

Abstract
Anti-mannotriose (Man3) antibodies were previously isolated from a Keio phage library displaying human single chain variable fragments (scFvs) using a neoglycolipid, Man3- dipalmitoylphosphatidylethanolamine. Of three genes constructed, the 5A3 clone was expressed in mouse myeloma NS0 cells as a conjugate with human IgG1 Fc (scFv-Fc) and characterized (Sakai, K., Shimizu, Y., Chiba, T., Matsumoto-Takasaki, A., Kusada, Y., Zhang, W., Nakata, M., Kojima, N., Toma, K., Takayanagi, A., Shimizu, N., and Fujita-Yamaguchi, Y. (2007) Biochemistry 46, 253–262; Zhang, W., Matsumoto-Takasaki, A., Kusada, Y., Sakaue, H., Sakai, K., Nakata, M., and Fujita-Yamaguchi, Y. (2007) Biochemistry 46, 263–270). Similarly, anti-Lex phages were screened from the same library with lacto-N-fucopentaose III (LNFPIII; Lex)-dipalmitoylphosphatidylethanolamine. Of five phage clones isolated, two scFv genes were constructed to express scFv-Fc proteins in NS0 cells. As was experienced with anti-Man3 scFv-Fc clones, only one anti-LNFPIII clone, 1F12, was successfully produced and purified as an scFv-Fc protein. Although anti-LNFPIII 1F12 and anti-Man3 5A3 scFv-Fc proteins were secreted into media, a decline in scFv-Fc production was observed with both stable clones during early passages. Transient expression of anti-LNFPIII and anti-Man3 scFv-Fc genes in COS-7 cells and subsequent analyses of scFv-Fc protein expression revealed accumulation of translated proteins in the endoplasmic reticulum for scFv-Fc proteins derived from clones that did not survive as stable clones. This report describes the following: (i) isolation of anti-LNFPIII scFv genes; (ii) purification of anti-LNFPIII scFv-Fc proteins from stably and transiently expressed cells; and (iii) extracellular or intracellular localization of two anti-LNFPIII and three anti-Man3 scFv-Fc proteins. The results suggest that expression of anti-Man3 and other anti-carbohydrate antibodies in mammalian cells is disadvantageous for cell growth.
Keywords: Antibodies, Carbohydrate, Carbohydrate-binding Protein, ER Stress, Glycoprotein Biosynthesis, Glycobiology, Anti-carbohydrate Antibodies, Antibody Production, Phage Display, Single Chain antibody
Introduction
Since the human genome project was completed, our understanding of structure-function relationships among proteins has made significant advances. In contrast, elucidating the role of carbohydrate modification on the functions of glycoproteins, glycolipids, and peptide glycans remains elusive. The project that was to produce a variety of carbohydrate-specific antibodies by the phage display technology was thus launched in 2003 with the hope that the resulting antibodies could not only provide tools for carbohydrate research but also be applied to diagnostic or therapeutic uses. In vitro production of anti-carbohydrate antibodies is seemingly a logical strategy because carbohydrate moieties are self-antigens by nature. Thus far, researchers have most often preferred to use phage display technologies because they have been successful to a certain extent, in producing antibodies against varieties of carbohydrate moieties of glycoconjugates (1–6). The phage display strategy readily provides genes encoding recombinant antibodies against antigens of interest, which then need to be introduced into prokaryotic or eukaryotic cells for translation of the obtained antibody genes to produce proteins. Unless other in vitro translation systems are employed, this technology cannot provide an absolute in vitro system for production of anti-carbohydrate antibodies.
Previously, we reported on producing and characterizing anti-mannotriose (Man3)4 antibodies (7, 8). The objectives of our first study (7) were to establish a new methodology so that single chain variable fragments (scFvs) against desired carbohydrate moieties could be readily isolated, and the objectives of our second paper (8) were to produce soluble scFv proteins in quantity so that purification and characterization of isolated scFvs could be readily accomplished. Two consecutive papers previously described the respective results of those efforts (7, 8). Briefly, 25 sequence-independent clones were isolated using a model neoglycolipid, mannotriose-dipalmitoylphosphatidylethanolamine (Man3-DPPE). Analyses of the resulting four phage antibodies and respective scFv protein preparations indicated good affinity and specificity for nonreducing terminal mannose residues (7). Purified antibody proteins must eventually be isolated from genes retrieved from phages to further characterize the antibodies against the antigen and to develop antibody diagnostics and therapeutics in the future. Our second paper (8) thus described the characterization of the isolated scFvs gene products after establishing expression, production, and purification of scFv proteins. That study demonstrated that production of an scFv-Fc protein with an affinity constant of 10−8 m for Man3-BSA, a high constant for an antibody against carbohydrates, is possible (8).
During attempts to establish expression systems for isolated anti-Man3 scFv genes, however, the extreme difficulty of producing anti-Man3 antibodies in mammalian cells became readily apparent. For example, three independent scFv genes, 1A4, 1G4, and 5A3, were isolated from the human scFv-displaying phage library with Man3-DPPE as an antigen with amino acid sequences and initial characterization as were described previously (7). Stable 1A4 and 1G4 scFv-Fc clones from these genes were never established in CHO or NS0 cells (8). Stable clones were established from 5A3 scFv-Fc gene-transfected NS0 cells, which were subsequently used for purification and characterization of the scFv gene product as the first anti-Man3 antibody (8). As later became apparent, however, such 5A3 clones stably expressing scFv-Fc no longer had the ability to produce anti-Man3 antibody during early passages, as described in this current study.
During the aforementioned work, anti-Lex (lacto-N-fucopentaose III, LNFPIII) phage antibodies were also isolated using the same phage library and another neoglycolipid, LNFPIII-DPPE, as an antigen for panning and screening. The first in vitro portion of isolation and initial characterization of isolated phage antibodies was performed without serious problems, which resulted in isolation of three anti-LNFPIII (Lex) scFv genes with differing scFv DNA sequences. The second part of the production, purification, and characterization of anti-LNFPIII (Lex) scFv-Fc proteins was, however, extremely problematic. Similar to the case of anti-Man3 scFv-Fc clones, scFv-Fc proteins were successfully produced and purified from one of two anti-LNFPIII scFv (Lex) genes isolated from the phage library. Although this 1F12 scFv-Fc protein was secreted into the media, production of 3F1 scFv-Fc proteins in NS0 cells was never accomplished. Furthermore, the decline in 1F12 scFv-Fc production was observed during early passages. These results suggested the possibility that expression of anti-Man3 and other anti-carbohydrate antibodies in mammalian cells is disadvantageous for these cells and could lead to eventual cell death.
This study describes not only the isolation and characterization of anti-LNFPIII (Lex) scFv genes and the purification of anti-LNFPIII (Lex) scFv gene products as scFv-Fc forms expressed in stable and transient expression systems, but it also describes the extracellular or intracellular localization of two anti-LNFPIII (Lex) and three anti-Man3 scFv-Fc proteins transiently expressed in COS-7 cells. This study thus provides insights into the pros and cons for use of phage display technology to produce anti-carbohydrate antibodies.
EXPERIMENTAL PROCEDURES
Materials
Escherichia coli strains, the suppressor strain TG1, and the nonsuppressor strain TOP10F′, Dulbecco's modified Eagle's medium (DMEM, high glucose), Lipofectamine 2000, and BODIPY FL C5-ceramide were purchased from Invitrogen. Helper phages M13KO7, anti-M13 antibodies, and protein A-Sepharose 4 Fast Flow were purchased from GE Healthcare. Bovine serum albumin (BSA), DPPE, dipalmitoylphosphatidylcholine, tunicamycin, and N-glycosidase F were purchased from Sigma. Mannotriose (Man3), lacto-N-tetraose, lacto-N-neotetraose, lacto-N-fucopentaose I (LNFPI; Fucα1–2Galβ1–4GlcNAcβ1–3Galβ1–4Glc), lacto-N-fucopentaose II (LNFPII; Galβ1–3[Fucα1–4]GlcNAcβ1–3Galβ1–4Glc), lacto-N-fucopentaose III (LNFPIII; Galβ1–4[Fucα1–3]GlcNAcβ1–3Galβ1–4Glc), LNFPIII-BSA, sialyl LNFPIII (sLex)-BSA, and Man3-BSA were obtained from Dextra Laboratories (Reading, UK). 2,2′-Azino-bis(3-ethylbenzthiazoline-6-sulfonic acid) (ABTS) was from Roche Diagnostics. The cDNA clone of human IgG1 Fc, consisting of the hinge, CH2, and CH3 regions, was originally from Dr. J. Schlom, Laboratory of Tumor Immunology and Biology, Division of Cancer Biology and Diagnosis, NCI, National Institutes of Health (Bethesda) (8). Plasmid pEE12.4 and a murine myeloma NS0 cell strain were obtained from Lonza Biologics (Slough, UK). Plasmid pCI-neo was purchased from Promega (Madison, WI). Goat anti-human IgG (Fc fragment-specific), horseradish peroxidase (HRP)-conjugated rabbit anti-human IgG secondary antibody, and Cy3-conjugated AffiniPure donkey anti-human IgG were from Jackson ImmunoResearch, Inc. (West Grove, PA). The DAB (3,3′-diaminobenzidine) detection kit was purchased from Vector Laboratories (Burlingame, CA). The EndFree plasmid maxi kit (catalog no. 12362) was obtained from Qiagen (Hilden, Germany). Anti-PDI antibody (catalog no. MA3-019) was purchased from Affinity BioReagents (Golden, CO).
Preparation of Neoglycolipids
Neoglycolipids were synthesized from carbohydrates (Man3, lacto-N-tetraose, lacto-N-neotetraose, LNFPI, LNFPII, or LNFPIII) and DPPE by reductive amination and purified by HPLC with a silica gel column as described previously (9, 10). The purity and structure of each neoglycolipid were confirmed with TLC and MALDI-TOF mass spectrometry.
Panning Procedures
The Keio phage display libraries representing over 1011 independent human scFvs, prepared as described previously (7, 11), were subjected to four rounds of panning similar to the procedures described for anti-Man3 scFvs (7), except that LNFPIII-DPPE was used as an antigen. Briefly, coating of wells with LNFPIII-DPPE was achieved by applying 50 μl of LNFPIII-DPPE (40 μg/ml methanol) and drying the solvent at 37 °C, followed by incubation overnight at 4 °C with 150 μl of Tris-HCl buffer, pH 7.4, containing 0.15 m NaCl (TBS) and 3% BSA (blocking buffer). Wells were rinsed twice with 50 μl of 0.2% Tween 20/TBS (TBS-T) and once with 200 μl of TBS. Fifty μl of phage suspension in TBS containing 0.1% Tween 20 and 1.4% BSA were added to the wells, which were then incubated at 37 °C for 60 min under continuous rotation. After wells were washed three times with 200 μl of TBS-T and twice with 200 μl of TBS, bound phages were eluted by addition of 50 μl of 100 mm triethylamine and incubation at 25 °C for 10 min and then neutralized by adding 100 μl of neutralizing solution (1 m Tris-HCl, pH 7.4, 3% BSA/TBS = 2:1, v/v) to wells of the control plate, which had been treated with the blocking buffer. Bound phages were further eluted by addition of 50 μl of 100 mm triethylamine and incubation at 25 °C for 20 min and were recovered in the neutralization solution as described above. Eluted phages were used to infect 100 μl of logarithmically growing E. coli TG1 at 37 °C for 1 h. Infected bacteria, grown on LB agar plates (inner diameter = 10 cm) containing 1 mm NaOH, 0.1% glucose, and carbenicillin (50 μg/ml) at 25 °C for 16 h, were scraped from the plates using a spreader after addition of 2 ml of LB-10 mm Tris-HCl, pH 7.5 (SBS), per plate. An aliquot of this suspension was inoculated into 40 ml of SBS containing carbenicillin and grown at 37 °C for 2 h with shaking. Phages were rescued after addition of 40 μl containing 3.5 × 109 plaque-forming units of helper phages M13KO07 and incubation at 37 °C for 1 h without shaking, followed by addition of kanamycin (25 μg/ml), chloramphenicol (10 μg/ml) and incubation at 25 °C for 40 h with rotation. Phage particles were concentrated by polyethylene glycol precipitation and dissolved in 400 μl of TBS, 400 μl of 3% BSA/TBS, and 40 μl of 10% Tween 20/TBS by incubation at 37 °C for 1 h. After centrifugation at 18,000 × g for 5 min at 4 °C, 800 μl of phage suspension were recovered and used for the 2nd panning. The 2nd, 3rd, and 4th pannings were carried out similar to the 1st panning except for washing conditions. Bacteria collected from single colonies after four rounds of panning were grown in 50 μl of SBS/carbenicillin in 96-well plates at 37 °C for 1 h with rotation, to which 50 μl of helper phages were added and incubated at 37 °C for 1 h with rotation. After an SBS/kanamycin/chloramphenicol mixture (50 μl/well) was added, phage suspensions were obtained by incubation at 25 °C for 16 h with rotation followed by centrifugation at 200 × g for 15 min at 4 °C. Fifty μl of the supernatants were added to wells containing 100 μl of 3% BSA/TBS, incubated at 37 °C for 1 h, and kept at 4 °C.
Screening of Phage Clones Expressing Antibodies Directed against LNFPIII-DPPE by ELISA
ELISA analysis of phage binding to LNFPIII-DPPE was performed on bacterial supernatants containing phages. Wells of a 96-well plate were coated with 1 μg of LNFPIII-DPPE/well as described above and blocked by incubation overnight at 4 °C with 150 μl of 3% BSA/TBS. Control plates were prepared as above without the antigen. Seventy five μl of phage suspensions were added to the wells and incubated at 37 °C for 1 h. The wells were washed five times with 200 μl of TBS-T. Bound phage antibodies were detected by incubation with HRP-conjugated anti-M13 antibody at 37 °C for 1 h, after which they were washed 10 times with 200 μl of TBS-T and once with 200 μl of TBS. Peroxidase activity was detected by reaction with ABTS/H2O2 for 30 min and termination with 1% oxalic acid. Absorbance was measured at 415 nm with a Bio-Rad Microplate Reader model 680 (Bio-Rad).
Colony PCR and Determination of DNA Sequences
scFv genes were amplified from respective E. coli TG1 colonies infected with phages by PCR with a primer set (forward primer Cm, 5′-TGTGATGGCTTCCATGTCGGCAGAATGCT-3′, and reverse primer g3, 5′-GCTAAACAACTTTCAACAGTCTATGCGGCAC-3′). After preheating at 94 °C for 2 min, PCR was carried out in 35 cycles under conditions of denaturing at 94 °C for 20 s, annealing at 53 °C for 20 s, and extension at 68 °C for 1 min. After purification and confirmation with 2% agarose gel electrophoresis, the resulting scFv genes were subjected to DNA sequencing. DNA sequences of scFvs were determined using a 3730 DNA analyzer (Applied Biosystem, Foster City, CA).
Construction of Recombinant Vectors of Signal-scFv-Human Fc
Two mammalian systems, GS/NS0 and neo/COS-7, were used for stable and transient expression, respectively, of scFv conjugated with the human IgG1 Fc domain. The construct included a consensus ribosome-binding sequence and the signal peptide from a murine κ light chain previously used for high level mammalian antibody secretion (12, 13). The signal peptide was followed by scFv and the human IgG1 Fc domain (see under “Materials”). To establish stable expression of scFv-Fc proteins in NS0 cells, the 1F12 and 3F1 scFv genes were inserted into a template plasmid signal human Fc-pEE12.4 (SFpEE), as described previously (8). Alternatively, the gene encoding 1F12, 3F1, 5A3, 1A4, or 1G4 scFv with the signal peptide sequence and human Fc domain was assembled by splice-overlap extension PCR and subcloned into the pCI-neo plasmid, as described previously (8), resulting in pCI-neo/signal scFv (1F12, 3F1, 5A3, 1A4, or 1G4)-Fc. For controls, expression vectors encoding 1H7 scFv-Fc, which has specificity for the insulin-like growth factor-I receptor (14), and Fc only were similarly prepared. All plasmids used for electroporation or lipofection into mammalian cells were purified with the EndFree plasmid maxi kit.
Cell Culture, Transfection, and Screening by Sandwich ELISA
The murine myeloma NS0 cell line was grown in DMEM supplemented with 10% fetal bovine serum (FBS) and 2 mm glutamine. Selective medium for human Fc-expressing NS0 cells consisted of glutamine-free DMEM, dialyzed FBS, and glutamine synthase supplement (JRH Biosciences, Lenexa, KS).
Forty μg of linearized 1F12 or 3F1/SFpEE DNA in sterile water was electroporated into 1 × 107 NS0 cells at conditions of 0.25 kV and 400 microfarad capacitance. Cells at 1.6 × 105 cells/ml were plated at 50 μl/well in 96-well plates and cultured in nonselective media. Twenty four hours after electroporation, 150 μl of selective medium were added to each well, and cells were allowed to recover and grow undisturbed for about 3 weeks until discrete surviving colonies appeared.
Supernatants of transformed cells that were able to grow under selective conditions were screened for scFv-Fc protein secretion by sandwich ELISA. Briefly, goat anti-human IgG (Fc fragment-specific, Jackson ImmunoResearch) diluted in phosphate-buffered saline (PBS) to a final concentration of 5 μg/ml was coated in wells of 96-well plates by adding 100 μl/well and incubating overnight at 4 °C. After blocking with 3% BSA in PBS, cell culture supernatants from different clones were added and incubated at 37 °C for 1 h. After washing with PBS, HRP-conjugated anti-human IgG (Invitrogen) diluted to 1:1,000 was added to the wells. The wells were washed with PBS and then incubated with ABTS. The color that developed in the wells of the plates was analyzed spectrophotometrically at 415 nm with the Bio-Rad Microplate Reader.
COS-7 cells (African green monkey SV40-transfected kidney fibroblast cell) were grown in DMEM supplemented with 10% FBS and penicillin/streptomycin (Sigma). For transient expression of 1F12 scFv-Fc gene, COS-7 cells grown to confluence in a 75-cm2 flask were cultured in 15 ml of Opti-MEM I (Invitrogen) for 2 h and then further cultured in the presence of 20 μg of pCI-neo/signal-1F12 scFv-Fc DNA and 70 μl of Lipofectamine 2000 (Invitrogen) for 4 h. Cells were cultured in fresh DMEM supplemented with 1% FBS for 72 h. Media (105 and 350 ml) were collected for purification of 1F12 scFv-Fc proteins.
Purification of scFv-Fc Antibody from Culture Media
The best clone (14F8 in Fig. 1) for 1F12 scFv-Fc, i.e. the one with the highest expression of antibody, was cultured on a large scale in a T175 flask (Corning) in selective medium. After culturing for 24–48 h, cell culture supernatants were collected and dialyzed against 1.5 m glycine-NaOH, pH 8.9, and then passed through a protein A column (5 ml). The column was washed with 10 column volumes of 1.5 m glycine-NaOH, pH 8.9, containing 3 m NaCl. Contaminating bovine IgG was eluted from the column with 100 mm citric acid buffer, pH 4.5. scFv-Fc proteins were then eluted from the column with 100 mm citric acid buffer, pH 3.0, and collected in 1.5-ml conical tubes containing 0.1 volume of 1 m Tris-HCl, pH 8.0.
FIGURE 1.

Screening of NS0 clones stably expressing 1F12 scFv-Fc proteins. Supernatants of transformed cells that were able to grow under selective conditions were screened for scFv-Fc protein secretion by sandwich ELISA as described under “Experimental Procedures.”
Alternatively, the 1F12 scFv-Fc protein was purified by protein A affinity chromatography with the following modifications. After culture supernatants were placed on the column equilibrated with 50 mm Tris-HCl, pH 7.5, and the column was washed with 5 column volumes of the same buffer. After contaminating bovine IgG was eluted from the column with 15 column volumes of 50 mm citric acid buffer, pH 5.0, scFv-Fc proteins were eluted from the column with 10 column volumes of 50 mm citric acid buffer, pH 3.0, containing 50 mm NaCl. The eluted scFv-Fc proteins were immediately neutralized by adding 0.17 volume of 1 m Tris-HCl, pH 9.0. The eluates were concentrated and dialyzed against PBS using Amicon Ultra-15 (Millipore) and analyzed by SDS-PAGE. For Western blot analyses, samples were transferred onto a PVDF membrane (GE Healthcare), and the scFv-Fc protein was visualized by reaction with rabbit anti-human IgG antibody conjugated with peroxidase using the DAB (3,3′-diaminobenzidine) detection kit. Protein concentration was measured according to the procedures used by Bradford with BSA as a standard.
ELISA
ELISA was used to evaluate the antigen binding activity of the purified 1F12 scFv-Fc protein. Briefly, 50 μl of the antigen, LNFPIII-BSA, in 0.05 m bicarbonate coating buffer, pH 9.6, (10 μg/ml) were added in wells of 96-well ELISA plates and incubated overnight at 4 °C. After blocking with 3% BSA/PBS, a series of diluted samples of 1F12 scFv-Fc was added in triplicate. After incubation at room temperature for 2 h, the bound scFv-Fc was detected with rabbit peroxidase-conjugated anti-human IgG antibody (1:7,000 dilution) using ABTS as a substrate. After incubation for 30 min in the dark, the reaction was terminated by adding 100 μl of 2% oxalic acid solution. Absorbance at 415 nm was measured using a plate reader. A group of different carbohydrate antigens conjugated with BSA (0.5 μg/well) was coated onto the ELISA plates to analyze the specificity of 1F12 scFv-Fc as described above.
Transient Expression of scFv-Fc Proteins in COS-7 Cells and Detection of Expressed scFv-Fc Proteins by Sandwich ELISA
Seventy six hours after lipofection of COS-7 cells with scFv-Fc genes encoding 3F1, 1F12, 5A3, 1A4, 1G4, or 1H7, the Fc gene only, and the vector (mock), expression of various scFv-Fc and control Fc proteins in media was examined by sandwich ELISA using anti-human IgG (Fc fragment-specific) antibody as described above.
To measure intracellular levels of scFv-Fc proteins, COS-7 cells 76 h after lipofection with genes encoding 3F1, 1F12, 5A3, 1A4, 1G4, or 1H7 scFv-Fc, Fc, and the vector (mock) were washed twice with PBS and treated with 5 ml of Cell Dissociation solution (Sigma) in a 75-cm2 flask. Cells were suspended by pipetting and recovered by centrifugation at 1000 × g for 1 min. Cells were suspended in PBS (1 × 106/1 ml PBS) with protease inhibitor mixture P8340 (Sigma) and sonicated on ice three times for 5 s. Clear supernatants were subjected to sandwich ELISA to quantify scFv-Fc protein expression levels as described above.
The scFv-Fc or Fc concentrations of both secreted and intracellular fractions were quantified by using human Fc fragment (Jackson ImmunoResearch) as a standard. The concentrations estimated were then calculated as micrograms of scFv-Fc or Fc/106 cells to readily compare secreted and intracellular amounts of scFv-Fc or Fc.
Transient Expression of scFv-Fc Proteins in COS-7 Cells Followed by Detection of Expressed scFv-Fc by Immunofluorescence Staining
COS-7 cells were grown to 70% confluence in wells of the Lab-Tek Chamber Slide System (Nunc, Roskilde, Denmark) and subjected to lipofection with scFv-Fc genes encoding 3F1, 1F12, 5A3, 1A4, or 1G4, Fc, and the vector (mock) by adding 0.4 μg/well as described above. Media were removed 16 h after lipofection, and the cells were washed three times with 200 μl/well of PBS. Cells were then fixed with 4% paraformaldehyde in PBS. The fixed cells were left in 5% BSA, 160 μl/well, containing 0.3% Triton X-100 at 25 °C for 20 min and then washed with PBS. Primary and secondary antibodies, anti-protein-disulfide isomerase (PDI) (Affinity BioReagents) and FITC-conjugated AffiniPure donkey anti-mouse IgG were used for staining of ER. BODIPY FL C5-ceramide (Invitrogen) was diluted with 1% BSA in PBS as indicated in Fig. 8 for staining of Golgi apparatuses. Cells were also treated with DAPI (Dojin Chemical, Kumamoto, Japan) and Cy3-conjugated AffiniPure donkey anti-human IgG (Jackson ImmunoResearch) for staining of nuclei and scFv-Fc, respectively. After they were washed with PBS and distilled water, cells were embedded in one drop of VECTASHIELD® (Vector Laboratories), covered with a NEO micro cover glass (Matsumi Glass, Osaka, Japan), and observed under a Keyence fluorescence microscope BZ-9000 (Keyence, Osaka, Japan).
FIGURE 8.
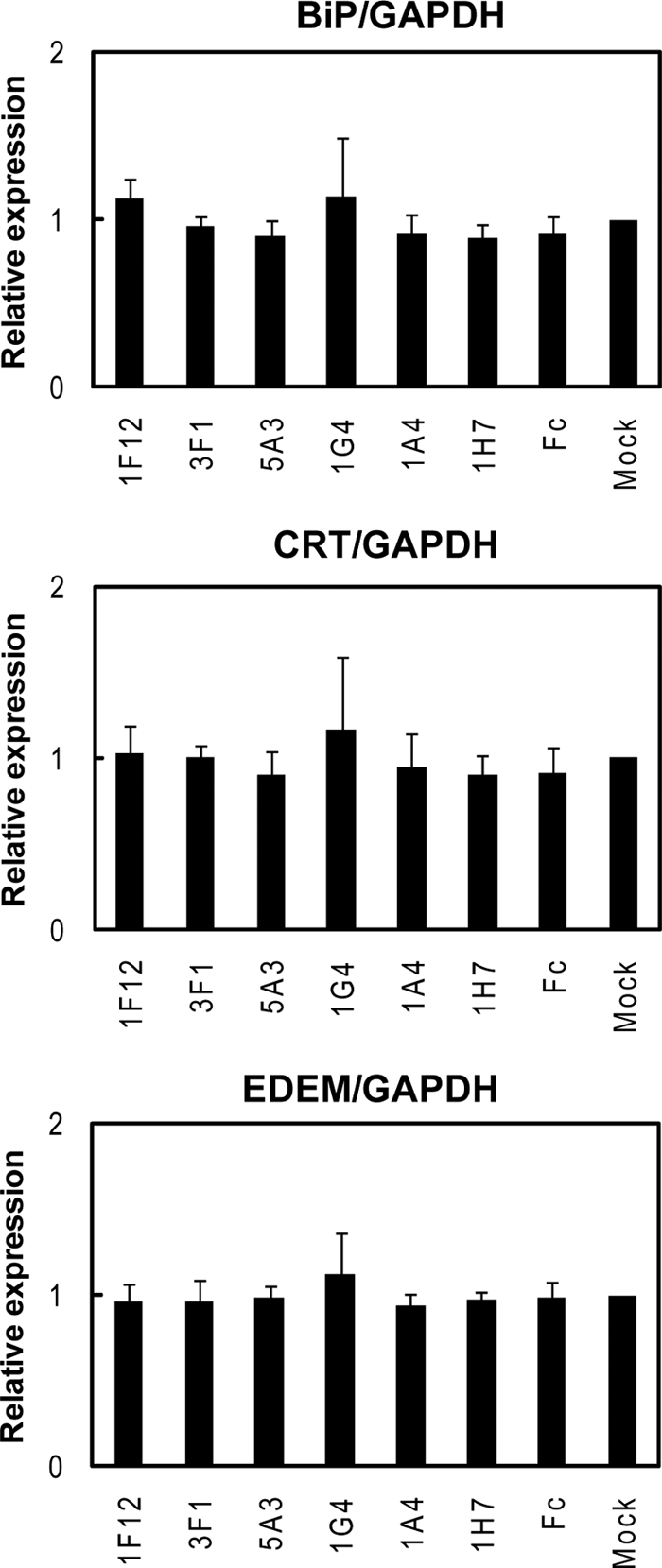
Induction of BiP, CRT, and EDEM expressions in scFv-Fc protein-producing COS-7 cells measured by RT-PCR. Real time PCR was performed as described under “Experimental Procedures.” Shown are the results using GAPDH mRNA as an endogenous control. The expression levels are normalized with the mock control. Basically, the same results were obtained with the experiments using β-actin as an endogenous control (data not shown).
Detection of ER Stress-inducible Genes by RT-PCR
COS-7 cells were transfected with each plasmid for expression of 1F12, 1A4, 3F1, 5A3, 1G4, and 1H7 scFv-Fc using Lipofectamine 2000 (Invitrogen) as described above. A plasmid encoding the Fc portion and a mock plasmid were used as controls. After 48 h, the transfected cells were harvested with PBS/EDTA and then washed twice with PBS. Poly(A)+ RNA was extracted from transfected cells using μMACS mRNA isolation kit (Miltenyi Biotec, Germany). Each cDNA was generated with SuperScript III first-strand synthesis system (Invitrogen) according to the manufacturer's instruction using 0.2 μg of poly(A)+ RNA. Quantitative real time PCR was performed on a Smart cycler real time PCR system (Takara Bio, Japan) using SYBR premix Ex Taq (Takara Bio). For each PCR, a reaction mixture without primers was included as a control. The amplification reaction was performed by a two-step PCR for 55 cycles with denaturation at 95 °C for 5 s, followed by extension and detection at 57 °C for 20 s. The relative RNA abundance of target gene transcripts was normalized against endogenous gene, glyceraldehyde-3-phosphate dehydrogenase (GAPDH), and β-actin, respectively. The primers for each gene are as follows: GAPDH-F, 5′-AGCCTCCCGCTTCGCTCTC-3′, and GAPDH-R, 5′-CCAGGCGCCCAATACGACC-3′; actin-F, 5′-GAGGCACTCTTCCAGCCTTC-3′, and actin-R, 5′-CGTACAGGTCTTTGCGGATG-3′; BiP-F, 5′-AACCGCTGAGGCTTATTTGG-3′, and BiP-R, 5′-TTTGGTTGCTTGGCGTTG-3′; CRT-F, 5′-AGTTCCGGCAAGTTCTACGG-3′, and CRT-R, 5′-TGGCCGACAGAGCATAAAAG-3′; EDEM-F, 5′-TTGACTCTTGTTGATGCATTGG-3′, and EDEM-R, 5′-CTTGGACGGTGGAATCTTTG-3′.
RESULTS
Screening of scFv-displaying Phages Directed against LNFPIII-DPPE
The Keio phage library displaying human scFvs as was used previously (7) was subjected to four rounds of panning against LNFPIII-DPPE. Of 1,470 clones screened by ELISA using LNFPIII-DPPE (1 μg/well) as an antigen, 100 clones were positive in ELISA with a signal/noise ratio of >4. Of these, five positive clones were identified to contain scFv inserts with an expected length. DNA sequencing of scFv inserts of the phage clones allowed deduction of the amino acid sequences of three clones, 1F12, 3F1, and 2A1 (Table 1). Of the three, the 1F12 and 3F1 clones were further characterized.
TABLE 1.
Deduced amino acid sequence alignment of VH and VL chains
VH and VL chain amino acids were aligned using the Kabat numbering system (28). The potential N-glycosylation site in the VH CDR2 of the 1F12 clone is in boldface type. VL is variable region of antibody light chain.

Production of scFv-Fc Proteins in Mouse Myeloma Cells
The GS/NS0 system was used for expression and production of scFv-Fc proteins as described previously (8, 14, 15). Of 1F12 and 3F1 scFv-Fc genes constructed in the pEE12.4 plasmid and introduced into NS0 cells, 76 and 4 clones expressing 1F12 and 3F1 scFv-Fc proteins, respectively, were originally identified (Table 2, although further details on 3F1 are described below). Fourteen 1F12-derived clones that survived in the selective medium were analyzed for Fc positivity by assaying culture supernatants with sandwich ELISA, which identified 11 positive clones (Fig. 1). The clone 14F8 that expressed the highest level of scFv-Fc was selected for large scale production and purification of anti-LNFPIII scFv-Fc proteins.
TABLE 2.
Production of stable clones in the GS/NS0 system and purification of scFv-Fc proteins from culture media
| Antigen | scFv-Fc proteins | No. of clones analyzed by sandwich ELISA | No. of Fc-positive clones initially identified | No. of stable clones obtained (no. of clones examined) | Expression levels in media | Source |
|---|---|---|---|---|---|---|
| μg/ml | ||||||
| LNFPIII | 1F12 | 97 | 76 | 1 (of 42) | 49.8 | This study |
| LNFPIII | 3F1 | 125 | 4 | 0 (of 4) | NAa | This study |
| Man3 | 1A4 | 98 | 0 | NA | NA | Zhang et al. (8) |
| Man3 | 1G4 | 80 | 0 | NA | NA | Zhang et al. (8) |
| Man3 | 5A3 | 25 | 6 | 2 (of 6) | 48.0 | Zhang et al. (8) |
| 1.3 and 0.63b | This study | |||||
| Fc only | 12 | 10 | 1 (of 1) | 120 | Zhang et al. (8) |
a NA means not applicable.
b Only 92 and 75 μg of 5A3 scFv-Fc proteins were purified from 145 and 60 ml of culture media, respectively, 3 years after original purification was performed (8).
Purification of the 1F12 scFv-Fc Protein from Culture Media
The scFv-Fc protein produced by the GS/NS0 system in media was purified by protein A-column chromatography. Four independent purifications were carried out following the original elution protocol. Initially, 8.5 mg of the 1F12 scFv-Fc protein were purified from 170 ml of culture media. The level of 1F12 scFv-Fc protein expression in the medium was thus calculated to be ∼50 μg/ml (Table 3A, Experiment 1). This preparation was used for analyses of LNFPIII binding by ELISA and surface plasmon resonance. As summarized in Table 3A, however, three consecutive purifications (Table 3A, Experiments 2–4) after the first purification resulted in significantly less recovery of 1F12 scFv-Fc proteins. Such a low level of production from late passage cells was later confirmed when an improved elution protocol was used to purify 1F12 scFv-Fc proteins from the medium that had been collected earlier and kept frozen (early passages) and from the newly collected medium (late passages) (Table 3B, Experiments 1 and 2, respectively). In these rounds of purification, 1F12 scFv-Fc proteins were eluted from a protein A affinity column using milder elution conditions to prevent possible inactivation of the 1F12 scFv-Fc protein (see below). Similar findings were also obtained with anti-Man3 5A3 scFv-Fc proteins, i.e. purification of 5A3 scFv-Fc proteins from late passages or frozen NS0 cells resulted in only a 2% level of original production (Table 2).
TABLE 3.
Production and purification of anti-LNFPIII (LeX) scFv-Fc proteins in the GS/NS0 system or transiently expressed in COS-7 cells
| Experiments | Culture media | scFv-Fc purified | Expression levels in media |
|---|---|---|---|
| ml | μg | μg/ml | |
| A. scFv-Fc proteins purified from early passages of the stable 1F12 clone | |||
| 1 | 170 | 8470 | 49.8 |
| 2 | 60 | 536 | 8.9 |
| 3 | 130 | 2300 | 17.6 |
| 4 | 150 | 700 | 4.6 |
| B. scFv-Fc proteins purified from early and later passages of the stable 1F12 clone via the alternative elution procedures | |||
| 1 (early passages) | 25 | 1480 | 59.2 |
| 2 (late passages) | 110 | 1670 | 15.2 |
| C. Transiently expressed 1F12 scFv-Fc proteins purified from the culture media of COS-7 cells via the alternative elution procedures | |||
| 1 | 105 | 280 | 2.69 |
| 2 | 350 | 530 | 0.51 |
Alternatively, 1F12 scFv-Fc proteins were purified from culture media of COS-7 cells transiently expressing scFv-Fc proteins by protein A affinity chromatography using milder elution procedures. Results of the two experiments are summarized in Table 3C. Although the expression levels were lower than the levels from NS0 cells stably expressing 1F12 scFv-Fc proteins, a reasonably high recovery rate was achieved by purification of scFv-Fc proteins transiently expressed by COS-7 cells.
Moreover, aglycosylated 1F12 scFv-Fc proteins were produced and purified by protein A affinity chromatography following the original elution procedures. The culture medium (130 ml) of NS0 cells grown at 37 °C for 18 h in the presence of 1 μg/ml tunicamycin yielded 30 μg of purified 1F12 scFv-Fc proteins, indicating a low level of expression (0.23 μg/ml) under these conditions (data not included in Table 3).
Purity and Structural Analysis of Purified 1F12 scFv-Fc Protein
The purified 1F12 scFv-Fc protein was analyzed by SDS-PAGE under nonreducing and reducing conditions. Fig. 2, A–C, summarizes the results of three rounds of purifications corresponding to Table 3, A–C, respectively. Because human IgG1 Fc contains a hinge region, the scFv-Fc produced by mammalian cells is expected to form a disulfide-linked dimer. Results clearly show that the 1F12 scFv-Fc protein is a dimer consisting of two monomers that contain the human IgG1 Fc domain.
FIGURE 2.

Analyses of various protein A affinity-purified 1F12 scFv-Fc preparations by SDS-PAGE and immunoblotting under nonreducing (DTT−) and reducing (DTT+) conditions. A, 1F12 scFv-Fc proteins purified from NS0 cells following the original elution procedures (Table 3A, Experiment 1) were analyzed by SDS-PAGE followed by Coomassie Brilliant Blue staining. B, 1F12 scFv-Fc proteins purified from NS0 cells following the alternative elution procedures (Table 3B, Experiment 1) were analyzed by SDS-PAGE followed by Coomassie Brilliant Blue staining (left, lanes 2 and 3) and immunoblotting with anti-human IgG antibody (right, lanes 2 and 3). C, 1F12 scFv-Fc proteins purified from COS-7 cells following the alternative elution procedures were analyzed by SDS-PAGE followed by Coomassie Brilliant Blue staining; lanes 2 and 4 (Table 3C, Experiment 1) and lanes 3 and 5 (Table 3C, Experiment 2) were analyzed under nonreducing and reducing conditions, respectively. Molecular weight markers (M) are shown in lane 1.
The molecular mass of the monomeric scFv-Fc estimated by SDS-PAGE under reducing conditions was larger than 53–55 kDa, the molecular mass expected based on the amino acid sequence (Fig. 2, A–C). This is possibly due to the post-translational modification with N-glycosylation of the Fc region as is observed with regular antibodies and/or structural effects during electrophoresis (16). In addition, one potential N-glycosylation site was identified in CDR2 of the VH domain of 1F12 scFv (Table 1A). To test whether additional N-glycosylation occurs or not, the purified 1F12 scFv-Fc and the purified anti-Man3 5A3 scFv-Fc proteins were subjected to N-glycosidase F digestion followed by SDS-PAGE analyses (Fig. 3A). The results clearly showed that after enzyme digestion both scFv-Fc proteins moved faster than the respective original undigested proteins did and that 1F12 scFv-Fc has a larger mass than 5A3 scFv-Fc, which is expected given the extra N-glycosylation in its VH domain. Furthermore, 1F12 scFv-Fc proteins were purified from medium in which 14F8 NS0 cells were grown in the presence of tunicamycin. The molecular mass of purified 1F12 scFv-Fc proteins from tunicamycin-treated cells was smaller than that of the original 1F12 scFv-Fc proteins and had an expected monomer size of 53–55 kDa (Fig. 3B). These results are consistent with the notion that N-glycosylation of scFv-Fc proteins is responsible for their observed molecular mass on SDS-polyacrylamide gels.
FIGURE 3.
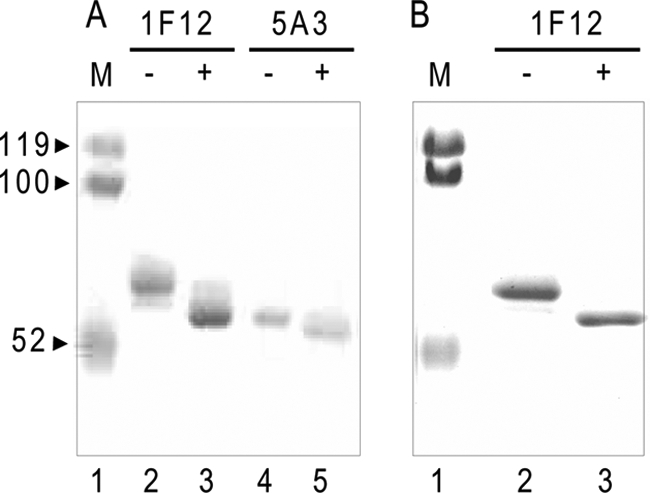
Comparison of original and aglycoforms of purified 1F12 and 5A3 scFv-Fc proteins. A, SDS-PAGE analyses of 1F12 and 5A3 scFv-Fc proteins before and after N-glycosidase F digestion. Both 1F12 and 5A3 scFv-Fc proteins digested with N-glycosidase F (lanes 3 and 5, respectively) were analyzed along with undigested scFv-Fc proteins (lanes 2 and 4, respectively). The gel was stained with Coomassie Brilliant Blue. B, comparison of purified 1F12 scFv-Fc proteins from NS0 cells grown in the presence (lane 3) and absence (lane 2) of tunicamycin. Shown is a Coomassie Brilliant Blue-stained SDS-polyacrylamide gel. Molecular weight markers (M) are shown in lane 1.
Binding Specificity of 1F12 scFv-Fc by ELISA
Initial binding studies, performed with the 1F12 scFv-Fc proteins purified following the original procedures, indicated that 1F12 scFv-Fc proteins had the highest binding affinity for LNFPIII-BSA among the neoglycolipids tested (Fig. 4). The binding of the 1F12 scFv-Fc protein to LNFPIII-BSA coated onto plastic plates (0.5 μg/well) is shown, in terms of scFv-Fc concentration, in Fig. 5A. Although binding to LNFPIII-BSA was confirmed with scFv-Fc proteins purified following the original procedures, the level of binding activity was not as high as expected because nonspecific binding to BSA also increased, resulting in a signal/noise ratio of nearly 2 (Fig. 5A). The binding activity is approximately one-tenth the binding activity of 5A3 scFv-Fc proteins to Man3-BSA as was reported previously (8). Although binding to LNFPIII-BSA was observed at concentrations of 0.28, 0.56, and 1.39 μm, SPR analyses of these preparations did not result in a successful determination of kinetic parameters (data not shown). These results strongly suggest that the scFv-Fc proteins purified following the original protocols may have lost significant binding activity during purification.
FIGURE 4.

Binding of protein A affinity-purified 1F12 scFv-Fc protein to different carbohydrate antigens conjugated with DPPE. ELISA was used to examine the binding specificity of 1F12 scFv-Fc purified following the original elution procedures using neoglycolipids.
FIGURE 5.
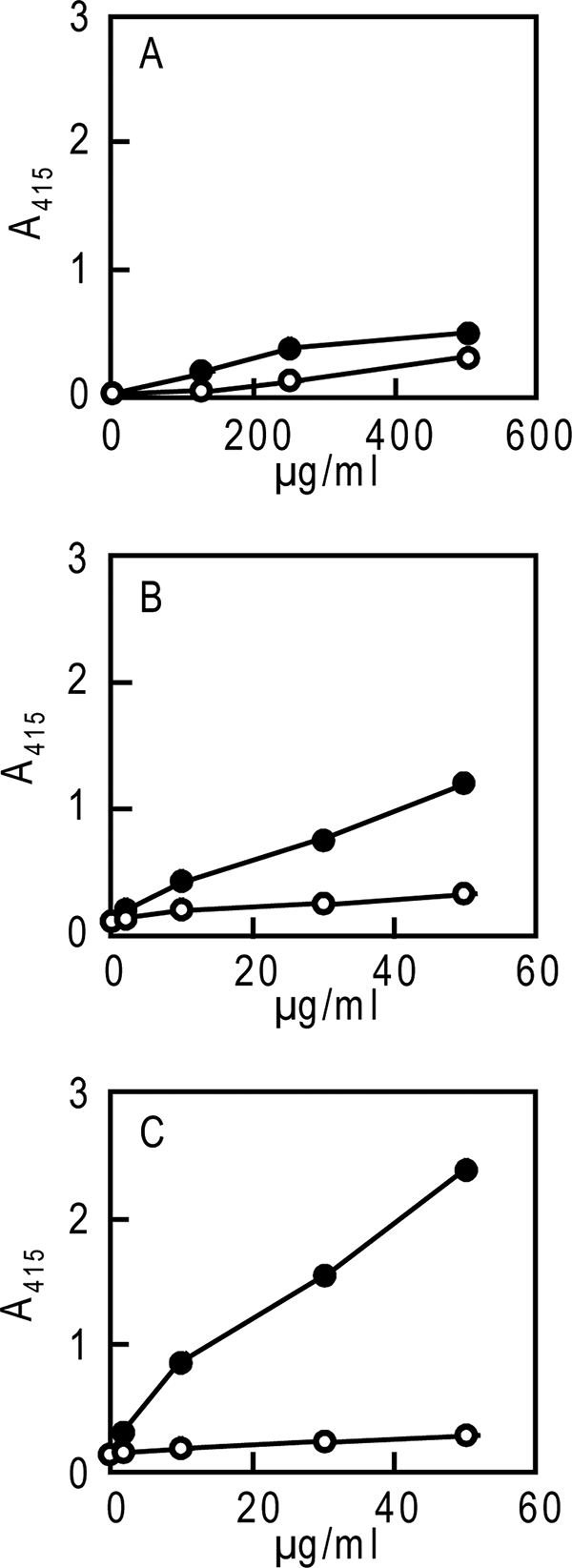
Binding of purified 1F12 scFv-Fc proteins to LNFPIII-BSA. 1F12 scFv-Fv proteins were purified from media of the NS0 stable cells following the original procedures or the alternative procedures (A and B, respectively) or were purified from media of COS-7 cells transiently transfected with pCI-neo/1F12 scFv-Fc following the alternative procedures (C). Binding of a series of diluted samples of purified 1F12 scFv-Fc proteins was measured by ELISA using LNFPIII-BSA (closed circle) or BSA (open circle) as described under “Experimental Procedures.”
Alternative elution procedures were thus introduced to purify more active 1F12 scFv-Fc proteins. Two sources of culture media were used for this purpose as follows: one from stable 14F8 NS0 cells and the other from COS-7 cells transiently expressing the scFv-Fc proteins. Purification results are summarized in Table 3, B and C, respectively. Results of SDS-PAGE analyses of purified scFv-Fc proteins are shown in Fig. 1, B and C, respectively. Both preparations had higher binding activity to LNFPIII-BSA than the BSA control (Fig. 5, B and C) in comparison with the activity obtained with 1F12 scFv-Fc proteins purified following the original procedures (Fig. 5A). This suggested that the alternative elution procedures were superior to the original procedures in terms of purifying active scFv-Fc proteins. Furthermore, comparison of stably and transiently expressed scFv-Fc proteins indicated that the latter had a higher binding activity than the former (Fig. 5, B and C).
Failure to Establish Stable NS0 Cell Lines for the 3F1 scFv-Fc Clone
As described above, 125 clones grown in selection medium were screened, and four positive clones expressing scFv-Fc proteins in media were identified by sandwich ELISA (Table 2). None of the four clones, however, survived to become stable cell lines expressing and secreting 3F1 scFc-Fc proteins. In contrast, initial screening of the 1F12 scFv-Fc gene-transfected NS0 cells resulted in more positive clones, and one stable clone was established.
Transient Expression of scFv-Fc in COS-7 Cells
Similar to the failure to establish stable clones for 3F1 scFv-Fc, as summarized in Table 2, stable clones secreting anti-Man3 1A4 and 1G4 scFv-Fc proteins were never obtained from 98 and 80 NS0 cells, respectively, although six positive clones secreting 5A3 scFv-Fc proteins were obtained from which two stable clones were originally established (8). The control Fc protein-expressing stable clones were obviously easy to establish because 10 of the 12 clones examined expressed Fc protein (Table 2). Furthermore, a previous study found that stable CHO cells expressing 1A4 or 1G4 scFv-Fc proteins were never established (8). To understand why achieving stable expression of scFv-Fc proteins for not only anti-LNFPIII 3F1 but also anti-Man3 1A4 and 1G4 is so difficult, COS-7 cells were transiently transfected with pCI-neo/signal-scFv genes. Expression levels of scFv-Fc proteins in both culture media and cell lysates were determined by sandwich ELISA. In the culture supernatants, Fc, 5A3, 1F12, and 1H7 scFv-Fc proteins were present as expected, whereas 3F1, 1G4, and 1A4 scFv-Fc proteins were not significantly detected (Fig. 6A). Note that 1F12 scFv-Fc protein secretion was significant but rather low in this particular experiment. Examination of their corresponding cell lysates, however, clearly revealed the expression of 3F1, 1G4, and 1A4 scFv-Fc proteins (Fig. 6B). The ratios of secreted/retained scFv-Fc proteins shown in Fig. 6 indicate that control 1H7 and Fc were secreted at higher ratios (secretion/accumulation), whereas the secretion ratios of 1F12 and 5A3 were modest. By contrast, such ratios for 3F1, 1G4, and 1A4 were less than 1, clearly demonstrating the majority of expressed scFv-Fc proteins were retained inside cells. These results suggested that although 1F12 and 5A3 scFv-Fc proteins are secreted in COS-7 cells, 3F1, 1G4, and 1A4 scFv-Fc proteins seem to be retained inside the cells after synthesis.
FIGURE 6.

Transient expression of scFv-Fc proteins in COS-7 cells. Seventy six hours after lipofection with scFv-Fc genes encoding 3F1, 1F12, 5A3, 1G4, 1A4, or 1H7, Fc gene only, and the vector (mock), expression of various scFv-Fc and control Fc proteins in media (A) was examined by sandwich ELISA using anti-human IgG (Fc fragment-specific) antibody as described under “Experimental Procedures.” The scFv-Fc or Fc concentrations in media were quantified by using human Fc fragment (Jackson ImmunoResearch) as a standard. The concentrations estimated were then calculated as micrograms of scFv-Fc or Fc/106 cells. To measure intracellular levels of scFv-Fc proteins (B), COS-7 cells, transfected with scFv-Fc genes as above were sonicated three times on ice for 5 s as described under “Experimental Procedures.” Clear supernatants were subjected to sandwich ELISA to quantify scFv-Fc protein expression levels and then calculated as micrograms of scFv-Fc or Fc/106 cells as described above. Ratios of secreted and intracellular amounts of scFv-Fc or Fc proteins (A/B; Secretion/Accumulation) are shown at the bottom.
Next, COS-7 cells after transfection of pCI-neo/signal-scFv genes were subjected to immunofluorescence staining. Expression of scFv-Fc proteins in all five clones, 3F1, 1F12, 5A3, 1G4, and 1A4, after the gene transfer was visualized in red by immunostaining with Cy3-conjugated anti-human IgG antibody, as seen in Fig. 7. ER and Golgi apparatuses were stained in green as described under “Experimental Procedures.” The results clearly demonstrated that 3F1, 1G4, and 1A4 scFv-Fc proteins were in fact synthesized in ER, but most of the expressed scFv-Fc proteins are retained in the ER, although some may be transferred to the Golgi apparatuses, which prevents the eventual secretion of the synthesized proteins into the media. In contrast, 1F12 and 5A3 scFv-Fc proteins, as well as Fc protein, were found to be colocalized in both ER and Golgi apparatuses, which is expected because these proteins were secreted into the media by moving through the ER/Golgi pathway.
FIGURE 7.

Colocalization of scFv-Fc or Fc proteins with ER. Left panel, COS-7 cells transfected with scFv-Fc genes encoding 3F1, 1F12, 5A3, 1A4, 1G4, or Fc and cultured for 16 h were fixed. Fixed cells were stained with anti-PDI antibody and FITC-conjugated AffiniPure donkey anti-mouse IgG to detect ER (green). Expressed scFv-Fc or Fc proteins were stained with Cy3-conjugated anti-human IgG (red). Yellow (orange when red is stronger than green) areas in ER/Fc Merge represent colocalized signals. Fixed cells were also treated with DAPI to stain nuclei (blue). Right panel, costaining of scFv-Fc or Fc proteins and Golgi apparatus. COS-7 cells transfected with scFv-Fc genes encoding 3F1, 1F12, 5A3, 1A4, 1G4, or Fc and cultured for 16 h were fixed. Fixed cells were stained with BODIPY FL C5-ceramide to detect Golgi apparatuses (green). Expressed scFv-Fc or Fc proteins were stained with Cy3-conjugated anti-human IgG (red). Shown are the Golgi/Fc Merge only. Fixed cells were also treated with DAPI (1:1,000 dilutions) to stain nuclei (blue). Dilutions used for primary and secondary antibodies were anti-PDI (1:100), FITC-conjugated AffiniPure donkey anti-mouse IgG (1:100), BODIPY FL C5-ceramide (1:200), and Cy3-conjugated anti-human IgG (1:500). The white bar indicates 20 μm.
Detection of ER Stress-inducible Gene Expression by RT-PCR
The above data (Fig. 6) showed that 1F12, 5A3, and 1H7 scFv-Fc proteins were highly secreted into the medium, compared with those of 1A4, 1G4, or 3F1. If scFv-Fc proteins expressed were not correctly folded in the ER, thereby preventing their secretion, several chaperones should be induced in the ER via unfolded protein response (17). To determine the expression of such ER stress-induced proteins, real time PCR was performed using 1F12-, 3F1-, 5A3-, 1G4-, 1A4-, and 1H7-scFv-Fc-transfected COS-7 cells. Experiments were performed for three ER stress-inducible proteins in duplicate using GAPDH or β-actin as an endogenous control. Because both results were similar, only the results with GAPDH as a control are shown in Fig. 8. BiP is a major protein of the Hsp70 family. BiP, which is localized in the ER, binds to various nascent and newly synthesized proteins to assist their folding as a chaperone (18). This protein is dramatically up-regulated when ER stress is induced by expression of unfolded or misfolded proteins (19). The expression of BiP in 1F12-, 3F1-, 5A3-, 1G4-, 1A4-, and 1H7-scFv-Fc-transfected COS-7 cells were, however, almost the same as mock-transfected cells (Fig. 8). Calreticulin (CRT), which is also a chaperone with sugar binding activity, and EDEM, which participates in the ER-associated degradation, are also induced with ER stress (20, 21). Again, these proteins showed the same expression levels among 1F12-, 3F1-, 5A3-, 1G4-, 1A4-, and 1H7-scFv-Fc-transfected COS-7 cells (Fig. 8). These results indicated that none of these cells showed ER stress caused by unfolded protein response.
DISCUSSION
Phage display technologies are thought to be excellent strategies to achieve in vitro production of antibodies against carbohydrates that are self-antigens. The phage display strategy actually consists of two major components. Panning and screening of phage libraries (component 1) can result in isolation of genes encoding recombinant antibodies against any antigens of interest in vitro. The isolated genes then need to be expressed in prokaryotic or eukaryotic cells to produce antibody proteins for further characterization of the respective genes obtained (component 2). Unless in vitro translation systems are also used in component 2, anti-carbohydrate antibodies cannot be fully produced in vitro. This study reports on the isolation of anti-LNFPIII (Lex) scFv genes, which was achieved along with the isolation of previously reported anti-Man3 scFv genes (7, 8). During the expression and characterization of the anti-LNFPIII (Lex) 1F12 scFv-Fc protein, however, two major problems were experienced with this particular scFv-Fc. Unlike the initial purification and characterization of anti-Man3 5A3 scFv-Fc, the 1F12 scFv-Fc protein had relatively low binding activity, and the established cells had less production of the gene product in early passages.
Comparing the binding activity of the anti-LNFPIII (Lex) 1F12 scFv-Fc and anti-Man3 5A3 scFv-Fc indicated that 1F12 scFv-Fc had ∼1/10th binding activity of 5A3 scFv-Fc (Fig. 5A in this study and Zhang et al. (8), respectively). The low binding activity of 1F12 scFv-Fc that was observed may have been caused by the elution procedures, which could have inactivated 1F12 scFv-Fc in particular. This seems to be the case because milder elution conditions resulted in purification of 1F12 scFv-Fc protein with 10 times greater binding activity (Fig. 5B).
An extra N-glycosylation site was found in the deduced amino acid sequence of CDR2 in the VH domain of 1F12 scFv (Table 2A). Glycosidase F digestion of 5A3 and 1F12 scFv-Fc proteins followed by SDS-PAGE revealed the fact that both 5A3 and 1F12 scFv-Fc proteins are glycosylated and that the extra N-glycosylation site in the 1F12 scFv-Fc protein appeared to be glycosylated. To test the possible effect of this extra N-glycosylation in CDR2 of the VH domain of 1F12 scFv on binding activity, an aglycoform of the 1F12 scFv-Fc protein was produced and purified. The purified aglyco 1F12 scFv-Fc protein exhibited much lower activity than that of the control glycosylated protein. The low activity may have been caused by the lack of N-glycosylation in the complementarity-determining region. Introduction of a point mutation at the N-glycosylation site is needed to confirm this finding.
The major problem encountered during this study was the decline in 1F12 scFv-Fc protein production by the stable NS0 cell line during rather early passages (Tables 3, A and B). A similar situation was also observed with anti-Man3 5A3 scFv-Fc-expressing NS0 cells, i.e. when cells that had been frozen for 3 years were used for production of anti-Man3 5A3 scFv-Fc proteins, the 5A3 clone produced only 2% of the original expression level (Table 2). In contrast to these anti-carbohydrate scFv-Fc proteins, anti-insulin-like growth factor-I receptor 1H7 scFv-Fc expressed in NS0 cells did not cause such problems, as noted previously (14). In fact, over 400 mg of the 1H7 scFv-Fc protein were continuously purified for use in in vivo studies (14, 22, 23). A previous study (8) also found that neither stable CHO nor NS0 cells expressing 1A4 or 1G4 scFv-Fc proteins were established, much like with the 3F1 scFv-Fc as is described here. In addition, transiently expressed scFv-Fc proteins that were purified had higher binding activity than those purified from stably expressing NS0 cells (Fig. 5C). This result is consistent with the hypothesis that stable expression of anti-carbohydrate antibodies is harmful to cells, leading to less production of anti-carbohydrate antibodies in early passages. This collective evidence suggests that anti-LNFPIII (Lex) scFv and anti-Man3 scFv proteins expressed in NS0 or CHO cells might be harmful to the host cells, leading to suppression of gene expression and eventual cell death.
To examine the subcellular localization of various scFv-Fc proteins, scFv-Fc constructs were transiently expressed in COS-7 cells. Biochemical characterization and immunofluorescence staining of COS-7 cells after transfection of the genes revealed that 3F1, 1G4, and 1A4 scFv-Fc proteins were, in fact, synthesized in the ER of COS-7 cells but were never secreted into the media, whereas 1F12 and 5A3 scFv-Fc proteins were synthesized and then readily secreted (Figs. 6 and 7). In the case of anti-Man3 scFvs, the likelihood is that biosynthesized anti-Man3 antibodies bind to N-glycan of glycoprotein precursors that contain high mannose moieties in the ER. This could possibly explain why 1A4 and 1G4 scFv-Fc expressing NS0 or CHO cells have not been isolated. In contrast, the 5A3 scFv-Fc protein, which should have an affinity for high mannose, somehow escaped from its retention in the ER. The affinity and specificity of 5A3, 1A4, and 1G4 must be studied further to reveal differences in the specificity and affinity of 5A3 scFv from 1A4 and 1G4 scFvs, which should provide insights into their different destinies after translation. In the case of anti-LNFPIII (Lex) 3F1 scFv-Fc-expressing COS-7 cells, however, retention of scFv-Fc proteins in the ER cannot be readily explained because the LNFPIII (Lex) epitope is not present in COS-7 cells (24, 25). Detailed analyses of the specificity and affinity of 1F12 and 3F1 scFvs for other carbohydrates, including Man3, represent an important next step.
The present and previous results demonstrated that two distinct sets of anti-carbohydrate scFvs were obtained. The first set of clones, e.g. 5A3 and 1F12 scFv-Fcs, can be expressed in and secreted as scFv-Fc proteins from mammalian cells, whereas the second set of clones, which includes 1A4, 1G4, and 3F1 scFv-Fcs, is expressed in the ER but cannot be secreted because they are retained in the ER. The second set of clones were scFv-Fcs from which no stable clones have been derived, suggesting that they are more harmful, due to their retention in the ER, to the host cells than the former set of clones (5A3 and 1F12). In addition, as described above, NS0 cells stably expressing 5A3 or 1F12 scFv-Fc were also found to lose their productivity in rather early passages, and frozen cells failed to recover and produce scFv-Fc proteins. Thus, stable production of anti-carbohydrate antibodies is extremely difficult for even the 1F12 and 5A3 scFv-Fc constructs. Such problems were not associated with anti-insulin-like growth factor-I receptor scFv-Fc production in the NS0 cell system (14, 22, 23). The following questions now need to be answered. (i) Are anti-carbohydrate antibodies in general detrimental to mammalian cell survival? (ii) Is this harmfulness caused by their retention in ER?
With regard to answering the questions, the causes of cell death need to be studied further. Retention of N-glycoproteins in the ER is one possible mechanism for anti-Man3 scFvs. This fails to readily explain why anti-Lex 3A1 or 1F12 scFv causes cell death unless its cross-reactivity to high mannose present in the ER plays a critical role. Another possible mechanism is so-called ER stress (17, 26), which may be caused by overexpression of scFv-Fc proteins in the ER. It is possible that although those scFv-Fc proteins are expressed in the ER as evidenced by immunostaining with anti-human IgG antibody, those proteins were misfolded in the ER, causing ER stress. Such a problem may be intrinsic to “in vitro” antibody production. The results shown in Fig. 8, however, ruled out this possibility.
The specificity and affinity of these scFvs must be analyzed further to ascertain differences between the first and second sets of scFvs in terms of their retention and secretion in mammalian cells. Expression of scFv proteins in prokaryotes appears to be a reasonable way to accomplish this goal. Expression and preparation of properly folded scFv proteins is not an easy task because overexpression usually results in formation of inclusion bodies and because refolding scFvs in an active form is extremely difficult particularly for molecules such as scFvs that have to be refolded with correct disulfide bonding. Following published procedures (27), the authors recently succeeded in purifying refolded scFvs against Man3 and T-antigen with high binding activity with some modifications.5 The specificity and affinity of scFv proteins produced in E. coli and purified are currently being analyzed by SPR and NMR analyses.
Acknowledgments
The initial part of work was carried out using the Keio phage library. We thank Drs. Atsushi Takayanagi and Nobuyoshi Shimizu, Keio University Medical School for providing the phage library.
This work was supported in part by a grant from the Core Research for Evolutional Science and Technology (CREST) Project, Japan Science and Technology Agency (JST), and Grant 22570125 from the Japan Society for the Promotion of Science.
A. Takasaki-Matsumoto, T. Koyama, T. Katagiri, S. Yamazaki, N. Yuasa, and Y. Fujita-Yamaguchi, manuscript in preparation.
- Man3
- mannotriose [Manα1–6(Manα1–3)Man]
- ABTS
- 2,2′-azino-bis(3-ethylbenzthiazoline-6-sulfonic acid)
- BiP
- IgH chain-binding protein
- CRT
- calreticulin
- DPPE
- dipalmitoylphosphatidylethanolamine
- EDEM
- ER degradation enhancing α-mannosidase-like protein
- LNFPIII
- lacto-N-fucopentaose III (Galβ1–4(Fucα1–3)GlcNAcβ1–3Galβ1-4Glc)
- PDI
- protein-disulfide isomerase
- scFv
- single chain variable fragment
- neo
- neomycin-resistant gene
- VH
- variable region of antibody heavy chain
- F
- forward
- R
- reverse
- ER
- endoplasmic reticulum.
REFERENCES
- 1.Deng S. J., MacKenzie C. R., Sadowska J., Michniewicz J., Young N. M., Bundle D. R., Narang S. A. (1994) J. Biol. Chem. 269, 9533–9538 [PubMed] [Google Scholar]
- 2.MacKenzie C. R., Hirama T., Deng S. J., Bundle D. R., Narang S. A., Young N. M. (1996) J. Biol. Chem. 271, 1527–1533 [DOI] [PubMed] [Google Scholar]
- 3.van Kuppevelt T. H., Dennissen M. A., van Venrooij W. J., Hoet R. M., Veerkamp J. H. (1998) J. Biol. Chem. 273, 12960–12966 [DOI] [PubMed] [Google Scholar]
- 4.Mao S., Gao C., Lo C. H., Wirsching P., Wong C. H., Janda K. D. (1999) Proc. Natl. Acad. Sci. U.S.A. 96, 6953–6958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lee K. J., Mao S., Sun C., Gao C., Blixt O., Arrues S., Hom L. G., Kaufmann G. F., Hoffman T. Z., Coyle A. R., Paulson J., Felding-Habermann B., Janda K. D. (2002) J. Am. Chem. Soc. 124, 12439–12446 [DOI] [PubMed] [Google Scholar]
- 6.Ravn P., Danielczyk A., Jensen K. B., Kristensen P., Christensen P. A., Larsen M., Karsten U., Goletz S. (2004) J. Mol. Biol. 343, 985–996 [DOI] [PubMed] [Google Scholar]
- 7.Sakai K., Shimizu Y., Chiba T., Matsumoto-Takasaki A., Kusada Y., Zhang W., Nakata M., Kojima N., Toma K., Takayanagi A., Shimizu N., Fujita-Yamaguchi Y. (2007) Biochemistry 46, 253–262 [DOI] [PubMed] [Google Scholar]
- 8.Zhang W., Matsumoto-Takasaki A., Kusada Y., Sakaue H., Sakai K., Nakata M., Fujita-Yamaguchi Y. (2007) Biochemistry 46, 263–270 [DOI] [PubMed] [Google Scholar]
- 9.Stoll M. S., Mizuochi T., Childs R. A., Feizi T. (1988) Biochem. J. 256, 661–664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shimizu Y., Nakata M., Matsunuma J., Mizuochi T. (2001) J. Chromatogr. B. Biomed. Sci. Appl. 754, 127–133 [DOI] [PubMed] [Google Scholar]
- 11.Sblattero D., Bradbury A. (2000) Nat. Biotechnol. 18, 75–80 [DOI] [PubMed] [Google Scholar]
- 12.Neumaier M., Shively L., Chen F. S., Gaida F. J., Ilgen C., Paxton R. J., Shively J. E., Riggs A. D. (1990) Cancer Res. 50, 2128–2134 [PubMed] [Google Scholar]
- 13.Wu A. M., Chen W., Raubitschek A., Williams L. E., Neumaier M., Fischer R., Hu S. Z., Odom-Maryon T., Wong J. Y., Shively J. E. (1996) Immunotechnology 2, 21–36 [DOI] [PubMed] [Google Scholar]
- 14.Li S. L., Liang S. J., Guo N., Wu A. M., Fujita-Yamaguchi Y. (2000) Cancer Immunol. Immunother. 49, 243–252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wu A. M., Tan G. J., Sherman M. A., Clarke P., Olafsen T., Forman S. J., Raubitschek A. A. (2001) Protein Eng. 14, 1025–1033 [DOI] [PubMed] [Google Scholar]
- 16.Ono K., Kamihira M., Kuga Y., Matsumoto H., Hotta A., Itoh T., Nishijima K., Nakamura N., Matsuda H., Iijima S. (2003) J. Biosci. Bioeng. 95, 231–238 [PubMed] [Google Scholar]
- 17.Ellgaard L., Helenius A. (2003) Nat. Rev. Mol. Cell Biol. 4, 181–191 [DOI] [PubMed] [Google Scholar]
- 18.Haas I. G. (1994) Experientia 50, 1012–1020 [DOI] [PubMed] [Google Scholar]
- 19.Gething M. J. (1999) Semin. Cell Dev. Biol. 10, 465–472 [DOI] [PubMed] [Google Scholar]
- 20.Mintz M., Vanderver A., Brown K. J., Lin J., Wang Z., Kaneski C., Schiffmann R., Nagaraju K., Hoffman E. P., Hathout Y. (2008) J. Proteome Res. 7, 2435–2444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hosokawa N., Wada I., Hasegawa K., Yorihuzi T., Tremblay L. O., Herscovics A., Nagata K. (2001) EMBO Rep. 2, 415–422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ye J. J., Liang S. J., Guo N., Li S. L., Wu A. M., Giannini S., Sachdev D., Yee D., Brünner N., Ikle D., Fujita-Yamaguchi Y. (2003) Horm. Metab. Res. 35, 836–842 [DOI] [PubMed] [Google Scholar]
- 23.Sachdev D., Li S. L., Hartell J. S., Fujita-Yamaguchi Y., Miller J. S., Yee D. (2003) Cancer Res. 63, 627–635 [PubMed] [Google Scholar]
- 24.Koda Y., Kimura H., Mekada E. (1993) Blood 9, 2915–2919 [PubMed] [Google Scholar]
- 25.Gersten K. M., Natsuka S., Trinchera M., Petryniak B., Kelly R. J., Hiraiwa N., Jenkins N. A., Gilbert D. J., Copeland N. G., Lowe J. B. (1995) J. Biol. Chem. 270, 25047–25056 [DOI] [PubMed] [Google Scholar]
- 26.Fribley A., Zhang K., Kaufman R. J. (2009) Methods Mol. Biol. 559, 191–204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Umetsu M., Tsumoto K., Hara M., Ashish K., Goda S., Adschiri T., Kumagai I. (2003) J. Biol. Chem. 278, 8979–8987 [DOI] [PubMed] [Google Scholar]
- 28.Kabat E. A., Wu T. T., Perry H., Gottesman K., Foeller C. (1991) Sequences of Proteins of Immunological Interest, 5th Ed., Vol. I, pp. 103–140 and 310–324, National Institutes of Health Publication No. 91-3242 National Institutes of Health, Bethesda [Google Scholar]