

Abstract
Steinernema carpocapsae is an insect parasitic nematode used in biological control, which infects insects penetrating by mouth and anus and invading the hemocoelium through the midgut wall. Invasion has been described as a key factor in nematode virulence and suggested to be mediated by proteases. A serine protease cDNA from the parasitic stage was sequenced (sc-sp-1); the recombinant protein was produced in an Escherichia coli system, and a native protein was purified from the secreted products. Both proteins were confirmed by mass spectrometry to be encoded by the sc-sp-1 gene. Sc-SP-1 has a pI of 8.7, a molecular mass of 27.3 kDa, a catalytic efficiency of 22.2 × 104 s−1 m−1 against N-succinyl-Ala-Ala-Pro-Phe-pNA, and is inhibited by chymostatin (IC 0.07) and PMSF (IC 0.73). Sc-SP-1 belongs to the chymotrypsin family, based on sequence and biochemical analysis. Only the nematode parasitic stage expressed sc-sp-1. These nematodes in the midgut lumen, prepared to invade the insect hemocoelium, expressed higher levels than those already in the hemocoelium. Moreover, parasitic nematode sense insect peritrophic membrane and hemolymph more quickly than they do other tissues, which initiates sc-sp-1 expression. Ex vivo, Sc-SP-1 was able to bind to insect midgut epithelium and to cause cell detachment from basal lamina. In vitro, Sc-SP-1 formed holes in an artificial membrane model (Matrigel), whereas Sc-SP-1 treated with PMSF did not, very likely because it hydrolyzes matrix glycoproteins. These findings highlight the S. carpocapsae-invasive process that is a key step in the parasitism thus opening new perspectives for improving nematode virulence to use in biological control.
Keywords: Basal Lamina, Enzyme Purification, Gene Expression, Parasite, Serine Protease, Nematode, Secreted/Excreted Products
Introduction
The nematode Steinernema carpocapsae is an obligate parasite of insects that forms a symbiotic association with the bacteria Xenorhabdus nematophila (1). This complex is able to parasitize a large number of insects that have significant economic impact, which emphasizes its use as a biological control agent (2, 3). Despite widespread use, the efficiency is quite variable among strains, mostly due to the ability of the infective stage to resist environmental conditions and the differential ability of each strain to parasitize insects successfully. The effectiveness of these nematodes has been improved by the search for more efficient strains, by selective inbreeding and by the construction of genetically modified organisms (4). So far, improvement based on a genetic modification was achieved in Steinernema feltiae (5) using a stress-resistant gene from yeast, due to the lack of virulence genes identified in steinernematids.
The nematode infective juvenile (IJ)6 is a resistant third juvenile that is free in the soil and seeks a suitable host in response to several cues, such as insect chemical and thermal signals (6). IJs enter the host through natural openings, mostly the mouth and anus, and cross the midgut to become established in the hemocoelium. The parasitic process may require the expression of novel genes encoding putative parasitic effectors (7). The expressed proteins would facilitate parasite penetration into the host hemocoelium and counteract insect defenses (8). The ability of the parasitic stage to surmount host barriers and colonize the insect hemocoelium has been considered mandatory for the success of this parasite (9). Experimental assays showed that IJs of virulent strains of S. carpocapsae are able to colonize about 50% of exposed Galleria mellonella larvae in as little as 6 h, and in a 12-h post-exposure, all insects have nematodes in their hemocoelium (10). Another relevant observation is that excreted/secreted products (ESP) from highly virulent strains contain larger amounts of proteases than those from less virulent strains (11). Taken together, these findings suggest that S. carpocapsae produces virulence factors containing proteases.
Proteolytic activity has been detected in the ESP of numerous human, animal, and plant parasitic nematodes. It has been assumed that proteases actively promote penetration of host barriers, thereby facilitating colonization (12). Serine proteases are among the most abundant proteases in the nematode ESP (13) and have been detected in the ESP of Schistosoma mansoni, Anisakis simplex, Onchocerca volvulus, and Trichinella spiralis (14–18). The presence of serine proteases in S. carpocapsae ESP was evidenced by the inhibition of proteolytic activity after treatment with specific serine protease inhibitors (11). Therefore, we decided to search for genes encoding serine proteases expressed during the nematode parasitic phase. In this study, we describe the cloning and sequencing of one such gene. We show that this gene is expressed only at the parasitic stage and that the encoded protease is present in ESP of the nematode parasitic stage. This protease hydrolyzes proteins in the basal lamina, disrupting the host barriers, thus suggesting it participates in the parasite-host interaction, helping invasion.
EXPERIMENTAL PROCEDURES
Nematode in Parasitic Phase
S. carpocapsae Breton strain was produced as reported previously (19). IJs were stored in tap water at 10 °C for 2–4 months for use in assays. The parasitic stage was obtained in vivo in parasitized larvae or in vitro by inducing recovery of IJs with 10% insect tissue homogenate (20).
Full-length cDNA Cloning of sc-sp-1
Parasitic stages were ground to a powder in liquid nitrogen, and total RNA was isolated with TRIzol (Invitrogen) and treated with RNase-free DNase (Roche Applied Science). First strand cDNAs were generated using the SuperScriptTM first strand kit (Invitrogen) following the manufacturer's instructions. PCR amplifications were done using the degenerate primer 5′-TIACNGCIGCICAYTGYKT-3′ (K = G or T; I = inosine; N = A, C, G or T; Y = C or T), based on conserved motifs present near the catalytic histidine of serine proteases, and the oligo(dT) anchor primer, by means of a touchdown PCR (20). The same anchor primer used for reverse transcription and the antisense primers 5′-GGCGTTTCCAGTAGCTTGGGCAAAGAAG-3′ and 5′-CCACACCTTTCCCGTCCTGAGCAACAAG-3′ were used for the first and second rounds of a nested PCR, respectively. Both PCR rounds were as follows: 94 °C for 5 min; 5 cycles of 94 °C for 5 s and 72 °C for 90 s; 5 cycles of 94 °C for 5 s; 70 °C for 10 s; 72 °C for 90 s; 30 cycles of 94 °C for 5 s, 68 °C for 10 s; and 72 °C for 90 s. Amplified cDNA fragments were TA-cloned into the vector pCR4-TOPO using the TOPO TA cloning kit (Invitrogen) and sequenced by Stabvida (Oeiras, Portugal).
Isolation of the sc-sp-1 Genomic Clone
IJs were suspended in lysis buffer (100 mm Tris-HCl, pH 8.5, 100 mm NaCl, 50 mm EDTA, 1% SDS, 1% 2-mercaptoethanol, and 100 μg/ml proteinase K), frozen for 1 h at −80 °C, then thawed, and incubated for 1 h at 65 °C. Genomic DNA was then extracted using the phenol/chloroform method (21). The sense 5′-ATGGGTCTGCTGCTTCTGGCTTTC-3′ and antisense 5′-TTAAGAGCAAGAGAAGGCGTTTCC-3′ primers were used to amplify a genomic sequence from the initial ATG to the stop codon. The PCR amplification was performed as described above and as follows: 94 °C for 5 min; 5 cycles of 94 °C for 30 s, 66 °C for 30 s, and 72 °C for 60 s; 5 cycles of 94 °C for 30 s, 63 °C for 30 s, and 72 °C for 60 s; 25 cycles of 94 °C for 15 s; 60 °C for 15 s, and 72 °C for 60 s. The amplified product was cloned and sequenced as above.
cDNA Sequence Analysis
The predicted amino acid sequence corresponding to the mature protein was used as a query in searches of GenBankTM at the National Center for Biotechnology Information (www.ncbi.nlm.nih), the Caenorhabditis elegans genome at WormBase, and the MEROPS protease data base at the Wellcome Trust Sanger Institute. Predictions of the signal sequence cleavage site and subcellular localization were performed using the SignalP and TargetP software, respectively (22). The sequence alignment was generated using ClustalW2 (23). SMART6 sequence analysis software (24) was used to predict PFAM domains in the protein sequence.
Northern Blot Analysis
IJs were incubated for 6 and 12 h with 10% (v/v) G. mellonella larvae homogenate to induce recovery. Nematodes were then separated, and total RNA was extracted. A total of 1 μg of RNA was size-fractionated on formaldehyde-denaturing agarose gel and then transferred to a positively charged nylon membrane (21). A 490-bp RNA probe was generated by in vitro transcription using the digoxigenin RNA labeling kit (Roche Applied Science) from a cDNA fragment amplified with the sense primer 5′-TGTCGGCCACAAACTAGGAGAGTCCAG-3′ and the antisense primer 5′-CCACACCTTTCCCGTCCTGAGCAACAAG-3′. The cDNA fragment was TA-cloned into the pGEM-T Easy vector (Promega) and linearized with PstI. Hybridization and washing steps at high stringency were done at 65 °C. Detection was carried out as described by the manufacturer's protocol.
Southern Blot Analysis
IJ genomic DNA (2.5 μg) was digested separately with HindIII, SalI, and XhoI, separated by electrophoresis, and transferred to a nylon membrane. The probe was generated by amplification of the 490-bp insert in the pGEM-T Easy vector (Promega) with M13 forward and reverse primers and labeled using a digoxigenin DNA labeling kit (Roche Applied Science). Hybridization and washing steps were carried out as above.
In Situ Hybridization of sc-sp-1
For mRNA in situ hybridization analysis, IJs were induced to recover for 12 h as described previously. The cDNA was extracted and amplified as described above and was digested with BamHI to give a 256-bp fragment; an RNA probe was then generated by in vitro transcription using the digoxigenin RNA labeling kit (Roche Applied Science), and whole-mount in situ hybridization was performed (25).
Expression Analysis of sc-sp-1
The expression of sc-sp-1 was investigated throughout the nematode life cycle developing in parasitized insects, using quantitative real time PCR (qRT-PCR). G. mellonella larvae parasitized with nematodes were dissected after chilling to harvest nematodes at each life cycle stage. In addition, to analyze the influence of insect tissue on sc-sp-1 expression, IJs were induced to recover with homogenates of G. mellonella larvae and, separately, with insect midgut, peritrophic membrane, fat bodies, and hemolymph. Insect tissues were excised by dissection of chilled insects. IJ recovery was performed in Tyrode's solution supplemented with homogenates of each insect tissue as described above. Ten nematodes per experimental situation, repeated at least three times, were used to extract RNA using an RNeasy micro kit (Qiagen). Reverse transcription was performed using Superscript First Strand III (Invitrogen). qRT-PCR was performed using SYBR Green master mix in an AB 7900 thermocycler (Applied Biosystem) with specific primers for sc-sp-1 (sense 5′-TGTTGCTCAGGACGGGAAAG-3′ and antisense 5′-CACAGAAGTAGGACGTGCGAAG-3′) and for endogenous control 18 S RNA (sense 5′-AAACGAAAGTCTTCCGGTTCC-3′ and antisense 5′-GGGTGAGTTTTCCCGTGTTG-3′). Differences in expression levels were tested for statistical significance using analysis of variance with a Tukey post hoc test using SPSS 15.0 software. The level of significance was set at p < 0.05, and the data are reported as means ± S.E. for all groups.
Native Sc-SP-1 Purification
ESP was obtained from the nematode parasitic stage induced with insect tissue homogenate and concentrated (20) and then fractionated using an Akta chromatography system (GE Healthcare) at 4 °C. Tree milligrams of ESP were loaded onto a gel filtration Superdex 200 column (Amersham Biosciences) equilibrated with 50 mm PB, pH 6.5, and proteins were eluted with a flow rate of 0.5 ml/min. Fractions were checked for activity by hydrolysis of the chromogenic peptide substrate N-succinyl-Ala-Ala-Pro-Phe-pNA (AAPF-pNA). The active fractions were pooled, applied to a 1-ml HiTrap S column (Amersham Biosciences) equilibrated with 50 mm PB, pH 6.5, and eluted with a linear gradient of 1 m NaCl. Fractions containing enzymatic activity were pooled, desalted using a Centricon 3K (Millipore) and applied to a Mono S 5/5 column (Amersham Biosciences), equilibrated, and eluted as in the HiTrap S conditions. The soluble protein concentration was spectrophotometrically determined at all purification steps using the BCA quantification kit (Pierce) and bovine serum albumin as standard. Fractions were collected and subsequently checked for purity by SDS-PAGE.
Expression and Purification of Recombinant Protein
A cDNA coding for the mature form of sc-sp-1 was PCR-amplified using the forward NdeI 5′-CATATGGTTTTGGGAGGTACCGAAGTC-3′ and reverse EcoRI 5′-GAATTCTTAAGAGCAAGAGAAGGCGTTTC-3′ primers and then cloned into the NdeI/EcoRI sites (underlined in primers) of the pET23a vector (Novagen) for expression in Escherichia coli strain BL21 (DE3). Cells grown in YT medium at 37 °C at A600 nm = 0.6 were induced by the addition of 1 mm IPTG and incubated for 3 h at 30 °C. Sc-SP-1 was obtained in inclusion bodies according to Ref. 26 with minor modifications. The cells were centrifuged; the pellet was resuspended in 60 ml of 50 mm Tris, pH 8.0, 5 mm EDTA, 6 mg of lysozyme, and incubated at 30 °C for 30 min. The solution was frozen and thawed, and 0.6 mg of deoxyribonuclease I and 100 mm MgCl2 was added and incubated at 4 °C for 1 h. Protein refolding was performed according to Ref. 27. Proteins were diluted into 1 liter of 50 mm Tris-HCl, 150 mm NaCl, 5 mm EDTA, 2 m urea, 1.25 mm reduced glutathione, 0.25 mm oxidized glutathione, pH 11, with stirring at 4 °C for 16 h and then desalted against 20 mm Tris-HCl, pH 7.2, 0.4 m urea at 4 °C for 16 h. Dialyzed supernatants were purified with HiTrap S column (Amersham Biosciences) as described for native protein. Purified recombinant Sc-SP-1 was collected and subsequently checked for purity by SDS-PAGE and activity by hydrolysis of the AAPF-pNA substrate. Authenticity was validated by N-terminal sequencing and MALDI-TOF/TOF analysis.
Antiserum Production
Polyclonal antibodies (>90% purity) were produced by immunization of a rabbit with gel slices of purified Sc-SP-1 at Genosphere Biotechnologies Co. (Paris, France).
SDS-PAGE, Zymogram, and Western Blot
SDS-PAGE was performed with a Mini-PROTEAN II gel system (Bio-Rad), using 12% polyacrylamide (28). Proteins were stained with colloidal Coomassie (29). The SDS-polyacrylamide gelatin zymogram was performed as described previously (30). To perform the Western blot, purified protein in SDS-polyacrylamide gels was electroblotted onto polyvinylidene difluoride membranes using a mini Trans-Blot Cell (Bio-Rad) (31). Membranes were then blocked in TBS (0.01 m Tris-HCl, pH 7.5, 0.1 m NaCl) containing 5% (w/v) BSA and 0.05% (v/v) Tween 20 for 30 min at room temperature and then incubated with rabbit Sc-SP-1 primary antibody (1:5,000 dilution) in blocking solution for 2 h at room temperature. Membranes were washed three times for 10 min in TBS and incubated with goat anti-rabbit IgG peroxidase conjugates (Sigma) diluted 1:4,000 in TBS. Antibodies were detected by incubation with tetramethylbenzidine peroxidase substrate (Sigma).
Two-dimensional Electrophoresis and N-terminal Amino Acid Sequence
Purified protein was resuspended in rehydration buffer (9.8 m urea, 4% (w/v) CHAPS, 2 mm tributylphosphine, 1% (v/v) ampholytes, pH 3–10) and applied to a 7-cm IPG strip (Bio-Rad). After active rehydration for 12 h at 20 °C, proteins were focused in a Protein IEF cell (Bio-Rad). Prior to the second dimension, the IPG was equilibrated with 130 mm DTT and 135 mm iodoacetamide. SDS-PAGE and gel staining were performed as above. The purified protein spot was electroblotted onto PVDF membranes (Millipore) for N-terminal amino acid sequencing, performed by automated Edman degradation at the Emory Microchemical Facility (Atlanta), and the peptide sequence was manually matched with the Sc-SP-1 sequence.
Mass Spectrometry Analysis
The polyacrylamide electrophoresis protein spot was digested in the gel (20). The peptide mixture was purified and concentrated by R2 pore microcolumns (32) and eluted directly to the MALDI plate with 0.5 μl of matrix α-cyano-4-hydroxycinnamic acid (5 mg/μl in 50% acetonitrile, 5% formic acid). The m/z spectra were acquired in a 4700 Proteomics Analyzer MALDI-TOF/TOF (Applied Biosystems) in both MS and MS/MS mode (33). Protein identification was achieved using a MASCOT search of the UniProtKB data base (downloaded 07/07/2009).
Enzymatic Assays
Endopeptidase activity against p-nitroanilide (p-NA) substrates (Sigma) was measured in reactions of 100 μl of 50 mm Tris-HCl, pH 8.0, containing 1 mm of substrate at 25 °C. p-NA released was measured in microtiter plates at 405 nm using an automated microtiter plate spectrophotometer (Bio-Rad). One unit of activity represented the hydrolysis of 1 μmol of p-NA/min under the assay conditions. The activity of 45 μm of the purified Sc-SP-1 over a range of 0.02–1 mm of AAPF-pNA was used to calculate kinetic constants. Rate constant, Vmax, and Michaelis constant, Km, were obtained by linear regression analysis of Lineweaver-Burk plot using Hyper32 software available on line. Experiments were performed in triplicate, and the kcat and kcat/Km values were calculated. Inhibition studies, the effect of temperature (from 15 to 65 °C), and optimal pH activity of purified protease Sc-SP-1 were carried out with 100 μm Sc-SP-1 and AAPF-pNA as substrate. The effect of pH on the activity was evaluated by measuring enzymatic activity at pH 6–10, with the following buffers: 100 mm PB, pH 6, 100 mm Tris-HCl, pH 7–9, and 100 mm glycine-NaOH, pH 10. For inhibition studies, Sc-SP-1 was incubated for 5 min with different protease-class selective inhibitors in different concentration ranges. Linear regression, using Probit 1.4 software (34), was performed to determine the 50% inhibition (IC50). Inhibition of enzyme activity was also tested in the presence of 100 mm DTT and 100 mm of 2-mercaptoethanol cysteine-reducing agents.
Histology and Binding Assays
Ex vivo assays in midguts from 4th instar larvae of G. mellonella were performed to analyze the effect of Sc-SP-1. Midguts of larvae were excised, sectioned transversely in 5-mm pieces, transferred to Hanks' balanced salt solution (Invitrogen) with 30 μg/ml purified Sc-SP-1, and incubated for 3 h at room temperature. For microscopic observations, midgut pieces were fixed in Carnoy's fixative and embedded in paraffin. Serial sections 7 μm thick were stained with Mayer's hematoxylin and eosin (35).
For binding assays, the midgut of insect larvae were fixed, sectioned, and mounted as above. Twenty micrograms of recombinant Sc-SP-1 was applied to the top of each section and incubated for 4 h at 4 °C. Detection of protein binding was performed according to Joshi et al. (36), with slight modifications. Incubation with rabbit anti-Sc-SP-1, 1:1,000 in TBS, was done for 16 h at 4 °C, followed by incubation with goat anti-rabbit peroxidase conjugate (Sigma) at 1:500 in TBS for 2 h at 4 °C and developed with the Sigma-Fast substrate (Sigma).
Degradation of Matrix Proteins
Basement membrane Matrigel (BD Biosciences) was used to mimic the basal membrane and to test the disruptive ability of Sc-SP-1, according to the manufacturer's protocol. Five μl of Sepharose S200 beads were soaked in 100 μm Sc-SP-1, in 100 μm Sc-SP-1 after incubation with 0.8 mm, and with 2 mm PMSF for 30 min. The control was performed under the same conditions, using beads soaked with α-chymotrypsin from bovine pancreas (Sigma). One microliter of beads was applied at the top of the membrane and incubated for 3 h at room temperature. The membrane was fixed with 5% (v/v) formaldehyde in PBS followed by dehydration with graded ethanol (50, 75, and 100% (v/v)) and coated with gold/palladium in a vacuum evaporator (JEOL JEE-400). Micrographs were obtained with a scanning electron microscope (JEOL JSM-5410).
Sc-SP-1 proteolytic activity was tested against the extracellular proteins collagen type IV, laminin, and fibronectin (Sigma). A total of 5 μg of each protein was incubated with 100 μm Sc-SP-1 at 15 °C for 20 min for laminin and fibronectin and for 20 min and 1 h for collagen. Controls were performed using purified Sc-SP-1 incubated with PMSF as above. Digestion profiles were analyzed by 8% SDS-PAGE.
RESULTS
cDNA and Genome Cloning
A degenerate RT-PCR strategy was designed to isolate cDNAs coding for serine proteases in the parasitic stage of the entomopathogenic nematode S. carpocapsae. The clone sc-sp-1 was initially amplified from cDNA as a 735-bp cDNA fragment and then extended to a full-length cDNA fragment of 980 bp, including an open reading frame of 831 bp. The 3′-untranslated region consisted of 83 nucleotides, including a stop codon (TAA), a single putative polyadenylation signal (AATAAA), and a poly(A) tail. The final products of the 5′-rapid amplification of cDNA ends encompassed the initial Met and a 5′-untranslated region of 66 nucleotides.
PCR primers flanking the 5′ and 3′ ends of the sc-sp-1 open reading frame were used to amplify a single DNA fragment of 1,169 bp corresponding to the genomic clone (Fig. 1). A six exon/five intron structure was identified, and all five introns follow the GT-AG rule (37). The intron sizes range from 43 to 46 nucleotides with the exception of the second intron, which has 159 nucleotides. The first intron in the sc-sp-1 genomic sequence separates the signal peptide and propeptide from the catalytic domain coding sequences. The four other introns split the mature active enzyme coding sequence. The three amino acids comprising the charge relay system are located on two different exons as follows: His57 and Asp102 in exon 2 and Ser195 in exon 5. Introns 3 and 5 shifted two codons corresponding to Phe146 and Asp226 in the 5′ direction without changing the open reading frame after splicing. Southern blotting was consistent with a single copy gene in this organism (Fig. 2).
FIGURE 1.

Gene, full-length cDNA, and predicted amino acid sequences of sc-sp-1. Nucleotide sequences are shown for the six exons (uppercase) and adjoining intron regions (lowercase). The stop codon is indicated by an asterisk, and the putative polyadenylation site is underlined. The signal peptide is boxed, and the propeptide is boxed and in boldface. The residues of the catalytic triad are circled.
FIGURE 2.

Southern blot analysis of sc-sp-1. Genomic DNA from S. carpocapsae was digested with HindIII, SalI, or XhoI and probed with a digoxigenin-labeled cDNA fragment. SalI and XhoI do not cleave the cDNA, but HindIII cleaves the cDNA at position 698. DNA size markers are indicated in kb.
Analysis of the Predicted Amino Acid Sequence
Comparison of the predicted amino acid sequence from the open reading frame with protein sequences in general databases unambiguously identified Sc-SP-1 as a member of the peptidase family S1; the chymotrypsin family contains the catalytic triad His, Asp, and Ser found in all members of subclan PA(S). The mature protein consists of a 253-amino acid polypeptide with a predicted molecular mass of 27.3 kDa and an isoelectric point of 8.68. sc-sp-1 contains a predicted signal peptide of 15 residues, a short 7-residue propeptide, and a single trypsin domain with a catalytic triad His57/Asp102/Ser195 (Fig. 1). By homology with other serine protease sequences, amino acid residues that appear to contribute to protease specificity for P1 hydrophobic residues were identified as Gly189, Gly216, and Asp226 (38). Six cysteines were identified in the primary sequence, two of them being unusually located at the C-terminal extension of the protease domain, whereas the highly conserved Cys191 and Cys220, corresponding to the third pair, were absent (39). Another domain between positions 32 and 140 of the mature protein was predicted to have a bulb-type mannose-specific lectin (B_lectin domain) homology. This domain contains a 3-fold internal repeat and the consensus sequence motif QXDXNXVXY, and it is involved in α-d-mannose recognition. ClustalW2 analysis showed that the Sc-SP-1 sequence has 31, 23, 22, and 20% identity with serine protease sequences from the parasitic nematodes Brugia malayi, Trichinella spiralis, Heterodera glycines, and Gnathostoma spinigerum, respectively (see supplemental Fig. S1).
sc-sp-1 Expression in the Parasitic Stage
The expression of sc-sp-1 was tested by Northern blot analysis using an antisense-specific RNA probe from the cloned cDNA, labeled with digoxigenin. sc-sp-1 mRNA was detected in L3-induced nematodes but not in uninduced nematodes (Fig. 3A). Expression of the sc-sp-1 gene analyzed by in situ hybridization was only detected in induced nematodes and in cells localized in the posterior part of the esophageal bulb (Fig. 3B). Following this observation, the expression of sc-sp-1 was checked at every stage of the nematode life cycle by qRT-PCR. This analysis showed that sc-sp-1 was highly expressed in nematode parasitic stages, particularly in parasitic stages inside the midgut (L3-mg) and at lower levels in those inside the hemocoelium (L3-hm). The fourth larval stage (L4) expressed much lower amounts than the previous stage, decreasing to insignificant amounts in adults and first and second larval stages (L1 and L2) (p < 0.05). Free living resistant stages (IJ) did not express sc-sp-1 at all (Fig. 4, A and B). To investigate how the expression of this gene is related to the host, the effects of insect tissues on the expression of sc-sp-1 was tested by qRT-PCR. Although all tissues tested were able to induce sc-sp-1 expression, the induction over time was quite different. In fact, at the parasitic stage, significantly (p = 0.03) higher amounts of expressed sc-sp-1 were obtained after 3 h of induction with hemolymph and peritrophic membrane in comparison with fat bodies, gut epithelium, and total insect homogenate (Fig. 2C). However, after 6 h of induction, no significant differences (p > 0.12) were detected in the level of expression in nematodes induced with the different tissues. The differential expression of sc-sp-1 suggests that nematodes sense insect tissues at different time points.
FIGURE 3.
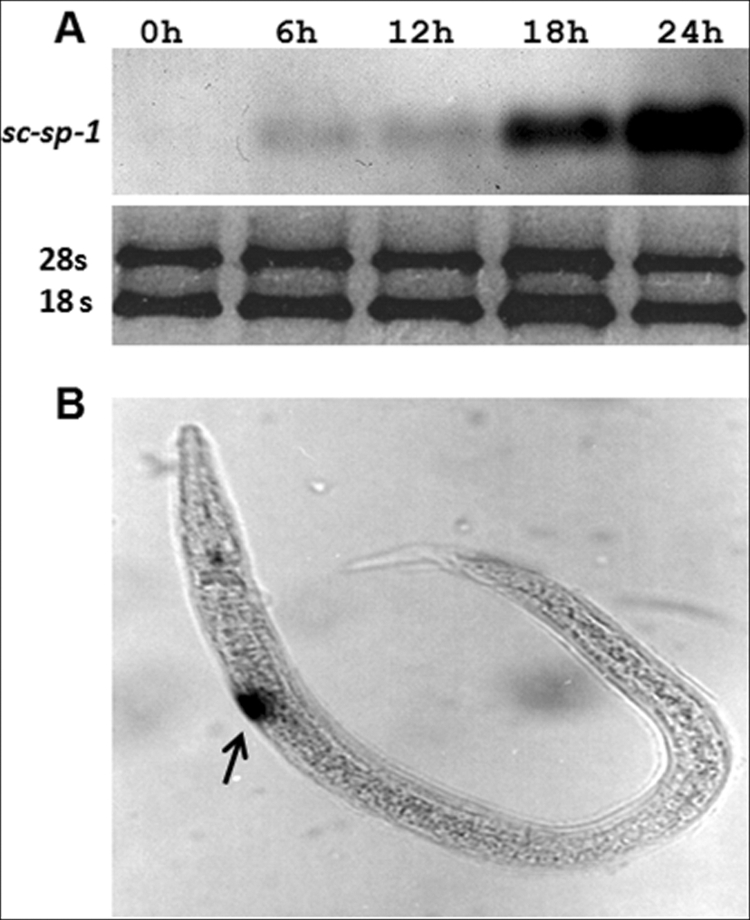
Detection of expression and in situ hybridization of sc-sp-1 gene. The sc-sp-1 mRNA was detected using a digoxigenin-labeled RNA probe with total RNA of noninduced (0 h) and induced nematodes for 6, 12, 18, and 24 h. The loaded RNA was normalized using 28 S and 18 S rRNAs (A). The same probe was used to localize sc-sp-1 expression in induced nematodes (B). Arrow indicates location of expression.
FIGURE 4.

Analyses of sc-sp-1 gene expression during nematode life cycle and after the induction with insect tissues. The diagram represents the free living juvenile (black box), the recovery (L3-mg), and invasive process through midgut wall to hemocoelium (L3-hm) and the parasite developmental stages inside insect hemocoelium (A). sc-sp-1 expression was quantified by real time PCR in free living nematode and in the successive stages of nematodes in the parasitic phase (B). The expressions of sc-sp-1 in free living stage induced to recovery with different tissue homogenates of G. mellonella larvae were also determined (C). Columns represent a mean of three measures. Values in columns with asterisk are significantly different (p < 0.05). Bars represent S.E. IJ, free living stage; L1–L4, nematode larval stages; fb, fat bodies; h, insect homogenates; hm, hemolymph; mg, midgut; mp, peritrophic membrane.
Recombinant Protein
A full-length cDNA of sc-sp-1 was cloned and expressed in E. coli. Bacterial transformation was confirmed by PCR. After refolding, the recombinant protein was purified and analyzed for validation. The N-terminal sequence obtained by Edman degradation, VLGGTEVPVGKYPF, was completely identical to the deduced sequence of Sc-SP-1, and MALDI-TOF/TOF analysis resulted in a successful identification with 12 peptides matching Sc-sp-1 (10 with MS/MS spectra) corresponding to 54% sequence coverage and a significant score of 768 (p < 0.05) (see supplemental Table S1). Moreover, the molecular mass of the recombinant protein agrees with the expected value of 27.3 kDa.
Native Protein
Proteolytic activity was detected in ESPs from the nematode parasitic stage induced with insect homogenate. To purify the detected protease, 100 ml of ESP were concentrated and used in three successive chromatographies, and the chromatographic fractions were checked by hydrolysis of the AAPF-pNA substrate (see supplemental Fig. S2). ESP sample with 224 units/mg of activity was applied onto Superdex 200 column, and a peak with a calculated molecular mass around 30 kDa and 495 units/mg of activity was collected. This peak was applied to a HiTrap SP column and a peak with 1957 units/mg of activity was recovered in fractions eluted with 0.25 m NaCl. This fraction was desalted and loaded onto a Mono S column, and a peak with 3416 units/mg of activity was eluted at 0.35 m NaCl (Table 1). This fraction was shown to correspond to a single protein in SDS-PAGE and to a single band of hydrolysis in the zymogram, both with a calculated molecular mass of around 27 kDa. The same molecular weight and a pI around 8.7 were calculated for the single spot visualized by two-dimensional electrophoresis (Fig. 5, A–C). Purified native protein analyzed by N-terminal sequencing gave rise to the amino acid sequence VLGGTEVPVGKYPFFVR, which corresponds to the deduced amino acids from the 5′ sc-sp-1 mature sequence. The analysis by MALDI-TOF/TOF yields 11 peptides that match Sc-SP-1, 9 with MS/MS spectra, corresponding to 51% sequence coverage and a significant score of 708 (p < 0.05). Moreover, pure native protein was recognized by antibodies raised against recombinant Sc-SP-1 (Fig. 5D). These findings support the statement that the purified protease is encoded by the sc-sp-1 cDNA and that Sc-SP-1 is produced in ESP of the nematode parasitic stage. An intact mass of 27,313.94 Da was determined by mass spectrometry (Fig. 5E), in agreement with the predicted value.
TABLE 1.
Summary of Sc-SP-1 secreted products purification
AAPF-pNA was used for enzyme quantification.
| Purification steps | Total volume | Total protein | Total activitya | Specific activityb | Purification factor |
|---|---|---|---|---|---|
| μl | mg | units | units/mg | ||
| ESPsc | 500 | 3.1 | 693 | 224 | |
| Superdex 200 | 500 | 0.6 | 297 | 495 | 2.2 |
| HiTrap SP | 500 | 0.14 | 247 | 1957 | 8.0 |
| Mono S | 500 | 0.06 | 205 | 3416 | 15.2 |
a Micromoles of nitroaniline released per min are indicated.
b Specific activity is estimated as the units of enzyme/mg of protein.
c ESP corresponds to concentrated excreted-secreted products.
FIGURE 5.

Biochemical characterization of Sc-SP-1. The Mono Q-purified fraction of native protease was analyzed in 12% SDS-PAGE (A), in two-dimensional electrophoresis (B), in a zymogram with 0.05% gelatin in SDS-PAGE (C), in an immunoblot against recombinant Sc-SP-1 (D), and the Sc-SP-1 molecular mass determined in a linear mode of MALDI-TOF-TOF (E). MW, molecular weight markers (kDa); pI, isoelectric points.
Biochemical Characterization
Native Sc-SP-1 was able to hydrolyze substrates with hydrophobic residue at P1 such as AAPF-pNA and N-methoxyl-Ala-Ala-Pro-Met-pNA (AAPM-pNA), thus indicating its preference for chymotrypsin substrates (Table 2). The kinetic parameters of Sc-SP-1 against AAPF-pNA were a Km of 2.12 ± 0.30 mm, a Vmax of 1.32 ± 0.08 mm s−1, a kcat of 28.9 s−1, and a catalytic efficiency (kcat/Vmax) of 22.2 × 104 s−1 m−1. Against AAPM-pNA were obtained a Km of 1.61 ± 0.17 mm, a Vmax of 0.68 ± 0.02 mm s−1, and a kcat of 15.11 s−1, values that are about two times less than against AAPF-pNA. In contrast, Sc-SP-1 showed very weak activity toward specific substrates for trypsins (N-benzoyl-Phe-Val-Arg-pNA) and elastases (N-succinyl-Ala-Ala-Pro-Leu-pNA). Recombinant Sc-SP-1 presents a Km of 0.81 mm ± 0.06, a Vmax of 0.076 ± 0.01 mm s−1, and a kcat of 1.6 against AAPF-pNA and a Km of 0.81 mm ± 0.06, a Vmax of 0.02 ± 0.004 mm s−1, and a kcat of 1.68 against AAPM-pNA (see supplemental Fig. S3). Native and recombinant Sc-SP-1 are pH- and temperature-dependent with a maximum of activity at pH of 8 and 8.5 and 45 °C (see supplemental Fig. S4).
TABLE 2.
Substrate specificity of native Sc-SP-1
| Substrate | Activity |
|---|---|
| unitsa | |
| N-Succinyl-Ala-Ala-Pro-Phe-pNA | 218 |
| N-Methoxyl-Ala-Ala-Pro-Met-pNA | 198 |
| N-Succinyl-Ala-Ala-Pro-Leu-pNA | 52 |
| N-Benzoyl-Phe-Val-Arg-pNA | 39 |
| N-Succinyl-Gly-Gly-Arg-pNA | 0 |
| Nα-Benzoyl-l-arginine 4-NA | 0 |
| N-Succinyl-Gly-Phe-pNA | 0 |
| N-Benzoyl-Pro-Phe-Arg-pNA | 0 |
a One unit of protease activity was defined as micromoles of nitroaniline released per min under the assay conditions.
The proteolytic activity of native Sc-SP-1 was inhibited by chymostatin, N-tosyl-l-phenylalanylchloromethyl ketone, PMSF, and benzamidine with IC50 of 0.07, 0.73, 7.91, and 15.40 mm, respectively (Table 3), thus also indicating it is a chymotrypsin-like serine protease. In sc-sp-1, six cysteine residues were predicted, two of them unusually located, and thus we investigated whether the treatment with reducing agents could affect enzymatic activity. Unexpectedly, the treatment with 100 mm DTT and with 100 mm 2-mercaptoethanol caused an increase of 20 and 14% in proteolytic activity against AAPF-pNA, respectively.
TABLE 3.
Fifty percent inhibition of native Sc-SP-1 using selective inhibitors
The following abbreviations are used: TPCK, toluenesulfonylphenylalanine chloromethyl ketone; E64, l-transepoxysuccinyl-leucylamido-[4-guanidino]butane.
| Inhibitor | IC50a | TRb |
|---|---|---|
| mm | mm | |
| Chymostatin | 0.07 (0.59–0.08)c | 0.05–4.0 |
| PMSF | 0.73 (0.61–0.95) | 0.05–4.0 |
| TPCK | 7.91 (6.50–8.85) | 1.0–20.0 |
| Benzamidine | 15.40 (13.80–17.12) | 1.0–20.0 |
| EDTA | NId | 1.0–20.0 |
| Antithrombin III | NI | 1.0–10 |
| Aprotinin | NI | 1.0–10 |
| Phosphoramidon | NI | 1.0–10 |
| E64 | NI | 0.1–1.0 |
| Leupeptin | NI | 0.1–1.0 |
| Pepstatin | NI | 1.0–10 |
a Probit 1.4 analysis program was used to calculate 50% inhibition (IC50).
b TR indicates the tested concentration range used.
c This indicates the 95% confidence limit.
d NI indicates not inhibited within the tested concentration range.
Biological Activity
Insect midgut treated for 3 h with Sc-SP-1 presented clear signs of tissue disarrangements. The most evident alterations were detachment of epithelial cells from the basal lamina with a few cells detached into the lumen and formation of bubbles in the apical part of the epithelium (Fig. 6, A and B). Moreover, Sc-SP-1 bound to the lumen side of gut epithelium in the susceptible host G. mellonella (Fig. 6, C and D), thus leading us to investigate how Sc-SP-1 may act in the basal lamina. Using an artificial membrane model (Matrigel) and beads soaked with Sc-SP-1, we show that Matrigel treated with 100 μm Sc-SP-1 presents clear signs of digestion. These signs are reduced in the membrane treated with Sc-SP-1 partially inhibited with 0.8 mm PMSF (IC50) and disappeared in treatments with Sc-SP-1 inhibited with 2 mm PMSF (IC100). The beads soaked in bovine α-chymotrypsin did not cause any disruption in the membrane (Fig. 7, A–D). The data suggest that hole formation was an Sc-SP-1 attribute dependent on catalytic activity rate. Because this artificial membrane mainly consists of laminin (56%), collagen IV (31%), and proteoglycans, including fibronectin (8%), we investigated which component was hydrolyzed by Sc-SP-1. Hydrolysis of fibronectin and laminin was confirmed after 20 min at 15 °C of controlled digestion and of collagen IV under a longer incubation time and at higher temperature. Sc-SP-1 treated with PMSF did not cause digestion of any of these proteins (Fig. 7E).
FIGURE 6.

Micrographs showing the activity of Sc-SP-1 in insect midgut epithelium. Effect of 30 μg of purified Sc-SP-1 in 4-mm-thick pieces of midgut (A), nontreated midgut (B), immunodetection of Sc-SP-1 in sections of midgut probed with anti-Sc-SP-1 antibodies (C), and control sections probed with secondary antibody alone (D) is shown.
FIGURE 7.

Activity of Sc-SP-1 in a basal membrane model (Matrigel). Activity was caused by 100 μm Sc-SP-1 immobilized in Sepharose S200 beads applied on the basal membrane (A), by 100 μm Sc-SP-1 preincubated with the 0.8 mm PMSF (B), by 100 μm Sc-SP-1 preincubated with the 2 mm PMSF (C), and activity of 10 mm of bovine α-chymotrypsin (D). Pattern of digestion of basal membrane proteins was incubated with 100 μm Sc-SP-1 (E). MW, molecular weight markers (kDa); Fn, fibronectin; Lam, laminin; Coll, collagen type IV; C, control; D, digested products. The arrows indicate digested products.
DISCUSSION
Our work shows that S. carpocapsae secretes a serine protease that is likely to help nematodes invade insect tissues. This protease has a molecular mass of 27,313.94 Da, as determined by mass spectrometry, and a pI of 8.7 in native and recombinant forms. Biochemical and sequence analysis proved that this protein belongs to the chymotrypsin family. In animal parasitic nematodes, a large number of proteases involved in the invasive process have been identified (40). The relevance of these proteases to the parasitic process is supported by the fact they have been implicated as the most relevant antigens to protect against some nematode infections in vertebrates (41). Among expressed proteases, serine proteases are the most represented in nematode ESPs (13). In fact, they were identified in ESPs from Ancylostoma caninum, T. spiralis, and T. pseudospiralis (41–43). In S. carpocapsae, preliminary assays also pointed out the presence of serine proteases in ESPs. This finding led us to search for genes encoding serine proteases during the S. carpocapsae parasitic stage, based on a strategy derived from degenerate oligonucleotides designed in accordance with the sequence of amino acids flanking the active site histidine residue (His57) of nematode serine proteases. A full-length cDNA with an open reading frame of 831 nucleotides was isolated, and PCR primers flanking the 5′ and 3′ ends of the sc-sp-1 cDNA were used to amplify the corresponding gene from genomic DNA, resulting in the cloning of the sc-sp-1 gene. Three putative introns of 43, 159, and 44 bp and two of 46 bp were found to interrupt the coding sequence, all of which followed the GT-AG rule (37). The first intron in the sc-sp-1 gene was found at the end of the sequence encoding the zymogen peptide, similar to the second intron found in crustacean chymotrypsin-like genes (44). The four other introns split the mature enzyme coding sequence. The three amino acids that include the charge relay system are located in two different exons. His57 and Asp102 are in the second exon, showing the same structure as the chymotrypsin genes of insects (45), and the third catalytic residue Ser195 is in the fifth exon.
Independently of intron number in vertebrate and invertebrate serine protease genes, the ending of a particular intron is commonly conserved six nucleotides before the Ser195 codon (44). This feature is also conserved in the sc-sp-1 gene. The position of the fifth intron in sc-sp-1 is located in the same position as the sixth intron in the gene encoding the trypsin-like protease protein 3 of C. elegans (GenBankTM accession number AAB09110.4) before the Asp226 and Gly226 codons, respectively. On the other hand, none of the trans-spliced leader sequences present in the C. elegans genome were found in the sc-sp-1 gene. The Sc-SP-1 protease lacks a negatively charged carboxylate at position 189, which is present at the base of the substrate-binding pocket for all known trypsins. Instead, Sc-SP-1 contains a glycine residue at the equivalent position and an additional Gly216 residue lining the entry of the pocket, corresponding to chymotrypsin-like enzymes. The negatively charged residue is rearranged as an Asp residue at position 226, replacing the smaller residues (Ala or Gly) found in vertebrate chymotrypsins (46), which may imply that Sc-SP-1 has a somewhat different substrate preference. Similar to most invertebrate serine proteases, the presence of six cysteine residues indicates three disulfide linkages (39). Nevertheless, two of them are located close to the carboxylic end of the protein and therefore we presume close to the well conserved Cys42–Cys58 and Cys168–Cys182 disulfide bonds. Nevertheless, the Sc-SP-1 activity was not affected by reducing agents.
Our work demonstrates that Sc-SP-1 is part of the excreted/secreted products of the parasitic stage of S. carpocapsae. The analysis of the mode of action for Sc-SP-1 supports our hypothesis that this protein is involved in invasion of the insect gut wall. The gut wall is a physical barrier that opposes pathogen invasion very efficiently in insects (47). The first component of this wall is the peritrophic membrane that is a mesh of proteoglycans that avoid pathogen contact with cells. The basal lamina, which is composed of collagen, elastin, glucosaminoglycans, and glycoproteins, serves as a filter to protect adjacent tissues (48). Our assays show that Sc-SP-1 is highly efficient at destroying both peritrophic and basal lamina. Additionally, the columnar cells detach after treatment with Sc-SP-1 that binds to the host basal lamina. Using a matrix membrane model to mimic basal lamina, which has been used before to study pathogen host tissue invasion (49) and invasive tumor cells (50), we proved that Sc-SP-1 is able to open holes in the matrix gel membrane, very likely creating passages by which nematodes can reach the insect hemocoelium. We suggest that Sc-SP-1 hydrolyzes fibronectin and laminin, and to a lesser extent collagen IV, which are the most abundant proteins of the basal lamina. The interaction of Sc-SP-1 with basal lamina glycoproteins, such as fibronectin and laminin, can be explained by the predicted B_lectin domain that was identified in sc-sp-1. This domain is known to enable binding with glycoproteins, thus mediating a wide variety of biological processes, particularly host-pathogen interactions (51).
The involvement of Sc-SP-1 in the parasitic process was also in accordance with the expression analysis of the encoding gene during the nematode life cycle. Nematodes in the arrested stage do not express sc-sp-1; however, this gene was overexpressed in recovered nematodes, particularly those in the gut lumen preparing to invade the insect hemocoelium. In other parasitic nematodes, the role for serine proteases in tissue invasion was inferred based on expression analysis (52–54). In vitro assays showed that, although the resistant stage does not express sc-sp-1, the nematodes initiate expression shortly after stimulation with insect tissues. Peritrophic membrane and hemolymph induce sc-sp-1 expression more rapidly than other tissues such as fat bodies and gut epithelium. This immediate response of the nematode is probably related to the route it normally uses to reach the insect hemocoelium, first contacting the peritrophic membrane that lines the gut lumen and then with various hemolymph components, which represent the first lines of defense. The Sc-SP-1 expression time frame and its ability to destroy proteins in the midgut, particularly those of the basal lamina, are in agreement with the development of parasitism.
The identification of Sc-SP-1 adds to our knowledge of protease family members in S. carpocapsae such as Sc-SP-3, which has been shown to cause cell death (20), and to Sc-Trypsin and Sc-Chymotrypsin, which have been shown to correlate with insect immune depression (55, 56). The identification of these serine proteases in excreted/secreted products from the S. carpocapsae parasitic stage strongly contributes to our understanding of the pathogenic process in this insect parasitic nematode. Furthermore, the involvement of Sc-SP-1 in host invasion makes this gene a useful candidate to improve nematodes to use in the biological control of insects.
Supplementary Material
Acknowledgment
We thank J. Medeiros for the expertise in S.E.
This work was supported in part by Project POCTI/BSE/41630/2001 founded by Fundação para a Ciência e Tecnologia and Project 223/06 founded by Fundação Lusa Americana para o Desenvolvimento.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1–S4 and Table S1.
The nucleotide sequence(s) reported in this paper has been submitted to the GenBankTM/EBI Data Bank with accession number(s) AY968052 and AY600457.
The amino acid sequence of this protein can be accessed through NCBI Protein Database under NCBI accession number AAT27470.
- IJ
- Infective juvenile
- ESP
- excreted/secreted products
- PB
- phosphate buffer
- pNA
- p-nitroanilide
- qRT
- quantitative real time.
REFERENCES
- 1.Akhurst R. J., Dunphy G. B. (1993) in Parasites and Pathogen of Insects (Beckage N. E., Thompson S. N., Federici B. A. eds) Vol. 2, pp. 1–23, Academic Press, New York [Google Scholar]
- 2.Klein M. G. (1990) in Entomopathogenic Nematodes in Biological Control (Gaugler R., Kaya H. K. eds) pp. 195–214, CRC Press, Inc., Boca Raton, FL [Google Scholar]
- 3.Begley J. W. (1990) in Entomopathogenic Nematodes in Biological Control (Gaugler R., Kaya H. K. eds) pp. 233–246, CRC Press, Inc., Boca Raton, FL [Google Scholar]
- 4.Burnell A. (2002) in Entomopathogenic Nematology (Gaugler R. ed) pp. 241–264, CABI Publishing, Oxon, UK [Google Scholar]
- 5.Vellai T., Molnár A., Lakatos L., Banfalvi T., Fodor A., Sáringer G. (1999) in COST 819 Entomopathogenic Nematodes: Survival of Entomopathogenic Nematodes (Glazer I., Richardson P., Boemare N., Coudert F. eds) pp. 105–119, EUR 18855 EN, Office for Official Publications of the EC, Luxembourg [Google Scholar]
- 6.Grewal P. S., Lewis E. E., Gaugler R., Campbell J. F. (1994) Parasitology 108, 207–215 [Google Scholar]
- 7.Hao Y. J., Montiel R., Abubucker S., Mitreva M., Simões N. (2010) Mol. Biochem. Parasitol. 169, 79–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dowds B. C., Peters A. (2002) in Entomopathogenic Nematology (Gaugler R. ed) pp. 79–98, CABI Publishing, Oxon, UK [Google Scholar]
- 9.Ishibashi N., Kondo E. (1990) in Entomopathogenic Nematodes in Biological Control (Gaugler R., Kaya H. K. eds) pp. 139–150CRC Press, Inc., Boca Raton, FL [Google Scholar]
- 10.Simões N., Rosa J. S. (1996) Biocontrol Sci. Technol. 6, 403–411 [Google Scholar]
- 11.Simões N., Caldas C., Rosa J. S., Bonifassi E., Laumond C. (2000) J. Invertebr. Pathol. 75, 47–54 [DOI] [PubMed] [Google Scholar]
- 12.Dzik J. M. (2006) Acta Biochim. Pol. 53, 33–64 [PubMed] [Google Scholar]
- 13.Trap C., Boireau P. (2000) Vet. Res. 31, 461–471 [DOI] [PubMed] [Google Scholar]
- 14.McKerrow J. H., Pino-Heiss S., Lindquist R., Werb Z. (1985) J. Biol. Chem. 260, 3703–3707 [PubMed] [Google Scholar]
- 15.Sakanari J. A., McKerrow J. H. (1990) J. Parasitol. 76, 625–630 [PubMed] [Google Scholar]
- 16.Haffner A., Guilavogui A. Z., Tischendorf F. W., Brattig N. W. (1998) Exp. Parasitol. 90, 26–33 [DOI] [PubMed] [Google Scholar]
- 17.Todorova V. K. (2000) Folia Parasitol. 47, 141–145 [DOI] [PubMed] [Google Scholar]
- 18.Romaris F., North S. J., Gagliardo L. F., Butcher B. A., Ghosh K., Beiting D. P., Panico M., Arasu P., Dell A., Morris H. R., Appleton J. A. (2002) Mol. Biochem. Parasitol. 122, 149–160 [DOI] [PubMed] [Google Scholar]
- 19.Bedding R. A., Molyneux A. S., Akhurst R. J. (1983) Exp. Parasitol. 55, 249–257 [DOI] [PubMed] [Google Scholar]
- 20.Toubarro D., Lucena-Robles M., Nascimento G., Costa G., Montiel R., Coelho A. V., Simões N. (2009) Int. J. Parasitol. 39, 1319–1330 [DOI] [PubMed] [Google Scholar]
- 21.Sambrook J., Fritsch E. F., Maniatis T. (1989) Molecular Cloning: A Laboratory Manual, 2nd Ed., pp. 9.31–9.58, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 22.Emanuelsson O., Brunak S., von Heijne G., Nielsen H. (2007) Nat. Protoc. 2, 953–971 [DOI] [PubMed] [Google Scholar]
- 23.Larkin M. A., Blackshields G., Brown N. P., Chenna R., McGettigan P. A., McWilliam H., Valentin F., Wallace I. M., Wilm A., Lopez R., Thompson J. D., Gibson T. J., Higgins D. G. (2007) Bioinformatics 23, 2947–2948 [DOI] [PubMed] [Google Scholar]
- 24.Letunic I., Doerks T., Bork P. (2009) Nucleic Acids Res. 37, D229–D232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mochii M., Yoshida S., Morita K., Kohara Y., Ueno N. (1999) Proc. Natl. Acad. Sci. U.S.A. 96, 15020–15025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fahey E. M., Chaudhuri J. B., Binding P. (2000) J. Chromatogr. B. Biomed. Sci. Appl. 737, 225–235 [DOI] [PubMed] [Google Scholar]
- 27.Zhang L., Wang J., Yu M., Ru B. (2004) Comp. Biochem. Physiol. C Toxicol. Pharmacol. 137, 115–122 [DOI] [PubMed] [Google Scholar]
- 28.Laemmli U. K. (1970) Nature 227, 680–685 [DOI] [PubMed] [Google Scholar]
- 29.Heinemeyer J., Lewejohann D., Braun H. P. (2007) Methods Mol. Biol. 355, 343–352 [DOI] [PubMed] [Google Scholar]
- 30.Lantz M. S., Ciborowski P. (1994) Methods Enzymol. 235, 563–594 [DOI] [PubMed] [Google Scholar]
- 31.Matsudaira P. (1987) J. Biol. Chem. 262, 10035–10038 [PubMed] [Google Scholar]
- 32.Gobom J., Nordhoff E., Mirgorodskaya E., Ekman R., Roepstorff P. (1999) J. Mass Spectrom. 34, 105–116 [DOI] [PubMed] [Google Scholar]
- 33.Santos R., Costa G., Franco C., Gomes-Alves P., Flammang P., Coelho A. V. (2009) Marine Biotechnol. 11, 686–698 [DOI] [PubMed] [Google Scholar]
- 34.Hubert J. J., Carter E. M. (1990) PROBIT: A Program in PASCAL for Univariate Probit Analysis with Exact Confidence Limits for LC50, Statistical Series, 1990–222, Department of Mathematics and Statistics, University of Guelph, Guelph, Ontario, Canada [Google Scholar]
- 35.Martoja R., Martoja-Pierson M. (1970) Técnicas de Histologia Animal (Toray Masson. ed) p. 341, Barcelona, Spain [Google Scholar]
- 36.Joshi M. C., Sharma A., Kant S., Birah A., Gupta G. P., Khan S. R., Bhatnagar R., Banerjee N. (2008) J. Biol. Chem. 283, 28287–28296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mount S. M. (1982) Nucleic Acids Res. 10, 459–472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gudmundsdottir E., Spilliaert R., Yang Q., Craik C. S., Bjarnason J. B., Gudmundsandttir A. (1996) Comp. Biochem. Physiol. B 113, 795–801 [DOI] [PubMed] [Google Scholar]
- 39.Perona J. J., Craik C. S. (1995) Protein Sci. 4, 337–360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Davis E. L., Hussey R. S., Baum T. J. (2004) Trends Parasitol. 20, 134–141 [DOI] [PubMed] [Google Scholar]
- 41.Dalton J. P., Brindley P. J., Knox D. P., Brady C. P., Hotez P. J., Donnelly S., O'Neill S. M., Mulcahy G., Loukas A. (2003) Int. J. Parasitol. 33, 621–640 [DOI] [PubMed] [Google Scholar]
- 42.Hawdon J. M., Jones B. F., Hoffman D. R., Hotez P. J. (1996) J. Biol. Chem. 271, 6672–6678 [DOI] [PubMed] [Google Scholar]
- 43.Trap C., Fu B., Le Guerhier F., Liu M., Le Rhun D., Romand T., Perret C., Blaga R., Boireau P. (2006) Parasitol. Res. 98, 288–294 [DOI] [PubMed] [Google Scholar]
- 44.Cwiklinski K., Meskill D., Robinson M. W., Pozio E., Appleton J. A., Connolly B. (2009) Vet. Parasitol. 159, 268–271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sellos D., Van Wormhoudt A. (1999) Biochim. Biophys. Acta 1432, 419–424 [DOI] [PubMed] [Google Scholar]
- 46.Mazumdar-Leighton S., Broadway R. M. (2001) Insect Biochem. Mol. Biol. 31, 633–644 [DOI] [PubMed] [Google Scholar]
- 47.Vallet-Gely I., Lemaitre B., Boccard F. (2008) Nat. Rev. Microbiol. 6, 302–313 [DOI] [PubMed] [Google Scholar]
- 48.Whitten M. M., Shiao S. H., Levashina E. A. (2006) Parasite Immunol. 28, 121–130 [DOI] [PubMed] [Google Scholar]
- 49.Rocha-Azevedo B. D., Jamerson M., Cabral G. A., Marciano-Cabral F. (2010) Exp. Parasitol. 126, 79–84 [DOI] [PubMed] [Google Scholar]
- 50.Kleinman H. K., Martin G. R. (2005) Semin. Cancer Biol. 15, 378–386 [DOI] [PubMed] [Google Scholar]
- 51.Ramachandraiah G., Chandra N. R. (2000) Proteins Struct. Funct. Genet. 39, 358–364 [PubMed] [Google Scholar]
- 52.Gomez Gallego S., Loukas A., Slade R. W., Neva F. A., Varatharajalu R., Nutman T. B., Brindley P. J. (2005) Parasitol. Int. 54, 123–133 [DOI] [PubMed] [Google Scholar]
- 53.Zhan B., Hotez P. J., Wang Y., Hawdon J. M. (2002) Mol. Biochem. Parasitol. 120, 291–296 [DOI] [PubMed] [Google Scholar]
- 54.Williamson A. L., Lustigman S., Oksov Y., Deumic V., Plieskatt J., Mendez S., Zhan B., Bottazzi M. E., Hotez P. J., Loukas A. (2006) Microbiology 74, 961–967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Balasubramanian N., Hao Y. J., Toubarro D., Nascimento G., Simões N. (2009) Int. J. Parasitol. 39, 975–984 [DOI] [PubMed] [Google Scholar]
- 56.Balasubramanian N., Toubarro D., Simões N. (2010) Parasite Immunol. 32, 165–175 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.