cells, increased numbers of plasma cells, and increased production of IgM autoantibodies [23].
In addition to C1q, it has also been shown that the loss of C4 or complement receptors 1 and 2 (CR1, CR2) facilitates autoimmunity directly by inducing loss of B cell tolerance and subsequent production of autoreactive B cells [25, 26]. Furthermore, complement proteins are known regulators of T cell proliferation [4, 27, 28]. Certain subpopulations of T cells are inhibited directly by C1q, preventing inappropriate self-attack [9, 29].
In total, the protean capabilities of various complement proteins in cellular clearance and modulation of the adaptive immune response demonstrate critical roles in the prevention of autoimmune disease. The continuous activation of complement proteins maintains tissue turnover as a physiologic event rather than a pathogenic one.
The complement cascade and cellular growth and regeneration
Complement proteins regulate hematopoietic stem cell migration, homing, and engraftment
The continuous activation of complement proteins and split products necessitates that healthy cells possess mechanisms to protect themselves from accidental destruction. In non-nucleated cells such as erythrocytes, inhibitory proteins, including decay-accelerating factor (DAF), CD59, and CR1-related gene y (Crry), prevent such destruction [30, 31]. Nucleated cells employ ion pumps that reverse MAC-induced solute shifts, utilize ecto- and endo-proteases that cleave complement components, and remove MACs from their membrane surfaces [2, 32].
Unlike the defenses maintained by mature erythrocytes and leukocytes against accidental complement destruction, the interaction of complement proteins with immature blood cells suggests a vital role in hematopoiesis, including mobilization and localization of progenitor cells. As a fundamental component of innate immunity, complement proteins become activated in cellular injury, and it has been suggested that hematopoietic stem cells (HSCs) likely play an important and concomitant role in this response to injury [33].
Extensive research into complement protein C3a has revealed a wealth of information on its roles in bone marrow engraftment and HSC mobilization. Progenitor cells including myeloid, erythroid, and megakaryocytic blasts, as well as human CD34+ cells, were all found to express the C3a receptor (C3aR) [34]. When stimulated by C3a secreted from bone marrow, THP-1, Jurkat, and PB-1 cell lines did not demonstrate a chemotactic response. However, when stimulated in the presence of stromal-cell derived factor 1 (SDF-1), all cells demonstrated increased chemotaxis in response to C3a [34]. Human CD34+ cells and their progenitors exhibited similar behavior: C3a increased SDF-1 mediated chemotaxis in mature CD34+ cells and CD34+ progenitors committed along all hematopoietic lineages [34]. These synergistic effects of C3a and SDF-1 suggest that C3a enhances the SDF-1-CXCR4-dependent axis in HSCs. Notably, the authors found that C3a had no influence over HSC survival or proliferation [34].
C3a also appears to be important in HSC engraftment. C3a aids SDF-1 mediated adhesion of human CD34+ cells to vascular cell adhesion molecule 1 (VCAM-1), and when primed with C3a, bone marrow Sca-1+ cells show an increased speed of engraftment in irradiated mice [34]. Studies of granulocyte colony-stimulating factor (G-CSF), an established mobilizer of HSCs [33], further delineate the role of C3a. In C3−/− and C3aR−/− mice, G-CSF significantly increased mobilization of stem cells when compared to wild type mice; however, when given a C3aR antagonist, wild type mice also showed increased mobilization [35]. Additionally, wild type mice given C3aR−/− HSCs were more sensitive to G-CSF mobilization versus C3aR−/− mice given wild type HSCs, again indicating the importance of C3a/C3aR interaction in engraftment, likely through increased sensitization to the effects of SDF-1 [35]. Similarly decreased HSC engraftment was observed when C3−/− mice were transplanted with wild type HSCs [36].
These results, however, contrast with those of Honczarenko et al. [37] showing that human and mouse bone marrow progenitor cells and B lineage cells demonstrate enhanced SDF-1 mediated migration and engraftment in the presence of C3a independent of the C3a receptor. They found no evidence of the C3a receptor on bone marrow stem cells and B lineage cells, and could not abrogate the chemotactic effects of C3a using a C3a receptor inhibitor or a C3aR−/− mouse [37]. They instead discovered that C3a binds SDF-1 and enhances its affinity for CXCR4 [37], presumably increasing or enhancing activity of this axis. As the authors note, apparent discrepancies in the presence of C3aR on human CD34+ cells and HSCs may be due to the manner in which they were collected and treated [37].
To clarify this seemingly discrepant data and the importance of the C3a receptor in HSC behavior, Wysoczynski et al. studied the role of the C3a/C3aR interaction in HSC retention. When compared to wild type murine HSCs, C3aR−/− HSCs demonstrate delayed bone marrow homing and engraftment in lethally irradiated mice [38]. Human HSCs exhibit similar behavior: when exposed to C3aR antagonist and then transplanted into NOD/SCID mice, defective colony growth was observed following recovery and plating [38]. They also found that C3a stimulates matrix metalloprotease-9 (MMP-9) production by wild type HSCs at the mRNA and protein levels, but not in C3aR−/− HSCs. This is believed to be important due to the role of MMP-9 in facilitating HSC migration through extracellular matrix and into tissues such as bone marrow [34].
These most recent data reconcile a dual role for C3a in HSC behavior that is both dependent and independent of the C3a receptor. It appears that the C3a/C3aR interaction is critical to HSC engraftment through mechanisms involving tissue migration and adhesion, whereas homing and chemotaxis are primed via SDF-1-CXCR4 sensitization by C3a independent of its receptor.
Interestingly, complement protein C5 appears to have an opposite effect on HSCs compared to C3a: C5−/− mice demonstrate greatly reduced HSC mobilization in response to G-CSF and zymosan, an activator of the alternative pathway [39]. Interestingly, murine HSCs do not express C5a receptor (C5aR), nor does C5a directly enhance SDF-1 mediated chemotaxis like C3a [40]. Instead, it appears that C5a indirectly induces HSC egress via granulocytes. C5a stimulates release of their proteases, which in turn interrupt pathways such as SDF-1-CXCR4 that help sequester HSCs in bone marrow [33, 40]. Released proteases also appear to enhance the response of HSCs to SDF-1 gradients, promoting their release from bone marrow into the peripheral circulation [40].
Further interest in the interaction between complement proteins and hematopoietic stem cells has centered on the identification and behavior of AA4. This murine stem cell antigen is believed to be homologous to human C1qRP, a receptor for C1q that is expressed in phagocytic myeloid progenitors, endothelial cells, and platelets [41]. In the adult mouse, AA4 expression has been identified in lung, heart, and bone marrow; in the fetus, expression occurs in vascular endothelial cells, aorta-associated hematopoietic clusters, and in all fetal liver hematopoietic stem cells [42]. Given its homology to mouse AA4, C1qRP may play a similar developmental role in humans, including the homing of hematopoietic progenitors to the fetal liver, and endothelial cell migration, adhesion, and formation of intercellular junctions [42].
Complement proteins aid cartilage turnover and bone development
A well-delineated sequence of events comprises endochondral bone formation in which three distinct zones of resting, proliferating and hypertrophic chondrocytes develop. However, the factors governing their growth and the subsequent vascular and mesenchymal cell invasion that immediately precedes matrix remodeling and bony deposition remain incompletely understood. Complement protein induction of cartilage and extracellular matrix degradation has been appreciated for several decades [43], but more recent research has yielded information about the distribution of complement proteins in areas of cartilage and bone and their putative function.
C1s possesses the ability to degrade Type 1 and 2 collagen [44]. Immunohistochemical analysis of the primary ossification center of the human femur revealed that C1s co-localizes with MMP-9 throughout this region, including hypertrophic chondrocytes, mesenchymal cells and the extracellular matrix [44]. C1s can also cleave the zymogen of MMP-9 and activate its protease activity [44]. Their co-localization implies a coordinated role in the turnover of cartilage to bone [44]. Another component of the C1 complex, C1q, also appears important in osteogenesis. C1q levels increase with differentiation of hypertrophic chondrocytes in hamster models of osteogenesis [45, 46].
The alternative pathway may also be involved in bone formation. Among fetal rat tibias and femurs, C3 and factor B are distributed in the resting and proliferating zones, properdin in the resting and hypertrophic zones, and C5 and C9 in the hypertrophic zones [43]. Their differential localization intimates participation in bone formation and remodeling, vascular invasion, and extracellular matrix remodeling [7, 43]. Taken together with evidence of classical pathway involvement, complement proteins can stimulate turnover of cartilage to bone.
Complement also participates in vitamin D3 regulation of bone. Vitamin D3 is known to induce bone production of C3. Murine osteoblastic cells and bone marrow derived stromal cells (ST2) release C3 in response to D3 in vitro [47, 48], and vitamin D3 additionally induces murine bone production of C3 in vivo [49]. Among bone marrow cultures grown with vitamin D3, C3 can be found in macrophage-like mononuclear cells, progenitor cells that can form mature osteoclasts [50]. These cells also express receptors for C3. When exposed to anti-C3 receptor antibodies in the presence of vitamin D3, formation of osteoclasts is inhibited [50]. Furthermore, while vitamin D3 binding appears to inhibit macrophage colony stimulating factor (M-CSF), a growth factor critical in osteoclast development, C3 co-exposure combats this inhibitory effect in vitro and potentiates the effects of M-CSF [50].
Complement proteins mediate limb and lens regeneration in urodeles
Urodele amphibians are known for their remarkable ability to regenerate injured tissues, including their limbs, tail, eye structures, and some internal organs [51]. A recent series of fascinating studies strongly implicates the complement cascade as a mediator of these regenerative pathways.
In response to injury, a complicated progression occurs in which mature cell types present at the site of injury undergo dedifferentiation, multiplication, and re-differentiation into the blastema, a stem-cell like structure [52]. Blastema cells are responsible for repopulating and reforming old structures by redifferentiation into mature tissues such as muscle, bone, and connective tissue. In one of the earliest studies on the subject, the C3 gene was discovered to be specifically expressed by urodele blastema cells at the transcript and protein level as evidenced by in situ hybridization and detection of antibodies against C3 [52]. Furthermore, C3 protein was present in blastema cells of myogenic lineage [52], confirming earlier experiments demonstrating diverse complement protein expression by myoblast cell cultures [53, 54]. C3 localizes to regenerating tissues and not normal tissue, suggesting a primary role in mediating regeneration [52]. It has been postulated that complement activation attracts a coordinated infiltration of macrophages responsible for protection from further injury-related damages [52]. Because the complement cascade interacts with numerous extracellular structural proteins responsible for cell anchoring and intercellular connections, including integrins, laminin, and fibronectin, it is possible that complement proteins facilitate regeneration of intercellular connections and ensuing cellular communication [52].
C5 is also an important factor in newt limb regeneration. While C3 shows robust immunohistochemical staining within the blastema and some staining in wound epithelium, C5 is absent from the blastema but strongly present in wound epithelium at all stages of limb regeneration [51]. The unique distributions of C3 and C5 are thought to arise from their differential roles in the regenerative process. This specificity is similarly seen in the newt eye: both proteins are present in the cornea and iris, but C5 alone is located in the lens vesicle [51]. Neither C3 nor C5 are present in the intact lens. Interestingly, while both C3 and C5 mRNA were detected in the regenerating lens vesicle by in situ hybridization, only C5 translation to protein was appreciated, indicating translational or post-translational modifications that differentially localize it to specialized areas [51].
Complement proteins facilitate liver regeneration
The human liver is one of few human tissues capable of near complete organogenesis in response to a variety of endogenous and exogenous insults [55, 56]. Initiation of liver regeneration is multifactorial, as growth factors, hormones, and immune mediators have all been implicated [56]. Interestingly, stem cells do not have an exclusive role in liver regeneration, despite the liver’s ability to repopulate all cell types intrinsic to the healthy organ, including hepatocytes, biliary epithelial cells, fenestrated endothelium, Kupffer cells and cells of Ito [56].
Several studies predating the established link between the complement cascade and liver regeneration suggested that complement proteins might play a role. One group of researchers found that Kupffer cells and hepatic stellate cells secrete prostanoids in response to C5a/C5aR binding on their surface, causing an increase in glucose production by adjacent hepatocytes and a local decrease in blood flow [57, 58]. This process is considered important in the provision of fuel necessary for the rapid metabolism seen in liver regeneration [7]. Another group highlighted the role of C5a in defense against liver injury by demonstrating that C5aR mRNA and protein is induced in hepatocytes in response to exogenous IL-6 in rats [59]. By responding to inflammation with functional C5a receptors, these hepatocytes are believed to prime themselves for defense against injury by allowing more direct facilitation of C5a induced glucose production [59]. Ultimately, the complexity of IL-6 actions and synthesis makes it difficult to conclude the exact nature of its inflammatory properties and its effects on the complement cascade.
The liver is the body’s primary site of complement protein synthesis, and a growing body of evidence now definitively demonstrates that complement proteins C3a and C5a are critical to liver regeneration following injury. Following experimental liver injury induced by carbon tetrachloride (CCl4), a well-characterized hepatotoxin that induces liver necrosis and apoptosis, C5−/− mice showed significantly diminished liver regeneration compared to wild type, C5 sufficient mice [55]. The hepatocytes in C5−/− mice demonstrated significantly delayed entry into S-phase of the cell cycle, decreased mitotic activity, persistent necrosis, and decreased proliferation. Restoration of murine C5 or C5a restored the regenerative response [55]. To corroborate the involvement of the C5a/C5aR interaction in promoting regeneration, the authors also demonstrated that antagonist blockade of C5aR in wild type mice diminishes liver regeneration [55]. A related study demonstrated the presence of C5a receptor on regenerating hepatocytes, noting its levels were greatly upregulated following hepatectomy or cell culture induced stress [60]. Furthermore, C5a binding to C5aR induced expression of mRNA for hepatocyte growth factor (HGF) and its corresponding c-MET receptor [60], proteins known to induce mitosis and liver regeneration [56].
C3 has also been shown to play a critical role in liver regeneration. For example, one study found that C3−/− mice and C3aR−/− mice exposed to CCl4 exhibited greatly reduced hepatic regeneration [61]. Among C3−/− mice, systemic introduction of C3a restored hepatic regeneration, reducing the possibility of downstream C5 effects given that these mice are intrinsically deficient in the C3b fragment needed to form the C5 convertase [61]. C3aR−/− mice displayed delayed hepatocyte S-phase entry and received no benefit from systemic C3a reconstitution, both suggesting that C3a exerts its regenerative effects through C3aR. Interestingly, deposition of C3 split products in injured liver occurred in a biphasic pattern following CCl4 exposure: C3a deposition peaked within the first 3 h, while C3b deposition peaked much later, typically between 24 and 48 h [61]. The authors postulate that C3a may therefore have a priming effect on injured liver that activates critical cell cycle pathways including the STAT-3 and NF-κB transcription factors [61, 62]. Subsequent C3b deposition likely aids the clearance of dead hepatic cells [61].
Recent evidence indicates that complement proteins aid the survival of proliferating liver cells in addition to acting as stimuli for healing and regeneration. Mice doubly deficient in C3 and C5 (C3/C5−/−) demonstrate marked hepatocyte apoptosis following partial hepatectomy. While reconstitution of C3 or C5 attenuates this injury, reconstitution of both almost completely prevents apoptotic injury [63]. This phenomenon may be due in part to the role of complement in promoting inflammatory cytokines that arise in the setting of liver injury and aid anti-apoptotic signalling [63]. For example, loss of serum IL-6 induction in C3/C5−/− mice undergoing partial hepatectomy interrupts cell signaling through the JAK1 pathway via loss of JAK1 phosphorylation and decreased activation of STAT-3 [63]. Additionally, C3−/− mice demonstrate lower activity of the PI3K/Akt pathway, causing a decrease in mammalian targets of rapamycin (mTOR) signaling [63, 64], another pathway strongly associated with cellular proliferation [65].
Conclusion
Abundant evidence indicates that the complement cascade can no longer solely be considered an effector of innate immunity. In addition to performing a variety of physiologic housekeeping functions, complement proteins clearly respond to tissue injury with induction of cellular proliferation and regeneration across a diverse array of organ systems, as evidenced by in vitro and in vivo research. These observations provide a previously unrecognized mechanism for tissue healing. As our understanding of the complement cascade’s capabilities increases, so too does the promise of therapeutic interventions that enhance the beneficial effects of complement proteins and attenuate their negative ones.
Acknowledgments
Martin Rutkowski received a grant from the Doris Duke Charitable Foundation. Dr. Sughrue received a National Research Service Award from the National Institutes of Health. Ari Kane received a grant from the Howard Hughes Medical Institute. Dr. Parsa was partially funded by the Reza and Georgianna Khatib Endowed Chair in Skull Base Tumor Surgery.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
References
- 1.Dempsey PW, Allison ME, Akkaraju S, Goodnow CC, Fearon DT. C3d of complement as a molecular adjuvant: bridging innate and acquired immunity. Science. 1996;271:348–350. doi: 10.1126/science.271.5247.348. [DOI] [PubMed] [Google Scholar]
- 2.Cole DS, Morgan BP. Beyond lysis: how complement influences cell fate. Clin Sci (Lond) 2003;104:455–466. doi: 10.1042/CS20020362. [DOI] [PubMed] [Google Scholar]
- 3.Walport MJ. Complement. First of two parts. N Engl J Med. 2001;344:1058–1066. doi: 10.1056/NEJM200104053441406. [DOI] [PubMed] [Google Scholar]
- 4.Kemper C, Atkinson JP. T-cell regulation: with complements from innate immunity. Nat Rev Immunol. 2007;7:9–18. doi: 10.1038/nri1994. [DOI] [PubMed] [Google Scholar]
- 5.Thomas A, Gasque P, Vaudry D, Gonzalez B, Fontaine M. Expression of a complete and functional complement system by human neuronal cells in vitro. Int Immunol. 2000;12:1015–1023. doi: 10.1093/intimm/12.7.1015. [DOI] [PubMed] [Google Scholar]
- 6.Walport MJ. Complement. Second of two parts. N Engl J Med. 2001;344:1140–1144. doi: 10.1056/NEJM200104123441506. [DOI] [PubMed] [Google Scholar]
- 7.Mastellos D, Lambris JD. Complement: more than a ‘guard’ against invading pathogens? Trends Immunol. 2002;23:485–491. doi: 10.1016/S1471-4906(02)02287-1. [DOI] [PubMed] [Google Scholar]
- 8.D’Ambrosio AL, Pinsky DJ, Connolly ES. The role of the complement cascade in ischemia/reperfusion injury: implications for neuroprotection. Mol Med. 2001;7:367–382. [PMC free article] [PubMed] [Google Scholar]
- 9.Markiewski MM, Lambris JD. The role of complement in inflammatory diseases from behind the scenes into the spotlight. Am J Pathol. 2007;171:715–727. doi: 10.2353/ajpath.2007.070166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rose KL, Paixao-Cavalcante D, Fish J, Manderson AP, Malik TH, Bygrave AE, et al. Factor I is required for the development of membranoproliferative glomerulonephritis in factor H-deficient mice. J Clin Invest. 2008;118:608–618. doi: 10.1172/JCI32525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pickering MC, Cook HT, Warren J, Bygrave AE, Moss J, Walport MJ, et al. Uncontrolled C3 activation causes membranoproliferative glomerulonephritis in mice deficient in complement factor H. Nat Genet. 2002;31:424–428. doi: 10.1038/ng912. [DOI] [PubMed] [Google Scholar]
- 12.Pickering MC, Warren J, Rose KL, Carlucci F, Wang Y, Walport MJ, et al. Prevention of C5 activation ameliorates spontaneous and experimental glomerulonephritis in factor H-deficient mice. Proc Natl Acad Sci USA. 2006;103:9649–9654. doi: 10.1073/pnas.0601094103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Manderson AP, Botto M, Walport MJ. The role of complement in the development of systemic lupus erythematosus. Annu Rev Immunol. 2004;22:431–456. doi: 10.1146/annurev.immunol.22.012703.104549. [DOI] [PubMed] [Google Scholar]
- 14.Wouters D, Voskuyl AE, Molenaar ET, Dijkmans BA, Hack CE. Evaluation of classical complement pathway activation in rheumatoid arthritis: measurement of C1q–C4 complexes as novel activation products. Arthritis Rheum. 2006;54:1143–1150. doi: 10.1002/art.21729. [DOI] [PubMed] [Google Scholar]
- 15.Storch MK, Piddlesden S, Haltia M, Iivanainen M, Morgan P, Lassmann H. Multiple sclerosis: in situ evidence for antibody- and complement-mediated demyelination. Ann Neurol. 1998;43:465–471. doi: 10.1002/ana.410430409. [DOI] [PubMed] [Google Scholar]
- 16.Botto M, Dell’Agnola C, Bygrave AE, Thompson EM, Cook HT, Petry F, et al. Homozygous C1q deficiency causes glomerulonephritis associated with multiple apoptotic bodies. Nat Genet. 1998;19:56–59. doi: 10.1038/ng0598-56. [DOI] [PubMed] [Google Scholar]
- 17.Gaipl US, Beyer TD, Heyder P, Kuenkele S, Bottcher A, Voll RE, et al. Cooperation between C1q and DNase I in the clearance of necrotic cell-derived chromatin. Arthritis Rheum. 2004;50:640–649. doi: 10.1002/art.20034. [DOI] [PubMed] [Google Scholar]
- 18.Korb LC, Ahearn JM. C1q binds directly and specifically to surface blebs of apoptotic human keratinocytes: complement deficiency and systemic lupus erythematosus revisited. J Immunol. 1997;158:4525–4528. [PubMed] [Google Scholar]
- 19.Mitchell DA, Pickering MC, Warren J, Fossati-Jimack L, Cortes-Hernandez J, Cook HT, et al. C1q deficiency and autoimmunity: the effects of genetic background on disease expression. J Immunol. 2002;168:2538–2543. doi: 10.4049/jimmunol.168.5.2538. [DOI] [PubMed] [Google Scholar]
- 20.Taylor PR, Carugati A, Fadok VA, Cook HT, Andrews M, Carroll MC, et al. A hierarchical role for classical pathway complement proteins in the clearance of apoptotic cells in vivo. J Exp Med. 2000;192:359–366. doi: 10.1084/jem.192.3.359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Muller-Eberhard HJ. Molecular organization and function of the complement system. Annu Rev Biochem. 1988;57:321–347. doi: 10.1146/annurev.bi.57.070188.001541. [DOI] [PubMed] [Google Scholar]
- 22.Manderson AP, Pickering MC, Botto M, Walport MJ, Parish CR. Continual low-level activation of the classical complement pathway. J Exp Med. 2001;194:747–756. doi: 10.1084/jem.194.6.747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ferry H, Potter PK, Crockford TL, Nijnik A, Ehrenstein MR, Walport MJ, et al. Increased positive selection of B1 cells and reduced B cell tolerance to intracellular antigens in c1q-deficient mice. J Immunol. 2007;178:2916–2922. doi: 10.4049/jimmunol.178.5.2916. [DOI] [PubMed] [Google Scholar]
- 24.Ferry H, Jones M, Vaux DJ, Roberts IS, Cornall RJ. The cellular location of self-antigen determines the positive and negative selection of autoreactive B cells. J Exp Med. 2003;198:1415–1425. doi: 10.1084/jem.20030279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen Z, Koralov SB, Kelsoe G. Complement C4 inhibits systemic autoimmunity through a mechanism independent of complement receptors CR1 and CR2. J Exp Med. 2000;192:1339–1352. doi: 10.1084/jem.192.9.1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Prodeus AP, Goerg S, Shen LM, Pozdnyakova OO, Chu L, Alicot EM, et al. A critical role for complement in maintenance of self-tolerance. Immunity. 1998;9:721–731. doi: 10.1016/S1074-7613(00)80669-X. [DOI] [PubMed] [Google Scholar]
- 27.Strainic MG, Liu J, Huang D, An F, Lalli PN, Muqim N, et al. Locally produced complement fragments C5a and C3a provide both costimulatory and survival signals to naive CD4+ T cells. Immunity. 2008;28:425–435. doi: 10.1016/j.immuni.2008.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lalli PN, Strainic MG, Yang M, Lin F, Medof ME, Heeger PS. Locally produced C5a binds to T cell-expressed C5aR to enhance effector T-cell expansion by limiting antigen-induced apoptosis. Blood. 2008;112:1759–1766. doi: 10.1182/blood-2008-04-151068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen A, Gaddipati S, Hong Y, Volkman DJ, Peerschke EI, Ghebrehiwet B. Human T cells express specific binding sites for C1q. Role in T cell activation and proliferation. J Immunol. 1994;153:1430–1440. [PubMed] [Google Scholar]
- 30.Molina H, Miwa T, Zhou L, Hilliard B, Mastellos D, Maldonado MA, et al. Complement-mediated clearance of erythrocytes: mechanism and delineation of the regulatory roles of Crry and DAF. Decay-accelerating factor. Blood. 2002;100:4544–4549. doi: 10.1182/blood-2002-06-1875. [DOI] [PubMed] [Google Scholar]
- 31.Sun X, Funk CD, Deng C, Sahu A, Lambris JD, Song WC. Role of decay-accelerating factor in regulating complement activation on the erythrocyte surface as revealed by gene targeting. Proc Natl Acad Sci USA. 1999;96:628–633. doi: 10.1073/pnas.96.2.628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Scolding NJ, Morgan BP, Houston WA, Linington C, Campbell AK, Compston DA. Vesicular removal by oligodendrocytes of membrane attack complexes formed by activated complement. Nature. 1989;339:620–622. doi: 10.1038/339620a0. [DOI] [PubMed] [Google Scholar]
- 33.Lee H, Ratajczak MZ. Innate immunity: a key player in the mobilization of hematopoietic stem/progenitor cells. Arch Immunol Ther Exp (Warsz) 2009;57:269–278. doi: 10.1007/s00005-009-0037-6. [DOI] [PubMed] [Google Scholar]
- 34.Reca R, Mastellos D, Majka M, Marquez L, Ratajczak J, Franchini S, et al. Functional receptor for C3a anaphylatoxin is expressed by normal hematopoietic stem/progenitor cells, and C3a enhances their homing-related responses to SDF-1. Blood. 2003;101:3784–3793. doi: 10.1182/blood-2002-10-3233. [DOI] [PubMed] [Google Scholar]
- 35.Ratajczak J, Reca R, Kucia M, Majka M, Allendorf DJ, Baran JT, et al. Mobilization studies in mice deficient in either C3 or C3a receptor (C3aR) reveal a novel role for complement in retention of hematopoietic stem/progenitor cells in bone marrow. Blood. 2004;103:2071–2078. doi: 10.1182/blood-2003-06-2099. [DOI] [PubMed] [Google Scholar]
- 36.Ratajczak MZ, Reca R, Wysoczynski M, Kucia M, Baran JT, Allendorf DJ, et al. Transplantation studies in C3-deficient animals reveal a novel role of the third complement component (C3) in engraftment of bone marrow cells. Leukemia. 2004;18:1482–1490. doi: 10.1038/sj.leu.2403446. [DOI] [PubMed] [Google Scholar]
- 37.Honczarenko M, Ratajczak MZ, Nicholson-Weller A, Silberstein LE. Complement C3a enhances CXCL12 (SDF-1)-mediated chemotaxis of bone marrow hematopoietic cells independently of C3a receptor. J Immunol. 2005;175:3698–3706. doi: 10.4049/jimmunol.175.6.3698. [DOI] [PubMed] [Google Scholar]
- 38.Wysoczynski M, Reca R, Lee H, Wu W, Ratajczak J, Ratajczak MZ. Defective engraftment of C3aR−/− hematopoietic stem progenitor cells shows a novel role of the C3a–C3aR axis in bone marrow homing. Leukemia. 2009;23:1455–1461. doi: 10.1038/leu.2009.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Reca R, Cramer D, Yan J, Laughlin MJ, Janowska-Wieczorek A, Ratajczak J, et al. A novel role of complement in mobilization: immunodeficient mice are poor granulocyte-colony stimulating factor mobilizers because they lack complement-activating immunoglobulins. Stem Cells. 2007;25:3093–3100. doi: 10.1634/stemcells.2007-0525. [DOI] [PubMed] [Google Scholar]
- 40.Lee HM, Wu W, Wysoczynski M, Liu R, Zuba-Surma EK, Kucia M, et al. Impaired mobilization of hematopoietic stem/progenitor cells in C5-deficient mice supports the pivotal involvement of innate immunity in this process and reveals novel promobilization effects of granulocytes. Leukemia. 2009;23:2052–2062. doi: 10.1038/leu.2009.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nepomuceno RR, Tenner AJ. C1qRP, the C1q receptor that enhances phagocytosis, is detected specifically in human cells of myeloid lineage, endothelial cells, and platelets. J Immunol. 1998;160:1929–1935. [PubMed] [Google Scholar]
- 42.Petrenko O, Beavis A, Klaine M, Kittappa R, Godin I, Lemischka IR. The molecular characterization of the fetal stem cell marker AA4. Immunity. 1999;10:691–700. doi: 10.1016/S1074-7613(00)80068-0. [DOI] [PubMed] [Google Scholar]
- 43.Andrades JA, Nimni ME, Becerra J, Eisenstein R, Davis M, Sorgente N. Complement proteins are present in developing endochondral bone and may mediate cartilage cell death and vascularization. Exp Cell Res. 1996;227:208–213. doi: 10.1006/excr.1996.0269. [DOI] [PubMed] [Google Scholar]
- 44.Sakiyama H, Inaba N, Toyoguchi T, Okada Y, Matsumoto M, Moriya H, et al. Immunolocalization of complement C1s and matrix metalloproteinase 9 (92 kDa gelatinase/type IV collagenase) in the primary ossification center of the human femur. Cell Tissue Res. 1994;277:239–245. [PubMed] [Google Scholar]
- 45.Nakagawa K, Sakiyama H, Fukazawa T, Matsumoto M, Takigawa M, Toyoguchi T, et al. Coordinated change between complement C1s production and chondrocyte differentiation in vitro. Cell Tissue Res. 1997;289:299–305. doi: 10.1007/s004410050876. [DOI] [PubMed] [Google Scholar]
- 46.Toyoguchi T, Yamaguchi K, Nakagawa K, Fukusawa T, Moriya H, Sakiyama H. Change of complement C1s synthesis during development of hamster cartilage. Cell Tissue Res. 1996;285:199–204. doi: 10.1007/s004410050637. [DOI] [PubMed] [Google Scholar]
- 47.Sato T, Hong MH, Jin CH, Ishimi Y, Udagawa N, Shinki T, et al. The specific production of the third component of complement by osteoblastic cells treated with 1 alpha, 25-dihydroxyvitamin D3. FEBS Lett. 1991;285:21–24. doi: 10.1016/0014-5793(91)80715-F. [DOI] [PubMed] [Google Scholar]
- 48.Hong MH, Jin CH, Sato T, Ishimi Y, Abe E, Suda T. Transcriptional regulation of the production of the third component of complement (C3) by 1 alpha, 25-dihydroxyvitamin D3 in mouse marrow-derived stromal cells (ST2) and primary osteoblastic cells. Endocrinology. 1991;129:2774–2779. doi: 10.1210/endo-129-5-2774. [DOI] [PubMed] [Google Scholar]
- 49.Jin CH, Shinki T, Hong MH, Sato T, Yamaguchi A, Ikeda T, et al. 1 alpha, 25-dihydroxyvitamin D3 regulates in vivo production of the third component of complement (C3) in bone. Endocrinology. 1992;131:2468–2475. doi: 10.1210/en.131.5.2468. [DOI] [PubMed] [Google Scholar]
- 50.Sato T, Abe E, Jin CH, Hong MH, Katagiri T, Kinoshita T, et al. The biological roles of the third component of complement in osteoclast formation. Endocrinology. 1993;133:397–404. doi: 10.1210/en.133.1.397. [DOI] [PubMed] [Google Scholar]
- 51.Kimura Y, Madhavan M, Call MK, Santiago W, Tsonis PA, Lambris JD, et al. Expression of complement 3 and complement 5 in newt limb and lens regeneration. J Immunol. 2003;170:2331–2339. doi: 10.4049/jimmunol.170.5.2331. [DOI] [PubMed] [Google Scholar]
- 52.Del Rio-Tsonis K, Tsonis PA, Zarkadis IK, Tsagas AG, Lambris JD. Expression of the third component of complement, C3, in regenerating limb blastema cells of urodeles. J Immunol. 1998;161:6819–6824. [PubMed] [Google Scholar]
- 53.Legoedec J, Gasque P, Jeanne JF, Fontaine M. Expression of the complement alternative pathway by human myoblasts in vitro: biosynthesis of C3, factor B, factor H and factor I. Eur J Immunol. 1995;25:3460–3466. doi: 10.1002/eji.1830251238. [DOI] [PubMed] [Google Scholar]
- 54.Legoedec J, Gasque P, Jeanne JF, Scotte M, Fontaine M. Complement classical pathway expression by human skeletal myoblasts in vitro. Mol Immunol. 1997;34:735–741. doi: 10.1016/S0161-5890(97)00093-X. [DOI] [PubMed] [Google Scholar]
- 55.Mastellos D, Papadimitriou JC, Franchini S, Tsonis PA, Lambris JD. A novel role of complement: mice deficient in the fifth component of complement (C5) exhibit impaired liver regeneration. J Immunol. 2001;166:2479–2486. doi: 10.4049/jimmunol.166.4.2479. [DOI] [PubMed] [Google Scholar]
- 56.Michalopoulos GK, Frances MC. Liver regeneration. Science. 1997;276:60–66. doi: 10.1126/science.276.5309.60. [DOI] [PubMed] [Google Scholar]
- 57.Puschel GP, Nolte A, Schieferdecker HL, Rothermel E, Gotze O, Jungermann K. Inhibition of anaphylatoxin C3a- and C5a- but not nerve stimulation- or Noradrenaline-dependent increase in glucose output and reduction of flow in Kupffer cell-depleted perfused rat livers. Hepatology. 1996;24:685–690. doi: 10.1002/hep.510240335. [DOI] [PubMed] [Google Scholar]
- 58.Schieferdecker HL, Pestel S, Puschel GP, Gotze O, Jungermann K. Increase by anaphylatoxin C5a of glucose output in perfused rat liver via prostanoids derived from nonparenchymal cells: direct action of prostaglandins and indirect action of thromboxane A(2) on hepatocytes. Hepatology. 1999;30:454–461. doi: 10.1002/hep.510300229. [DOI] [PubMed] [Google Scholar]
- 59.Schieferdecker HL, Schlaf G, Koleva M, Gotze O, Jungermann K. Induction of functional anaphylatoxin C5a receptors on hepatocytes by in vivo treatment of rats with IL-6. J Immunol. 2000;164:5453–5458. doi: 10.4049/jimmunol.164.10.5453. [DOI] [PubMed] [Google Scholar]
- 60.Daveau M, Benard M, Scotte M, Schouft MT, Hiron M, Francois A, et al. Expression of a functional C5a receptor in regenerating hepatocytes and its involvement in a proliferative signaling pathway in rat. J Immunol. 2004;173:3418–3424. doi: 10.4049/jimmunol.173.5.3418. [DOI] [PubMed] [Google Scholar]
- 61.Markiewski MM, Mastellos D, Tudoran R, De Angelis RA, Strey CW, Franchini S, et al. C3a and C3b activation products of the third component of complement (C3) are critical for normal liver recovery after toxic injury. J Immunol. 2004;173:747–754. doi: 10.4049/jimmunol.173.2.747. [DOI] [PubMed] [Google Scholar]
- 62.Strey CW, Markiewski M, Mastellos D, Tudoran R, Spruce LA, Greenbaum LE, et al. The proinflammatory mediators C3a and C5a are essential for liver regeneration. J Exp Med. 2003;198:913–923. doi: 10.1084/jem.20030374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Markiewski MM, De Angelis RA, Strey CW, Foukas PG, Gerard C, Gerard N, et al. The regulation of liver cell survival by complement. J Immunol. 2009;182:5412–5418. doi: 10.4049/jimmunol.0804179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hong F, Nguyen VA, Shen X, Kunos G, Gao B. Rapid activation of protein kinase B/Akt has a key role in antiapoptotic signaling during liver regeneration. Biochem Biophys Res Commun. 2000;279:974–979. doi: 10.1006/bbrc.2000.4044. [DOI] [PubMed] [Google Scholar]
- 65.Corradetti MN, Guan KL. Upstream of the mammalian target of rapamycin: do all roads pass through mTOR? Oncogene. 2006;25:6347–6360. doi: 10.1038/sj.onc.1209885. [DOI] [PubMed] [Google Scholar]