

Abstract
HER-2/neu overexpression in tumor cells caused abnormalities of MHC class I surface expression due to impaired expression of components of the antigen-processing machinery (APM) including the low molecular weight proteins, the transporter associated with antigen processing (TAP), and the chaperone tapasin, whereas the expression of MHC class I heavy chain as well as β2-microglobulin was only marginally affected. This oncogene-mediated deficient APM component expression could be reverted by interferon-γ treatment, suggesting a deregulation rather than structural alterations as underlying molecular mechanisms. To determine the level of regulation, the transcriptional activity of APM components was analyzed in HER-2/neu− and HER-2/neu+ cells. All major APM components were transcriptionally down-regulated in HER-2/neu+ when compared with HER-2/neu− cells, which was accompanied by a reduced binding of RNA polymerase II to the APM promoters. Site-directed mutagenesis of the p300- and E2F-binding sites in the APM promoters did not reconstitute the oncogene-mediated decreased transcription rate with the exception of tapasin, which was restored in HER-2/neu+ cells to levels of wild type tapasin promoter activity in HER-2/neu− fibroblasts. The E2F-directed control of tapasin expression was further confirmed by chromatin immunoprecipitation analyses showing that E2F1 and p300 bind to the tapasin and APM promoters in both cell lines. Moreover, siRNA-mediated silencing of E2F1 was associated with an increased tapasin expression, whereas transient overexpression of E2F1 launch a reduced tapasin transcription, suggesting that E2F1 is an essential transcription factor for tapasin.
Keywords: Chromatin Immunoprecipitation (ChIP), E2F Transcription Factor, p300, RNA Polymerase II, Transcription Regulation, LMP2, TAP1, TAP2, Antigen-processing Machinery, Tapasin
Introduction
The expression of multiple components of the antigen-processing machinery (APM)2 is a prerequisite for constitutive MHC class I surface expression and necessary for recognition of non-self antigens by CD8+ cytotoxic T lymphocytes (1–3). Abnormalities in the MHC class I surface expression have been described in tumors of distinct origin that allow their escape from immune cell recognition (4–8). These defects often result in decreased immunogenicity of tumors, disease progression, and reduced survival of patients (9, 10). Recently, the molecular mechanisms underlying altered MHC class I surface expression have been attributed to impaired expression of the low molecular weight proteins (LMP)-2, -7, and -10, the proteasome activator (PA)28, the transporter associated with antigen processing (TAP) 1 and 2, β2-microglobulin (β2-m), and the MHC class I heavy chain (HC).
Despite the identification of structural alterations in β2-m, HC, and TAP, which are caused by deletions, mutations, loss of heterozygosity, and/or recombinations, their occurrence is rare (11–14). Because in most cases MHC class I APM defects could be restored by treatment with proinflammatory and inflammatory cytokines such as tumor necrosis factor (TNF)-α and type I and type II interferons (IFN), their impaired expression appears to be mainly due to deregulation rather than sequence abnormalities (15, 16). Epigenetic, transcriptional, and/or posttranscriptional control of several APM components has been described in malignant cells (7). In particular mRNA and/or protein of major APM components, such as TAP1, TAP2, LMP2, LMP7, tapasin, β2-m, and/or HC, was often simultaneously suppressed upon malignant or viral transformation (17–27). In addition, methylation and altered histone acetylation were found with a low frequency for β2-m, HC, and TAP1 in tumors and were associated with a decreased MHC class I surface expression (8, 28–32).
So far, detailed information about the control of tapasin expression in murine and human tumor cells is scarce. Tapasin expression has been demonstrated to be down-regulated in breast cancer (16), melanoma (33, 34), colorectal carcinoma (17, 19, 35), cervical cancer (36), small cell and non-small cell lung carcinoma (10), fibrosarcoma (37), as well as bladder cancer (38). In human colorectal cancers, tapasin expression is even more strongly affected and more frequently lost than TAP1, LMP2, and LMP7 (17). Thus, tapasin expression might represent a key event in overcoming immune surveillance in these tumors (35).
Tapasin harbors a central role in the assembly of the peptide-loading complex by stabilizing TAP expression, determining peptide selection, and maintaining an appropriate MHC class I redox status. Although several components of the peptide-loading complex are required for optimal assembly and transport of MHC class I molecules to the surface, TAP and tapasin are essential for an optimized peptide loading to empty MHC class I molecules and their successive presentation to cytotoxic CD8+ T lymphocytes (39). Studies on the transcriptional regulation mainly focused on HC, β2-m, TAP1, and LMP2 (40–42) and identified different common transcription factors, which are involved in constitutive and IFN-γ induced protein expression. These include (i) CREB for constitutive expression and the IFN-sensitive response element for an IFN-γ-induced expression of the HC (40, 43–46) and the bidirectional TAP1/LMP2 promoter (8, 41) and (ii) the cooperating transcription factors IRF2 and STAT1 for constitutive expression as well as IRF1 and STAT1 for the IFN-γ-mediated enhanced TAP1/LMP2 expression (41, 42, 47–51). A similar regulation pattern was observed for murine TAP2, which is controlled by CREB and IRF for constitutive and/or IFN-γ-controlled activity (52, 53).
Both the human and the murine tapasin promoters contain one IFN-sensitive response element and two IFNγ-activated sequence motif consensus sequences in the 5′-UTR of the gene (54, 55). A study by Herrmann et al. (62) identified NF-κB as a basal transcription factor and a shared IRF1/2-binding site mediating IFN-γ inducibility of tapasin expression in murine cells. Recently, a cytokine-independent up-regulation of APM components by the CD40L-mediated transactivation of TAP1, TAP2, tapasin, LMP2, and LMP10 genes via NF-κB was observed, resulting in a de novo IRF1 synthesis accompanied by activating IRF1 in a STAT1-independent manner (56).
Despite the transcriptional and posttranscriptional down-regulation of APM components in tumor cells, detailed information about the molecular mechanisms or factors involved in this process is limited or controversially discussed (11, 38, 57, 58). In addition, there exists little information about transcription factors modulating the MHC class I APM component expression. Although promyelocytic leukemia protein has been suggested as a general regulator of APM component expression, its role in controlling this pathway is controversially discussed (59, 60). In contrast, the antagonistic transcription factor B lymphocyte-induced maturation protein-1 (PR domain-containing 1 (PRDM1)) has recently been shown to repress the IFN-γ-dependent transcription of ERAP1, tapasin, MECL1, and LMP7, resulting in a failure to up-regulate MHC class I surface antigens in human cell lines (61). Therefore, the aim of this study was to identify factors involved in the HER-2/neu-mediated down-regulation of MHC class I APM components using an in vitro model of oncogenic transformation. We here show for the first time an important role for E2F1 in the transcriptional regulation of tapasin in untransformed and HER-2/neu-transformed fibroblasts.
EXPERIMENTAL PROCEDURES
Cell Culture
The murine HER-2/neu− fibroblast cell line NIH3T3 was purchased from the American Type Culture Collection (ATCC), and the HER-2/neu+-overexpressing NIH3T3 cells were kindly provided by H. Bernhard (University Hospital of the Technical University, Munich, Germany) and have been described elsewhere in detail (62). Both cell lines were routinely maintained in Eagle's modified essential medium (Lonza, Cologne, Germany) supplemented with 10% (v/v) fetal bovine serum (Vienna, Austria), 2 mm l-glutamine, 100 units/ml penicillin, and 100 μg/ml streptomycin at 37 °C in 5% (v/v) CO2 humidified air.
cDNA Synthesis, qRT-PCR, and Cloning Procedures
cDNA was synthesized using RevertAidTM H Minus First Strand cDNA synthesis kit (Fermentas, St. Ingbert, Germany), and qRT-PCR was performed with target-specific primers (supplemental Table 1) by using Platinum® SYBR® Green qPCR SuperMix-UDG (Invitrogen, Karlsruhe, Germany) for 40 cycles; 95 °C, 15 s; 60 °C, 30 s. Relative mRNA expression levels for specific genes were normalized to that of the eukaryotic elongation factor (EF)-1α. The transcription levels of untreated HER-2/neu− cells were set to one, and respective expression levels in transiently transfected cells and HER-2/neu+ cells were calculated.
Protein Extraction and Western Blot Analysis
Proteins were extracted from 1–4 × 106 cells, and protein concentration was determined with the Pierce BCA protein assay kit (Fisher Scientific, Schwerte, Germany). 30 μg of protein/lane was separated on 8% SDS-PAGE, transferred onto nitrocellulose membranes (Schleicher & Schuell, Dassel, Germany), and stained with Ponceau S. Immunodetection was performed with specific primary antibodies directed against tapasin, E2F1 (sc-251x, Santa Cruz Biotechnology, Heidelberg, Germany), Rbl2 (retinoblastoma-like 2) (Pharmingen, Heidelberg, Germany), and GAPDH (Cell Signaling, Frankfurt, Germany). The staining was detected with horseradish peroxidase (HRP)-conjugated secondary antibodies (DAKO, Hamburg, Germany). The membranes were washed, and protein bands were visualized with a LumiliteTM (Roche Diagnostics, Mannheim, Germany) and exposed to a CCD camera (Eastman Kodak Co., Berlin, Germany).
Flow Cytometry
The monoclonal antibodies (mAbs) used for flow cytometry were the phycoerythrin-labeled anti-H-2Ld/q (Cedarlane Laboratories Ltd., Hornby, Canada) and the respective phycoerythrin-labeled isotype mouse immunoglobulin (Beckman-Coulter, Krefeld, Germany). Direct immunofluorescence analysis of MHC class I antigens was performed as described previously (63) using a FACSCanto II (BD Biosciences, Heidelberg, Germany). The results are expressed as mean specific fluorescence intensity obtained from at least three independent experiments.
Generation of APM Promoter Constructs and the E2F1 Expression Vector
TAP1/LMP2, TAP2, and tapasin promoter sequences were amplified from genomic DNA and cloned into pGL3 vector containing luciferase (luc) as a reporter gene (Promega, Mannheim, Germany), thereby generating the different pAPM-luc constructs. In addition, a series of TAP1, TAP2, and tapasin promoter transcription factor-binding mutants (mut-pAPM-luc) was generated from wild type (WT) pAPM-luc constructs by site-directed mutagenesis using a mutagenesis kit (Stratagene, Waldbronn, Germany) according to the manufacturer's instructions. Primer sequences for the WT APM promoter amplification and the respective transcription factor-binding site mutagenesis are listed in supplemental Table 1. All PCRs were performed with 15–35 cycles and at an annealing temperature of 55–60 °C. The E2F1 expression vector was generated by amplification of the E2F1-specific cDNA from murine fibroblasts using E2F1-specific adapter primers (supplemental Table 1) and cloned into the pCMV expression vector (64). The structural integrity of the WT and mutant (mut) pAPM-luc promoter constructs as well as of the E2F1 expression vector (CMV-E2F1) was verified by direct sequencing using vector-specific primers.
Determination of Promoter Activity
1 × 105 cells were seeded into six-well plates. 24 h after seeding, cells were cotransfected with either 2 μg of pAPM-luc or 2 μg of mut-pAPM-luc, respectively, together with pSVβ-galactosidase (gal) vector (Promega), which served as a control using Lipofectamine 2000 in OptiMEM (Invitrogen) according to the manufacturer's instructions. 6 h later, cells were washed and cultured in complete medium at 37 °C with 5% (v/v) CO2 until use. After 48 h, transfected cells were harvested, and luc activity was determined using the luc assay system according to the manufacturer's instructions (Promega). The transfection efficiency was evaluated by determination of the β-gal activity using Galacto-Light (Tropix, Bedford, MA) according to the instruction manual. Relative luc activity was calculated as (mean luc activity/mean β-gal activity) − (mean luc activity of pGL3/mean β-gal activity). The transfections were carried out in triplicate experiments and were independently repeated at least five times. The data are represented as mean of triplicates with variations among replicates of <5%.
Transfection of Cells with CMV-E2F1 and E2F1 siRNA
1 × 105 cells were seeded into six-well plates 1 day prior to transfection. To suppress or overexpress E2F1, cells were transfected either with 200 pmol of siRNA using 10 μl of Lipofectamine 2000 (Invitrogen) in OptiMEM (Invitrogen) or with 2 μg of the CMV-E2F1 plasmid and the respective mock control, using 10 μl of Effectene (Qiagen, Hilden, Germany) according to the manufacturer's instructions. SiRNA specific for mouse E2F1 (supplemental Table 1) and an unspecific nonsense siRNA control purchased from Invitrogen were used. Cells were harvested 48–72 h after transfection for isolation of RNA and protein.
Chromatin Immunoprecipitation (ChIP)
ChIP analyses were performed with the ChIP-IT express kit (Active Motif, Rixensart, Belgium) according to the manufacturer's instructions. Briefly, 5 × 107 cells were fixed with 1% formaldehyde at room temperature for 10 min, nuclei were lysed by using a Dounce homogenizer, and chromatin was enzymatically digested. ChIP was performed overnight at 6 °C with 6.3 μg of DNA, 2 μg of control IgG, RNA polymerase II (RNA pol II) (Active Motif), E2F1 (Santa Cruz Biotechnology), and p300- (Millipore, Eschborn, Germany) specific antibody and captured using protein G magnetic beads (Active Motif). After reversion of cross-links for 4 h at 65 °C, protein was digested with proteinase K. The isolated DNA was employed as template for PCR amplifications using Taq DNA polymerase (Qiagen) and the following amplification conditions: an initial melting step at 94 °C for 3 min, 36 (primers for EF1α as positive control (Active Motif)) to 40 amplification cycles (primers for tapasin, TAP1, LMP2, and TAP2) at 94 °C for 20 s, 59 °C or 60 °C for 30 s, and 72 °C for 30 s, and a final elongation step for 5 min at 72 °C followed by a hold cycle at 10 °C. 8 μl of each PCR reaction was separated on a 2% agarose gel and visualized by UV illumination following ethidium bromide staining.
3′-Biotin Labeling of Oligonucleotides, Preparation of Nuclear Extracts, and Electromobility Shift Assay (EMSA)
Single-stranded DNA sequences containing the respective E2F-binding sites of the tapasin promoter (supplemental Table 1) were labeled with biotin-11-dUTP using the biotin 3′-end DNA labeling kit containing the terminal deoxynucleotide transferase (Fisher Scientific). The reaction was stopped by adding EDTA, and terminal deoxynucleotide transferase was extracted with phenol:chloroform:isoamyl alcohol (25:24:1). Labeled antiparallel single-stranded DNA fragments were denatured at 95 °C for 5 min and slowly annealed at their melting temperature for 30 min.
Crude nuclear extracts from 1 × 107 cells were isolated as described earlier (65) with minor modifications. The EMSAs were performed using the LightShift chemiluminescent EMSA kit (Fisher Scientific) according to the manufacturer's instructions. 10 μg of crude nuclear extract was incubated with 100 pg of the biotinylated oligonucleotide (supplemental Table 1). The binding reaction was stopped after 30 min by adding 5-fold loading buffer. For competition experiments, a 100-fold amount of unlabeled oligonucleotide was added 5 min prior to the reaction. The samples were run on 6% Tris-borate-EDTA gels, subsequently blotted onto nylon membranes (Pall, Dreieich, Germany) using a semidry blotting procedure and UV cross-linked, followed by the streptavidin detection of biotinylated oligonucleotides.
Non-radioactive Southwestern Blotting
For Southwestern blotting, 120 μg of nuclear protein/lane was separated under denaturing conditions on 8% polyacrylamide gels and blotted onto nitrocellulose membranes (Schleicher & Schuell). After Ponceau S staining, the membranes were washed with distilled water and blocked with buffer A (10 mm HEPES, 5% BSA, pH 7.5) for 2 h. Then, membranes were rinsed in buffer B (10 mm HEPES, 0.25% BSA, 50 mm NaCl, 1 mm EDTA, 1 mm DTT, pH 7.9) supplemented with 10 μg/ml denatured salmon sperm DNA (Invitrogen) for 30 min before the biotinylated dsDNA oligonucleotide was added for 1 h (66). Subsequently, membranes were washed four times with buffer B containing 250 mm NaCl, pH 7.5. Bound biotinylated oligonucleotides were detected with streptavidin-HRP conjugate (Fisher Scientific), and excess of conjugate was removed by four washing steps in buffer D (10 mm HEPES, 1% BSA, 50 mm NaCl, 1 mm EDTA, pH 7.5). Detection was performed with a LumiliteTM (Roche Applied Science) according to the manufacturer's protocol and a CCD camera (Kodak).
RESULTS
HER-2/neu-mediated Down-regulation of MHC class I Surface Expression
Recently, HER-2/neu overexpression has been associated with a reduced MHC class I surface antigen expression (63, 67). Using a murine model of HER-2/neu transformation, the MHC class I surface expression of HER-2/neu− and HER-2/neu+ cells was determined by flow cytometry (Fig. 1A). HER-2/neu overexpression caused a dramatic decrease of MHC class I surface expression, which was accompanied by an impaired transcription of tapasin, TAP1, TAP2, and LMP2 (Fig. 1B) and a reduced protein expression of major APM components as representatively shown for tapasin (Fig. 1C).
FIGURE 1.

HER-2/neu-mediated reduced MHC class I surface molecules associated with low tapasin expression. A, flow cytometric analysis of H-2Lq surface antigen expression. 1 × 105 cells were subjected to flow cytometry as described under “Experimental Procedures” using an H-2Lq and H-2Ld cross-reactive mAb and a respective murine isotype control. The upper histograms show the staining with isotype control antibody, and the lower histograms display the mean specific fluorescence intensity (MFI) of H-2Lq on the cell surface of HER-2/neu− cells (left) and HER-2/neu+ cells (right). B, analysis of the tapasin protein expression in HER-2/neu− and HER-2/neu+ cells. 30 μg of protein extract/lane was separated by SDS-PAGE under denaturating and reducing conditions, and protein expression was assessed by Western blot analysis as described under “Experimental Procedures” using an anti-tapasin-specific antibody. Staining of the same blot with GAPDH as a housekeeping gene served as control. Error bars indicate S.D. C, reduced APM transcription in HER-2/neu+ cells. Quantitative RT-PCR analyses of tapasin, TAP1, and TAP2 were performed with oligo(dT)-primed cDNA from HER-2/neu− cells (black bars) and of HER-2/neu+ cells (white bars) and normalized to EF-1α transcription. Transcriptional levels from HER-2/neu− cells were used as control and set to 1, whereas the transcripts of HER-2/neu+ cells (white bars) were directly compared. Data shown represent three independently analyzed biological samples.
Transcriptional Control of APM Components
To investigate whether the HER-2/neu-mediated impaired APM component expression is caused by transcriptional down-regulation, the pAPM-luc constructs containing the bidirectional WT TAP1/LMP2, WT TAP2, or WT tapasin promoter, respectively, and the pSVβ-gal vector serving as a control were transiently cotransfected into HER-2/neu− and HER-2/neu+ cells, and luc activity was determined 48 h later. When compared with HER-2/neu− cells, the different APM promoter activities were strongly reduced in HER-2/neu+ transfectants, but to a different extent varying from 64% for TAP1, 84% for LMP2, 62% for TAP2, and 75% for tapasin (Fig. 2A). These results were in line with the decreased mRNA levels of APM components in HER-2/neu+ cells, suggesting that deficient MHC class I surface expression in HER-2/neu transformants is due to a transcriptional down-regulation of the various components.
FIGURE 2.

Deficient APM promoter activity in HER-2/neu+ cells correlated with reduced RNA pol II-initiated transcription in HER-2/neu+ cells. A, WT APM-luc promoters or APM-mut-luc promoters and the pGL3/Enhancer vector, which served as a control (data not shown), were transiently cotransfected with the β-gal vector into HER-2/neu− and HER-2/neu+ cells 48 h prior to the determination of the luc activity using a reporter gene assay as described under “Experimental Procedures.” The data were normalized to β-gal activity. For comparison, the WT APM promoter activities in HER-2/neu− cells were set to 100%, and the respective APM promoter activities in HER-2/neu+ cells were calculated. Error bars indicate S.D. rel luc activity, relative luc activity. B, immunoprecipitated chromatin of HER-2/neu− and HER-2/neu+ cells was used as template in PCR. Dilutions (1:10) of input DNA served as controls. Sequences of the APM promoters (TAP1, LMP2, TAP2, and tapasin) and the EF-1α promoter were amplified from RNA pol II-ChIP of HER-2/neu− cells and from HER-2/neu+ cells, but not from isotype control IgG-ChIP. A non-template control was included.
The impaired APM transcription was confirmed by determination of the RNA pol II-driven transcription initiation at the respective promoters in HER-2/neu− versus HER-2/neu+ cells using ChIP assays. In accordance with the promoter assays and RT-PCR analyses, a reduced initiation of TAP1, LMP2, TAP2, and tapasin, but not of EF-1α transcription, was detected in HER-2/neu+ transformants when compared with HER-2/neu− cells (Fig. 2B).
Enhanced Activity in E2F and p300 Tapasin Promoter Mutants
To determine the transcription factors involved in the control of APM expression, site-directed mutagenesis was performed to specifically eliminate the transcription factor-binding sites (TFBS) for p300 and E2F located within the tapasin promoter at positions −309 (p300), −229, and −146 (E2F), in the TAP1 promoter at positions −459 and −103 (E2F), and in the TAP2 promoter at positions −1248, −648, −320 (p300), and −85 (E2F). In untransformed fibroblasts, an increased activity of the E2F(−229, −146) and p300(−309) mut-tapasin-luc constructs was found, which ranged between 130% for p300(−309) and for E2F(−146) and 140% for E2F(−229) mut promoters (Fig. 3A). In addition, mutation of these different p300- and E2F1-specific TFBS reconstituted the tapasin promoter activity in HER-2/neu+ cells to levels higher than that of untransformed fibroblasts with an increased activity ranging between 174% for the p300(−309), 192% for the E2F(−229),and 136% for the E2F(−146) mut-tapasin-luc constructs. Thus, the p300 and E2F-binding sites may negatively interfere with the constitutive tapasin promoter activity.
FIGURE 3.

Restoration of deficient APM promoter activity in HER-2/neu+ cells by selective mutations of TFBS. A, tapasin-mut-luc promoters. B and C, TAP1-mut-luc promoters (B) and TAP2-mut-luc promoters (C) and the pGL3/Enhancer vector, which served as a control (data not shown), were transiently cotransfected with the β-gal vector into HER-2/neu− and HER-2/neu+ cells 48 h prior to the determination of the luc activity using a reporter gene assay as described under “Experimental Procedures.” The data were normalized to β-gal activity. For comparison, the WT APM promoter activities in HER-2/neu− cells were set to 100%, and the respective APM promoter activities in HER-2/neu+ cells were calculated. Error bars in all panels indicate S.D. rel luc activity, relative luc activity.
In contrast, mutations in the E2F motifs of the TAP1 and TAP2 promoter sequences either had no effect or reduced the promoter activity. Knock-out of the E2F(−459, −103) TFBS reduced the TAP1 promoter activity to 76% of the WT TAP1 promoter, whereas E2F(−103) did not affect the promoter activity (Fig. 3B). Mutations of the TFBS p300 at positions −648 and −320 strongly decreased the TAP2 promoter activity in both cell lines ranging from 40% for p300(−320) to 61% for p300(−648) in untransformed cells, which were at 73 and 89%, respectively, even more pronounced in HER-2/neu transfectants. In contrast, when compared with the WT TAP2 promoter activity, an 20% or 25% increase of mutTAP2-luc activity due to a mutation in the E2F1 TFBS at position −85 was found in HER-2/neu− and HER-2/neu+ cells, respectively, when compared with the WT TAP2 promoter, suggesting an at least partial reconstitution of the WT TAP2 promoter activity in HER-2/neu-transformed fibroblasts. Based on these results, we postulate that the proximal E2F site of the TAP2 promoter might be involved in the regulation of TAP2 expression.
Detection of E2F1 Binding to the Tapasin Promoter
To confirm binding activity of E2F1 to the tapasin promoter, ChIP was performed using an E2F1-specific mAb. Tapasin promoter-specific PCR products were amplified from immunoprecipitated chromatin of HER-2/neu− and HER-2/neu+ cells, suggesting that E2F1 binds to the tapasin promoter in both cell lines (Fig. 4A). The presence of E2F1 was further detected at the TAP1 and TAP2 promoters in both cell lines, thereby confirming the qRT-PCR and promoter results. Binding of E2F1 to the LMP2 promoter was demonstrated in HER-2/neu−, but not in HER-2/neu+ cells. Furthermore, ChIP analyses revealed p300 binding to the tapasin and LMP2 promoter in untransformed and oncogene-transformed fibroblasts but with a lower extent in HER-2/neu+ cells. However, p300 binding was only detected at the TAP2 promoter of HER-2/neu− but not of HER-2/neu+ cells (Fig. 4B).
FIGURE 4.

Identification of E2F1 and p300 as tapasin-binding proteins in HER-2/neu− and HER-2/neu+ cells. A and B, immunoprecipitated chromatin of HER-2/neu− and HER-2/neu+ cells was used as template in PCR. Dilutions (1:10) of input DNA served as controls. Sequences of the APM promoters (TAP1, LMP2, TAP2, and tapasin) were amplified from E2F1- and p300-specific ChIP assays, but not from control IgG-ChIP. A non-template control was included.
E2F1 as a Transcriptional Repressor in Mouse Fibroblasts
To further investigate the role of E2F1 in tapasin regulation, short siRNAs directed against E2F1 transcription factor and a nonsense control were transiently transfected into HER-2/neu− cells. As expected, E2F1-specific siRNA, but not nonsense siRNA, down-regulated E2F1 transcription. This siRNA-mediated down-regulation of E2F1 was accompanied by an increased tapasin expression, thereby demonstrating an E2F1-directed transcriptional control of tapasin. The data were further confirmed by Western blot analysis using anti-E2F1-specific and anti-tapasin-specific mAb (68) (Fig. 5A), demonstrating reduced E2F1 protein levels in the siRNA-transfected fibroblasts when compared with control cells, which were accompanied by an induction of tapasin expression. These results are in accordance with the increased tapasin promoter activity of the E2F(−146) mutant in HER-2/neu− cells (Fig. 3A) and were further supported by E2F1 overexpression in HER-2/neu− cells, leading to a more than 130-fold increase of E2F1 transcripts (Fig. 5B) and a 6-fold cyclin E1 (CCNE1) transcription after 24 h (Fig. 5C) (69), which was associated with an ∼50% decrease of tapasin transcription 72 h after transfection (Fig. 5D).
FIGURE 5.

Identification of E2F1 as modulator of tapasin transcription. A, siRNA-mediated inhibition of E2F1 transcription was investigated using siRNA directed against mouse E2F1. 20 μg of protein from untreated, nonsense control- or siRNA-transfected HER-2neu− cells was separated and transferred to nitrocellulose. E2F1-siRNA caused a reduced E2F1 protein expression in non-transformed fibroblasts accompanied by an increase of tapasin. Staining of the Western blot with an anti-GAPDH antibody served as loading control. B–D, transient overexpression of E2F1 mediates tapasin suppression. To validate the specificity of the E2F1 expression levels, untreated HER-2/neu− cells were transfected with CMV vector backbone or with the recombinant CMV-E2F1 expression vector. RNA was isolated at different time points: 24 (B and C) and 72 h (D) after transfection and transcription levels for E2F1 (B), CCNE1 (C), and tapasin (D) were determined. EF-1α served as reference gene. A 135-fold induction of E2F1 was observed at 24 h, initiating a 6-fold induction of the CCNE1 gene transcription and mediating a half-maximal reduction of tapasin 72 h after transfection.
Detection of Different E2F-binding Proteins in HER-2/neu+ Cells
To further investigate the role of E2F-binding sites in modulating tapasin expression, gel shift analyses using respective tapasin promoter sequences of both distal and proximal E2F-binding sites were performed. Comparative analyses of crude nuclear extracts of HER-2/neu− and HER-2/neu+ cell lines demonstrated binding complexes to the oligonucleotide representing the proximal, but not the distal, E2F-binding site, which may display differentially sized transcription factors interacting with the respective tapasin promoter sequence (Fig. 6A). The specificity of the binding reaction was determined by using a 100-fold excess of unlabeled oligonucleotide resulting in a total loss of binding affinity in HER-2/neu+ extracts, although a residual binding in nuclear extracts from untransformed cells remained. Using the same biotinylated oligonucleotide, Southwestern blots were performed, and equally sized proteins with a molecular mass of ∼38 kDa were detected in both cell extracts (Fig. 6B).
FIGURE 6.
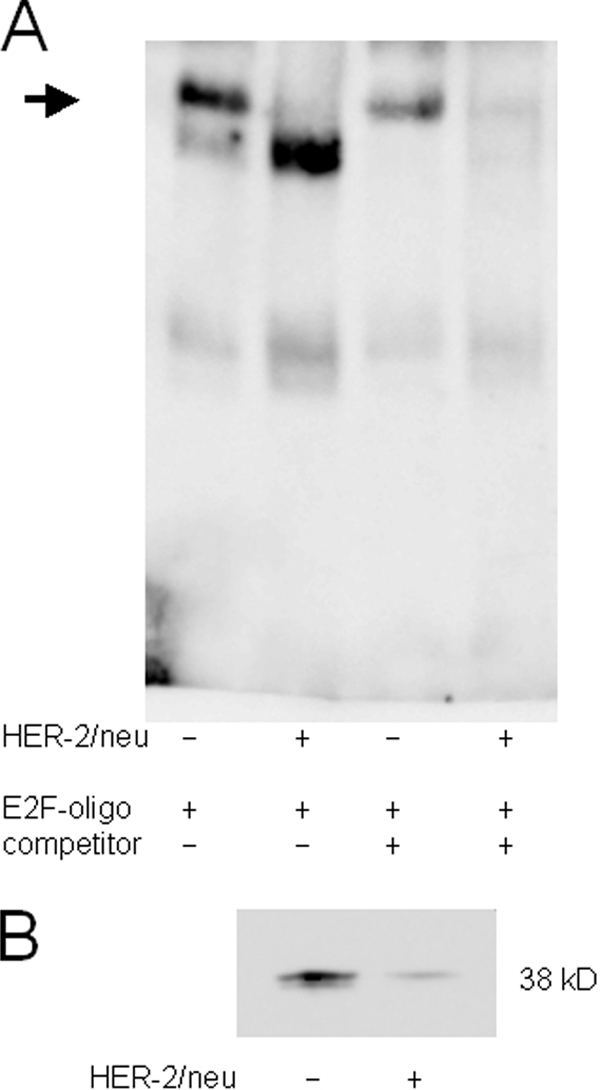
Different binding properties for distal E2F motif in the tapasin promoter. A, electrophoretic mobility shift assay for the analysis of the proximal E2F(−146)-binding site. EMSA was performed as described under “Experimental Procedures” using a biotin-labeled oligonucleotide spanning the tapasin promoter sequence from −146 to −139 (supplemental Table 1). All lanes contain 100 pg of labeled oligonucleotide. The arrow indicates different E2F binding properties in crude nuclear extracts of HER-2/neu− and HER-2/neu+ cells. The specific signal in HER-2/neu+ cells disappeared by adding a 100-fold excess of the respective unlabeled oligonucleotide, whereas the signal intensity is reduced in HER-2/neu− cells. B, a distinct 38-kDa protein bound to the labeled E2F probe in Southwestern blot of nuclear extracts from HER-2/neu− and HER-2/neu+ cells. The Southwestern blot was probed with the biotinylated oligonucleotide containing the proximal E2F(−146)-binding site from the tapasin promoter. An increased amount of labeled oligonucleotide bound to proteins from nuclear extracts of HER-2/neu− cells when compared with HER-2/neu+ cells.
DISCUSSION
A detailed analysis of the APM components in HER-2/neu− versus HER-2/neu+ cells revealed a strong coordinated down-regulation of the expression of several APM components, with the exception of β2-m and HC (63, 67). However, the molecular mechanisms involved in the impaired APM component expression in the HER-2/neu+ cells have not yet been identified.
By investigating the TAP1, LMP2, TAP2, and tapasin promoter activity, a transcriptional down-regulation of the respective APM promoters was found in HER-2/neu-overexpressing fibroblasts when compared with non-transformed cells varying from 38% (TAP2) to 84% (LMP2). These data were further confirmed by qRT-PCR analyses of HER-2/neu− versus HER-2/neu+ fibroblasts. Based on these results, we postulated that the mRNA levels of APM components could be modulated by (i) an instability of transcription initiation due to the absence of the enhancing transcription factor CBP/p300, which might be limited in oncogene-transformed cells (8), or the presence of negatively acting suppressors; (ii) a chromatin-mediated physical barrier in HER-2/neu+ cells, thereby reducing the access of the RNA pol II complex to the tapasin promoter; or (iii) the absence of transcription elongation factors, which might control the constitutive transcription level in a gene- or locus-specific manner (70–72).
To address these questions, the role of TFBS controlling the TAP1/LMP2, TAP2, and tapasin promoter activity was determined using TFBS KO mutants generated by site-directed mutagenesis (62). Mutations of the E2F and p300 TFBS caused a reconstitution of the tapasin promoter activity in HER-2/neu+ cells, suggesting the involvement of E2F1 and p300 in the transcriptional down-regulation of tapasin in oncogenic transformants. In contrast, E2F is not crucial for the HER-2/neu-mediated down-regulation of TAP1 transcription, whereas the E2F-binding site at position −85 plays an important role for TAP2 transcription. These data were further validated by investigating the access of RNA pol II complexes to the APM promoters using ChIP. The decreased binding of RNA pol II to the APM promoters in HER-2/neu+ cells mirrored the decreased transcription level of tapasin, TAP1, LMP2, and TAP2 in these cells. These results are in accordance with a recent report by Jefferies and co-workers (8) demonstrating an association of a weak TAP1 promoter activity with reduced recruitment of CBP to the TAP1 promoter in TAP1-deficient cells.
To clarify the role of the histone-acetylating protein p300, a slightly reduced attachment of p300 to tapasin and LMP2 but not to the TAP2 promoter in HER-2/neu+ cells when compared with untransformed cells was found, suggesting a correlation between RNA pol II and p300 access to these promoters. Because the binding of E2F1–3 to promoters is associated with the acquisition of the histone H3 and H4 acetylation (73, 74), E2F1 ChIP assays were performed to clarify the role of E2F1 in APM component transcription.
Indeed, E2F1 was able to bind to the WT tapasin, TAP1, and TAP2 promoters. The amplicons obtained match perfectly the amplicon pattern from ChIP analyses of RNA pol II and p300, suggesting a coordinated transcription initiation complex engulfing RNA pol II, p300, and E2F1. Recently, the specificity of E2F activation and repression has been investigated for the Cdc2 and Cdc6 genes, demonstrating both positive and negative effects on the transcription for both proximal and distal E2F-binding sites depending on the E2F transcription factor (75–77).
In an attempt to explore the function of E2F1 regarding the regulation of tapasin expression, E2F1 was either inhibited by E2F1-specific siRNA or overexpressed. Although E2F1 silencing was directly accompanied by an increased tapasin transcription and translation in HER-2/neu− cells, the transient E2F1 overexpression resulted in a reduced tapasin transcription. Moreover, differently sized DNA-binding proteins in crude nuclear extracts of WT and oncogene-transformed cells were found at the proximal E2F(−146)-binding site of the tapasin promoter in EMSA, which were completely abolished by unlabeled competitor. However, two or more proteins with distinct molecular weights might bind to this region, which might be explained by the broad homologies in the DNA binding capacity of the different members of E2F transcription factor family (78–83). Because different E2F family members have been shown to bind with a distinct specificity to similar target genes (76, 84, 85), we performed Southwestern blots to verify our EMSA results. A signal with an estimated size of 38 kDa was detected, which was more pronounced in crude nuclear extracts of HER-2/neu− cells.
Using different experimental approaches, we here demonstrate for the first time a selective transcriptional control of tapasin expression in both untransformed and HER-2/neu-transformed cells. In this model system, E2F1 and p300 are in particular involved in the control of tapasin transcription, but the role of these transcription factors and their recruitment to promoters involved in modulation of the immunogenicity of tumor cells await further investigations (86–89).
Supplementary Material
Acknowledgment
We thank S. Magdeburg for excellent secretarial help.
This work was supported by the Deutsche Forschungsgemeinschaft (DFG) Grant SE-589/11-1, 11-2 and Grant SE-581/9-2.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Table 1.
- APM
- antigen-processing machinery
- β2-m
- β2-microglobulin
- HC
- MHC class I heavy chain
- LMP
- low molecular weight protein
- luc
- luciferase
- mut
- mutant
- RNA pol II
- RNA polymerase II
- TAP
- transporter associated with antigen processing
- TF
- transcription factor
- TFBS
- transcription factor-binding site
- CREB
- cAMP-response element-binding protein
- CBP
- CREB-binding protein
- IRF
- interferon regulatory transcription factor
- qRT-PCR
- quantitative RT-PCR
- mut
- mutant.
REFERENCES
- 1.Garbi N., Tanaka S., van den Broek M., Momburg F., Hämmerling G. J. (2005) Immunol. Rev. 207, 77–88 [DOI] [PubMed] [Google Scholar]
- 2.Jensen P. E. (2007) Nat. Immunol. 8, 1041–1048 [DOI] [PubMed] [Google Scholar]
- 3.Raghavan M., Del Cid N., Rizvi S. M., Peters L. R. (2008) Trends Immunol. 29, 436–443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Aptsiauri N., Carretero R., Garcia-Lora A., Real L. M., Cabrera T., Garrido F. (2008) Cancer Immunol. Immunother. 57, 1727–1733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rescigno M., Avogadri F., Curigliano G. (2007) Biochim Biophys. Acta 1776, 108–123 [DOI] [PubMed] [Google Scholar]
- 6.Restifo N. P., Esquivel F., Kawakami Y., Yewdell J. W., Mulé J. J., Rosenberg S. A., Bennink J. R. (1993) J. Exp. Med. 177, 265–272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Seliger B. (2008) Cancer Immunol. Immunother. 57, 1719–1726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Setiadi A. F., David M. D., Seipp R. P., Hartikainen J. A., Gopaul R., Jefferies W. A. (2007) Mol. Cell Biol. 27, 7886–7894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Garrido C., Algarra I., Maleno I., Stefanski J., Collado A., Garrido F., Garcia-Lora A. M. (2010) Cancer Immunol. Immunother. 59, 13–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lou Y., Vitalis T. Z., Basha G., Cai B., Chen S. S., Choi K. B., Jeffries A. P., Elliott W. M., Atkins D., Seliger B., Jefferies W. A. (2005) Cancer Res. 65, 7926–7933 [DOI] [PubMed] [Google Scholar]
- 11.Cabrera C. M., Jiménez P., Cabrera T., Esparza C., Ruiz-Cabello F., Garrido F. (2003) Tissue Antigens 61, 211–219 [DOI] [PubMed] [Google Scholar]
- 12.Chang C. C., Ogino T., Mullins D. W., Oliver J. L., Yamshchikov G. V., Bandoh N., Slingluff C. L., Jr., Ferrone S. (2006) J. Biol. Chem. 281, 18763–18773 [DOI] [PubMed] [Google Scholar]
- 13.Ferrone S., Marincola F. M. (1995) Immunol. Today 16, 487–494 [DOI] [PubMed] [Google Scholar]
- 14.Maleno I., López-Nevot M. A., Cabrera T., Salinero J., Garrido F. (2002) Cancer Immunol. Immunother. 51, 389–396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Epperson D. E., Arnold D., Spies T., Cresswell P., Pober J. S., Johnson D. R. (1992) J. Immunol. 149, 3297–3301 [PubMed] [Google Scholar]
- 16.Seliger B., Schreiber K., Delp K., Meissner M., Hammers S., Reichert T., Pawlischko K., Tampé R., Huber C. (2001) Tissue Antigens 57, 39–45 [DOI] [PubMed] [Google Scholar]
- 17.Atkins D., Breuckmann A., Schmahl G. E., Binner P., Ferrone S., Krummenauer F., Störkel S., Seliger B. (2004) Int. J. Cancer 109, 265–273 [DOI] [PubMed] [Google Scholar]
- 18.Bennett E. M., Bennink J. R., Yewdell J. W., Brodsky F. M. (1999) J. Immunol. 162, 5049–5052 [PubMed] [Google Scholar]
- 19.Delp K., Momburg F., Hilmes C., Huber C., Seliger B. (2000) Bone Marrow Transplant 25, Suppl. 2, S88–S95 [DOI] [PubMed] [Google Scholar]
- 20.Griffioen M., Steegenga W. T., Ouwerkerk I. J., Peltenburg L. T., Jochemsen A. G., Schrier P. I. (1998) Mol. Immunol. 35, 829–835 [DOI] [PubMed] [Google Scholar]
- 21.Hansen T. H., Bouvier M. (2009) Nat. Rev. Immunol. 9, 503–513 [DOI] [PubMed] [Google Scholar]
- 22.Rotem-Yehudar R., Groettrup M., Soza A., Kloetzel P. M., Ehrlich R. (1996) J. Exp. Med. 183, 499–514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Seliger B., Maeurer M. J., Ferrone S. (1997) Immunol. Today 18, 292–299 [DOI] [PubMed] [Google Scholar]
- 24.Seliger B., Maeurer M. J., Ferrone S. (2000) Immunol. Today 21, 455–464 [DOI] [PubMed] [Google Scholar]
- 25.Smahel M., Sobotková E., Bubeník J., Símová J., Zák R., Ludviková V., Hájková R., Kovarík J., Jelínek F., Povýsil C., Marinov J., Vonka V. (2001) Br. J. Cancer 84, 374–380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vertegaal A. C., Kuiperij H. B., Houweling A., Verlaan M., van der Eb A. J., Zantema A. (2003) J. Biol. Chem. 278, 139–146 [DOI] [PubMed] [Google Scholar]
- 27.Vertuani S., Triulzi C., Roos A. K., Charo J., Norell H., Lemonnier F., Pisa P., Seliger B., Kiessling R. (2009) Cancer Immunol. Immunother. 58, 653–664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Campoli M., Ferrone S. (2008) Oncogene 27, 5869–5885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Khan A. N., Gregorie C. J., Tomasi T. B. (2008) Cancer Immunol. Immunother. 57, 647–654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sers C., Kuner R., Falk C. S., Lund P., Sueltmann H., Braun M., Buness A., Ruschhaupt M., Conrad J., Mang-Fatehi S., Stelniec I., Krapfenbauer U., Poustka A., Schäfer R. (2009) Int. J. Cancer 125, 1626–1639 [DOI] [PubMed] [Google Scholar]
- 31.Setiadi A. F., Omilusik K., David M. D., Seipp R. P., Hartikainen J., Gopaul R., Choi K. B., Jefferies W. A. (2008) Cancer Res. 68, 9601–9607 [DOI] [PubMed] [Google Scholar]
- 32.Ye Q., Shen Y., Wang X., Yang J., Miao F., Shen C., Zhang J. (2009) Tissue Antigens 75, 30–39 [DOI] [PubMed] [Google Scholar]
- 33.Dissemond J., Kothen T., Mörs J., Weimann T. K., Lindeke A., Goos M., Wagner S. N. (2003) Arch. Dermatol. Res. 295, 43–49 [DOI] [PubMed] [Google Scholar]
- 34.Seliger B., Ritz U., Abele R., Bock M., Tampé R., Sutter G., Drexler I., Huber C., Ferrone S. (2001) Cancer Res. 61, 8647–8650 [PubMed] [Google Scholar]
- 35.Cabrera C. M., López-Nevot M. A., Jiménez P., Garrido F. (2005) Int. J. Cancer 113, 611–618 [DOI] [PubMed] [Google Scholar]
- 36.Mehta A. M., Jordanova E. S., Corver W. E., van Wezel T., Uh H. W., Kenter G. G., Jan Fleuren G. (2009) Genes Chromosomes Cancer 48, 410–418 [DOI] [PubMed] [Google Scholar]
- 37.Garcia-Lora A., Martinez M., Algarra I., Gaforio J. J., Garrido F. (2003) Int. J. Cancer 106, 521–527 [DOI] [PubMed] [Google Scholar]
- 38.Romero J. M., Jiménez P., Cabrera T., Cózar J. M., Pedrinaci S., Tallada M., Garrido F., Ruiz-Cabello F. (2005) Int. J. Cancer 113, 605–610 [DOI] [PubMed] [Google Scholar]
- 39.Garbi N., Tan P., Diehl A. D., Chambers B. J., Ljunggren H. G., Momburg F., Hämmerling G. J. (2000) Nat. Immunol. 1, 234–238 [DOI] [PubMed] [Google Scholar]
- 40.Agrawal S., Kishore M. C. (2000) J. Hematother Stem. Cell Res. 9, 795–812 [DOI] [PubMed] [Google Scholar]
- 41.Min W., Pober J. S., Johnson D. R. (1996) J. Immunol. 156, 3174–3183 [PubMed] [Google Scholar]
- 42.Wright K. L., White L. C., Kelly A., Beck S., Trowsdale J., Ting J. P. (1995) J. Exp. Med. 181, 1459–1471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gobin S. J., van Zutphen M., Woltman A. M., van den Elsen P. J. (1999) J. Immunol. 163, 1428–1434 [PubMed] [Google Scholar]
- 44.Gobin S. J., Wilson L., Keijsers V., Van den Elsen P. J. (1997) J. Immunol. 158, 3587–3592 [PubMed] [Google Scholar]
- 45.van den Elsen P. J., Holling T. M., Kuipers H. F., van der Stoep N. (2004) Curr. Opin. Immunol. 16, 67–75 [DOI] [PubMed] [Google Scholar]
- 46.van den Elsen P. J., Peijnenburg A., van Eggermond M. C., Gobin S. J. (1998) Immunol. Today 19, 308–312 [DOI] [PubMed] [Google Scholar]
- 47.Brucet M., Marqués L., Sebastián C., Lloberas J., Celada A. (2004) Genes Immun. 5, 26–35 [DOI] [PubMed] [Google Scholar]
- 48.Chatterjee-Kishore M., Kishore R., Hicklin D. J., Marincola F. M., Ferrone S. (1998) J. Biol. Chem. 273, 16177–16183 [DOI] [PubMed] [Google Scholar]
- 49.Dovhey S. E., Ghosh N. S., Wright K. L. (2000) Cancer Res. 60, 5789–5796 [PubMed] [Google Scholar]
- 50.Kishi F., Suminami Y., Monaco J. J. (1993) Gene 133, 243–248 [PubMed] [Google Scholar]
- 51.Rouyez M. C., Lestingi M., Charon M., Fichelson S., Buzyn A., Dusanter-Fourt I. (2005) J. Immunol. 174, 3948–3958 [DOI] [PubMed] [Google Scholar]
- 52.Arons E., Kunin V., Schechter C., Ehrlich R. (2001) J. Immunol. 166, 3942–3951 [DOI] [PubMed] [Google Scholar]
- 53.Guo Y., Yang T., Liu X., Lu S., Wen J., Durbin J. E., Liu Y., Zheng P. (2002) Int. Immunol. 14, 189–200 [DOI] [PubMed] [Google Scholar]
- 54.Abarca-Heidemann K., Friederichs S., Klamp T., Boehm U., Guethlein L. A., Ortmann B. (2002) Immunol. Lett 83, 197–207 [DOI] [PubMed] [Google Scholar]
- 55.Herberg J. A., Sgouros J., Jones T., Copeman J., Humphray S. J., Sheer D., Cresswell P., Beck S., Trowsdale J. (1998) Eur. J. Immunol. 28, 459–467 [DOI] [PubMed] [Google Scholar]
- 56.Moschonas A., Kouraki M., Knox P. G., Thymiakou E., Kardassis D., Eliopoulos A. G. (2008) Mol. Cell Biol. 28, 6208–6222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Facoetti A., Nano R., Zelini P., Morbini P., Benericetti E., Ceroni M., Campoli M., Ferrone S. (2005) Clin. Cancer Res. 11, 8304–8311 [DOI] [PubMed] [Google Scholar]
- 58.Raffaghello L., Prigione I., Bocca P., Morandi F., Camoriano M., Gambini C., Wang X., Ferrone S., Pistoia V. (2005) Oncogene 24, 4634–4644 [DOI] [PubMed] [Google Scholar]
- 59.Bruno S., Ghiotto F., Fais F., Fagioli M., Luzi L., Pelicci P. G., Grossi C. E., Ciccone E. (2003) Blood 101, 3514–3519 [DOI] [PubMed] [Google Scholar]
- 60.Zheng P., Guo Y., Niu Q., Levy D. E., Dyck J. A., Lu S., Sheiman L. A., Liu Y. (1998) Nature 396, 373–376 [DOI] [PubMed] [Google Scholar]
- 61.Doody G. M., Stephenson S., McManamy C., Tooze R. M. (2007) J. Immunol. 179, 7614–7623 [DOI] [PubMed] [Google Scholar]
- 62.Herrmann F., Trowsdale J., Huber C., Seliger B. (2003) Immunogenetics 55, 379–388 [DOI] [PubMed] [Google Scholar]
- 63.Herrmann F., Lehr H. A., Drexler I., Sutter G., Hengstler J., Wollscheid U., Seliger B. (2004) Cancer Res. 64, 215–220 [DOI] [PubMed] [Google Scholar]
- 64.Jung D., Hilmes C., Knuth A., Jaeger E., Huber C., Seliger B. (1999) Scand. J. Immunol. 50, 242–249 [DOI] [PubMed] [Google Scholar]
- 65.Dignam J. D., Lebovitz R. M., Roeder R. G. (1983) Nucleic Acids Res. 11, 1475–1489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Handen J. S., Rosenberg H. F. (1997) Front Biosci. 2, c9–c11 [DOI] [PubMed] [Google Scholar]
- 67.Choudhury A., Charo J., Parapuram S. K., Hunt R. C., Hunt D. M., Seliger B., Kiessling R. (2004) Int. J. Cancer 108, 71–77 [DOI] [PubMed] [Google Scholar]
- 68.Ortmann B., Copeman J., Lehner P. J., Sadasivan B., Herberg J. A., Grandea A. G., Riddell S. R., Tampé R., Spies T., Trowsdale J., Cresswell P. (1997) Science 277, 1306–1309 [DOI] [PubMed] [Google Scholar]
- 69.Morris E. J., Ji J.-Y., Yang F., Di Stefano L., Herr A., Moon N.-S., Kwon E.-J., Haigis K. M., Näär A. M., Dyson N. J. (2008) Nature 455, 552–556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ares M., Jr., Proudfoot N. J. (2005) Cell 120, 163–166 [DOI] [PubMed] [Google Scholar]
- 71.Brès V., Yoh S. M., Jones K. A. (2008) Curr. Opin. Cell Biol. 20, 334–340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Fujita T., Piuz I., Schlegel W. (2009) FEBS Lett. 583, 2893–2898 [DOI] [PubMed] [Google Scholar]
- 73.Takahashi Y., Rayman J. B., Dynlacht B. D. (2000) Genes Dev. 14, 804–816 [PMC free article] [PubMed] [Google Scholar]
- 74.Taubert S., Gorrini C., Frank S. R., Parisi T., Fuchs M., Chan H. M., Livingston D. M., Amati B. (2004) Mol. Cell Biol. 24, 4546–4556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Freedman J. A., Chang J. T., Jakoi L., Nevins J. R. (2009) Oncogene 28, 2873–2881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Schlisio S., Halperin T., Vidal M., Nevins J. R. (2002) EMBO J. 21, 5775–5786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zhu W., Giangrande P. H., Nevins J. R. (2004) EMBO J. 23, 4615–4626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Dimova D. K., Dyson N. J. (2005) Oncogene 24, 2810–2826 [DOI] [PubMed] [Google Scholar]
- 79.Rabinovich A., Jin V. X., Rabinovich R., Xu X., Farnham P. J. (2008) Genome Res. 18, 1763–1777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Adams P. D., Kaelin W. G., Jr. (1995) Semin. Cancer Biol. 6, 99–108 [DOI] [PubMed] [Google Scholar]
- 81.Bandara L. R., Buck V. M., Zamanian M., Johnston L. H., La Thangue N. B. (1993) EMBO J. 12, 4317–4324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Helin K., Wu C. L., Fattaey A. R., Lees J. A., Dynlacht B. D., Ngwu C., Harlow E. (1993) Genes Dev. 7, 1850–1861 [DOI] [PubMed] [Google Scholar]
- 83.Krek W., Livingston D. M., Shirodkar S. (1993) Science 262, 1557–1560 [DOI] [PubMed] [Google Scholar]
- 84.Giangrande P. H., Hallstrom T. C., Tunyaplin C., Calame K., Nevins J. R. (2003) Mol. Cell Biol. 23, 3707–3720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Wells J., Graveel C. R., Bartley S. M., Madore S. J., Farnham P. J. (2002) Proc. Natl. Acad. Sci. U.S.A. 99, 3890–3895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Nevins J. R. (1992) Nature 358, 375–376 [DOI] [PubMed] [Google Scholar]
- 87.Blais A., Dynlacht B. D. (2007) Curr. Opin. Cell Biol. 19, 658–662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ogawa H., Ishiguro K., Gaubatz S., Livingston D. M., Nakatani Y. (2002) Science 296, 1132–1136 [DOI] [PubMed] [Google Scholar]
- 89.Pupa S. M., Howard C. M., Invernizzi A. M., De Vecchi R., Giani C., Claudio P. P., Colnaghi M. I., Giordano A., Ménard S. (1999) Oncogene 18, 651–656 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.