

Abstract
Efficiency of translation termination relies on the specific recognition of the three stop codons by the eukaryotic translation termination factor eRF1. To date only a few proteins are known to be involved in translation termination in eukaryotes. Saccharomyces cerevisiae Tpa1, a largely conserved but uncharacterized protein, has been described to associate with a messenger ribonucleoprotein complex located at the 3′ end of mRNAs that contains at least eRF1, eRF3, and Pab1. Deletion of the TPA1 gene results in a decrease of translation termination efficacy and an increase in mRNAs half-lives and longer mRNA poly(A) tails. In parallel, Schizosaccharomyces pombe Ofd1, a Tpa1 ortholog, and its partner Nro1 have been implicated in the regulation of the stability of a transcription factor that regulates genes essential for the cell response to hypoxia. To gain insight into Tpa1/Ofd1 function, we have solved the crystal structure of S. cerevisiae Tpa1 protein. This protein is composed of two equivalent domains with the double-stranded β-helix fold. The N-terminal domain displays a highly conserved active site with strong similarities with prolyl-4-hydroxylases. Further functional studies show that the integrity of Tpa1 active site as well as the presence of Yor051c/Ett1 (the S. cerevisiae Nro1 ortholog) are essential for correct translation termination. In parallel, we show that Tpa1 represses the expression of genes regulated by Hap1, a transcription factor involved in the response to levels of heme and oxygen. Altogether, our results support that Tpa1 is a putative enzyme acting as an oxygen sensor and influencing several distinct regulatory pathways.
Keywords: Protein Chemical Modification, Transcription Factors, Translation Release Factors, X-ray Crystallography, Yeast Genetics, Oxygen Sensing, Post-translational Modification, Prolyl-4-hydroxylase, Translation Termination
Introduction
Protein synthesis is a complex process performed by ribosomes, translation factors, and amino-acyl tRNAs that act synergistically to translate mRNAs into corresponding proteins. The mechanism of translation of mRNAs into protein can be divided into three major steps; they are initiation, elongation, and termination. In eukaryotes, translation initiation relies on the formation of a stable closed-loop structure bringing the 5′ m7G cap and the 3′ poly(A) tail of a single mRNA in proximity and requires at least 11 initiation factors (1, 2). During elongation, the mRNA codon present in the ribosomal A site is recognized by a cognate amino-acyl tRNA associated with elongation factor 1A. The nascent polypeptide chain is then transferred from the tRNA present in the P-site to the amino acid of the A-site tRNA. Subsequently, elongation factor 2 induces translocation of peptidyl-tRNA from the A- to the P-site allowing the next elongation step to proceed. The entrance of one of the three stop codons (UAA, UAG, or UGA) in the A-site triggers translation termination. In contrast with other codons, stop codons are not recognized by cognate tRNAs but by proteins known as class I release factors (RF16 and RF2 in bacteria and eRF1 in eukaryotes). These are also directly involved in the hydrolysis of the ester bond connecting the newly synthesized polypeptide chain to the tRNA located in the ribosomal P-site (3–5). The activity of class I RFs is stimulated by the class II RFs GTPases: RF3 in bacteria and eRF3 in eukaryotes (3–6). The eRF1 and eRF3 proteins form a stable complex in the absence of the ribosome (7, 8). The GTP hydrolysis activity of eRF3 is dependent on ribosome-bound eRF1 (9), but the eRF3 precise role remained obscure until recently. In vitro reconstitution of eukaryotic translation system indicated that binding of the eRF1·eRF3·GTP ternary complex to pre-termination complexes causes a structural rearrangement of the ribosome. This results in GTP hydrolysis by eRF3 followed by eRF1-induced hydrolysis of peptidyl-tRNA (6).
Translation termination relies on the efficient and specific recognition of stop codons (UGA, UAA, and UAG) by eRF1 despite the presence of near-cognate tRNAs (such as the tRNATrp that recognizes the UGG codon) that could induce amino acid misincorporation and the synthesis of C-terminal extensions. The efficiency of stop codon recognition is primarily influenced by the stop codon itself and the first downstream nucleotide, commonly referred to as the tetranucleotide termination signal (10). Several trans-acting factors influencing the accuracy of this process have also been identified (11). Among these, the interaction of eRF3 with the poly(A)-binding protein (Pab1), which itself associates with the 3′-poly(A) tail present on almost all eukaryotic mRNAs, has been proposed to participate to normal translation termination and to mRNA stabilization (12, 13). The glutamine side chain from the strictly conserved GGQ motif from class I release factors has also been shown to be post-translationally modified both in prokaryotes and in eukaryotes, thereby affecting the efficiency of translation termination in vivo, at least in Escherichia coli (14–17). More recently, the deletion of the uncharacterized Saccharomyces cerevisiae open reading frame YER049W has been shown to result in increased readthrough of stop codons, longer poly(A) tails, and increased mRNAs half-lives (18). The YER049W gene product, named Tpa1, was co-immunoprecipitated with translation termination factors eRF1 and eRF3 as well as with the poly(A)-binding protein Pab1, suggesting that it could be part of a messenger ribonucleoprotein complex associated at the mRNA 3′ end. Sequence analysis suggested that Tpa1 belongs to the 2-oxoglutarate-Fe(II) dioxygenase family (19). Dioxygenase enzymes catalyze oxidation reactions involving dioxygen and use 2-oxoglutarate (2OG) as co-substrate and ferrous ion (Fe2+) as cofactor. They are involved in various biological processes such as fatty acid metabolism, antibiotic biosynthesis, nucleic acid repair, collagen biosynthesis, histone Lys/Arg demethylation, and transcriptional regulation of the hypoxic response (20, 21). Recently, the Ofd1 protein, a Tpa1 ortholog from Schizosaccharomyces pombe, has been implicated with Nro1 (for negative regulator of Ofd1) in the regulation of low oxygen gene expression (22, 23). In S. pombe, the Sre1 transcription factor regulates the expression of genes involved in cholesterol and lipid biosynthesis (24, 25). Under low oxygen levels, the membrane-bound Sre1 protein is proteolytically cleaved, thus releasing its N-terminal transcription factor domain (26, 27). As a consequence, the transcription factor domain enters the nucleus and activates the expression of genes essential for hypoxic growth. In the absence of oxygen, the Ofd1 C-terminal domain was reported to interact with Nro1, and the Sre1 N-terminal domain is stabilized. In the presence of oxygen, the Ofd1 N-terminal dioxygenase domain would act as an oxygen sensor, activating its own C-terminal domain and preventing its interaction with Nro1, thus enhancing degradation of the Sre1 N-terminal domain.
Based on these two independent studies realized in S. cerevisiae and S. pombe, the precise Tpa1/Ofd1 function remains obscure. On the one hand, no clear ortholog of S. pombe Sre1 protein (homologous to human sterol regulatory element-binding protein (SREBP)) has been found in S. cerevisiae. On the other hand, a S. cerevisiae ortholog of Nro1 (Yor051c) does exist and was reported to interact with Tpa1 (28). Therefore, Tpa1/Ofd1 may have a role that is organism-dependent, and/or it may act on several distinct pathways. We present here the results of structural, biochemical, and genetic studies that improve our understanding of the function of Tpa1, a protein conserved from fungi to human.
EXPERIMENTAL PROCEDURES
Cloning, Expression, and Purification of Tpa1
To overexpress Tpa1 in E. coli, its coding sequence was amplified from genomic DNA with oligonucleotides OBS2151 and OBS2152 (supplemental Table 1) and inserted in the BspE1 site of a modified pET vector, yielding pBS3077.
The protein has been expressed using E. coli Rosetta (DE3) pLysS strain (Novagen) at 23 °C in 2×YT medium (BIO101 Inc.) supplemented with kanamycin at 50 μg/ml. At A600 = 0.8, the transcription of the plasmid was induced during 20 h by the addition of 50 μg/ml isopropyl 1-thio-β-d-galactopyranoside. Cells were harvested by centrifugation, resuspended in 35 ml of 20 mm Tris-HCl, pH 7.5, 200 mm NaCl, 5 mm β-mercaptoethanol (buffer A), and stored at −20 °C. Cell lysis was performed by sonication. The His-tagged protein was purified on a nickel-nitrilotriacetic acid column (Qiagen) followed by gel filtration chromatography on a SuperdexTM 200 HL 16/60 column (GE Healthcare) equilibrated in buffer A.
Protein labeling with SeMet was conducted as described (29). The SeMet protein was purified using the same protocol as the native protein.
Size-exclusion Chromatography-Multiangle Laser Light Scattering Measurements
Sample injection, chromatography, and detection were carried out using a Triple Detector Array (TDA302) system coupled in line to a GPCmax chromatographic system (Viscotek). A sample of 100 μl of Tpa1 (6.2 mg/ml) was injected at a flow rate of 0.5 ml/min on a SuperdexTM 200 HR 10/30 column (GE Healthcare) equilibrated in buffer A. Elution was followed by a UV-visible spectrophotometer, a differential refractometer, a 7° low angle light scattering detector, a 90° right angle light scattering detector, and a differential pressure viscometer. The data were collected and processed with the program OmniSEC (Viscotek). Mr was directly calculated from the absolute light scattering measurements. The dn/dc value (0.168) for Tpa1 has been determined experimentally using the refractive index at four different concentrations of Tpa1.
Structure Determination
Crystals of the native and SeMet protein were grown from a mixture in a 1:1 ratio of 15 mg/ml protein solution in buffer A and crystallization liquor containing 0.1 m Hepes, pH 7.5, 44% methylpentanediol, and 0.1 m Mg(NO3)2 at 18 °C over 2 months. The crystals were directly flash-cooled in liquid nitrogen.
Native crystal (crystal 1) diffracted to 2.8 Å of resolution on beam line ID14-EH2 at the European Synchrotron Radiation Facility (Grenoble, France). This crystal belongs to space group C2221 with one molecule in the asymmetric unit (a = 81.2 Å, b = 104.8 Å, c = 205.6 Å, α = β = γ = 90°). SeMet crystals suffered from serious diffraction anisotropy (6 Å in one direction and 3.5–2.5 Å in the other one), but datasets could be recorded at the European Synchrotron Radiation Facility to 4 Å resolution on beam line ID23-EH1 (crystal 2) and 3.3 Å on beam line BM30A (crystal 3). The diffraction data were collected at the selenium edge (λ = 0.9796Å). These crystals belong to space group P212121 with four molecules in the asymmetric unit (a = 105.7 Å, b = 167.9 Å, c = 196.9 Å, α = β = γ = 90°).
The structure was determined by the single wavelength anomalous dispersion method using the anomalous signal of selenium (Se) atoms. Data were processed with the XDS package (30). The program SHELXD was used to find an initial set of 32 selenium sites (36 were expected according to the protein sequence and cell content) from the crystal 2 dataset in the 30–5.5 Å resolution range (31). Refinement of the selenium sites and phasing were carried out with the program SHARP using the SAD datasets collected on crystals 2 and 3 (32). Additional sites were included in the refinement after inspection of residual maps. The final substructure model comprises all expected selenium atoms. Phase refinement and extension were carried out via density modification and NCS averaging with the program RESOLVE (33). The experimental phases allowed initial automatic building by BUCCANEER (34). Iterative cycles of manual rebuilding using COOT (35) followed by refinement with PHENIX (36) led to a first almost-complete model. This model was then used for molecular replacement using native data collected on crystal 1 with MOLREP (37) and finally submitted to a new series of manual rebuilding with COOT and refinement with PHENIX. The statistics for data collection and refinement are summarized in Table 1. Due to the absence of electron density, the following regions were omitted from the final model: 1–25, 94–110, 269–275, 306–330, 455–459, 562–584, and 635–644. The atomic coordinates and structure factors have been deposited into the Brookhaven Protein Data Bank under accession numbers (3MGU).
TABLE 1.
| Native | SeMet 1 | SeMet 2 | |
|---|---|---|---|
| Data collection | |||
| Space group | C2221 | P21212 | P21212 |
| Unit cell parameters | a = 81.2 Å b = 104.8 Å c = 205.6 Å | a = 105.7 Å b = 168.2 Å c = 197.3 Å | a = 106.2 Å b = 169.0 Å c = 197.8 Å |
| Redundancy | 3.58 (3.64) | 4.64 (4.52) | 3.82 (3.76) |
| Resolution range (Å) | 50–2.8 (2.9–2.8) | 50–3.4 (3.5–3.4) | 50–3.7 (3.8–3.7) |
| Completeness (%) | 97.0 (92.5) | 97.0 (93.2) | 98.1 (94.6) |
| I/σ(I) | 6.76 (2.13) | 8.29 (2.88) | 6.13 (2.52) |
| Rsym (%) | 15.3 (50.1) | 16.2 (57.1) | 19.1 (54.0) |
| Refinement | |||
| Resolution (Å) | 46.7-2.8 | ||
| R/Rfree (%) | 23.9/29.6 | ||
| Geometry statistics | |||
| r.m.s.d. bonds (Å) | 0.006 | ||
| r.m.s.d. angles (°) | 1.077 | ||
| Average B-factor (Å2) | 52.10 | ||
| Ramachandran plot (Procheck) | |||
| Most favored (%) | 84.7 | ||
| Additionally allowed (%) | 15.2 | ||
Strains and Media
S. cerevisiae strains YJW618 (MATa leu2-3,112 his3-11,15 trp1-1 ura3-1 ade1-14 PSI+) and YJW619 (as YJW618 but tpa1::LEU2) were obtained from D. Bedwell (19). YOR051C was deleted in YJW618 and YJW619 with a Candida glabrata HIS3 cassette amplified with OBS3866/OBS3867, giving strains BSY2236 and BSY2237, respectively. Cells were grown at 30 °C in standard media. Cell treatment with guanidine hydrochloride to test the role of the [PSI−] factor was done as described previously (38).
The plasmid, pBS3820, containing the wild type TPA1 gene was constructed by amplifying the TPA1 ORF with upstream and downstream sequences from genomic DNA using oligonucleotides OBS3527 and OBS3528 and inserting it between the BamH1 and Not1 sites of the pRS424 (TRP1, 2μ) vector (39). The H159A/D161N allele was constructed by site-directed mutagenesis with primers OBS3687 and OBS3688 (supplemental Table 1) yielding pBS3793. Domain deletions pBS3794 and pBS3795 were constructed by PCR and cloning using oligonucleotides OBS3738-OBS3741. The TAP tag was introduced by cloning using oligonucleotides OBS3822-OBS3824, OBS3831, OBS3832, and OBS3844, yielding pBS3796–3799.
Stop Codon Readthrough Measurements
Yeast strains were transformed with reporters containing the coding sequences of the Renilla and firefly luciferase separated either by a stop codon (TAA) or a sense codon (CAA) as described (18). Three transformants were pooled and grown in selective liquid media to A600 0.5–0.8 for enzymatic assays. 10 μl of yeast culture was used to measure the luminescence with the dual luciferase reporter assay reagent (Promega) in a Berthold luminometer. The percent readthrough is calculated from the ratio of firefly luciferase produced by the stop codon-containing construct to the firefly luciferase by the CAA-containing construct, normalized in each case to the level of Renilla luciferase produced in the same cells. Five biological experiments were performed with, in each case, technical duplicates. Differences reported were statistically significant (Mann-Whitney U test, 95% confidence interval).
β-Galactosidase Assay
Reporters, provided by B. Guiard (40), contained Hap1-driven UAS elements from the CYC1 or CYB2 genes, driving expression of β-galactosidase. Yeast strains were transformed with these reporters, and three transformants were pooled to perform β-galactosidase assays. Cells were grown to an A600 of 0.5–0.8. 1 ml of culture is used. Assays were performed as previously described with three biological replicate and for each assay one technical duplicate (41).
Protein Analyses
Proteins extracts (42) were fractionated on 8% SDS-PAGE, and TAP TAG proteins were detected with PAP (Sigma) and an ECL kit (GE Healthcare).
RESULTS
The structure of full-length S. cerevisiae Tpa1 has been solved using the single-wavelength anomalous diffraction signal of SeMet-substituted protein crystals and refined to 2.8 Å resolution using diffraction data from native protein crystals. The resulting density maps allowed us to rebuild 567 amino acids of 644. The missing residues correspond to the 25 N-terminal residues or the 9 C-terminal residues or belong to 5 loops for which no electron density was observed. The native protein crystallized in space group C2221 with one molecule in the asymmetric unit, whereas the SeMet-labeled protein crystals grown under the same conditions belong to space group P21212 with four Tpa1 copies in the asymmetric unit (one unit cell parameter is twice longer than for the native crystals). As a consequence, the crystal packing is identical between both crystal forms, and the Tpa1 protomers are organized as two identical dimers (buried surface 2000 Å2). This suggests that Tpa1 can form homodimers in solution (see below).
Tpa1 is composed of two stacked domains forming a cylinder of ∼90 Å length and 45 Å diameter (Fig. 1A). The N-terminal domain (Tpa1N) encompasses amino acids 26–260 (residues 1–25 are not visible in the electron density maps), whereas the C-terminal domain (Tpa1C) encompasses amino acid 293–635 (residues 636–644 could not be modeled due to the lack of electron density). Both domains are packed together and connected by a linker (amino acids 261–292) that partially folds as α-helix.
FIGURE 1.

S. cerevisiae Tpa1 structure. A, a ribbon representation of the Tpa1 protein is shown. B, a topology diagram is shown. The same color code as in panel A is used. C, a stereo view of Tpa1N active site (light brown) superposed on PHD2cat (yellow) is shown. The 2OG co-substrate has been modeled into the active site cavity by superposing the structure of a DSBH protein obtained in the presence of 2OG onto the Tpa1N domain. Black and red spheres depict the iron atom and water molecules observed in the crystal structure, respectively. D, a surface representation of the Tpa1 protein is shown.. Tpa1N (wheat) and Tpa1C (slate) are joined by a linker (gray). The iron atom is represented as a black sphere. E, a surface representation of residue conservation mapped at the surface of Tpa1 is shown. Coloring is from blue (highly conserved) to gray (low conservation). F, representation of electrostatic potential at the Tpa1 surface is shown.
The N-terminal Catalytic Domain
Sequence analysis has suggested that S. cerevisiae Tpa1 contains an N-terminal 2OG-Fe(II) dioxygenase domain from the double-stranded β-helix (DSBH) family based on the conservation of three characteristic motifs in the N-terminal part of the protein; a HXD dyad (where X is any amino acid) near the N terminus, a His toward the C terminus as well as an Arg or Lys farther downstream (19). Basically, the DSBH-fold consists of eight β-strands forming a β-sandwich structure composed of two four-stranded antiparallel sheets (21). The β-sandwich is partitioned between a minor (β-strands 2 if present, 7, 4, and 5) and a major sheet (β-strands 1, 8, 3, and 6) that very often contains additional strands. As proposed, the Tpa1N domain adopts a DSBH-fold consisting of a β-sandwich of two antiparallel β-sheets with five α-helices packed onto the major sheet (Fig. 1B). This latter is composed of five β-strands (order 1, 8, 3, 6, and A). Two additional strands (βB and βC) are located at the entrance of the β-sandwich cavity.
The DSBH-fold is shared by a wide variety of Fe2+/2OG-dependent dioxygenases, and many crystal structures have been described for this architecture (21, 43). Structural alignments with Tpa1N reveal similarity with prolyl-4-hydroxylases (r.m.s.d. of 2.3–2.5 Å over 150–170 Cα atoms and 15–19% sequence identity), phytanoyl CoA dioxygenase (r.m.s.d. of 2.6 Å over 140 Cα atoms and 15% sequence identity), DNA/RNA repair proteins AlkB/ABH3 (r.m.s.d. of 2.6 Å over 135–140 Cα atoms and 10–15% sequence identity), and isopenicillin N synthase (r.m.s.d. of 2.7–2.9 Å over 150 Cα atoms and 10% sequence identity). However, the closest structural homolog is human PHD2cat, a prolyl-4-hydroxylase that acts as an oxygen-sensing component and post-translationally hydroxylates the hypoxia-inducible transcription factor HIF1α in the presence of oxygen, thus leading to its degradation by the proteasome (r.m.s.d. of 2.5 Å over 153 Cα atoms and 19% sequence identity (44)). Analysis of the amino acid side chains strictly conserved between Tpa1N and PHD2cat reveals that most are clustered within the cavity located between both β-sheets that corresponds to PHD2cat enzyme active site (Fig. 1C). First of all, the HXD…H triad (positions 159, 161, and 227 in Tpa1, respectively) superposes perfectly with the characteristic HXD…H Fe2+ binding motif observed in PHD2cat. The 159HXD161 dyad is located within the loop connecting strands β2 and β3, whereas the second histidine (His227) originates from strand β7. In typical DSBH enzymes, these three residues form a triad, leaving three coordination sites on the octahedral Fe(II) center for the binding of 2OG and dioxygen. During refinement, a strong residual electron density peak was observed in close proximity to His159, Asp161, and His227 of Tpa1, exactly at the position corresponding to the iron atom in PHD2cat structure. Element analysis confirmed the presence of iron in the protein samples (ratio 1 iron atom for 2 Tpa1; data not shown). This metal is present at the entrance of a highly conserved cavity known to bind the 2OG co-substrate. All but two residues forming the 2OG binding pocket are strictly conserved between Tpa1N and PHD2cat (Fig. 1C, differences are Leu156 and Gln242 in Tpa1N versus Tyr310 and Thr387 in PHD2cat). At the bottom of this cavity, the positively charged side chains from Arg238 from β8 as well as Tyr173 are well positioned to interact with the carboxyl side chain of the 2OG co-substrate. Similarly, the side chains from Tyr150, Ile171, Leu189, and Val229 of Tpa1N form the wall of the 2OG binding site and structurally match with identical residues from PHD2cat. Altogether, these elements strongly suggest that the Tpa1N domain displays a putative active site configuration that is close to that from PHD2cat.
Further inspection of amino acid sequences of the Tpa1/Ofd1 family clearly shows that the iron atom is located in the middle of a highly conserved groove delineated by side chains from Asp144, Ser146, Asp160, Ile163, and Arg166 on one side and from the β-hairpin made by strands βB and βC (Lys82, Asp86, Ile87, Tyr88, and Gln92) on the other side (Fig. 1, D–E). This groove might be extended by residues 94–102 (connecting strand βC to helix α4), which are well conserved in fungi but undefined in electron density maps and, hence, absent from the final model. It is very likely that this loop will become ordered upon substrate binding. In addition, the rather negative electrostatic potential of this crevice suggests that it could interact with a molecule or peptide containing positive charges (Fig. 1F). In summary, the strong structural similarity between Tpa1N and human HIF prolyl-4-hydroxylase combined with the characteristics of the substrate binding groove suggest that Tpa1N has a prolyl-4-hydroxylase activity on Pro and Arg/Lys-containing peptides that remains to be identified.
The C-terminal Domain and Its Association with Tpa1N
Contrary to the Tpa1N domain, amino acid sequence analysis did not provide convincing predictions for the three-dimensional structure of the Tpa1C domain. Unexpectedly, this domain (residues 293–635) adopts a DSBH very similar to Tpa1N (r.m.s.d. of 2.5 Å over 153 Cα atoms despite only 13% sequence identity; Fig. 2A), suggesting an internal duplication event. The minor sheet of Tpa1C is only made of three strands (β7′, 4′, and 5′) as the antiparallel hydrogen bond pairing of residues corresponding to strand β2′ and the neighboring strand β7′ are not maintained (Fig. 1, A and B). The major sheet is composed of seven antiparallel β strands (order A′, 6′, 3′, 8′, 1′, C′, and B′). Seven of the eight α-helices are packed against one face of the major sheet. Two helices (αA′ and αB′) are specific of this domain, whereas the remaining five (α1′ to α5′) are structural matches of the corresponding helices (α1 to α5) from Tpa1N. Despite its structural resemblance with Tpa1N, the HXD…H catalytic triad is absent in Tpa1C and is replaced by residues Thr515, Cys517, and Glu610, which are not conserved in Tpa1 orthologs (Fig. 2A and supplemental Fig. S1). In addition, the cavity of the Tpa1C domain that corresponds to the active site in enzymes with a DSBH-fold is not lined by conserved residues and is largely occluded by the linker region (residues 261–290) connecting both Tpa1 domains. Altogether, these elements argue that this domain is very unlikely to be associated with any enzymatic activity.
FIGURE 2.

A, a stereo view representation of the superposition of Tpa1N (light brown) on Tpa1C (blue) is shown. B, a surface representation of residue conservation mapped at the surface of the Tpa1C domain is shown. Coloring is from blue (highly conserved) to gray (low conservation). C, Tpa1C interacting surface with Tpa1N is shown. D, a ribbon representation of the Tpa1 homodimer observed in the crystal is shown. One monomer is shown in blue, and the other one is in green. Tpa1N and Tpa1C are in light and dark colors, respectively. E, surface representation of residue conservation mapped at the surface of Tpa1 homodimer is shown (same color code as Fig. 2A). F, size-exclusion chromatography as followed by a triple detector array of Tpa1 is shown. For clarity only, the UV absorption (280 nm) for the eluted sample (left y axis) and molecular mass calculated from light scattering (right y axis, logarithmic scale) are shown. mAU, milliabsorbance units. G, shown is a surface representation of Tpa1 active site (same color code as Fig. 2A) with the HIFα CODD (yellow) and (Ser-Pro)5 (pink) peptides shown in ribbon. The hydroxylated proline is shown in sticks. The iron atom is shown as a black sphere. H, the representation is the same as panel G. Tpa1N and Tpa1C are colored gray and beige, respectively. The βB-βC β-hairpin is shown in green.
Tpa1C has a large and highly conserved surface area that mainly contacts Tpa1N domain (buried surface area of 1200 Å2; Fig. 2, B and C) and, hence, is not accessible. Contrary to Tpa1C, the region from Tpa1N engaged in interdomain interaction does not display significant sequence conservation. Both domains interact via orthogonal packing of their minor sheets (Fig. 1, A and B). This interaction positions the α-helical layers from both domains at the opposite sides of Tpa1. Tpa1C is located below the entrance of the Tpa1N active site and contributes to the formation of the conserved groove that runs across the Tpa1N active site (Fig. 1, D and E). In addition, one face of Tpa1C displays a negative electrostatic potential and is juxtaposed to the Tpa1N conserved and negatively charged groove (Fig. 1F). This results in a large surface with negative potential that could be implicated in binding to a positively charged partner.
As stated above, analysis of the crystal packing reveals a tightly packed crystallographic dimer that buries 2000 Å2 surface area, suggesting that Tpa1 forms dimers (Fig. 2, D and E). Dimer formation occurs between Tpa1C domains and is mediated by poorly conserved residues from helices αA′, α1′, αC′, α4′, and α5′, strand β1′, and from the loops connecting α-helix to αA′, αB′ to αC′, and β1′ to β2′. The core of this interface is mainly hydrophobic, with some polar residues involved in its periphery. Analysis of the quaternary structure of Tpa1 in solution using size exclusion chromatography coupled online to a triple detection array (Viscotek) indicated a molecular mass of 143.3 kDa close to the value expected for the homodimer (150 kDa compared with 75 kDa for the monomer, Fig. 2F). The presence of Tpa1 homodimers in vivo remains to be determined. However, it is noteworthy that among the three residues from S. cerevisiae Tpa1 that have been identified by in-depth phosphoproteome analysis (45), one (Ser293) is located within a loop (connection between α-helix to αA′) involved in this homodimeric interface. In our structure, this residue is not modified, but the protein has been produced in E. coli. Hence, we cannot exclude that the phosphorylation of Ser293 from Tpa1 will affect its quaternary structure and potentially its function.
To our knowledge this is the first example of a Fe(II)-2OG dioxygenase family with two DSBH domains in tandem. Cupin superfamily members (phosphomannose isomerase (46), oxalate decarboxylase (47), quercetin-2,3-dioxygenase (48), and the human transcription cofactor pirin (49)) also have repeated DSBH domains, but they are not 2OG-dependent enzymes. Tpa1 differs from these proteins as Tpa1 domains are independent, whereas in pirin and related proteins, the domains are connected to each other by a strand-swapping mechanism; the N-terminal fragment contributing a β-strand to the C-terminal domain and vice versa.
Modeling of Tpa1 Bound to a “Substrate” Peptide
The strong structural similarity of Tpa1 with prolyl-4-hydroxylases suggests Tpa1 is an enzyme that post-translationally modifies proline residues in the presence of oxygen. Among these enzymes, some display broad substrate specificity as exemplified by collagen and algal prolyl-4-hydroxylases that modify the central proline of the X-Pro-Gly motif at several positions within collagen and proteins from the lumen of plant endoplasmic reticulum (50, 51). On the contrary, the human HIF1α prolyl-4-hydroxylase (PHD2 or EGLN1) selectively modifies Pro564 from HIF1α in the presence of oxygen, thereby leading to its degradation by the proteasome (52). Very recently, the mode of substrate binding to prolyl-4-hydroxylases was revealed by two high resolution crystal structures. The first one corresponds to the complex between algal prolyl-4-hydroxylase bound to a peptide corresponding to the (Ser-Pro)5 motif (r.m.s.d. with Tpa1N of 2.4 Å over 152 Cα atoms, 15% sequence identity (53)). The second is the crystal structure of PHD2cat in complex with a 19-residues peptide (region 556–574 containing the targeted Pro564) from the C-terminal oxygen degradation domain (CODD) from HIF1α (r.m.s.d. with Tpa1N of 2.5 Å over 170 Cα atoms, 19% sequence identity (54)). The CODD peptide binds in an extended form to a well defined cleft of the PHD2cat active site. Using these complexes as templates, we modeled peptides within the Tpa1N putative active site (Fig. 2G). In both models (with the exception of the C-terminal part of the CODD peptide), no major steric hindrance between the peptides and Tpa1 was observed. The peptides fit into the highly conserved long groove containing the putative Tpa1N active site, and the hydroxylated proline is positioned close to the metal from the active site (distance between the Pro C4 atoms and the iron atom ≈ 4–5 Å). In both cases, the peptides have the same directionality within the active site groove (i.e. the N-terminal extremity from both peptides is located at the same end of the groove). The main difference between the Tpa1-peptide model structure and the prolyl-4-hydroxylase-substrate peptide complexes resides in the conformation of the β-hairpin made by strands βB and βC. In Tpa1, this region (residues 81–91) is in an open conformation and forms one side of the conserved groove (Fig. 2H). It is also engaged in a large interaction with Tpa1C. In the PHD2cat apo-structure, this region has an open conformation as observed for Tpa1N, whereas upon CODD peptide binding, it folds back onto the substrate and clamps Pro564 in the active site. This region undergoes the largest conformational changes upon substrate binding (54). Similar conformational changes occur upon peptide binding for the corresponding β-hairpin of algal prolyl-4-hydroxylase (53). Whether rearrangements of the Tpa1 β-hairpin take place upon substrate binding is hard to predict in the absence of any information on Tpa1 substrates. Rearrangements of this β-hairpin very likely would affect the interface between both Tpa1 domains. This would result in a different orientation of these domains, thereby exposing to solvent part of the highly conserved region of Tpa1C that interacts with Tpa1N.
Mutation of Conserved Residues in the Putative Tpa1 Catalytic Site Promote Stop Codon Readthrough
The implication of Tpa1 in translational readthrough, mRNA decay, and poly(A) tail length control (18) offered a possibility to test whether the putative catalytic residues of Tpa1 are important for its function. Similarly, the two domains of Ofd1, the S. pombe homolog of Tpa1, were shown to have different functions in the oxygen-dependent degradation of the active form of the Sre1 transcription factor (22). It was, thus, also interesting to test the role(s) of the two domains of Tpa1. For this purpose, we concentrated on the level of stop codon readthrough using reporter plasmids encoding two luciferases (Renilla and firefly) separated either by a UAA stop codon or by CAA encoding glutamine (Fig. 3A (18)). These constructs were introduced in a wild type yeast strain or a strain deleted for TPA1 (tpa1Δ). In addition, these plasmids were introduced in the tpa1Δ strain simultaneously with a plasmid encoding either the wild type TPA1 gene or mutant alleles. Stop codon readthrough was first assayed by measuring the relative activities of the two types of luciferase produced by these cells, with 100% readthrough being taken as the ratio observed for the construct with a CAA codon. Consistent with the previous report (18), deletion of the TPA1 gene resulted in a roughly 3-fold increase of the readthrough of the UAA stop codon (Fig. 3, B and C). Introduction of a plasmid-borne copy of the wild type TPA1 gene essentially corrected this defect, restoring low readthrough of the stop codon (Fig. 3B). To inactivate the catalytic activity of Tpa1, we substituted simultaneously His159 in alanine and Asp161 in asparagine. Substitution of either one of the equivalent residues in the related protein hABH3 totally abrogates its DNA repair activity (55). Interestingly, a plasmid encoding the H159A/D161N allele of the TPA1 gene failed to complement the tpa1Δ strain in the stop codon readthrough assay (Fig. 3B). The effect was not specific for luciferase reporter, as it was also observed by scoring the cell color and growth on media without adenine that report expression of the UGA-containing ade1-14 allele (supplemental Fig. S2). Similarly, plasmids encoding either the Tpa1N or Tpa1C domains failed to restore translation termination efficiency to a wild type level (Fig. 3C and supplemental Fig. S2). To exclude that mutation of the putative Tpa1 catalytic center or deletion of one or the other of its domains destabilized the resulting protein (which would have provided a straightforward explanation for their apparent lack of activity), the levels of the wild type and mutant proteins fused to an epitope tag were measured by Western blot (Fig. 3D). This demonstrated that the three mutants were expressed to a level similar to the wild type protein and were equally stable (even though a slightly lower level of the protein containing Tpa1C alone was detected). We conclude that the observed phenotypes do not result from lack of Tpa1. Thus, it appears that both domains of Tpa1 as well as its putative catalytic activity are required to favor stop codon recognition in yeast cells.
FIGURE 3.

Requirement of both Tpa1 domains and of conserved residues in its putative catalytic center for stop codon readthrough. A, shown is the structure of the reporter used to measure stop codon readthrough. Synthesis of mRNAs encoding both the Renilla and firefly luciferases is driven by a Pgk-1 promoter (P PGK). The two ORFs are separated either by a CAA codon encoding glutamine or by the UAA stop codon. Percent of stop codon readthrough is calculated as the ratio of firefly luciferase to Renilla luciferase for the stop codon-containing construct divided by the ratio of firefly luciferase to Renilla luciferase for the construct encoding glutamine ×100. B, the percent of UAA readthrough in wild type PSI+ cells (YJW618 strain transformed with the empty vector) or in the isogenic strain deleted for TPA1 (YJW619 strain) carrying either an empty vector, a plasmid encoding wild type TPA1 gene, or the Tpa1 H159A/D161N mutant is plotted. S.D. from five experiments are indicated. C, the experiments were the same as in B, except that plasmids encoding either Tpa1N or Tpa1C were used. D, shown is Western blot analysis of total cellular protein extracted from cells expressing Tpa1-TAP, Tpa1N-TAP, Tpa1-H159A/D161N-TAP, or Tpa1C-TAP.
Ett1, the S. cerevisiae Nro1 Homolog Also Affects Stop Codon Readthrough
Recently, a factor, Nro1, was shown to interact with Ofd1 in S. pombe and proposed to inhibit the ability of Ofd1 to degrade the active form of the Sre1 transcription factor (23). It is noteworthy that, using TAP purification (56), the Nro1 homolog in S. cerevisiae, Yor051c, was independently found to interact with Tpa1 (homologous to Ofd1) (28). To strengthen the parallel between these distantly related yeast species, we constructed a S. cerevisiae strain lacking the YOR051C gene and a strain in which both the TPA1 and YOR051C genes had been simultaneously inactivated. Both strains were transformed with the reporter plasmids, and the level of stop codon trans-reading was assayed (Fig. 4A). This demonstrated that, similarly to TPA1, YOR051C gene deletion increased stop codon readthrough. Moreover, the effects of TPA1 and YOR051C deletions were not additive, suggesting that they act in the same pathway. Altogether, these data support the idea that, as for Ofd1 and Nro1 in S. pombe, Yor051c and Tpa1 activities are linked in S. cerevisiae. However, although Nro1 was suggested to antagonize Ofd1 activity, we observed similar, rather than opposite effects of Tpa1 and Yor051c proteins on stop codon trans-reading. Due to this difference with S. pombe Nro1, we therefore propose to name the S. cerevisiae YOR051C gene product Ett1 for enhancer of translation termination 1.
FIGURE 4.

Ett1 affects stop codon readthrough. A, readthrough of the stop codon in the wild type strain (YJW618 strain), in the isogenic strain lacking either TPA1 or ETT1, or in both genes was measured as described in Fig. 3. B, increased readthrough of the UGA codon present in the ade1-14 allele in strains lacking TPA1, ETT1 is demonstrated by reduced accumulation of the red pigment in these cells on complete synthetic media (CSM) and proportional increased growth on complete synthetic media without adenine (CSM-ADE). Readthrough is decreased in PSI− cells (obtained after treatment with guanidine hydrochloride), but the effect of TPA1 and ETT1 deletion is still clearly detectable, indicating that Tpa1 and Ett1 do not act through the control of eRF3 aggregation. This is consistent with previously reported results (18). For this assay equivalent numbers of cells of the yeast strains of the indicated genotype were grown on complete synthetic media (CSM) or CSM without adenine (CSM-ADE) media for 3 days at 30 °C (using this strain background, at this time point, suppression of the ade1-14 mutation by PSI+ on these media was sufficiently limited to allow scoring of the suppression of ade1-14, resulting from the deletion of TPA1 or ETT1). Note that both in PSI+ and PSI− context, deletion of ETT1 had a lower effect on readthrough using both assays compared with the deletion of TPA1.
Tpa1 has already been shown to control stop codon readthrough independently of the status of the PSI factor that varies according to the aggregation state of the eRF3 proteins (18). To ascertain that this is the case and test whether this holds true for Ett1, we cured PSI+ cells by guanidine hydrochloride treatment and assayed the expression of ade1-14 through accumulation of derived red pigment and growth in media lacking adenine in PSI+ and [PSI−] strains carrying the TPA1 or ETT1 deletion or wild type as a control (Fig. 4B). Deletion of TPA1 and ETT1 genes enhanced stop codon readthrough in both PSI+ and [PSI−] contexts. We conclude that the effect of these proteins on stop codon readthrough does not occur exclusively through control of eRF3 aggregation. Quantitatively, for luciferase production, accumulation of red pigment, and growth on media without adenine, stop codon readthrough was lower in the ett1Δ strain compared with the tpa1Δ strain (Fig. 4, A and B). Both experiments reflect a general effect of Tpa1 and Ett1 on translation termination.
Tpa1 Deletion Affects Expression of Some Genes Responsive to Oxygen Concentration
Because Ofd1 regulates expression of a transcription factor mediating the response to oxygen concentration in S. pombe (22) and given the structural evidence implicating Tpa1 in an oxygen-dependent reaction, we asked whether expression of genes known to be affected by oxygen concentration is influenced by the presence of Tpa1. Genes under the control of Mga2 (57) and Hap1 (58) were tested for this purpose. Although we did not detect a significant difference of expression of a β-galactosidase reporter driven by a Mga2-sensitive promoter between wild type and tpa1Δ strains (data not shown), we observed significant changes of expression of reporters in which transcription was driven by activating sequences binding Hap1, such as the CYC1 UAS1 and the CYB2 UAS1 (Fig. 5). In both cases, β-galactosidase levels were increased roughly 4-fold in the tpa1Δ strain compared with the wild type isogenic control. Because Mga2-dependent promoters were not affected in this assay, this result suggests that Tpa1 represses specifically Hap1 activity by an unknown mechanism.
FIGURE 5.
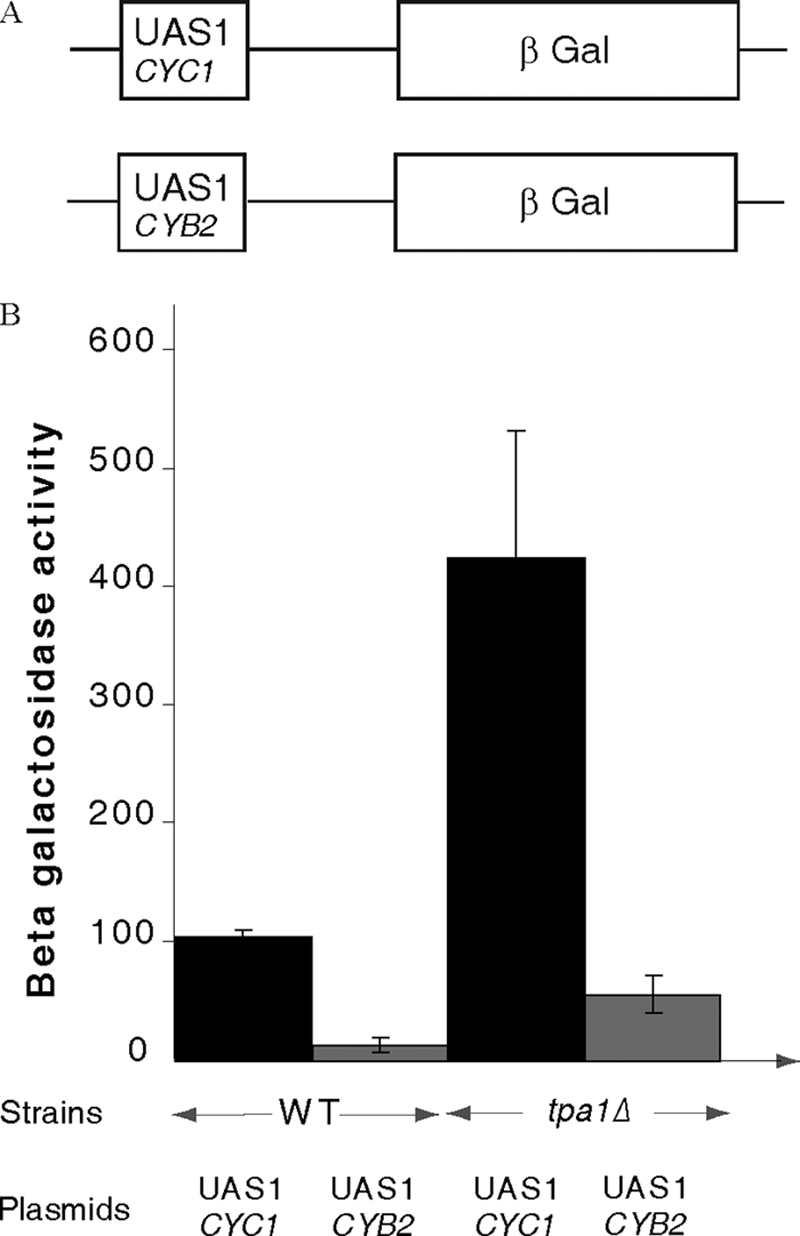
Tpa1 is required for full activity of Hap1-dependent promoters. A, structure of the two reporters used for this assay is shown. B, the levels of β-galactosidase activity measured for the reporter with the CYC1 UAS1 or the CYB2 UAS1 in either the wild type or the tpa1Δ strains are plotted with the standard deviation from three experiments.
DISCUSSION
In eukaryotes, the detection of the three mRNA stop codons in the A-site of the ribosome is ensured by two interacting translation termination factors: eRF1 and eRF3. Upon binding to the ribosome, this complex triggers hydrolysis of the ester bond connecting the polypeptide chain to the tRNA located in the ribosomal P-site, thereby releasing the newly synthesized protein. Accurate translation termination requires efficient recognition of stop codons (UAA, UAG, or UGA) by eRF1. Tpa1, a previously uncharacterized protein was recently shown to affect this process (18). In parallel, Ofd1, the S. pombe homolog of Tpa1, was implicated in the oxygen-dependent regulation of Sre1, the sterol regulatory element-binding protein from fission yeast (22). Due to the fact that independent studies in different organisms on the role of the Tpa1/Ofd1 orthologs yielded non-overlapping results, no unifying view on the Tpa1/Ofd1system has yet been proposed. To progress on this issue, we initiated a structure-based functional analysis of S. cerevisiae Tpa1. We have solved the crystal structure of Tpa1 to 2.8 Å resolution. As suggested for S. pombe Ofd1, Tpa1 is composed of two domains. Interestingly, both domains adopt the same DSBH-fold found in 2OG-Fe(II) dioxygenases (Figs. 1, A and B, and 2A (43)). The N-terminal domain (Tpa1N) harbors the catalytic residues (HXD…H triad) present in all members of this class of enzymes (Figs. 1C and 2A), whereas they are absent in the structurally very similar C-terminal domain. The three invariant residues are localized in the middle of a highly conserved groove of the N-terminal domain (Fig. 1E). The conservation of both folding and catalytically important residues between Tpa1 and prolyl-4-hydrolases (such as human PHD2 and algal prolyl-4-hydroxylase (44, 53) Fig. 1C) strongly suggests that Tpa1 possesses a prolyl-4-hydroxylase enzymatic activity.
Tpa1 and Ett1 Are Involved in Correct Translation Termination
We further investigated the involvement of Tpa1 in the control of stop codon readthrough, poly(A) tail length and mRNA stability (18). In our hands, we were not able to detect significant effects of Tpa1 deletion on poly(A) tail length and mRNA stability. However, consistent with the previous results of Keeling et al. (18), the deletion of TPA1 gene resulted in a roughly 3-fold increase of the readthrough of the UAA stop codon (Fig. 3, B and C). We also demonstrated that this effect was not gene-specific as tpa1Δ partly suppressed the ade1-14 (UGA stop codon) mutation both for growth in media lacking adenine and for the resulting accumulation of the red pigment (Fig. 4B). Wild type levels of stop codon readthrough were restored by transformation of the tpa1Δ strain with a plasmid encoding wild type Tpa1. Next, we tested whether the Tpa1 active site is involved in translation termination efficiency by replacing the invariant HXD motif from the triad involved in metal binding by the AXN sequence. This mutant did not complement the tpa1Δ to restore translation termination efficiency, proving that the integrity of Tpa1 putative active site is necessary for this process, at least at UAA and UGA stop codons. Because Hughes and Espenshade (22) suggested that the N-terminal domain of Ofd1 regulates, in an oxygen-dependent manner, the activity of its C-terminal domain on the degradation of Sre1N, we tested the role of the respective deletions of the Tpa1N and Tpa1C domains on stop codon readthrough. Both Tpa1 domains are required for translation termination to occur at a wild type level (Fig. 3C). Finally, in S. pombe, Nro1 was shown to interact with Ofd1, leading to the inhibition of its effect on Sre1N degradation (23). Interestingly, large scale comprehensive purification in yeast S. cerevisiae using TAP purification identified the Nro1 homolog (Yor051c) as a Tpa1 partner (28). Disruption of the YOR051C/ETT1 gene results in increased readthrough of stop codons present in the luciferase and ade1-14 reporters compared with wild type cells. Yet this effect is quantitatively less important than the one observed in tpa1Δ strains. In addition, the combined deletion of both TPA1 and ETT1 genes did not further diminish translation termination efficiency compared with individual deletions, supporting a concomitant action. These data support the idea that Ett1 and Tpa1 act in the same pathway in S. cerevisiae. The involvement of Tpa1 in correct translation termination sustains co-immunoprecipitation results, showing that Tpa1 interacts with translation termination factors eRF1 and eRF3 as well as with poly(A) binding protein Pab1 in S. cerevisiae (18). Tpa1 is very likely to be a prolyl-4-hydroxylase acting as an oxygen sensor (52), and the integrity of its active site is crucial to reduce stop codon readthrough. Therefore, we suspect that in the presence of oxygen, Tpa1 hydroxylates a proline residue in a specific target protein(s). Because it needs dioxygen as co-substrate, Tpa1 should be unable to hydroxylate its substrate during hypoxia. As a consequence, this could increase the level of inaccurate translation termination. Whether Tpa1 affects readthrough by directly modifying eRF1, eRF3, or Pab1 or another protein controlling these factors remains to be investigated. However, consistent with the results reported by Bedwell and co-workers (18), we observed that stop codon suppression still occurred both in PSI+ and PSI− strains, indicating that it does not occur through control of eRF3 aggregation.
Ett1, the S. cerevisiae Nro1 ortholog, has been identified in a genetic-wide screen aimed at identifying genes from S. cerevisiae that affect replication of a positive-strand RNA virus (Brome mosaic virus (BMV)) (59). Among the nearly 100 genes whose deletion either enhanced or reduced viral replication, many are involved in transcription, RNA processing, translation, and protein degradation. Deletion of ETT1 gene resulted in enhancement of viral replication, suggesting that Ett1 inhibits replication. This is not the first time that proteins from yeast, but also human, are identified to affect both RNA virus replication and translation. This is for instance the case for yeast proteins Pat1, Lsm1, and Lsm6, functioning in mRNA decapping. These proteins are involved in BMV RNA translation and are recruited from translation complexes to RNA replication complexes (60, 61). Similarly, in human cells, translation and replication of hepatitis C virus RNA depends on Pat1, Rck/P54 (Dhh1 in yeast), and the Lsm1–7 complex (62). However, it remains to be determined whether or not there is a functional link between the roles of Ett1 in inhibition of BMV replication and in enhancement of stop codon recognition or whether the effect of Ett1 on BMV results from altered termination of translation of a specific BMV replication factor.
Tpa1 Is Involved in Regulation of Hap1 Transcription Factor Activity
The precise identification of the cellular function of Tpa1 is complicated by the observation that the orthologous Ofd1 is implicated in an apparently completely different process, i.e. the oxygen-dependent regulation of Sre1, the fission yeast (or SREBP (22)). The S. pombe SREBP transcription factor is anchored at the endoplasmic reticulum membrane, but upon sterol depletion it is cleaved, and its N-terminal domain enters the nucleus to activate transcription of genes encoding sterol biosynthesis enzymes, as observed for its mammalian counterpart (24, 27). In addition, in response to low oxygen levels, Sre1 stimulates transcription of genes required for adaptation to hypoxia. In budding yeast, several transcription factors (Upc2, Ecm22, Mga2, Hap1, Rox1, …) have been implicated in the response to sterol depletion and/or hypoxia, but none present significant sequence homology with members of the SREBP family (57, 58, 63, 64). We have, therefore, investigated the effect of the deletion of TPA1 on genes controlled by Mga2 and Hap1 as representative of transcription factors affecting the response to oxygen concentration. Mga2 is an endoplasmic reticulum membrane protein activated during hypoxia by processing of the full-length protein into a smaller soluble transcription factor domain that is addressed to the nucleus similarly to S. pombe Sre1 and mammalian SREBP (57, 64, 65). Hap1 is a transcription factor involved in the complex regulation of gene expression in response to levels of heme and oxygen (66, 67). Our choice to study Hap1 was also motivated by the observation that the deletion of TPA1 gene was shown to rescue the synthetic lethality phenotype of a Δhap1Δupc2Δecm22 triple mutant (68).
No effect of the deletion of TPA1 on the expression of reporters regulated by the Mga2 transcription factor was detected (data not shown). In contrast, deletion of TPA1 generated significant changes (4-fold increase) in transcription of two reporters driven by the CYC1 UAS1 or the CYB2 UAS1 (Fig. 5). Expression of both CYC1 and CYB2 is stimulated by these sequences in the presence of oxygen through the Hap1 transcription factor (69–71). Our analysis, therefore, suggests that Tpa1 represses specifically Hap1 activity by an unknown mechanism.
The functional link between Tpa1 and Hap1 is reminiscent of that between Ofd1 and Sre1N (22). Interestingly, another prolyl-4-hydroxylase, human PHD2, has been implicated in the control of the activity of a family of transcription factors; that is, HIF, by hydroxylation of conserved proline residues that targets HIF factors to ubiquitin-mediated degradation (for review, see Ref. 72). We have experimental evidence that in S. cerevisiae, Tpa1 also influences the activity of a transcription factor, Hap1. Hap1 is structurally unrelated to SREBPs and HIF. However, Hap1 is degraded into smaller fragments in the absence of oxygen, whereas only the unprocessed form is observed in the presence of oxygen (58). We have observed that Tpa1 represses the activity of Hap1 (Fig. 5). Tpa1 functions very likely as an oxygen sensor through its N-terminal PHD domain, but the role of Tpa1 in modulating the activity of Hap1 remains to be clarified.
Conclusion
The resolution of the crystal structure of Tpa1 revealed that this protein contains two DSBH domains and that it is a putative prolyl-4-hydroxylase whose active site is located in its N-terminal domain. Tpa1 probably acts as an oxygen sensor system, and our genetic experiments show that it is involved in two unrelated cellular processes. First, Tpa1 reduces stop codon readthrough, and its enzymatic activity is critical for this function. Second, Tpa1 participates in the regulation of the transcription of genes by the Hap1 transcription factor and represses Hap1. Whether the implication of Tpa1 in these two pathways is mediated by a unique substrate protein that will further act on distinct proteins involved in these two processes or by at least two substrates implicated in each process remains to be elucidated.
Supplementary Material
Acknowledgments
We are indebted to K. Blondeau for help with SeMet labeling and V. Henriot, B. Bonneau, and D. Rentz for general work including competent cells, cloning, minipreps, and media. We thank D. M. Bedwell, C. E. Martin, M. Globerg, and B. Guiard for plasmids and yeast strains.
Note Added in Proof
While we were finalizing this article, the crystal structure of S. cerevisiae was described by Kim et al. ((2010) Nucleic Acids Res. 38, 2099–2110). In addition to the structure of the protein in a binary complex with Fe(III), they solved the structure of a ternary complex with Fe(III) and 2OG. No significant differences exist between all these structures. These authors also present results on poly(rA) binding by Tpa1.
This work was supported by La Ligue contre le Cancer “Equipe Labelisée 2008” and the CNRS. This work was also supported by the CNRS and Agence Nationale pour la Recherche Grant ANR-06-BLAN-0075-02 (to M. G.).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Table 1 and Figs. S1 and S2.
The atomic coordinates and structure factors (code 3MGU) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/).
- RF
- release factor
- 2OG
- 2-oxoglutarate
- DSBH
- double-stranded β-helix
- HIF1α
- hypoxia-inducible transcription factor
- CODD
- C-terminal oxygen degradation domain
- BMV
- Brome mosaic virus
- SREBP
- sterol regulatory element-binding protein
- SeMet
- selenomethionine
- r.m.s.d.
- root mean square deviation.
REFERENCES
- 1.Gallie D. R. (1991) Genes Dev. 5, 2108–2116 [DOI] [PubMed] [Google Scholar]
- 2.Pestova T., Lorsch J. R., Hellen C. U. T. (2007) in Translational Control in Biology and Medicine (Mathews M. B., Sonenberg N., Hershey J. W. B. eds) pp 87–128, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 3.Frolova L., Le Goff X., Rasmussen H. H., Cheperegin S., Drugeon G., Kress M., Arman I., Haenni A. L., Celis J. E., Philippe M. (1994) Nature 372, 701–703 [DOI] [PubMed] [Google Scholar]
- 4.Kisselev L. L., Buckingham R. H. (2000) Trends Biochem. Sci. 25, 561–566 [DOI] [PubMed] [Google Scholar]
- 5.Kisselev L., Ehrenberg M., Frolova L. (2003) EMBO J. 22, 175–182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Alkalaeva E. Z., Pisarev A. V., Frolova L. Y., Kisselev L. L., Pestova T. V. (2006) Cell 125, 1125–1136 [DOI] [PubMed] [Google Scholar]
- 7.Stansfield I., Jones K. M., Kushnirov V. V., Dagkesamanskaya A. R., Poznyakovski A. I., Paushkin S. V., Nierras C. R., Cox B. S., Ter-Avanesyan M. D., Tuite M. F. (1995) EMBO J. 14, 4365–4373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhouravleva G., Frolova L., Le Goff X., Le Guellec R., Inge-Vechtomov S., Kisselev L., Philippe M. (1995) EMBO J. 14, 4065–4072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Frolova L., Le Goff X., Zhouravleva G., Davydova E., Philippe M., Kisselev L. (1996) RNA 2, 334–341 [PMC free article] [PubMed] [Google Scholar]
- 10.Bonetti B., Fu L., Moon J., Bedwell D. M. (1995) J. Mol. Biol. 251, 334–345 [DOI] [PubMed] [Google Scholar]
- 11.von der Haar T., Tuite M. F. (2007) Trends Microbiol. 15, 78–86 [DOI] [PubMed] [Google Scholar]
- 12.Amrani N., Ganesan R., Kervestin S., Mangus D. A., Ghosh S., Jacobson A. (2004) Nature 432, 112–118 [DOI] [PubMed] [Google Scholar]
- 13.Cosson B., Couturier A., Chabelskaya S., Kiktev D., Inge-Vechtomov S., Philippe M., Zhouravleva G. (2002) Mol. Cell. Biol. 22, 3301–3315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Graille M., Heurgué-Hamard V., Champ S., Mora L., Scrima N., Ulryck N., van Tilbeurgh H., Buckingham R. H. (2005) Mol Cell 20, 917–927 [DOI] [PubMed] [Google Scholar]
- 15.Heurgué-Hamard V., Champ S., Engström A., Ehrenberg M., Buckingham R. H. (2002) EMBO J. 21, 769–778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Heurgué-Hamard V., Graille M., Scrima N., Ulryck N., Champ S., van Tilbeurgh H., Buckingham R. H. (2006) J. Biol. Chem. 281, 36140–36148 [DOI] [PubMed] [Google Scholar]
- 17.Mora L., Heurgué-Hamard V., de Zamaroczy M., Kervestin S., Buckingham R. H. (2007) J. Biol. Chem. 282, 35638–35645 [DOI] [PubMed] [Google Scholar]
- 18.Keeling K. M., Salas-Marco J., Osherovich L. Z., Bedwell D. M. (2006) Mol. Cell. Biol. 26, 5237–5248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Aravind L., Koonin E. V. (2001) Genome Biol 2, RESEARCH0007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chang B., Chen Y., Zhao Y., Bruick R. K. (2007) Science 318, 444–447 [DOI] [PubMed] [Google Scholar]
- 21.Clifton I. J., McDonough M. A., Ehrismann D., Kershaw N. J., Granatino N., Schofield C. J. (2006) J. Inorg. Biochem. 100, 644–669 [DOI] [PubMed] [Google Scholar]
- 22.Hughes B. T., Espenshade P. J. (2008) EMBO J. 27, 1491–1501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee C. Y., Stewart E. V., Hughes B. T., Espenshade P. J. (2009) EMBO J. 28, 135–143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Espenshade P. J., Hughes A. L. (2007) Annu. Rev. Genet. 41, 401–427 [DOI] [PubMed] [Google Scholar]
- 25.Todd B. L., Stewart E. V., Burg J. S., Hughes A. L., Espenshade P. J. (2006) Mol. Cell. Biol. 26, 2817–2831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hughes A. L., Lee C. Y., Bien C. M., Espenshade P. J. (2007) J. Biol. Chem. 282, 24388–24396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hughes A. L., Todd B. L., Espenshade P. J. (2005) Cell 120, 831–842 [DOI] [PubMed] [Google Scholar]
- 28.Krogan N. J., Cagney G., Yu H., Zhong G., Guo X., Ignatchenko A., Li J., Pu S., Datta N., Tikuisis A. P., Punna T., Peregrín-Alvarez J. M., Shales M., Zhang X., Davey M., Robinson M. D., Paccanaro A., Bray J. E., Sheung A., Beattie B., Richards D. P., Canadien V., Lalev A., Mena F., Wong P., Starostine A., Canete M. M., Vlasblom J., Wu S., Orsi C., Collins S. R., Chandran S., Haw R., Rilstone J. J., Gandi K., Thompson N. J., Musso G., St Onge P., Ghanny S., Lam M. H., Butland G., Altaf-Ul A. M., Kanaya S., Shilatifard A., O'Shea E., Weissman J. S., Ingles C. J., Hughes T. R., Parkinson J., Gerstein M., Wodak S. J., Emili A., Greenblatt J. F. (2006) Nature 440, 637–643 [DOI] [PubMed] [Google Scholar]
- 29.Hendrickson W. A., Horton J. R., LeMaster D. M. (1990) EMBO J. 9, 1665–1672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kabsch W. (1993) J. Appl. Crystallogr. 26, 795–800 [Google Scholar]
- 31.Schneider T. R., Sheldrick G. M. (2002) Acta Crystallogr. D Biol. Crystallogr. 58, 1772–1779 [DOI] [PubMed] [Google Scholar]
- 32.Bricogne G., Vonrhein C., Flensburg C., Schiltz M., Paciorek W. (2003) Acta Crystallogr. D Biol. Crystallogr. 59, 2023–2030 [DOI] [PubMed] [Google Scholar]
- 33.Terwilliger T. C. (1999) Acta Crystallogr. D Biol. Crystallogr. 55, 1863–1871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cowtan K. (2006) Acta Crystallogr. D Biol. Crystallogr. 62, 1002–1011 [DOI] [PubMed] [Google Scholar]
- 35.Emsley P., Cowtan K. (2004) Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 36.Adams P. D., Grosse-Kunstleve R. W., Hung L. W., Ioerger T. R., McCoy A. J., Moriarty N. W., Read R. J., Sacchettini J. C., Sauter N. K., Terwilliger T. C. (2002) Acta Crystallogr. D Biol. Crystallogr. 58, 1948–1954 [DOI] [PubMed] [Google Scholar]
- 37.Vagin A., Teplyakov A. (1997) J. Appl. Crystallogr. 30, 1022–1025 [Google Scholar]
- 38.Eaglestone S. S., Ruddock L. W., Cox B. S., Tuite M. F. (2000) Proc. Natl. Acad. Sci. U.S.A. 97, 240–244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Christianson T. W., Sikorski R. S., Dante M., Shero J. H., Hieter P. (1992) Gene 110, 119–122 [DOI] [PubMed] [Google Scholar]
- 40.Ramil E., Agrimonti C., Shechter E., Gervais M., Guiard B. (2000) Mol. Microbiol. 37, 1116–1132 [DOI] [PubMed] [Google Scholar]
- 41.Dziembowski A., Ventura A. P., Rutz B., Caspary F., Faux C., Halgand F., Laprévote O., Séraphin B. (2004) EMBO J. 23, 4847–4856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kushnirov V. V. (2000) Yeast 16, 857–860 [DOI] [PubMed] [Google Scholar]
- 43.Hausinger R. P. (2004) Crit. Rev. Biochem. Mol. Biol. 39, 21–68 [DOI] [PubMed] [Google Scholar]
- 44.McDonough M. A., Li V., Flashman E., Chowdhury R., Mohr C., Liénard B. M., Zondlo J., Oldham N. J., Clifton I. J., Lewis J., McNeill L. A., Kurzeja R. J., Hewitson K. S., Yang E., Jordan S., Syed R. S., Schofield C. J. (2006) Proc. Natl. Acad. Sci. U.S.A. 103, 9814–9819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Albuquerque C. P., Smolka M. B., Payne S. H., Bafna V., Eng J., Zhou H. (2008) Mol. Cell. Proteomics 7, 1389–1396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cleasby A., Wonacott A., Skarzynski T., Hubbard R. E., Davies G. J., Proudfoot A. E., Bernard A. R., Payton M. A., Wells T. N. (1996) Nat. Struct. Biol. 3, 470–479 [DOI] [PubMed] [Google Scholar]
- 47.Just V. J., Stevenson C. E., Bowater L., Tanner A., Lawson D. M., Bornemann S. (2004) J. Biol. Chem. 279, 19867–19874 [DOI] [PubMed] [Google Scholar]
- 48.Steiner R. A., Kalk K. H., Dijkstra B. W. (2002) Proc. Natl. Acad. Sci. U.S.A. 99, 16625–16630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pang H., Bartlam M., Zeng Q., Miyatake H., Hisano T., Miki K., Wong L. L., Gao G. F., Rao Z. (2004) J. Biol. Chem. 279, 1491–1498 [DOI] [PubMed] [Google Scholar]
- 50.Cassab G. I. (1998) Annu. Rev. Plant Physiol. Plant Mol. Biol. 49, 281–309 [DOI] [PubMed] [Google Scholar]
- 51.Myllyharju J. (2003) Matrix Biol. 22, 15–24 [DOI] [PubMed] [Google Scholar]
- 52.Schofield C. J., Ratcliffe P. J. (2004) Nat. Rev. Mol. Cell Biol. 5, 343–354 [DOI] [PubMed] [Google Scholar]
- 53.Koski M. K., Hieta R., Hirsilä M., Rönkä A., Myllyharju J., Wierenga R. K. (2009) J. Biol. Chem. 284, 25290–25301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chowdhury R., McDonough M. A., Mecinović J., Loenarz C., Flashman E., Hewitson K. S., Domene C., Schofield C. J. (2009) Structure 17, 981–989 [DOI] [PubMed] [Google Scholar]
- 55.Sundheim O., Vågbø C. B., Bjørås M., Sousa M. M., Talstad V., Aas P. A., Drabløs F., Krokan H. E., Tainer J. A., Slupphaug G. (2006) EMBO J. 25, 3389–3397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Puig O., Caspary F., Rigaut G., Rutz B., Bouveret E., Bragado-Nilsson E., Wilm M., Séraphin B. (2001) Methods 24, 218–229 [DOI] [PubMed] [Google Scholar]
- 57.Jiang Y., Vasconcelles M. J., Wretzel S., Light A., Martin C. E., Goldberg M. A. (2001) Mol. Cell. Biol. 21, 6161–6169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hon T., Dodd A., Dirmeier R., Gorman N., Sinclair P. R., Zhang L., Poyton R. O. (2003) J. Biol. Chem. 278, 50771–50780 [DOI] [PubMed] [Google Scholar]
- 59.Kushner D. B., Lindenbach B. D., Grdzelishvili V. Z., Noueiry A. O., Paul S. M., Ahlquist P. (2003) Proc. Natl. Acad. Sci. U.S.A. 100, 15764–15769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Díez J., Ishikawa M., Kaido M., Ahlquist P. (2000) Proc. Natl. Acad. Sci. U.S.A. 97, 3913–3918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Noueiry A. O., Diez J., Falk S. P., Chen J., Ahlquist P. (2003) Mol. Cell. Biol. 23, 4094–4106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Scheller N., Mina L. B., Galão R. P., Chari A., Giménez-Barcons M., Noueiry A., Fischer U., Meyerhans A., Díez J. (2009) Proc. Natl. Acad. Sci. U.S.A. 106, 13517–13522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Davies B. S., Rine J. (2006) Genetics 174, 191–201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hoppe T., Matuschewski K., Rape M., Schlenker S., Ulrich H. D., Jentsch S. (2000) Cell 102, 577–586 [DOI] [PubMed] [Google Scholar]
- 65.Jiang Y., Vasconcelles M. J., Wretzel S., Light A., Gilooly L., McDaid K., Oh C. S., Martin C. E., Goldberg M. A. (2002) Eukaryot. Cell 1, 481–490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Pfeifer K., Kim K. S., Kogan S., Guarente L. (1989) Cell 56, 291–301 [DOI] [PubMed] [Google Scholar]
- 67.Zhang L., Hach A. (1999) Cell. Mol. Life Sci. 56, 415–426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Valachovic M., Bareither B. M., Shah Alam Bhuiyan M., Eckstein J., Barbuch R., Balderes D., Wilcox L., Sturley S. L., Dickson R. C., Bard M. (2006) Genetics 173, 1893–1908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lodi T., Guiard B. (1991) Mol. Cell. Biol. 11, 3762–3772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Pfeifer K., Arcangioli B., Guarente L. (1987) Cell 49, 9–18 [DOI] [PubMed] [Google Scholar]
- 71.Pfeifer K., Prezant T., Guarente L. (1987) Cell 49, 19–27 [DOI] [PubMed] [Google Scholar]
- 72.Kaelin W. G. (2005) Annu. Rev. Biochem. 74, 115–128 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.