

Abstract
Nitrosative stress has been implicated in a large number of neurological disorders. The molecular mechanisms underlying the neuronal injury associated with this stimulus, however, are not clearly understood. Emerging evidence suggests that the liberation of intracellular zinc as well as over-activation of potassium channels may be two important components of nitrosative stress-induced neuronal death.
Keywords: Nitric oxide, Peroxynitrite, Metallothionein, Potassium channel, Zinc, p38, Apoptosis, Kv2.1
Introduction
Nitric oxide (NO) has long been known to have a multitude of contrasting effects on biological systems and is an important neuronal cellular messenger (Garthwaite et al. 1988; Garthwaite and Boulton 1995). NO has also been associated with N-methyl-d-aspartate (NMDA) neurotoxicity, neuronal death in stroke, as well as being implicated in a variety of neurological disorders (Dawson and Dawson 1995; Aizenman et al. 1998). Emerging evidence indicates, however, that many of the toxic effects of NO may, in fact, be mediated by peroxynitrite (Estevez and Jordan 2002). Peroxynitrite can have a number of deleterious effects in cells, including oxidation of protein and non-protein sulfhydryls, induction of membrane lipid peroxidation, as well as inhibition of the electron transport chain (Radi et al. 1991, 1994). In addition to these effects, peroxynitrite can react with proteins to form nitrotyrosines, inhibiting the phosphorylation of tyrosine residues and thereby interfering with intracellular signal transduction pathways (Brito et al. 1999). Indeed, elevated levels of nitrotyrosine have been detected in a variety of disease states, including amyotrophic lateral sclerosis, Parkinson’s disease and Alzheimer’s disease (Groves et al. 1999). In the present review we discuss the possible mechanism by which nitrosative stress induces neuronal death, and summarize findings pertaining to NO-derived-species-induced release of intracellular zinc, and the link between this process and the role of potassium channels in neuronal apoptosis. We conclude by describing the effects of peroxynitrite on neuronal voltage-gated potassium channel function.
Zinc toxicity in the CNS
Zinc has been shown to be directly toxic to neurons (Yokoyama et al. 1986; Choi et al. 1988; Koh 2001). In nearly all previous studies, Zn2+-mediated neuronal cell death was dependent upon influx of the cation into cells, a process facilitated by concurrent membrane depolarization (Weiss et al. 1993). Indeed, Zn2+-induced injury can be blocked by cell-impermeant metal chelators (Koh et al. 1996), and by antagonists of Ca2+-permeable voltage- and glutamate-gated ion channels, which are potential entry routes for this ion (Weiss et al. 1993; Koh and Choi 1994; Sensi et al. 1997). As a substrate for the Na+-Ca2+ exchanger, Zn2+ may also enter neurons via this route during periods of energetic dysfunction and collapsing ion gradients (Sensi et al. 1997). Cellular influx of synaptically released Zn2+ has been suggested to contribute to the neuronal damage associated with ischemia and epilepsy, as “chelatable,” and presumably free Zn2+ is present in synaptic vesicles (Frederickson 1989). In turn, vesicular Zn2+ can be released from neurons upon depolarization (Assaf and Chung 1984; Howell et al. 1984; Aniksztejn et al. 1987; Vogt et al. 2000) and translocate from presynaptic release sites into postsynaptic neurons (Sloviter 1985; Koh et al. 1996; Frederickson et al. 1988; Tonder et al. 1990; Koh et al. 1996; Lee et al. 2000; but see Kay 2003).
The mechanism by which Zn2+ induces cell death is unresolved. Zn2+ is redox inactive under most physiological conditions (Berg and Shi 1996), and, as such, is thought to be relatively non-toxic in and of itself (Cohen and Duke 1984). Unlike iron or copper, Zn2+ does not catalyze directly the formation of reactive oxygen species. Hence, several possible mechanisms have been proposed to account for the toxic actions of Zn2+ following its translocation from the extracellular space into the cytoplasm. Manev et al. (1997) and Sensi et al. (1999, 2000, 2003) have suggested that Zn2+ can enter mitochondria and induce the release of oxygen-derived free radicals by disrupting mitochondrial integrity. In contrast, Dineley et al (2003) have suggested that the effects of Zn2+ on mitochondrial membrane potential and the ensuing dysfunction do not require influx of the metal into the mitochondria. Nonetheless, Zn2+ has been shown to affect several key components of mitochondrial function (Kleiner 1974), including inhibition of complex III (Link and von Jagow 1995) and α-ketoglutarate dehydrogenase (Brown et al. 2000), in addition to inducing permeability transition (Wudarczyk et al. 1999). Furthermore, recent work by Sheline et al. (2000) has suggested that Zn2+ can inhibit glycolysis and deplete NAD+. Each of these studies points to a critical role for mitochondrial dysfunction in Zn2+ toxicity, yet the specific cell signaling pathways that contribute to Zn2+-induced injury remain ill defined.
Zn2+-induced cellular injury has been associated with both necrosis and apoptosis (Kim et al. 1999a, 1999b). The activation of these apparently disparate cell death pathways most likely correlates to the intensity of Zn2+ exposure (Manev et al. 1997; Kim et al. 1999a, 1999b). Other factors, such as activation of Zn2+-dependent transcription factors (Atar et al. 1995; Park and Koh 1999) and protein kinases (Csermely et al. 1988; Noh et al. 1999; McLaughlin et al. 2001), as well as the release of apoptogens such as ceramide (Spence et al. 1989; Hannun 1994; Brugg et al. 1996; Schissel et al. 1996), or activation of death induction proteins (Park et al. 2000; Kim and Koh 2002), probably influence the process by which neurons die following Zn2+ exposure. Interestingly, in some systems Zn2+ can also inhibit apoptosis (Zalewski et al. 1993; Fraker and Telford 1997; Perry et al. 1997; Chai et al. 1999; Ho et al. 2000; Schrantz et al. 2001), and cellular depletion of this metal can be deleterious to cells (Ahn et al. 1998; Adler et al. 1999; Virag and Szabo 1999). As such, the balance between the physiological and pathophysiological actions of Zn2+ closely resembles that of another important ion, calcium (Lipton and Kater 1989; Toescu 1998). Indeed, Zn2+ may be sequestered by intracellular organelles (Manev et al. 1997; Sensi et al. 1999, 2000), bound to metal chaperone proteins such as metallothionein (MT; Aschner et al. 1996), and regulated by plasma membrane transporters (McMahon and Cousins 1998), all of which contribute to the maintenance intracellular Zn2+ homeostasis in a manner akin to that observed with intracellular Ca2+ (Colvin et al. 2003). Additionally, as has been suggested for Ca2+ (Choi 1992), Zn2+ is likely to trigger a variety of molecular signaling pathways depending upon the intensity and sub-cellular localization of the stimulus, as well as the energetic status of the cell (Weiss et al. 2000).
Liberation of intracellular zinc
Zinc is the second most abundant metal in the body, after iron (Takeda 2000). Of the total zinc content in the brain, only 10% is free or “chelatable,” and contained within synaptic vesicles (Frederickson 1989). Rather than existing as a free ion in the cytoplasm, non-vesicular cellular Zn2+ is complexed tightly to cysteine residues of proteins such as MT, and Zn2+ finger-containing transcription factors (Frederickson 1989; Berg and Shi 1996). It has been shown, however, that Zn2+ can be liberated from these stores by oxidative stimuli, NO-related species, as well as other metals such as Cd2+ (Fliss and Menard 1991; Kroncke et al. 1994; Berendji et al. 1997; Cuajungco and Lees 1998; Maret and Vallee 1998; Jiang et al. 1998; Aravindakumar et al. 1999; Aizenman et al. 2000; Pearce et al. 2000; Katakai et al. 2001; St Croix et al. 2002; Lee et al. 2003; Zhang et al. 2003; Hara and Aizenman 2004). Maret and Vallee (1998) have argued that MT-bound zinc can be released readily by mild thiol oxidants, in particular by disulfides such as oxidized glutathione (Maret 1994, 1995). This is due to the fact that MT itself has a relatively low redox potential (−366 mV; Maret and Vallee 1998). Hence, even though MT has a relatively high affinity for Zn2+ (Kd 1.4×10−13 M; Kagi 1993), thiol oxidants may act as cellular signals that induce the transfer of Zn2+ from MT to other proteins with lower affinities for this metal (Jiang et al. 1998).
Incubation of MT with the thiol oxidant 2,2′-dithiodipyridine (DTDP) results in the oxidation of all 20 cysteines contained in the protein and release of all 7 zinc atoms in cell-free assays (Maret and Vallee 1998). We have utilized DTDP to induce the release of zinc from intracellular metalloproteins in cortical neurons in vitro (Aizenman et al. 2000). Importantly, DTDP-liberated Zn2+ induces neuronal apoptosis via a p38 kinase-dependent process (McLaughlin et al. 2001). In that process, the intracellular release of Zn2+ is followed, sequentially, by the production of reactive oxygen species, p38 activation, enhancement of K+ currents, and, finally, caspase activation (McLaughlin et al. 2001). This enhancement of K+ currents has been shown to be a crucial element in several apoptotic paradigms (Yu 2003).
Potassium channel-mediated neuronal apoptosis
The elevation of extracellular potassium blocks apoptosis in neurons originating from a variety of central and peripheral locations. In mouse neocortical neurons, apoptosis induced by serum deprivation, or exposure to staurosporine is accompanied by amplification of a voltage-gated potassium current (Yu et al. 1997). Surprisingly, apoptosis can be prevented by the potassium channel blocker tetraethylamonium (TEA) (Yu et al. 1997). The same study also showed that exposure to the K+ ionophore valinomycin, or the potassium channel opener cromakalin can induce apoptosis. Electrophysiological measurements from cortical neurons undergoing apoptosis in other settings have also shown enhancement of K+ currents. Neuron cultures exposed to ceramide have enhanced delayed rectifier K+ currents before subsequent caspase activation and apoptosis (Yu et al. 1999). The application of TEA is neuroprotective against ceramide-induced death. In a model for amyloid β-peptide (Aβ) toxicity, a cholinergic septal cell line (SN56) exposed to Aβ exhibited a sizeable increase in TEA-sensitive outward potassium currents (Colom et al. 1998). In a separate study, cultured murine cortical neurons showed an enhancement in delayed-rectifier K+ currents after Aβ exposure (Yu et al. 1998). Co-treatment with TEA proved to be protective in both of these studies. Alterations in potassium channel activity have also been seen in transient cerebral ischemia models. Whole-cell voltage-clamp recordings from rat hippocampal slices have demonstrated that a significant increase in potassium currents occurs in CA1 neurons 6–8 h after carotid artery occlusion (Chi and Xu 2000). We have shown that primary neuronal cultures exposed to the sulfhydryl oxidizing agent DTDP also have augmented potassium currents and that this enhancement is required for the ensuing apoptotic cascade (McLaughlin et al. 2001). Thus it seems that neuronal injury provoked by various apoptotic stimuli including oxidative stress is associated with amplified potassium currents. However, the role of potassium currents in the apoptotic cascade as well as the mechanism for their augmentation has yet to be defined clearly.
It has been proposed that the enhancement of potassium currents during the apoptotic process leads to a decrease in the concentration of this cation in the cytoplasm (Yu et al. 1999) and it is the decrease of intracellular potassium that acts as an apoptotic signal. Indeed, K+ is the predominant intracellular cation and it is conceivable that the cell shrinkage associated with apoptosis is due to the loss of cellular K+. Evidence for this hypothesis can be seen in a variety of non-neuronal models of apoptosis. Walev et al. (1995) have demonstrated that the K+ depletion in monocytes results in the activation of caspases. Cidlowski and colleagues have estimated that intracellular [K+] falls from 140 mM to 35–50 mM in lymphocytes undergoing apoptosis (Bortner and Cidlowski 1999). They also determined that the activation of caspase-3 as well as the activation of the apoptotic enzyme nuclease is facilitated by such a reduction in [K+]. Conversely, apoptotic nuclease activity is completely inhibited at the normal physiological concentration of K+ (150 mM). The relevance of potassium-dependent inhibition of apoptotic enzymes was also shown in the same study by sorting apoptotic thymocytes into normal and shrunken subpopulations by flow cytometry. This sorting demonstrated that caspase activation and nuclease activity were restricted to shrunken cells with low potassium content. A similar observation was made in an earlier study using dexamethasone-treated mouse lymphoma cell line (S49 Neo) cultures (Bortner and Cidlowski 1996). This study as well demonstrated that only shrunken apoptotic cells with a low [K+] have caspase activity and degraded DNA. Even though these studies show possible implications of intracellular potassium loss in promoting cell death, they have yet to be demonstrated in neuronal models of apoptosis.
Another issue is the identity of the potassium channel or channels that are involved in this process. A number of functionally distinct voltage-gated potassium channel types have been identified in mammalian neurons, including low- and high-conductance, Ca2+-dependent channels, inward rectifiers, transient A-type channels, and the delayed rectifier. We, as well as others, have observed that the enhanced currents are outward, have slow kinetics, and are sensitive to TEA, consistent with a delayed rectifier channel. However, certain studies have also described alterations in a transient outward current that is sensitive to 4-aminopyridine (4AP). Yu et al. (1997) have observed an increase in this fast inactivating current after serum deprivation and a decrease in this current after staurosporine treatment in cortical cultures. Chi and Xu (2000) also have shown an increase in amplitude for the transient current in CA1 pyramidal neurons after transient forebrain ischemia. This reported alteration is consistent with the observation that the excitability of CA1 neurons progressively decreases after an ischemic insult. It does not seem likely, however, that changes in the rapidly inactivating K current are involved in the apoptotic death pathway. Blocking this current with 4AP during any neuronal death paradigm associated with enhanced K currents has been reported to be ineffective as a protective strategy. Other studies have reported that M-type potassium channel blockers, such as linopiridine, promote survival in rat sympathetic neurons deprived of nerve growth factor (Xia et al. 2002). This effect, as well as the antiapoptotic effect of elevated extracellular potassium in sympathetic neurons, seems to be mediated by an increase in intracellular Ca2+ through L-type voltage-dependent Ca2+ channels (Galli et al. 1995). Work in central neurons has shown that TEA and elevated extracellular potassium is protective independently of changes in intracellular Ca2+ (Yu et al. 1997). Given these results, it seems that the alteration of a delayed rectifier channel is what is crucial in a variety of apoptotic pathways. Recently, the specific channel mediating the enhanced potassium currents associated with neuronal apoptosis has been identified by our group (Pal et al. 2003). We have shown that disruption of Kv2.1-encoded potassium channels prevents the current enhancements after treatment with toxic doses of DTDP or staurosporine. Disruption of these channels also precludes the ensuing cell death after treatment with these apoptogens. It thus seems that Kv2.1-encoded potassium channels provide the main exit route for K+ in cortical neurons undergoing apoptosis.
Peroxynitrite mediated enhancement of potassium currents
3-Morpholinosyndnomine (SIN-1) decomposes in solution to release both NO and superoxide (Holm et al. 1998). Three h after treatment of cortical neurons with 1 mM SIN-1, a robust potentiation occurs in potassium currents with relatively slow kinetics, consistent with that of delayed rectifier channels (Fig. 1a). This increase in steady-state amplitude is not accompanied by any change in voltage dependence (Fig. 1b), as reported previously (Yu et al. 1997; Pal et al. 2003), or by any significant change in the activation time constant (step to +5 mV from a holding potential of −70 mV: 9.6±2.7 ms n=9, control; 13.5±1.5 ms n=14, SIN-1; means±SEM). On the basis of the extremely fast reaction rate between NO and superoxide (2×1010 M−1 s−1; Kissner et al. 1998) we assumed that the effect of SIN-1 is mediated by peroxynitrite. To confirm this, we co-treated neurons with 1 mM SIN-1 and 25 μM 5,10,15,20-tetrakis(4-sulfonatophenyl)porphyrinato iron [III] (FeTPPS), a ferric porphyrin complex that can isomerize peroxynitrite to nitrate very rapidly (Misko et al. 1998). Co-treatment with FeTPPS completely blocked any current enhancement produced by SIN-1 (Fig. 1c). Treatment with FeTPPS alone had no effect on potassium. We also determined whether the direct application of peroxynitrite itself could mimic the effects of SIN-1 on potassium currents. Cultures were thus treated continuously with 150 μM peroxynitrite and whole-cell recordings obtained 3 h after initiation of treatment. Peroxynitrite produced a consistent and sizeable potentiation in potassium currents (Fig. 2), indistinguishable from the effects produced by SIN-1. These results show that neuronal exposure to peroxynitrite can result in sizeable changes in delayed rectifier potassium currents.
Fig. 1a–c.

3-Morpholinosydnonimine (SIN-1)-induced changes in potassium channel function. a Whole-cell potassium currents obtained in two separate cortical neurons after 180 min exposure to either vehicle (control) or 1 mM SIN-1. Currents were evoked by a series of voltage steps to +35 mV from a holding potential of −70 mV. b Current/voltage relationship of a single vehicle-treated neuron (closed circles) and a neuron exposed to 1 mM SIN-1 (open circles). c Whole-cell potassium currents were obtained from cortical neurons following 180 min exposure to vehicle (n=4), 1 mM SIN-1 alone (n=7), or SIN-1 in the presence of 25 μM 5,10,15,20-tetrakis(4-sulfonatophenyl)porphyrinato iron [III] (FeTPPS, n=6). Potassium currents were evoked by a voltage step to +5 mV from a holding potential of −70 mV. Current amplitudes were normalized to cell capacitance. Mean±SEM; *P<0.001 vs. control (ANOVA followed by a Dunnett multiple comparison test)
Fig. 2.
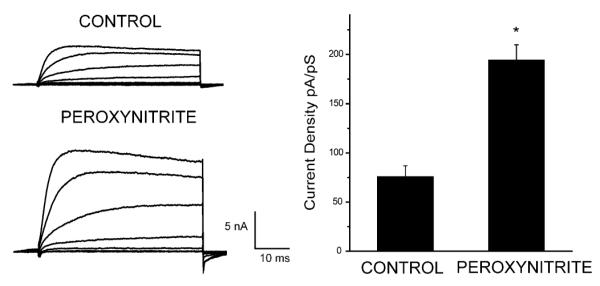
Peroxynitrite enhances potassium channel currents. Whole-cell potassium currents were obtained from cortical neurons following 180 min exposure to vehicle (n=4), or peroxynitrite (100 μM, n=6). Current amplitudes were normalized to cell capacitance. Potassium currents were evoked with a series of voltage step to +35 mV from a holding potential of −70 mV. Bars represent mean (±SEM) potassium current density (to +5 mV) for both treatment groups; *P<0.001 vs. control (two-tailed Student’s t-test)
To establish whether zinc release is also involved in the potassium current changes mediated by peroxynitrite, we determined whether the zinc chelator N,N,N’,N’-tetrakis(2-pyridylmethyl)ethylenediamine (TPEN) inhibits the current enhancements induced by SIN-1. Co-exposure to 1 mM SIN-1 and 10 μM TPEN completely prevented the current enhancement (Fig. 3), indicating that zinc chelation does in fact prevent the signal transduction pathway involved in the peroxynitrite-induced amplification of potassium currents. To rule out the possibility that the origin of the liberated zinc was extracellular, we repeated these experiments with a zinc chelator that is not membrane permeable. Tricine, at a concentration previously shown to be able to buffer high levels of extracellular zinc (10 mM; Paoletti et al 1997), was ineffective in blocking the SIN-1 induced enhancement of potassium currents (Fig. 3). This indicates that the source of the zinc mediating this effect is, in fact, intracellular.
Fig. 3.
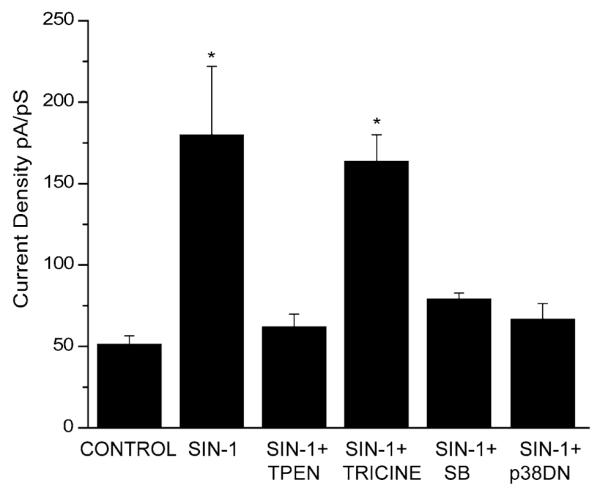
Role of Zn2+ and p38 and in peroxynitrite-induced current enhancement. Neurons were exposed for 180 min to vehicle (n=4), 1 mM SIN-1 (n=6), and 1 mM SIN-1 in the presence of the zinc chelators N,N,N’,N’-tetrakis(2-pyridylmethyl)ethylenediamine (TPEN, 10 μM, n=6), tricine (10 mM, n=6), or the p38 inhibitor SB 239063 (20 μM, n=8). Recordings were also made in neurons expressing a p38 dominant-negative construct (p38DN). Potassium currents were evoked by voltage steps to +5 mV from a holding potential of −70 mV. Current amplitudes were normalized to cell capacitance. Means±SEM; *P<0.005 vs. control (ANOVA followed by a Dunnett multiple comparisons test)
Recently, we have also tested a highly selective and neuroprotective inhibitor of p38, SB 239063 (Underwood et al. 2000; McLaughlin et al. 2001; Legos et al. 2002), to determine whether p38 activation is required for SIN-1 induced changes in potassium channel activity, similar to the cascade initiated by DTDP (McLaughlin et al. 2001). Neurons co-treated for 3 h with SIN-1 and SB 239063 (20 μM) showed no enhancement in whole cell potassium currents (Fig. 3). To confirm these results, we disrupted endogenous p38α function with the use of a dominant-negative (DN) expression construct. Recordings were obtained from neurons expressing the p38 DN and green fluorescent protein (GFP) and treated with SIN-1. Disruption of p38 function with this method was sufficient to prevent the current enhancement produced by SIN-1 (Fig. 3). In contrast, non-transfected neurons on the same cover-slip (Pal et al. 2003) still displayed increases in current density. To test for the possibility that the overexpression of p38 protein itself may have interfered with potassium channel conductance, we recorded from neurons transfected with wild-type p38 and GFP. Under these circumstances we observed no changes in current density. Thus, p38 function appears to be necessary for changes in potassium channel activity stimulated by peroxynitrite.
These results demonstrate that peroxynitrite can up-regulate voltage-gated potassium channel activity in cultured cortical neurons. This enhancement occurs through the participation of intracellular zinc, and p38. Similar results were reported very recently by Bossy-Wetzel et al. (2004), confirming the existence of the zinc-p38-K channel neuronal cell death axis described in our earlier work with thiol oxidants (Aizenman et al. 2000; McLaughlin et al. 2001). The mechanism by which zinc activates p38 after peroxynitrite exposure still needs further elucidation, although recent reports have shown that mobilized intracellular zinc can affect mitochondrial function (Dineley et al. 2003; Sensi et al. 2003), which may in turn lead to p38 activation (Tobiume et al. 2001; McLaughlin et al. 2001). One can thus envision a scenario in which peroxynitrite triggers the release of intracellular zinc, causing mitochondrial impairment and p38 activation, leading to the enhancement of voltage-gated potassium currents; a pathway that is virtually identical to that induced by oxidants such as DTDP (Aizenman et al. 2000; McLaughlin et al. 2001).
Perspectives
The enhancement of potassium channel activity following an apoptotic stimulus may prove to be a widespread phenomenon in neuronal injury. Following the identification of Kv2.1-encoded channels as the species responsible for the potentiation of delayed rectification following oxidative injury (Pal et al. 2003), a large-scale, high-throughput screening in a yeast system very recently identified a novel lead neuroprotective compound that blocks Kv2.1-mediated currents and neuronal apoptosis in the low micromolar range (Zaks-Makhina et al. 2004). Hence, selective pharmacological targeting of the potassium channels responsible for the enhanced currents may provide a novel therapeutic strategy in a wide number of neurodegenerative disorders.
Acknowledgements
The authors wish to thank I.J. Reynolds for helpful suggestions and K.A. Hartnett for technical help. SB 239063 was kindly provided by F.C. Barone (GlaxoSmithKline, Philadelphia, PA, USA); the p38-containing vectors (pCMV5-DNp38α and pcDNA3-Flag-p38α) were the gift of J. Han (Scripps Institute, La Jolla, CA, USA). This work was supported by NIH grant NS43277 to EA, and by an American Heart Association Predoctoral Fellowship to SP.
References
- Adler M, Shafer H, Hamilton T, Petrali JP. Cytotoxic actions of the heavy metal chelator TPEN on NG108-15 neuroblastoma-glioma cells. Neurotoxicology. 1999;20:571–582. [PubMed] [Google Scholar]
- Ahn YH, Kim YH, Hong SH, Koh JY. Depletion of intracellular zinc induces protein synthesis-dependent neuronal apoptosis in mouse cortical culture. Exp Neurol. 1998;154:47–56. doi: 10.1006/exnr.1998.6931. [DOI] [PubMed] [Google Scholar]
- Aizenman E, Brimecombe JC, Potthoff WK, Rosenberg PA. Why is the role of nitric oxide in NMDA receptor function and dysfunction so controversial? Prog Brain Res. 1998;118:53–71. doi: 10.1016/s0079-6123(08)63200-8. [DOI] [PubMed] [Google Scholar]
- Aizenman E, Stout AK, Hartnett KA, Dineley KE, McLaughlin B, Reynolds IJ. Induction of neuronal apoptosis by thiol oxidation: putative role of intracellular zinc release. J Neurochem. 2000;75:1878–1888. doi: 10.1046/j.1471-4159.2000.0751878.x. [DOI] [PubMed] [Google Scholar]
- Aniksztejn L, Charton G, Ben-Ari Y. Selective release of endogenous zinc from the hippocampal mossy fibers in situ. Brain Res. 1987;404:58–64. doi: 10.1016/0006-8993(87)91355-2. [DOI] [PubMed] [Google Scholar]
- Aravindakumar CT, Ceulemans J, De Ley M. Nitric oxide induces Zn2+ release from metallothionein by destroying zinc-sulphur clusters without concomitant formation of S-nitrosothiol. Biochem J. 1999;344:253–258. doi: 10.1042/0264-6021:3440253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aschner M. The functional significance of brain metallothioneins. FASEB J. 1996;10:1129–1136. doi: 10.1096/fasebj.10.10.8751715. [DOI] [PubMed] [Google Scholar]
- Assaf SY, Chung SH. Release of endogenous Zn2+ from brain tissue during activity. Nature. 1984;308:734–736. doi: 10.1038/308734a0. [DOI] [PubMed] [Google Scholar]
- Atar D, Backx PH, Appel MM, Gao WD, Marban E. Excitation-transcription coupling mediated by zinc influx through voltage-dependent calcium channels. J Biol Chem. 1995;270:2473–2477. doi: 10.1074/jbc.270.6.2473. [DOI] [PubMed] [Google Scholar]
- Berendji D, Kolb-Bachofen V, Meyer KL, Grapenthin O, Weber H, Wahn V, Kroncke KD. Nitric oxide mediates intracyto-plasmic and intranuclear zinc release. FEBS Lett. 1997;405:37–41. doi: 10.1016/s0014-5793(97)00150-6. [DOI] [PubMed] [Google Scholar]
- Berg JM, Shi Y. The galvanization of biology: a growing appreciation for the roles of zinc. Science. 1996;271:1081–1085. doi: 10.1126/science.271.5252.1081. [DOI] [PubMed] [Google Scholar]
- Bortner CD, Cidlowski JA. Absence of volume regulatory mechanisms contributes to the rapid activation of apoptosis in thymocytes. Am J Physiol. 1996;271:C950–C961. doi: 10.1152/ajpcell.1996.271.3.C950. [DOI] [PubMed] [Google Scholar]
- Bortner CD, Cidlowski JA. Caspase independent/dependent regulation of K+, cell shrinkage, and mitochondrial membrane potential during lymphocyte apoptosis. J Biol Chem. 1999;274:21953–21962. doi: 10.1074/jbc.274.31.21953. [DOI] [PubMed] [Google Scholar]
- Bossy-Wetzel E, Talantova MV, Lee WD, Scholzke MN, Harrop A, Mathews E, Gotz T, Han J, Ellisman MH, Perkins GA, Lipton SA. Crosstalk between nitric oxide and zinc pathways to neuronal cell death involving mitochondrial dysfunction and p38-activated K+ channels. Neuron. 2004;41:351–365. doi: 10.1016/s0896-6273(04)00015-7. [DOI] [PubMed] [Google Scholar]
- Brito C, Naviliat M, Tiscornia AC, Vuillier F, Gualco G, Dighiero G, Radi R, Cayota AM. Peroxynitrite inhibits T lymphocyte activation and proliferation by promoting impairment of tyrosine phosphorylation and peroxynitrite-driven apoptotic death. J Immunol. 1999;162:3356–3366. [PubMed] [Google Scholar]
- Brown AM, Kristal BS, Effron MS, Shestopalov AI, Ullucci PA, Sheu KF, Blass JP, Cooper AJ. Zn2+ inhibits alpha-ketoglutarate-stimulated mitochondrial respiration and the isolated alpha-ketoglutarate dehydrogenase complex. J Biol Chem. 2000;275:13441–13447. doi: 10.1074/jbc.275.18.13441. [DOI] [PubMed] [Google Scholar]
- Brugg B, Michel PP, Agid Y, Ruberg M. Ceramide induces apoptosis in cultured mesencephalic neurons. J Neurochem. 1996;66:733–739. doi: 10.1046/j.1471-4159.1996.66020733.x. [DOI] [PubMed] [Google Scholar]
- Chai F, Truong-Tran AQ, Ho LH, Zalewski PD. Regulation of caspase activation and apoptosis by cellular zinc fluxes and zinc deprivation: a review. Immunol Cell Biol. 1999;77:272–278. doi: 10.1046/j.1440-1711.1999.00825.x. [DOI] [PubMed] [Google Scholar]
- Chi XX, Xu ZC. Differential changes of potassium currents in CA1 pyramidal neurons after transient forebrain ischemia. J Neurophysiol. 2000;84:2834–2843. doi: 10.1152/jn.2000.84.6.2834. [DOI] [PubMed] [Google Scholar]
- Choi DW. Excitotoxic cell death. J Neurobiol. 1992;23:1261–1276. doi: 10.1002/neu.480230915. [DOI] [PubMed] [Google Scholar]
- Choi DW, Yokoyama M, Koh J. Zinc neurotoxicity in cortical cell culture. Neuroscience. 1988;24:67–79. doi: 10.1016/0306-4522(88)90312-0. [DOI] [PubMed] [Google Scholar]
- Cohen JJ, Duke RC. Glucocorticoid activation of a calcium-dependent endonuclease in thymocyte nuclei leads to cell death. J Immunol. 1984;132:38–42. [PubMed] [Google Scholar]
- Colom LV, Diaz ME, Beers DR, Neely A, Xie WJ, Appel SH. Role of potassium channels in amyloid-induced cell death. Neurochemistry. 1998;70:1925–1934. doi: 10.1046/j.1471-4159.1998.70051925.x. [DOI] [PubMed] [Google Scholar]
- Colvin RA, Fontaine CP, Laskowski M, Thomas D. Zn2+ transporters and Zn2+ homeostasis in neurons. Eur J Pharmacol. 2003;479:171–185. doi: 10.1016/j.ejphar.2003.08.067. [DOI] [PubMed] [Google Scholar]
- Csermely P, Szamel M, Resch K, Somogyi J. Zinc can increase the activity of protein kinase C and contributes to its binding to plasma membranes in T lymphocytes. J Biol Chem. 1988;263:6487–6490. [PubMed] [Google Scholar]
- Cuajungco MP, Lees GJ. Nitric oxide generators produce accumulation of chelatable zinc in hippocampal neuronal perikarya. Brain Res. 1998;799:118–129. doi: 10.1016/s0006-8993(98)00463-6. [DOI] [PubMed] [Google Scholar]
- Dawson VL, Dawson TM. Physiological and toxicological actions of nitric oxide in the central nervous system. Adv Pharmacol. 1995;34:323–342. doi: 10.1016/s1054-3589(08)61095-9. [DOI] [PubMed] [Google Scholar]
- Dineley KE, Votyakova TV, Reynolds IJ. Zinc inhibition of cellular energy production: implication for mitochondria and neurodegeneration. J Neurochem. 2003;85:563–570. doi: 10.1046/j.1471-4159.2003.01678.x. [DOI] [PubMed] [Google Scholar]
- Estevez AG, Jordan J. Nitric oxide and superoxide, a deadly cocktail. Ann NY Acad Sci. 2002;962:207–211. doi: 10.1111/j.1749-6632.2002.tb04069.x. [DOI] [PubMed] [Google Scholar]
- Fliss H, Menard M. Hypochlorous acid-induced mobilization of zinc from metalloproteins. Arch Biochem Biophys. 1991;287:175–179. doi: 10.1016/0003-9861(91)90403-6. [DOI] [PubMed] [Google Scholar]
- Fraker PJ, Telford WG. A reappraisal of the role of zinc in life and death decisions of cells. Exp Biol Med. 1997;215:229–236. doi: 10.3181/00379727-215-44132. [DOI] [PubMed] [Google Scholar]
- Frederickson CJ. Neurobiology of zinc and zinc-containing neurons. Int Rev Neurobiol. 1989;31:145–238. doi: 10.1016/s0074-7742(08)60279-2. [DOI] [PubMed] [Google Scholar]
- Frederickson CJ, Hernandez MD, Goik SA, Morton JD, McGinty JF. Loss of zinc staining from hippocampal mossy fibers during kainic acid induced seizures: a histofluorescence study. Brain Res. 1988;446:383–386. doi: 10.1016/0006-8993(88)90899-2. [DOI] [PubMed] [Google Scholar]
- Galli C, Meucci O, Scorziello A, Werge TM, Calissano P, Schettini G. Apoptosis in cerebellar granule cells is blocked by high KCl, forskolin, and IGF-1 through distinct mechanisms of action: the involvement of intracellular calcium and RNA synthesis. J Neurosci. 1995;15:1172–1179. doi: 10.1523/JNEUROSCI.15-02-01172.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garthwaite J, Boulton CL. Nitric oxide signalling in the central nervous system. Annu Rev Physiol. 1995;57:683–706. doi: 10.1146/annurev.ph.57.030195.003343. [DOI] [PubMed] [Google Scholar]
- Garthwaite J, Charles SL, Chess-Williams R. Endothelium-derived relaxing factor release on activation of NMDA receptors suggests role as intercellular messenger in the brain. Nature. 1988;336:385–388. doi: 10.1038/336385a0. [DOI] [PubMed] [Google Scholar]
- Groves JT. Peroxynitrite: reactive, invasive and enigmatic. Curr Opin Chem Biol. 1999;3:226–235. doi: 10.1016/S1367-5931(99)80036-2. [DOI] [PubMed] [Google Scholar]
- Hannun YA. The sphingomyelin cycle and the second messenger function of ceramide. J Biol Chem. 1994;269:3125–3128. [PubMed] [Google Scholar]
- Hara H, Aizenman E. A molecular technique for detecting the liberation of intracellular zinc in cultured neurons. J Neurosci Methods. 2004 doi: 10.1016/j.jneumeth.2004.02.018. (In press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho LH, Ratnaike RN, Zalewski PD. Involvement of intracellular labile zinc in suppression of DEVD-caspase activity in human neuroblastoma cells. Biochem Biophys Res Commun. 2000;268:148–154. doi: 10.1006/bbrc.2000.2090. [DOI] [PubMed] [Google Scholar]
- Holm P, Kankaanranta H, Metsa-Ketela T, Moilanen E. Radical releasing properties of nitric oxide donors GEA 3162, SIN-1 and S-nitroso-N-acetylpenicillamine. Eur J Pharmacol. 1998;346:97–102. doi: 10.1016/s0014-2999(98)00009-0. [DOI] [PubMed] [Google Scholar]
- Howell GA, Welch MG, Frederickson CJ. Stimulation-induced uptake and release of zinc in hippocampal slices. Nature. 1984;308:736–738. doi: 10.1038/308736a0. [DOI] [PubMed] [Google Scholar]
- Jiang LJ, Maret W, Vallee BL. The glutathione redox couple modulates zinc transfer from metallothionein to zinc-depleted sorbitol dehydrogenase. Proc Natl Acad Sci USA. 1998;95:3483–348. doi: 10.1073/pnas.95.7.3483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kägi JHR. Evolution, structure and chemical activity of class I metallothioneins: an overview. In: Suzuki KT, Imura N, Kimura M, editors. Metallothionein III: biological roles and medical implications. Birkhäuser; Basel: 1993. pp. 29–55. [Google Scholar]
- Katakai K, Liu J, Nakajima K, Keefer LK, Waalkes MP. Nitric oxide induces metallothionein (MT) gene expression apparently by displacing zinc bound to MT. Toxicol Lett. 2001;119:103–108. doi: 10.1016/s0378-4274(00)00301-5. [DOI] [PubMed] [Google Scholar]
- Kay AR. Evidence for chelatable zinc in the extracellular space of the hippocampus, but little evidence for synaptic release of Zn. J Neurosci. 2003;23:6847–6855. doi: 10.1523/JNEUROSCI.23-17-06847.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YH, Koh JY. The role of NADPH oxidase and neuronal nitric oxide synthase in zinc-induced poly (ADP-ribose) polymerase activation and cell death in cortical culture. Exp Neurol. 2002;177:407–418. doi: 10.1006/exnr.2002.7990. [DOI] [PubMed] [Google Scholar]
- Kim EY, Koh JY, Kim YH, Sohn S, Joe E, Gwag BJ. Zn2+ entry produces oxidative neuronal necrosis in cortical cell cultures. Eur J Neurosci. 1999a;11:327–334. doi: 10.1046/j.1460-9568.1999.00437.x. [DOI] [PubMed] [Google Scholar]
- Kim YH, Kim EY, Gwag BJ, Sohn S, Koh JY. Zinc-induced cortical neuronal death with features of apoptosis and necrosis: mediation by free radicals. Neuroscience. 1999b;89:175–182. doi: 10.1016/s0306-4522(98)00313-3. [DOI] [PubMed] [Google Scholar]
- Kissner R, Nauser T, Bugnon P, Lye PG, Koppenol WH. Formation and properties of peroxynitrite as studied by laser flash photolysis, high-pressure stopped-flow technique, and pulse radiolysis. Chem Res Toxicol. 1998;10:1285–1292. doi: 10.1021/tx970160x. [DOI] [PubMed] [Google Scholar]
- Kleiner D. The effect of Zn2+ ions on mitochondrial electron transport. Arch Biochem Biophys. 1974;165:121–125. doi: 10.1016/0003-9861(74)90148-9. [DOI] [PubMed] [Google Scholar]
- Koh JY. Zinc and diseases of the brain. Mol Neurobiol. 2001;24:99–106. doi: 10.1385/MN:24:1-3:099. [DOI] [PubMed] [Google Scholar]
- Koh JY, Choi DW. Zinc toxicity on cultured cortical neurons: involvement of N-methyl-d-aspartate receptors. Neuroscience. 1994;60:1049–1057. doi: 10.1016/0306-4522(94)90282-8. [DOI] [PubMed] [Google Scholar]
- Koh JY, Suh SW, Gwag BJ, He YY, Hsu CY, Choi DW. The role of zinc in selective neuronal death after transient global cerebral ischemia. Science. 1996;272:1013–1016. doi: 10.1126/science.272.5264.1013. [DOI] [PubMed] [Google Scholar]
- Kroncke KD, Fehsel K, Schmidt T, Zenke FT, Dasting I, Wesener JR, Bettermann H, Breunig KD, Kolb-Bachofen V. Nitric oxide destroys zinc-sulfur clusters inducing zinc release from metallothionein and inhibition of the zinc finger-type yeast transcription activator LAC9. Biochem Biophys Res Commun. 1994;200:1105–1110. doi: 10.1006/bbrc.1994.1564. [DOI] [PubMed] [Google Scholar]
- Lee JY, Park J, Kim YH, Kim DH, Kim CG, Koh JY. Induction by synaptic zinc of heat shock protein-70 in hippocampus after kainate seizures. Exp Neurol. 2000;161:433–441. doi: 10.1006/exnr.1999.7297. [DOI] [PubMed] [Google Scholar]
- Lee JY, Kim JH, Palmiter RD, Koh JY. Zinc released from metallothionein-III may contribute to hippocampal CA1 and thalamic neuronal death following acute brain injury. Exp Neurol. 2003;184:337–347. doi: 10.1016/s0014-4886(03)00382-0. [DOI] [PubMed] [Google Scholar]
- Legos JJ, McLaughlin B, Skaper SD, Strijbos PJ, Parsons AA, Aizenman E, Herin GA, Barone FC, Erhardt JA. The selective p38 inhibitor SB-239063 protects primary neurons from mild to moderate excitotoxic cell death. Eur J Pharmacol. 2002;447:37–42. doi: 10.1016/s0014-2999(02)01890-3. [DOI] [PubMed] [Google Scholar]
- Link TA, von Jagow G. Zinc ions inhibit the QP center of bovine heart mitochondrial bc1 complex by blocking a protonatable group. J Biol Chem. 1995;270:25001–25006. doi: 10.1074/jbc.270.42.25001. [DOI] [PubMed] [Google Scholar]
- Lipton SA, Kater SB. Neurotransmitter regulation of neuronal outgrowth, plasticity and survival. Trends Neurosci. 1989;12:265–270. doi: 10.1016/0166-2236(89)90026-x. [DOI] [PubMed] [Google Scholar]
- Manev H, Kharlamov E, Uz T, Mason RP, Cagnoli CM. Characterization of zinc-induced neuronal death in primary cultures of rat cerebellar granule cells. Exp Neurol. 1997;146:171–178. doi: 10.1006/exnr.1997.6510. [DOI] [PubMed] [Google Scholar]
- Maret W. Oxidative metal release from metallothionein via zinc-thiol/disulfide interchange. Proc Natl Acad Sci USA. 1994;91:237–241. doi: 10.1073/pnas.91.1.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maret W. Metallothionein/disulfide interactions, oxidative stress, and the mobilization of cellular zinc. Neurochem Int. 1995;27:111–117. doi: 10.1016/0197-0186(94)00173-r. [DOI] [PubMed] [Google Scholar]
- Maret W, Vallee BL. Thiolate ligands in metallothionein confer redox activity on zinc clusters. Proc Natl Acad Sci USA. 1998;95:3478–3482. doi: 10.1073/pnas.95.7.3478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaughlin B, Pal S, Tran MP, Parsons AA, Barone FC, Erhardt JA, Aizenman E. p38 activation is required upstream of potassium current enhancement and caspase cleavage in thiol oxidant-induced neuronal apoptosis. J Neurosci. 2001;21:3303–3311. doi: 10.1523/JNEUROSCI.21-10-03303.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMahon RJ, Cousins RJ. Regulation of the zinc transporter ZnT-1 by dietary zinc. Proc Natl Acad Sci USA. 1998;95:4841–486. doi: 10.1073/pnas.95.9.4841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misko TP, Highkin MK, Veenhuizen AW, Manning PT, Stern MK, Currie MG, Salvemini D. Characterization of the cytoprotective action of peroxynitrite decomposition catalysts. J Biol Chem. 1998;273:15646–15653. doi: 10.1074/jbc.273.25.15646. [DOI] [PubMed] [Google Scholar]
- Noh KM, Kim YH, Koh JY. Mediation by membrane protein kinase C of zinc-induced oxidative neuronal injury in mouse cortical cultures. J Neurochem. 1999;72:1609–1616. doi: 10.1046/j.1471-4159.1999.721609.x. [DOI] [PubMed] [Google Scholar]
- Pal S, Hartnett KA, Nerbonne JM, Levitan ES, Aizenman E. Mediation of neuronal apoptosis by Kv2.1-encoded potassium channels. J Neurosci. 2003;23:4798–4802. doi: 10.1523/JNEUROSCI.23-12-04798.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paoletti P, Ascher P, Neyton J. High-affinity zinc inhibition of NMDA NR1-NR2A receptors. J Neurosci. 1997;17:5711–5725. doi: 10.1523/JNEUROSCI.17-15-05711.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JA, Koh JY. Induction of an immediate early gene egr-1 by zinc through extracellular signal-regulated kinase activation in cortical culture: its role in zinc-induced neuronal death. J Neurochem. 1999;73:450–456. doi: 10.1046/j.1471-4159.1999.0730450.x. [DOI] [PubMed] [Google Scholar]
- Park JA, Lee JY, Sato TA, Koh JY. Co-induction of p75NTR and p75NTR-associated death executor in neurons after zinc exposure in cortical culture or transient ischemia in the rat. J Neurosci. 2000;20:9096–9103. doi: 10.1523/JNEUROSCI.20-24-09096.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearce LL, Gandley RE, Han W, Wasserloos K, Stitt M, Kanai AJ, McLaughlin MK, Pitt BR, Levitan ES. Role of metallothionein in nitric oxide signaling as revealed by a green fluorescent fusion protein. Proc Natl Acad Sci USA. 2000;97:477–482. doi: 10.1073/pnas.97.1.477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry DK, Smyth MJ, Stennicke HR, Salvesen GS, Duriez P, Poirier GG, Hannun YA. Zinc is a potent inhibitor of the apoptotic protease, caspase-3. A novel target for zinc in the inhibition of apoptosis. J Biol Chem. 1997;272:18530–18533. doi: 10.1074/jbc.272.30.18530. [DOI] [PubMed] [Google Scholar]
- Radi R, Beckman JS, Bush KM, Freeman BA. Peroxynitrite-induced membrane lipid peroxidation: the cytotoxic potential of superoxide and nitric oxide. Arch Biochem Biophys. 1991;288:481–487. doi: 10.1016/0003-9861(91)90224-7. [DOI] [PubMed] [Google Scholar]
- Radi R, Rodriguez M, Castro L, Telleri R. Inhibition of mitochondrial electron transport by peroxynitrite. Arch Biochem Biophys. 1994;308:89–95. doi: 10.1006/abbi.1994.1013. [DOI] [PubMed] [Google Scholar]
- Schissel SL, Schuchman EH, Williams KJ, Tabas I. Zn2+-stimulated sphingomyelinase is secreted by many cell types and is a product of the acid sphingomyelinase gene. J Biol Chem. 1996;271:18431–18436. doi: 10.1074/jbc.271.31.18431. [DOI] [PubMed] [Google Scholar]
- Schrantz N, Auffredou M-T, Bourgeade MF, Besnault L, Leca G, Vazquez A. Zinc-mediated regulation of caspases activity: dose-dependent inhibition or activation of caspase-3 in the human Burkitt lymphoma B cells (Ramos) Cell Death Diff. 2001;8:152–161. doi: 10.1038/sj.cdd.4400772. [DOI] [PubMed] [Google Scholar]
- Sensi SL, Canzoniero LM, Yu SP, Ying HS, Koh JY, Kerchner GA, Choi DW. Measurement of intracellular free zinc in living cortical neurons: routes of entry. J Neurosci. 1997;17:9554–9564. doi: 10.1523/JNEUROSCI.17-24-09554.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sensi SL, Yin HZ, Carriedo SG, Rao SS, Weiss JH. Preferential Zn2+ influx through Ca2+-permeable AMPA/ kainate channels triggers prolonged mitochondrial superoxide production. Proc Natl Acad Sci USA. 1999;96:2414–2419. doi: 10.1073/pnas.96.5.2414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sensi SL, Yin HZ, Weiss JH. AMPA/kainate receptor-triggered Zn2+ entry into cortical neurons induces mitochon-drial Zn2+ uptake and persistent mitochondrial dysfunction. Eur J Neurosci. 2000;12:3813–3818. doi: 10.1046/j.1460-9568.2000.00277.x. [DOI] [PubMed] [Google Scholar]
- Sensi SL, Ton-That D, Sullivan PG, Jonas EA, Gee KR, Kaczmarek LK, Weiss JH. Modulation of mitochondrial function by endogenous Zn2+ pools. Proc Natl Acad Sci USA. 2003;100:6157–6162. doi: 10.1073/pnas.1031598100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheline CT, Behrens MM, Choi DW. Zinc-induced cortical neuronal death: contribution of energy failure attributable to loss of NAD+ and inhibition of glycolysis. J Neurosci. 2000;20:3139–3146. doi: 10.1523/JNEUROSCI.20-09-03139.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sloviter RS. A selective loss of hippocampal mossy fiber Timm stain accompanies granule cell seizure activity induced by perforant path stimulation. Brain Res. 1985;330:150–153. doi: 10.1016/0006-8993(85)90017-4. [DOI] [PubMed] [Google Scholar]
- Spence MW, Byers DM, Palmer FB, Cook HW. A new Zn2+-stimulated sphingomyelinase in fetal bovine serum. J Biol Chem. 1989;264:5358–5363. [PubMed] [Google Scholar]
- St. Croix CM, Wasserloos KJ, Dineley KE, Reynolds IJ, Levitan ES, Pitt BR. Nitric oxide-induced changes in intracellular zinc homeostasis are mediated by metallothionein/thionein. Am J Physiol. 2002;282:L185–L192. doi: 10.1152/ajplung.00267.2001. [DOI] [PubMed] [Google Scholar]
- Takeda A. Movement of zinc and its functional significance in the brain. Brain Res Rev. 2000;34:137–148. doi: 10.1016/s0165-0173(00)00044-8. [DOI] [PubMed] [Google Scholar]
- Tobiume K, Matsuzawa A, Takahashi T, Nishitoh H, Morita K, Takeda K, Minowa O, Miyazono K, Noda T, Ichijo H. ASK1 is required for sustained activations of JNK/p38 MAP kinases and apoptosis. EMBO Rep. 2001;2:222–228. doi: 10.1093/embo-reports/kve046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toescu EC. Apoptosis and cell death in neuronal cells: where does Ca2+ fit in? Cell Calcium. 1998;24:387–403. doi: 10.1016/s0143-4160(98)90062-8. [DOI] [PubMed] [Google Scholar]
- Tonder N, Johansen FF, Frederickson CJ, Zimmer J, Diemer NH. Possible role of zinc in the selective degeneration of dentate hilar neurons after cerebral ischemia in the adult rat. Neurosci Lett. 1990;109:247–252. doi: 10.1016/0304-3940(90)90002-q. [DOI] [PubMed] [Google Scholar]
- Underwood DC, Osborn RR, Kotzer CJ, Adams JL, Lee JC, Webb EF, Carpenter DC, Bochnowicz S, Thomas HC, Hay DW, Griswold DE. SB 239063, a potent p38 MAP kinase inhibitor, reduces inflammatory cytokine production, airways eosinophil infiltration, and persistence. J Pharmacol Exp Ther. 2000;293:281–288. [PubMed] [Google Scholar]
- Virag L, Szabo C. Inhibition of poly(ADP-ribose) synthetase (PARS) and protection against peroxynitrite-induced cytotox-icity by zinc chelation. Br J Pharmacol. 1999;126:769–777. doi: 10.1038/sj.bjp.0702332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogt K, Mellor J, Tong G, Nicoll R. The actions of synaptically released zinc at hippocampal mossy fiber synapses. Neuron. 2000;26:187–196. doi: 10.1016/s0896-6273(00)81149-6. [DOI] [PubMed] [Google Scholar]
- Walev I, Reske K, Palmer M, Valeva A, Bhakdi S. Potassium-inhibited processing of IL-1 beta in human monocytes. EMBO J. 1995;14:1607–1614. doi: 10.1002/j.1460-2075.1995.tb07149.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss JH, Hartley DM, Koh JY, Choi DW. AMPA receptor activation potentiates zinc neurotoxicity. Neuron. 1993;10:43–49. doi: 10.1016/0896-6273(93)90240-r. [DOI] [PubMed] [Google Scholar]
- Weiss JH, Sensi SL, Koh JY. Zn2+: a novel ionic mediator of neural injury in brain disease. Trends Pharmacol Sci. 2000;21:395–401. doi: 10.1016/s0165-6147(00)01541-8. [DOI] [PubMed] [Google Scholar]
- Wudarczyk J, Debska G, Lenartowicz E. Zinc as an inducer of the membrane permeability transition in rat liver mitochondria. Arch Biochem Biophys. 1999;363:1–8. doi: 10.1006/abbi.1998.1058. [DOI] [PubMed] [Google Scholar]
- Xia S, Lampe PA, Deshmukh M, Yang A, Brown BS, Rothman SM, Johnson EM, Jr, Yu SP. Multiple channel interactions explain the protection of sympathetic neurons from apoptosis induced by nerve growth factor deprivation. J Neurosci. 2002;22:114–122. doi: 10.1523/JNEUROSCI.22-01-00114.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoyama M, Koh J, Choi DW. Brief exposure to zinc is toxic to cortical neurons. Neurosci Lett. 1986;71:351–355. doi: 10.1016/0304-3940(86)90646-4. [DOI] [PubMed] [Google Scholar]
- Yu SP. Regulation and critical role of potassium homeostasis in apoptosis. Prog Neurobiol. 2003;70:363–86. doi: 10.1016/s0301-0082(03)00090-x. [DOI] [PubMed] [Google Scholar]
- Yu SP, Yeh CH, Sensi SL, Gwag BJ, Canzoniero LM, Farhangrazi ZS, Ying HS, Tian M, Dugan LL, Choi DW. Mediation of neuronal apoptosis by enhancement of outward potassium current. Science. 1997;278:114–117. doi: 10.1126/science.278.5335.114. [DOI] [PubMed] [Google Scholar]
- Yu SP, Farhangrazi ZS, Ying HS, Yeh CH, Choi DW. Enhancement of outward potassium current may participate in beta-amyloid peptide-induced cortical neuronal death. Neurobiol Dis. 1998;5:81–88. doi: 10.1006/nbdi.1998.0186. [DOI] [PubMed] [Google Scholar]
- Yu SP, Yeh CH, Gottron F, Wang X, Grabb MC, Choi DW. Role of the outward delayed rectifier K+ current in ceramide-induced caspase activation and apoptosis in cultured cortical neurons. J Neurochem. 1999;73:933–941. doi: 10.1046/j.1471-4159.1999.0730933.x. [DOI] [PubMed] [Google Scholar]
- Zaks-Makhina E, Yonjung K, Aizenman E, Levitan ES. Novel neuroprotective K+ channel inhibitor identified by high-throughput screening in yeast. Mol Pharmacol. 2004;65:214–219. doi: 10.1124/mol.65.1.214. [DOI] [PubMed] [Google Scholar]
- Zalewski PD, Forbes IJ, Betts WH. Correlation of apoptosis with change in intracellular labile Zn(II) using zinquin [(2-methyl-8-p-toluenesulphonamido-6-quinolyloxy)acetic acid], a new specific fluorescent probe for Zn(II) Biochem J. 1993;296:403–408. doi: 10.1042/bj2960403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, Georgiev O, Hagmann M, Guenes C, Cramer M, Faller P, Vasak M, Schaffner W. Activity of metal-responsive transcription factor 1 by toxic heavy metals and H2O2 in vitro is modulated by metallothionein. Mol Cell Biol. 2003;23:8471–8485. doi: 10.1128/MCB.23.23.8471-8485.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]