

Abstract
5 S rRNA is an essential component of ribosomes. In eukaryotic cells, it is distinguished by particularly complex intracellular traffic, including nuclear export and re-import. The finding that in mammalian cells 5 S rRNA can eventually escape its usual circuit toward nascent ribosomes to get imported into mitochondria has made the scheme more complex, and it has raised questions about both the mechanism of 5 S rRNA mitochondrial targeting and its function inside the organelle. Previously, we showed that import of 5 S rRNA into mitochondria requires unknown cytosolic proteins. Here, one of them was identified as mitochondrial thiosulfate sulfurtransferase, rhodanese. Rhodanese in its misfolded form was found to possess a strong and specific 5 S rRNA binding activity, exploiting sites found earlier to function as signals of 5 S rRNA mitochondrial localization. The interaction with 5 S rRNA occurs cotranslationally and results in formation of a stable complex in which rhodanese is preserved in a compact enzymatically inactive conformation. Human 5 S rRNA in a branched Mg2+-free form, upon its interaction with misfolded rhodanese, demonstrates characteristic functional traits of Hsp40 cochaperones implicated in mitochondrial precursor protein targeting, suggesting that it may use this mechanism to ensure its own mitochondrial localization. Finally, silencing of the rhodanese gene caused not only a proportional decrease of 5 S rRNA import but also a general inhibition of mitochondrial translation, indicating the functional importance of the imported 5 S rRNA inside the organelle.
Keywords: Mitochondria, Mitochondrial Transport, RNA, RNA-binding Protein, RNA Folding, Chaperone, Rhodanese
Introduction
5 S rRNA is a highly conserved essential component of large ribosomal subunits of all living organisms, the only exceptions being mitochondria of some protists, yeast, and mammals where 5 S rRNA is not encoded by mtDNA nor was it detected in mitoribosomes (1). This ∼120-nucleotide-long RNA molecule is distinguished by complex structural organization (for review see Ref. 2) as well as by its crucial role in translation (3). But one of the most striking features of eukaryotic 5 S rRNA is the unusual scheme of their intracellular transport. Synthesized outside the nucleolus by RNA polymerase III, they take a special export/re-import pathway to reach and complement nascent large ribosomal subunits (4, 5). Although this pathway was described in detail only in Xenopus oocytes, the data exist that quite a similar scheme of intracellular movements may be found in mammalian somatic cells; thus, it appears to be common for all vertebrates (6, 7).
Surprisingly, an interesting bypass from this circuit was reported in mammals: a portion of cytoplasmic 5 S rRNA pool was found to be redirected into the mitochondrial matrix (8–10). Neither mechanism of this interception nor the function of 5 S rRNA inside mitochondria is understood. Although basic requirements for the 5 S rRNA import resemble those for internalization of mitochondrial precursor proteins (dependence on ATP hydrolysis, electrochemical potential across the inner membrane, and functional pre-protein import apparatus), the former mechanism stands apart because of the different nature of the imported molecule and the necessity of some unknown cytosolic protein factors (10). In our recent study (11), two distinct structural elements of 5 S rRNA were identified as signals of its mitochondrial localization (Fig. 1A). We suggested that these import determinants correspond to binding sites with protein factors responsible for the mitochondrial targeting of 5 S rRNA. It was also hypothesized that 5 S rRNA, once exported from the nucleus, becomes a target of competition between two pathways. The first pathway functions through binding to the ribosomal protein L5, which carries the RNA into the nucleolus where both of them become incorporated into nascent ribosomal particles (5). The second pathway is ensured by import factors that address 5 S rRNA into mitochondria (11). Thus, understanding how the intracellular distribution of 5 S rRNA is regulated requires characterization of the protein factors directing its mitochondrial import.
FIGURE 1.

Identification of rhodanese as a factor of 5 S rRNA mitochondrial import. A, secondary structure of human 5 S rRNA and its main functional sites (from Ref. 11). B, fractioning scheme used for purification and identification of 5 S rRNA mitochondrial import factors. Profiles of absorbance at 280 nm of fractions obtained in step-by-step fractioning of 25–50% (red) and 65–80% (blue) differential ammonium sulfate precipitation preparations of HepG2 proteins are presented. Fractions active in 5 S rRNA import into isolated human mitochondria in combination with their counterparts (50% fraction derivate + 80% fraction derivate) are highlighted. AU, absorbance units. C, imported into isolated human mitochondria, 32P-labeled 5 S rRNA was detected by denaturing gel electrophoresis followed by Typhoon scanning. Import assays were performed in the presence of HepG2 protein fractions obtained by differential ammonium sulfate (AS) precipitation (upper panel) and by subsequent chromatography fractioning (lower panel). 5–10 μg of protein/assay were used. 1% of input corresponds to 5 fmol of labeled 5 S rRNA. D, separation of the 14H/50AS fraction by SDS-PAGE (Coomassie) and the corresponding autoradiography after incubation with 32P-labeled 5 S rRNA as a probe for Northwestern (NW) analysis. The band corresponding to rhodanese is marked with an arrow. E, in vitro import of 5 S rRNA in the presence of purified rhodanese (15 pmol) and the 80AS fraction. F, dependence of the 5 S rRNA in vitro import on the presence of increasing amounts of native or urea-denatured rhodanese. G, inhibition of 5 S rRNA import into isolated human mitochondria by preincubation of total protein extract with increasing amounts of anti-rhodanese or anti-ALDH2 antibodies. w/o, without.
In this work, we identify one of these factors as mitochondrial thiosulfate sulfurtransferase, rhodanese (EC 2.8.1.1), a member of a vast protein superfamily with very diversified functions (12). We found that rhodanese binds to 5 S rRNA cotranslationally to form a tight complex where the protein is preserved in its enzymatically inactive conformation. Upon this interaction, 5 S rRNA shows certain functional traits characteristic of Hsp40-type cochaperones involved in mitochondrial precursor targeting. Because only one particular conformation of 5 S rRNA was found to possess such an activity, an elegant 5 S rRNA import mechanism based on a series of reciprocal chaperone events can be proposed. Identification of rhodanese as a 5 S rRNA import factor provided us with a tool to demonstrate, for the first time, the functional significance of the 5 S rRNA import for translational activity of mammalian mitochondria.
EXPERIMENTAL PROCEDURES
Fractioning of Total Protein Extract of Human Cells
HepG2 cells grown on the standard DMEM (13) were resuspended in a small volume of the NPMD buffer (20 mm sodium/phosphate buffer, pH 6.5, 150 mm NaCl, 1 mm MgCl2, 5 mm dithiothreitol, 0.2 mm PMSF, 1× proteinase inhibitor mixture; Roche Applied Science) and disrupted by sonication on ice. The resulting lysate was cleared twice at 16,000 × g for 10 min. 0.1% polyethyleneimine (pH 6, adjusted with HCl) was added to the supernatant to remove nucleic acids, and the supernatant was incubated on ice for 20 min and then centrifuged for 30 min at 16,000 × g. The resulting supernatant was subjected to the differential (0–25, 25–50, 50–65, and 65–80%) ammonium sulfate precipitation. The pellets were collected by centrifugation at 16,000 × g for 20 min, dialyzed overnight against the NPMD buffer, and filtered through 0.45-μm bacterial filters. The 25–50% (50AS) and the 65–80% (80AS) fractions were further subjected to gel filtration on HiPrep 16/60 Sephacryl S-200 high resolution. The column was pre-equilibrated with TBS buffer (50 mm Tris-HCl, pH 8, 150 mm NaCl). Fractions active in in vitro 5 S rRNA import assays were then loaded on the HiTrap Mono Q column (elution with 20 mm Tris-HCl, pH 7, 0.2 mm PMSF, and the linear NaCl gradient from 0 to 3 m). Resulting active fractions were analyzed by affinity chromatography on heparin-Sepharose (10 mm sodium/phosphate buffer, pH 7, 50 mm NaCl, elution with the linear NaCl gradient from 50 to 1500 mm).
5 S rRNA Construction
All the 5 S rRNA versions used are described in detail in Ref. 11. Corresponding PCR-generated genes were transcribed in vitro using the T7 RiboMAX Express large scale RNA production system (Promega), and the resulting transcripts were purified by denaturing gel electrophoresis.
Import of 5 S rRNA into Isolated Human Mitochondria
Human mitochondria were isolated from HepG2 cells as described previously (10, 14). The standard RNA import assay was performed as described previously (10). For this, mitochondria were incubated in the Import Mix (0.6 m sorbitol, 20 mm Hepes-KOH, 20 mm KCl, 2.5 mm MgCl2, 1 mm ATP, 5 mm dithiothreitol, 0.5 m phosphoenol pyruvate, pH 7.5) with [γ-32P]ATP-labeled 5 S rRNA in the presence of HepG2 cytosolic protein fractions and/or purified rhodanese at 37 °C for 10 min. Nonimported RNAs were digested with RNase A; mitochondrial RNAs were isolated and imported RNAs detected by denaturing PAGE followed by Typhoon-Trio (GE Healthcare) scanning and quantification. 1–5% of 5 S rRNA taken into the assay (“input”) were run in parallel for calculation of import efficiency. In all experiments, negative controls without mitochondria or proteins were performed. In some experiments, cytosolic protein preparation was preincubated with anti-rhodanese or anti-ALDH2 polyclonal rabbit antibodies (Santa Cruz Biotechnology) at 4 °C for 30 min.
Northwestern Blotting
5–10 μg of proteins were resolved by SDS-PAGE and electrotransferred on a nitrocellulose membrane. The membrane was then incubated in 0.1 m Tris-HCl, 20 mm KCl, 2.5 mm MgCl2, 0.1% Nonidet P-40, pH 7.5, at 4 °C for 1 h, washed several times with the same solution, and blocked in 10 mm Tris-HCl, pH 7.5, 5 mm Mg(CH3COO)2, 2 mm dithiothreitol, 5% BSA, 0.01% Triton X-100 for 5 min at 4 °C. Hereafter, the membrane was incubated at 37 °C for 2 h in the Import Mix containing 1 nm [γ-32P]ATP-labeled human 5 S rRNA. The membrane was then washed with the Import Mix without 5 S rRNA and autoradiographed using the PhosphorImager system (Fuji). The corresponding bands on the SDS gel were excised and analyzed by MALDI-TOF/Mascot.
Silencing of rhodanese genes was ensured by two consecutive transfections of HepG2 cells spaced by 3 days with a mixture of three cognate siRNAs (Santa Cruz Biotechnology) in accordance with the manufacturer's protocol. For control, cells were transfected with siRNA against luciferase mRNA, as described previously (15), or mock-transfected. Both transfected and mock-transfected cells were analyzed for rhodanese expression 3 and 13 days after the second transfection by Western blotting with rhodanese-directed polyclonal rabbit antibodies (Santa Cruz Biotechnology). Mitochondria were isolated and purified as described previously (14) and treated with RNase A and digitonin to remove nonspecifically attached RNAs. Total and mitochondrial RNAs were isolated with TRIzol reagent (Invitrogen) and analyzed by Northern blot hybridization.
For respiration tests, 103 of wild-type or siRNA-transfected cells were grown in parallel in DMEM or in DMEM where glucose was replaced by galactose (4 g/liter). Cells were periodically detached from the wells and counted in a standard hemocytometer chamber, and the number of living cells was controlled with trypan blue.
Analysis of products of mitochondrial translation in human cells was performed as described previously (15). For this, 0.5 × 106 semi-confluent cells were preincubated at 37 °C for 5 min in DMEM containing 0.5% dialyzed calf serum without methionine with 0.2 g/liter emetine, and 200 mCi/liter [35S]methionine was then added (>1000 Ci/mmol, Amersham Biosciences) and incubated for 30 min. Finally, 0.1 mm nonlabeled methionine was added to the medium and incubated for 10 min. Cells were then disrupted, and the translation products were analyzed by 10–20% gradient SDS-PAGE followed by autoradiography.
Rhodanese Manipulations
Bovine liver rhodanese (Sigma) was stored in 50 mm Tris-HCl, 20 mm dithiothreitol, 50 mm sodium thiosulfate, pH 7, at −80 °C.
Urea-denatured rhodanese was prepared as described previously (16). For this, the solution of the native enzyme (5 g/liter) was diluted with 3 volumes of 8 m urea, 150 mm KCl, 24 mm NaCl, 10 mm dithiothreitol in 1× TBE buffer and incubated at 25 °C for 1 h. The residual enzymatic activity was not higher than 4%.
The aggregation assay was performed as described previously (16). For this, 6 μl of urea-denatured rhodanese were rapidly diluted in 100 μl of 100 mm HEPES-NaOH, 150 mm KCl, 24 mm NaCl, 10 mm MgCl2, pH 7 (pre-equilibrated at 25 °C for 5 min), containing as additives various 5 S rRNA versions or other nucleic acids, and thoroughly mixed for exactly 15 s. Immediately, the kinetics of absorbance at 320 nm (reflecting growth of turbidity caused by large aggregates) was chased.
The reactivation of urea-denatured rhodanese was carried out as described previously (17). For this, denatured rhodanese was diluted with 200 mm β-mercaptoethanol, 50 mm Na2S2O3, 50 mm Tris-HCl, pH 7.8, to a concentration of 3.6 mg/liter and incubated at 25 °C. Aliquots of the reaction mixture were taken for enzymatic activity measurements. The activity of the equivalent amount of the native enzyme was taken as 100%.
Measurement of enzymatic activity (catalysis of sulfur transfer from thiosulfate to cyanide, followed by formation of the complex between Fe3+ and thiocyanate produced in the reaction to develop the coloration): 3–10 μl of rhodanese-containing solution were mixed with 100 μl of freshly prepared buffer, composed of 40 mm KH2PO4, 25 mm Na2S2O3, 50 mm KCN, and incubated at 25 °C for 10 min. The reaction was stopped by addition of 50 μl of 18% formaldehyde. Coloration was developed by mixing with 150 μl of freshly prepared reagent, containing 66.7 g/liter Fe(NO3)3·9H2O, 8.67% HNO3, and measured at 460 nm in 1-cm cuvettes. Thermo-inactivation of rhodanese was followed at 25 °C in 50 mm Tris-HCl, 10 mm MgCl2, pH 7.8, with final rhodanese concentration at 10 mg/liter.
UV absorbance spectra measurements were carried out with the NanoDrop-1000 (LabTech). For this, 0.25 μm rhodanese solutions in the buffer, containing 0.21 m sorbitol, 7 mm HEPES-KOH, 7 mm KCl, 0.875 mm MgCl2, 0.35 mm ATP, 1.75 mm dithiothreitol, 0.175 mm phosphoenolpyruvate, pH 7.5, + 0.05× phosphate buffer (Sigma) + 0.15× TBE buffer, were used. The denatured enzyme was diluted in the buffer to make the final concentrations of all components equal for all samples (the final concentration of urea being 1.25 m). All spectra were read against identical protein-free controls, in 20 s after beginning of aggregation (early stage) and in 15 min (aggregation completed). Mitochondrial import assay of 35S-labeled rhodanese synthesized in rabbit reticulocyte lysate was performed as described previously (18).
Electrophoretic Mobility Shift Assay
1–10 nm purified 32P-labeled RNA was denatured at 100 °C in the presence of either 1 mm EDTA or 5 mm MgCl2 and then slowly cooled down to 25 °C. It was then incubated with rhodanese in the Import Mix for 10–15 min. Resulting complexes were analyzed by electrophoresis in 8% PAAG,2 0.5× TBE, 5% glycerol in 0.5× TBE buffer at 10 V/cm at 4 °C followed by Typhoon-Trio (GE Healthcare) scanning and quantification. The standard Scatchard plot construction and analysis were described previously (19). Because the resulting curves are concave (indicating the presence of several binding sites, a consequence of the complex mixture of rhodanese forms produced after urea dilution (17, 20)), only estimation of average dissociation constants was performed.
Cotranslational binding of 5 S rRNA to rhodanese was studied as described previously (21). Human rhodanese cDNA was kindly provided by F. Broly. Synthesis of [35S]Met-labeled human rhodanese was carried out in TnT T7 Quick for PCR DNA (Promega) according to the manufacturer's protocol. 10 μm human 5 S rRNA labeled with ChromaTide Alexa Fluor 488-5-UTP (Invitrogen) was added to the translation mixture at the very beginning of synthesis (for cotranslational binding) or in 70 min after launching translation (post-translational binding). Translation products were analyzed by SDS gel and by native gradient 4–10% PAAG, 0.5× TBE, 0–30% glycerol electrophoresis (10 V/cm, 4 °C). Both RNA and protein signals were measured with Typhoon-Trio scanner.
Native PAGE of 5 S rRNA
T7 transcripts were purified on denaturing gel to homogeneity. 0.1–0.2 μg of the RNA was incubated at 100 °C for 1 min in 10 μl of either 1 mm MgCl2 (to obtain 5 S rRNAMg) or 1 mm EDTA (to obtain 5 S rRNAEDTA). Then the RNA was renatured by slow cooling down to 25 °C, mixed with 2 μl of 0.25% bromphenol blue, 0.25% xylene cyanol, 30% glycerol, and loaded on native 8% PAAG in 1× TBE buffer. Samples were run at 10 V/cm until the bromphenol blue was out. Positions of RNA bands were visualized by ethidium bromide staining.
In-gel Förster Resonance Energy Transfer (FRET)
Human 5 S rRNA was preassembled from three oligoribonucleotides (Integrated DNA Technologies) in a way that the extremity of the β-domain carried a Cy-3 moiety; the extremity of the γ-domain was labeled with fluorescein (Fig. 5B) and that of the α-domain bore the [32P]phosphate. Both fluorophores were attached to CC-dinucleotides; this environment was optimal for their mobility (22). Annealing was performed in two steps as follows: (i) oligonucleotides “2” and “3” (see Fig. 5, B and C) (0.4 μm each) were heated together to 85 °C in 40 mm NaCl, 0.5 mm MgCl2, or 1 mm EDTA and then left to cool down slowly to 50 °C; (ii) the resulting mixture was added to 1 μm oligonucleotide “1” (see Fig. 5, B and C), heated to 60 °C, and left to cool down slowly overnight. Forms obtained either in the presence (5 S rRNAMg) or in the absence (5 S rRNAEDTA) of Mg2+ were run in parallel in 8% PAAG, 0.5× TBE, 200 V at 4 °C, and then their fluorescent (fluorescein, excitation at 488 nm/emission at 520 nm; cyanin-3, excitation at 532 nm/emission at 580 nm) and radioactive (32P) signals were measured with the use of a Typhoon (Amersham Biosciences) in wet gel and quantified with ImageQuantTM. 5 S rRNA preassembled from the same oligonucleotides but without any fluorescence acceptor on the β-domain was used as control. The FRET efficiency (E) was calculated by fluorescein quenching, with the use of linearization of the standard Equation 1,
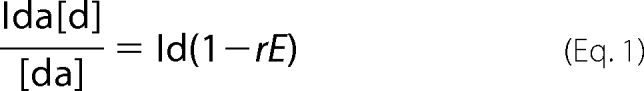 |
where Ida and Id are fluorescein fluorescence intensities in 5 S rRNAs with both donor and acceptor, and with donor alone, respectively; [da] and [d] are radioactive signals of 5 S rRNAs with both donor and acceptor, and with donor alone, respectively, r indicates efficiency of labeling with the donor fluorophore (r = 1, in our case). The distance between fluorophores (R) was calculated from Equation 2,
 |
where R0 indicates the Förster critical distance (for the pair fluorescein/cyanin-3 under these conditions it is usually ∼56 Å; see for example Ref. 23).
FIGURE 5.

Conformation of human 5 S rRNA in solution. A, conformations of 5 S rRNA with various loop A organizations (as proposed by Ref. 56) and the putative order of their relative mobilities in native gel. Domains are colored as follows: α, yellow; β, red; and γ, blue. B, secondary structure of human 5 S rRNA assembled from oligonucleotides labeled with the following: 1) 32P-phosphate; 2) cyanin-3; and 3) fluorescein. C, typical native gel used for separation of annealing products and in gel FRET measurements. Superposition of fluorescein (green), cyanin-3 (red), and 32P (blue) signals. D, example of in gel FRET measurement for human 5 S rRNAMg (in gel fluorescence signals and the autoradiography of the same gel are presented, see “Experimental Procedures” for more details) and linearization of experimental data used for calculation of FRET efficiencies in 5 S rRNAMg and 5 S rRNAEDTA. E, model of domain level tertiary structures of human 5 S rRNAMg and 5 S rRNAEDTA based on in gel FRET data (Table 2) and known geometric parameters (57).
RESULTS
Import of 5 S rRNA into Isolated Human Mitochondria Necessitates at Least Two Protein Factors
To search for proteins involved in the 5 S rRNA mitochondrial import, the total HepG2 cell protein extract was subjected to multistep fractioning (Fig. 1B). On every step, resulting fractions were tested for their capacity to direct import of radioactively labeled 5 S rRNA into isolated HepG2 mitochondria. None of the separate fractions was found to be import-competent. In contrast, 5 S rRNA readily accumulated in mitochondria when a particular combination of two distinct fractions was used (Fig. 1C), suggesting requirement of at least two soluble proteins for 5 S rRNA import into the organelles.
Rhodanese as a 5 S rRNA Import Factor
Because RNA import factors are assumed to recognize and to associate with their cargo, we checked the presence of 5 S rRNA-binding proteins in one of the final active fractions (14H/50AS) by Northwestern analysis (Fig. 1D). Two major proteins of ∼34 and ∼55 kDa were revealed. Among candidates identified in this fraction by MALDI-TOF/MS (Table 1), a 33.4-kDa enzyme, thiosulfate sulfurtransferase (rhodanese), was the best scored. The other protein able to interact with 5 S rRNA is probably another mitochondrial enzyme, aldehyde dehydrogenase or its cytosolic precursor.
TABLE 1.
Protein components of the fraction 14H/50AS identified by MALDI-TOF/MS as putatively capable of 5 S rRNA binding
| Protein | Mass | Localization | Probability-based Mowse score |
|---|---|---|---|
| kDa | |||
| Aldehyde dehydrogenase | 55 | Mitochondrial inner membrane | 86 |
| Precursor of aldehyde dehydrogenase | 57 | Cytosol (moves to mitochondria) | 84 |
| Annexin V | 36 | Cytosol, nucleus | 60 |
| Precursor of aldo-keto reductase 2 | 37 | Cytosol (moves to mitochondria) | 150 |
| Regucalcin | 34 | Nucleus, cytosol | 76 |
| Rhodanese | 33 | Mitochondrial matrix, cytosol | 185 |
| Glutathione S-transferase | 26 | Cytosol | 81 |
Rhodanese appeared to be of great interest because it is known to be synthesized on free cytosolic ribosomes and then imported into mitochondria (24, 25). Rhodanese homology superfamily embraces proteins involved in diverse processes, including cyanide detoxication, Fe/S cluster formations, and other redox reactions as well as intracellular transport and regulatory pathways (12). Thus, rhodanese seemed to be a valuable candidate for the role of 5 S rRNA mitochondrial import factor. To check this possibility, we performed a series of in vitro import assays, where purified bovine liver rhodanese (89.6% identical to the human one), combined with the 80AS fraction, was shown to be as active in directing 5 S rRNA into human mitochondria as the whole fraction 14H/50AS (Fig. 1E). The 5 S rRNA import efficiency increased in a saturable manner upon growth of rhodanese concentration, and vice versa, preincubation of the cytosolic protein extract with anti-rhodanese antibodies led to a significant decrease or even total loss of its ability to direct the import (Fig. 1, F and G). In the same experiment, antibodies specific to another putative 5 S rRNA-binding protein, aldehyde dehydrogenase, had no effect on the 5 S rRNA mitochondrial import (Fig. 1G). These data strongly suggest that rhodanese was indeed the 5 S rRNA import factor present in the 14H/50AS fraction.
Inhibition of Rhodanese Expression Affects the 5 S rRNA Import into Human Mitochondria and Mitochondrial Translation in Vivo
To validate rhodanese as putative 5 S rRNA import factor in vivo, we studied dependence between the expression level of the protein and the amount of 5 S rRNA inside mitochondria in living human cells. For this, expression of rhodanese genes was inhibited by transient transfection of HepG2 cells with specific siRNAs, and the effect of this inhibition on the 5 S rRNA import was studied (Fig. 2, A and B). Strong correlation between the intracellular level of rhodanese and the mitochondrial 5 S rRNA level was observed. The decrease of rhodanese expression led to a 4-fold drop of 5 S rRNA import (the minor amounts of remaining mitochondrial 5 S rRNA may reflect its longer turnover time than that of rhodanese). On the contrary, restoration of expression 13 days after transfection was marked by equally proportional growth of the 5 S rRNA level inside mitochondria. Neither transcription of mitochondrial DNA nor the overall cellular 5 S rRNA level was affected, indicating that the effect of the short term withdrawal of rhodanese on the 5 S rRNA import was specific. This result indicates that rhodanese is indeed essential for the 5 S rRNA import in vivo.
FIGURE 2.

Effect of rhodanese knockdown on the 5 S rRNA import into HepG2 mitochondria and mitochondrial translational activity. The Western blot of total cellular protein extracts (A), the corresponding Northern blot of total and mitochondrial RNAs (B), and the autoradiography of SDS gels with mitochondrial translation products (C) are provided. w/o siRNA, mock-transfected cells (showed no difference from ones transfected with siRNA against luciferase mRNA, data not shown); siRNA, cells transfected with rhodanese mRNA-directed siRNAs; siRNA + 13 day, recovery of rhodanese expression 13 days after the 2nd transfection. The correlation between the rhodanese expression, the 5 S rRNA mitochondrial level, and the general mitochondrial translation level is presented in the histogram. The values in mock-transfected cells were taken as 100%. Error bars represent the result of two independent experiments. D, growth curves of cultures transfected with rhodanese mRNA-directed siRNAs or nontransfected HepG2 cells, grown on glucose (Glc)- or galactose (Gal)-containing DMEM.
Intriguingly, those changes in the mitochondrial level of 5 S rRNA were found to be associated with proportional decrease/restoration of the general mitochondrial translational activity (Fig. 2C). Cells transfected with rhodanese mRNA-directed siRNAs also demonstrated significantly slower growth on the galactose-containing medium, indicating insufficient mitochondrial respiration activity (Fig. 2D). Because the function of rhodanese inside mitochondria does not seem to be associated with translation or formation of respiratory complexes (12), deletion of its putative orthologue (YOR251c) in yeast does not produce a respiration phenotype on galactose medium (26) (and, noteworthy, no 5 S rRNA import was found to exist in yeast cells (10)); this effect appears to be directly linked to the level of 5 S rRNA inside mitochondria. This is the first demonstration of functional importance of the imported 5 S rRNA for mitochondrial translation in mammalian cells.
Misfolded Rhodanese Forms a Stable Complex with 5 S rRNA
Until now, no data on RNA-binding properties of rhodanese have been reported. We proceeded with direct analysis of 5 S rRNA-rhodanese interaction, using the mobility shift assay. Surprisingly, only a weak association (apparent Kd >35 μm) of human 5 S rRNA with the native enzyme could be observed (data not shown). It is known that the de novo synthesized rhodanese, like most precursor proteins, is present in cytosol in an enzymatically inactive state with a changed conformation, and it acquires its activity only upon import into mitochondria (27–29). Therefore, we checked the misfolded rhodanese, obtained by denaturation with urea followed by dilution of the denaturant (16), and have found that it does bind 5 S rRNA specifically and with a very high affinity (apparent Kd value estimated by Scatchard analysis ∼20 pm; Fig. 3, A–C). Furthermore, the misfolded rhodanese also proved to be significantly more active than the native one in the 5 S rRNA in vitro import assay (Fig. 1F).
FIGURE 3.

Rhodanese-5 S rRNA interaction. A, mobility shift assay of complexes formed by human 5 S rRNA with urea-denatured rhodanese. The major rhodanese-5 S rRNA complex is marked with an arrow. The additional band may correspond to a minor complex (which was not always observed). B, Scatchard plot estimation of the dissociation constant for the rhodanese-5 S rRNA complex. C, mobility shift assays of rhodanese-5 S rRNA complexes formed in the presence of either nonspecific (yeast tRNA) or specific (nonlabeled human 5 S rRNA) competitors. Molar excess of competitors is marked above the autoradiographies. D, mobility shift assays of misfolded rhodanese complexed with a γ-domain (Δ(78–98)) or an α-domain (7G→U, 8G→A, and 9C→U) mutants of human 5 S rRNA. The total concentrations of rhodanese (in μm) are provided above. The major complex is marked with an arrow. E, Scatchard plot estimation of the dissociation constant for the complex of rhodanese with the γ-domain mutant of 5 S rRNA (Δ(78–98)). F, cotranslational binding of rhodanese to 5 S rRNA. The native gel (upper panel) shows the rhodanese-5 S rRNA complex formed during translation of rhodanese mRNAs in a rabbit reticulocyte system under various conditions. Uncomplexed rhodanese molecules remained on the top of the gel (data not shown). Lower panel, autoradiography of the same amounts of rabbit reticulocyte lysates separated by SDS-PAGE, showing the total amount of newly synthesized rhodanese.
Previously, we hypothesized that signals of mitochondrial localization of 5 S rRNA (Fig. 1A) may match its binding sites with import factors (11). This is why it was of particular interest to verify if they were involved in interaction with rhodanese. Indeed, deletion of the γ-domain site led to a dramatic (>200-fold) increase of the complex dissociation constant, whereas disruption of the α-domain site resulted in almost total loss of binding capacity (Fig. 3, D and E). These data suggest that the α-domain site functions as the main rhodanese-binding platform, whereas the γ-domain may contain a region of additional, less important contact needed for more tight binding. All the data show that rhodanese in its misfolded form recognizes particular sites of 5 S rRNA, needed for its mitochondrial localization, binds this RNA with high affinity, and thus can perform the function of 5 S rRNA carrier to mitochondria.
Rhodanese Binds 5 S rRNA Cotranslationally
The results above raised the question of physiologic relevance of 5 S rRNA interaction with misfolded rhodanese. One can expect that this kind of association may occur cotranslationally with the nascent protein. Indeed, it is known that rhodanese is bound by chaperones in a cotranslational manner, although it is still remaining in mainly an unfolded state, which can be critical for acquisition of its import-competent conformation (30). On the other hand, cotranslational binding of 5 S rRNA was demonstrated earlier as well: the ribosomal protein L5 synthesized in a mammalian in vitro translation system was found in the complex with 5 S rRNA only when the latter was present in the reaction medium from the very beginning of the assay (21). We performed a similar experiment to test the affinity of the de novo synthesized rhodanese to 5 S rRNA. For this, human 35S-labeled rhodanese was synthesized in a rabbit reticulocyte lysate system in the presence of fluorescently labeled 5 S rRNA, and the products of translation were analyzed by native gel electrophoresis. 36 ± 2% of the newly synthesized protein was found associated with 5 S rRNA when the latter was added at the very beginning of the assay (Fig. 3F). In contrast, addition of 5 S rRNA after completion of rhodanese synthesis resulted in a significantly lower binding (10 ± 1%). A small but notable amount of rhodanese (6 ± 1%) was reproducibly found in complex with 5 S rRNA naturally present in the reticulocyte lysate, even if no additional 5 S rRNA molecules were introduced into the system. Noteworthy, this was the only rhodanese-RNA complex formed under these conditions; even addition of an excess of Escherichia coli tRNAs did not change this pattern (Fig. 3F). These data prove that nascent rhodanese can specifically bind to 5 S rRNA.
5 S rRNA Functions as a Chaperone for Misfolded Rhodanese
Cotranslational interaction of unfolded rhodanese with 5 S rRNA reveals an interesting parallel with the action of chaperone systems responsible for precursor protein binding and their preservation in an import-competent conformation (30–32). On the other hand, it was shown earlier that the interaction of 5 S rRNA with the L5 protein occurs through the mechanism of “mutual induced fit”; both partners confer to each other functional conformations and thus operate as reciprocal chaperones (33). Another example of 5 S rRNA-assisted refolding, concerning the DnaK protein, was recently demonstrated in a bacterial system (34). This is why we suggested that the 5 S rRNA molecule may perform the function of chaperone upon its interaction with rhodanese as well.
To verify this hypothesis, the standard aggregation assays (16) were performed (Fig. 4A). Urea-denatured rhodanese rapidly diluted in a buffer without the denaturant begins to aggregate due to stochastic collapsing of exposed hydrophobic surfaces, resulting in growth of turbidity of the solution. On the contrary, in the presence of a chaperone preventing such a reaction, the process is suppressed. Because particular conformation is critical for activity of both RNA and protein chaperones (35–38), we tested two different forms of human 5 S rRNA, which can be obtained by refolding either in the presence (human 5 S rRNAMg) or in the absence (human 5 S rRNAEDTA) of magnesium and distinguished by their mobility in native gel (Fig. 4B). Interestingly, addition of only one of them, 5 S rRNAEDTA, resulted in a significant suppression of rhodanese aggregation, the other form remaining completely inert (Fig. 4A). Thus, this chaperone action was found to be not only 5 S rRNA-specific (because nonspecific RNA or DNA molecules failed in this task; Fig. 4A) but also conformation-specific.
FIGURE 4.

Chaperone activity of 5 S rRNA upon its interaction with misfolded rhodanese. A, rhodanese aggregation assay in the presence of various nucleic acids. B, mobility of human (H) and yeast (Y) 5 S rRNAs in 8% 1× TBE native gel. 5 S rRNAs were folded either in the absence of magnesium (1 mm EDTA) or in the presence of 5 mm MgCl2. C, time course reactivation of urea-denatured rhodanese in the absence and in the presence of 3 μm human 5 S rRNAEDTA. D, thermal inactivation of rhodanese in the absence of 5 S rRNA and in the presence of 3 μm 5 S rRNAEDTA or 5 S rRNAMg. E, scheme describing behavior of urea-denatured rhodanese in a solution without denaturant (e.g. in the aggregation assay, see A) in the absence (black arrows) and in the presence (blue arrows) of human 5 S rRNAEDTA (developed from Refs. 17, 20). F, UV spectra of rhodanese (0.25 μg/μl) in native, urea-denatured, and 5 S rRNA-complexed forms, 20 s and 15 min after beginning of aggregation. G, import of rhodanese into isolated human mitochondria. 35S-Labeled proteins were synthesized in the cell-free PCR-optimized TnT system (Promega) either in the absence or in the presence of 4 μm human 5 S rRNAEDTA. Rhodanese does not demonstrate visible mobility shift upon import, because no cleavage of its mitochondrial transport signal occurs (24).
Noteworthy, both yeast 5 S rRNA forms (yeast 5 S rRNAMg and yeast 5 S rRNAEDTA) demonstrate the mobility in native gel similar to that of human 5 S rRNAEDTA (Fig. 4B), suggesting that they acquire a closely related conformation. As expected, their addition to the aggregation assay resulted in an identical suppression curve (Fig. 4A). Human 5 S rRNA variants with mutations in rhodanese-binding sites (in - or γ-domains) were unable to suppress rhodanese aggregation even if folded in the absence of magnesium. This indicates that the observed chaperone effect was indeed dependent on specific 5 S rRNA binding to the misfolded rhodanese.
Intriguingly, the kinetics of rhodanese aggregation in the presence of human 5 S rRNAEDTA or yeast 5 S rRNA is far from being an ordinary one. Instead of a gradual slowdown without changes in the shape of the curve (indicating that rhodanese continues to aggregate even in the presence of a chaperone but with a lower rate), which is usually observed (39), in the presence of 5 S rRNA the curve becomes clearly biphasic. Its early segment corresponds to a rapid formation of aggregates upon dilution of the denaturant. Then, in 1.5–2 min, the aggregation stops abruptly without any significant growth of turbidity thereafter (Fig. 4A). This very particular pattern reflects aggregation associated with the refolding process (40). Although rhodanese is one of the most popular substrates for chaperone studies, only a few examples of such a kinetics have been reported to date. They concern the following cochaperones: mammalian and yeast DnaJ homologues Hdj1 and Hdj2 (41, 42), hTid-1L and hTid-1S (43), Ydj1p (44), and a Caenorhabditis elegans p97 homologue CDC48.2 (45). All of them were found to function as cochaperone components in Hsp40/Hsp70 systems, which are thought to be the first chaperones to bind to nascent proteins and preserve them in a misfolded but refolding-competent state (46–48). We found that the same is true for human 5 S rRNAEDTA, because it does not support reactivation of denatured rhodanese nor prevents the native protein from thermal inactivation (Fig. 4, C and D). These data suggest that human 5 S rRNA folds aggregation-prone rhodanese species into a nonaggregative but enzymatically inactive conformation, quite like known protein cochaperones (Fig. 4E).
This conclusion is strongly supported by analysis of UV spectra of rhodanese on different steps of the aggregation process (Fig. 4F), revealing parallels with the mechanism proposed earlier for the classic cochaperone action (46, 47). Rhodanese has 8 Trp and 8 Tyr residues mostly buried in the depth of the native globula (49). This is why its UV spectrum is particularly sensitive to any conformational changes (Fig. 4F). Thus, the urea-denatured form in 20 s after dilution in urea-free buffer (corresponding mostly to the first partially folded intermediate formed; Fig. 4E) shows significant hyperchromatism and a very particular filling of the absorption gap around 250 nm. This pattern becomes even more pronounced on the late stages of the aggregation process, which indicates that the molecule is extensively destructured. The behavior of the spectrum in the presence of human 5 S rRNAEDTA is dramatically different. The first phase (the rapid formation of aggregates; Fig. 4A) is marked with even higher hyperchromatism, suggesting that rhodanese is being preserved in almost a totally unfolded state, and does not collapse spontaneously (corresponds to Complex I on Fig. 4E). At the second phase (arrested aggregation; Fig. 4A), it acquires a very compact conformation that somewhat combines the features of both native and denatured states (corresponds to Complex II on Fig. 4E). All these data indicate that in the complex with 5 S rRNA rhodanese achieves a compact, nonaggregative state, which can still be clearly distinguished from the native conformation (Fig. 4E).
Taken together, these results enable us to conclude that a kind of functional mimicry of the protein cochaperone system by the 5 S rRNA molecule is manifested upon its interaction with rhodanese. Interestingly, specific interaction between bacterial 5 S rRNA and an Hsp70 (DnaK) has been reported (34, 50), continuing this intriguing comparison. Taking into account that the Hsp40/Hsp70 system was shown to be involved in the protein precursor import into mitochondria (51, 52), it appears probable that 5 S rRNA may thus exploit this pathway for its own targeting to mitochondria. Noteworthy, the contrary is not true because import of rhodanese into mitochondria was found to be independent of the 5 S rRNA concentration (Fig. 4G), and its targeting to the mitochondrial surface thus can be ensured by standard protein chaperone systems.
In-solution Conformations of Human 5 S rRNA
Only one form of human 5 S rRNA (5 S rRNAEDTA) was shown to possess a cochaperone-like activity upon its interaction with rhodanese (Fig. 4A). Although a more branched conformation can be expected for this form, regarding its slower mobility in native gel (Fig. 4B), its precise nature remains still enigmatic. It was suggested previously that such large scale conformational shifts of 5 S rRNA are likely to be associated with the loop A rearrangement (53–55). The loop A, being a three-way junction (Fig. 1A), defines the stacking mode of the three domains of the molecule, and thus it is above all responsible for the tertiary structure of 5 S rRNA (56). For 5 S rRNA, only two variants of geometry are possible as follows: (i) the classic, compact one (so-called “family C”), and (ii) the more branched, loose conformation (“family A”) (56), which should have different mobility in native gel (Fig. 5A).
Because the shift from family C to family A conformation should be accompanied by a substantial change of geometric parameters of the molecule, we decided to measure directly the distance between two most characteristic points of 5 S rRNA, the extremities of its major domains, in both forms. For this, we used the in gel FRET technique, which allows coupling of the FRET efficiency measurement to native gel electrophoresis (22). Thus, the identity and the conformation of molecule are better controlled, and the probability of a complex mixture of forms is largely decreased. Human 5 S rRNA molecules were assembled from three oligonucleotides in the way shown on Fig. 5B, either in the presence or in the absence of magnesium. Both mixtures were resolved by native PAGE (Fig. 5C), and FRET efficiencies were determined for each conformation (Fig. 5D and Table 2). Interestingly, although the distance between the fluorophores in the 5 S rRNAMg is in perfect agreement with data obtained earlier in other in-solution studies, that in the 5 S rRNAEDTA indicates a crucial rearrangement of the molecule that results in bringing together both extremities. Such a drastic conformational shift can be only explained by a cooperative reorganization of the loops A and B, as reflected in the model shown on Fig. 5E.
TABLE 2.
Results of in gel FRET analysis of human 5 S rRNA compared with other known 5 S rRNAMg measurements
| 5 S rRNA | FRET efficiency | Distance between extremities of domains-β and -γ | Ref. |
|---|---|---|---|
| Å | |||
| Human 5 S rRNAMg | <0 | >120 | This study |
| Human 5 S rRNAEDTA | 0.93 ± 0.03 | 36 ± 3 | This study |
| E. coli 5 S rRNA in solution | 162 (maximum dimension) | 67 | |
| 160 (maximum dimension) | 68 | ||
| 125 (maximum dimension) | 69 | ||
| 133 | 67 | ||
| 155 (extremely expanded) | 67 | ||
| Thermus flavus 5 S rRNA in solution | 120 ± 5 | 70 | |
| Xenopus laevis 5 S rRNA (theoretical model) | 118 | 71 | |
| E. coli 5 S rRNA in ribosome (Protein Data Bank code 1C2X) | 111 | 72 | |
| Thermus thermophilus 5 S rRNA in ribosome (Protein Data Bank code 1GIY) | 84 | 73 | |
| Haloarcula marismortui 5 S rRNA in ribosome (Protein Data Bank code 1JJ2) | 84 | 57 | |
| Deinococcus radiodurans 5 S rRNA in ribosome (Protein Data Bank code 1NKW) | 73 | 74 |
A very important difference between the two human 5 S rRNA forms should be emphasized. Whereas in the classic family C structure the major groove of the α-domain is inaccessible because of the contact with the β-domain (57), it becomes fully open in the family A form (Fig. 5E). Because it was shown previously that one of the signals of mitochondrial localization of 5 S rRNA resides in the proximal part of the α-domain (Fig. 1A) (11), and the same site is absolutely necessary for the 5 S rRNA-rhodanese interaction (Fig. 3D), it becomes clear why only the 5 S rRNAEDTA (family A), and not the 5 S rRNAMg (family C), is able to bind and to fold the denatured rhodanese (Fig. 4A). This result reveals the biologic significance of both the conformation-specific 5 S rRNA-rhodanese association and the α-domain-dependent targeting of 5 S rRNA into mitochondria.
DISCUSSION
RNA import into mitochondria is a quasi-universal phenomenon among eukaryotes. Nevertheless, unlike the mitochondrial pre-protein import pathway, the mechanisms cytosolic RNAs exploit to get into mitochondria seem to have evolved independently in different phyla, explaining the extreme diversity of physico-chemical requirements and protein factors involved (58). The overwhelming majority of data accumulated to date concerns numerous examples of tRNA import, for some of which protein factors implicated in mitochondrial targeting and translocation of cytosolic RNAs have been identified (10, 59–64). However, in mammalian cells, quite a particular case of 5 S rRNA import into mitochondria was reported (8, 9). Investigation of this pathway appears to be of fundamental interest because of the following: (i) its mechanism and biologic significance remain unclear and somewhat enigmatic, and (ii) it presents a unique opportunity to confront different RNA import mechanisms for a better understanding of laws governing such a kind of transport processes.
Previously, physico-chemical requirements and structural elements functioning as signals of mitochondrial localization of human 5 S rRNA were characterized (10, 11). In this study, we continue the dissection of this pathway by identification of one of protein factors implicated in 5 S rRNA mitochondrial targeting. Mitochondrial thiosulfate sulfurtransferase, rhodanese, traditionally thought to be involved in sulfur metabolism and cyanide detoxication (12), appears to have been recruited for an unrelated “second job,” participating in the pathway of 5 S rRNA import into mammalian mitochondria. Surprising though it may seem, one should not forget that the rhodanese homology domain superfamily embraces proteins of extremely diversified functions, RNA metabolism, and macromolecular transport included (12). Indeed, rhodanese possesses all traits necessary to accomplish such a task as 5 S rRNA mitochondrial targeting as follows: (i) synthesized in cytosol, it is then directed into mitochondrial matrix (24, 25); (ii) it has strong specific affinity to 5 S rRNA; (iii) the binding sites rhodanese exploits for this interaction match the signals of 5 S rRNA mitochondrial localization; and (iv) the clear concentration dependence of 5 S rRNA import on rhodanese in both in vitro and in vivo systems indicates its direct involvement in the pathway.
The detailed study of the 5 S rRNA-rhodanese interaction let us approach the mechanistic aspect of the 5 S rRNA targeting process. We show here that the binding can occur cotranslationally, and 5 S rRNA apparently behaves as a cochaperone for the nascent rhodanese. This finding enables us to draw two interesting parallels. First, the obvious similarity between 5 S rRNA-rhodanese and 5 S rRNA-ribosomal protein L5 interactions (33) suggests that the same and unique principle can be applied to both nucleolar and mitochondrial targeting of 5 S rRNA in mammalian cells; the 5 S rRNA actively changes the conformation of the protein upon cotranslational binding to ensure their concerted relocalization. This principle provides the mechanistic base for the “cross-roads hypothesis” proposed earlier, according to which the intracellular distribution of 5 S rRNA is a matter of direct competition between the L5 protein and mitochondrial import factors for the cargo (11). Second, the functional mimicry of protein cochaperones involved in mitochondrial targeting of pre-proteins that 5 S rRNA demonstrates upon its binding to rhodanese hints at the possibility of exploiting the more ancient protein precursor import pathway for the concomitant 5 S rRNA delivery into mammalian mitochondria. In line with earlier findings that the intact pre-protein translocation apparatus or at least some of its components are involved in RNA mitochondrial import in yeast (61), human (10), and plants (64), this result once again suggests that a close association between the two macromolecular import pathways does exist in these systems.
Recruitment of an apparently unrelated protein for RNA transport into mitochondria is not unprecedented however. Thus, enolase, known in the first place as a glycolytic enzyme, was shown to be an essential component of the tRNALys import vehicle in yeast (63). Found to possess the specific affinity to the tRNA and located in the vicinity of mitochondria, it can be compared to some extent with the part rhodanese plays in 5 S rRNA import into mammalian mitochondria. Still, a substantial difference is to be outlined; enolase, devoid of any mitochondrial localization signal, is obliged to donate the cargo RNA to another import factor, the precursor of mitochondrial lysyl-tRNA synthetase, whereas rhodanese seems to be well enough equipped to perform the role of carrier for 5 S rRNA on its own.
Intriguingly, by investigating the human 5 S rRNA chaperone activity, we have found that only a particular “more branched” conformation with the family A geometry is capable of productive binding to misfolded rhodanese. This conformation (the only one in which the main rhodanese-binding site is available for interaction) cannot exist in a magnesium-containing medium however. It suggests involvement of some upstream protein factor(s) needed to induce such a kind of structural rearrangement, the classic example of such a protein-dependent three-way junction reorganization was the binding of ribosomal protein S15 to 16 S rRNA (65). The necessity to induce an import-competent RNA conformation was also reported for the yeast tRNALys mitochondrial targeting system, where the function of RNA chaperone is ensured by enolase (63, 66). Identification of analogous chaperone factors (obviously, present in the 17Q/80AS fraction) is one of the next major challenges for the further 5 S rRNA import research.
Finally, identification of rhodanese as one of the 5 S rRNA import factors enabled us to approach the functional role of the imported molecule in the mitochondrial compartment of the mammalian cell. A robust correlation between the 5 S rRNA mitochondrial level and the general mitochondrial translational activity was established in experiments in vivo (Fig. 2). This intriguing finding, together with the previous report of equimolarity between 5 S rRNA molecules present in human mitochondria and mitochondrial ribosomes (10), suggests that the cytosolic 5 S rRNA can be intimately implicated in mitochondrial protein synthesis. The precise molecular function of 5 S rRNA (e.g. being a structural component of mitochondrial ribosome or involvement into its assembly) still awaits further elucidation.
Acknowledgments
We thank C. Lemaitre-Guillier for mass spectrometry analysis, E. Vinogradova for optimization of siRNA silencing procedure, and A. Keilbach for administrative help.
This work was supported by the CNRS, Université de Strasbourg, Moscow State University, Association Française Contre les Myopathies, Agence Nationale de la Recherche, Fondation pour la Recherche Médicale, Russian Foundation for Basic Research, and ARCUS (cooperation program Alsace-Russia-Ukraine).
- PAAG
- polyacrylamide gel.
REFERENCES
- 1.Koc E. C., Burkhart W., Blackburn K., Moyer M. B., Schlatzer D. M., Moseley A., Spremulli L. L. (2001) J. Biol. Chem. 276, 43958–43969 [DOI] [PubMed] [Google Scholar]
- 2.Smirnov A. V., Entelis N. S., Krasheninnikov I. A., Martin R., Tarassov I. A. (2008) Biochemistry 73, 1418–1437 [DOI] [PubMed] [Google Scholar]
- 3.Smith M. W., Meskauskas A., Wang P., Sergiev P. V., Dinman J. D. (2001) Mol. Cell. Biol. 21, 8264–8275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Guddat U., Bakken A. H., Pieler T. (1990) Cell 60, 619–628 [DOI] [PubMed] [Google Scholar]
- 5.Rudt F., Pieler T. (1996) EMBO J. 15, 1383–1391 [PMC free article] [PubMed] [Google Scholar]
- 6.Ogata K., Kurashashi A., Nishiyama C., Terao K. (1993) Eur. J. Biochem. 213, 1277–1282 [DOI] [PubMed] [Google Scholar]
- 7.Hanas J. S., Hocker J. R., Cheng Y. G., Lerner M. R., Brackett D. J., Lightfoot S. A., Hanas R. J., Madhusudhan K. T., Moreland R. J. (2002) Gene 282, 43–52 [DOI] [PubMed] [Google Scholar]
- 8.Yoshionari S., Koike T., Yokogawa T., Nishikawa K., Ueda T., Miura K., Watanabe K. (1994) FEBS Lett. 338, 137–142 [DOI] [PubMed] [Google Scholar]
- 9.Magalhães P. J., Andreu A. L., Schon E. A. (1998) Mol. Biol. Cell 9, 2375–2382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Entelis N. S., Kolesnikova O. A., Dogan S., Martin R. P., Tarassov I. A. (2001) J. Biol. Chem. 276, 45642–45653 [DOI] [PubMed] [Google Scholar]
- 11.Smirnov A., Tarassov I., Mager-Heckel A. M., Letzelter M., Martin R. P., Krasheninnikov I. A., Entelis N. (2008) RNA 14, 749–759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cipollone R., Ascenzi P., Visca P. (2007) IUBMB Life 59, 51–59 [DOI] [PubMed] [Google Scholar]
- 13.Dulbecco R., Freeman G. (1959) Virology 8, 396–397 [DOI] [PubMed] [Google Scholar]
- 14.Gaines G., Attardi G. (1984) Mol. Cell. Biol. 4, 1605–1617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kolesnikova O. A., Entelis N. S., Jacquin-Becker C., Goltzene F., Chrzanowska-Lightowlers Z. M., Lightowlers R. N., Martin R. P., Tarassov I. (2004) Hum. Mol. Genet. 13, 2519–2534 [DOI] [PubMed] [Google Scholar]
- 16.Silberg J. J., Hoff K. G., Vickery L. E. (1998) J. Bacteriol. 180, 6617–6624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Panda M., Gorovits B. M., Horowitz P. M. (2000) J. Biol. Chem. 275, 63–70 [DOI] [PubMed] [Google Scholar]
- 18.Fernandez-Silva P., Martinez-Azorin F., Micol V., Attardi G. (1997) EMBO J. 16, 1066–1079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Henis Y. I., Levitzki A. (1976) Eur. J. Biochem. 71, 529–532 [DOI] [PubMed] [Google Scholar]
- 20.Mendoza J. A., Rogers E., Lorimer G. H., Horowitz P. M. (1991) J. Biol. Chem. 266, 13587–13591 [PubMed] [Google Scholar]
- 21.Lin E., Lin S. W., Lin A. (2001) Nucleic Acids Res. 29, 2510–2516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Radman-Livaja M., Biswas T., Mierke D., Landy A. (2005) Proc. Natl. Acad. Sci. U.S.A. 102, 3913–3920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Norman D. G., Grainger R. J., Uhrín D., Lilley D. M. (2000) Biochemistry 39, 6317–6324 [DOI] [PubMed] [Google Scholar]
- 24.Miller D. M., Delgado R., Chirgwin J. M., Hardies S. C., Horowitz P. M. (1991) J. Biol. Chem. 266, 4686–4691 [PubMed] [Google Scholar]
- 25.Waltner M., Hammen P. K., Weiner H. (1996) J. Biol. Chem. 271, 21226–21230 [PubMed] [Google Scholar]
- 26.Giaever G., Chu A. M., Ni L., Connelly C., Riles L., Véronneau S., Dow S., Lucau-Danila A., Anderson K., André B., Arkin A. P., Astromoff A., El-Bakkoury M., Bangham R., Benito R., Brachat S., Campanaro S., Curtiss M., Davis K., Deutschbauer A., Entian K. D., Flaherty P., Foury F., Garfinkel D. J., Gerstein M., Gotte D., Güldener U., Hegemann J. H., Hempel S., Herman Z., Jaramillo D. F., Kelly D. E., Kelly S. L., Kötter P., LaBonte D., Lamb D. C., Lan N., Liang H., Liao H., Liu L., Luo C., Lussier M., Mao R., Menard P., Ooi S. L., Revuelta J. L., Roberts C. J., Rose M., Ross-Macdonald P., Scherens B., Schimmack G., Shafer B., Shoemaker D. D., Sookhai-Mahadeo S., Storms R. K., Strathern J. N., Valle G., Voet M., Volckaert G., Wang C. Y., Ward T. R., Wilhelmy J., Winzeler E. A., Yang Y., Yen G., Youngman E., Yu K., Bussey H., Boeke J. D., Snyder M., Philippsen P., Davis R. W., Johnston M. (2002) Nature 418, 387–391 [DOI] [PubMed] [Google Scholar]
- 27.De Duve C., Pressman B. C., Gianetto R., Wattiaux R., Appelmans F. (1955) Biochem. J. 60, 604–617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Eilers M., Schatz G. (1986) Nature 322, 228–232 [DOI] [PubMed] [Google Scholar]
- 29.Glick B., Schatz G. (1991) Annu. Rev. Genet. 25, 21–44 [DOI] [PubMed] [Google Scholar]
- 30.Ryan M. T., Naylor D. J., Høj P. B., Clark M. S., Hoogenraad N. J. (1997) Int. Rev. Cytol. 174, 127–193 [DOI] [PubMed] [Google Scholar]
- 31.Hardesty B., Kudlicki W., Odom O. W., Zhang T., McCarthy D., Kramer G. (1995) Biochem. Cell Biol. 73, 1199–1207 [DOI] [PubMed] [Google Scholar]
- 32.Kudlicki W., Odom O. W., Kramer G., Hardesty B., Merrill G. A., Horowitz P. M. (1995) J. Biol. Chem. 270, 10650–10657 [DOI] [PubMed] [Google Scholar]
- 33.DiNitto J. P., Huber P. W. (2003) J. Mol. Biol. 330, 979–992 [DOI] [PubMed] [Google Scholar]
- 34.Kim H. K., Choi S. I., Seong B. L. (2010) Biochem. Biophys. Res. Commun. 391, 1177–1181 [DOI] [PubMed] [Google Scholar]
- 35.Herschlag D. (1995) J. Biol. Chem. 270, 20871–20874 [DOI] [PubMed] [Google Scholar]
- 36.Kudlicki W., Coffman A., Kramer G., Hardesty B. (1997) Fold. Des. 2, 101–108 [DOI] [PubMed] [Google Scholar]
- 37.Semrad K., Green R., Schroeder R. (2004) RNA 10, 1855–1860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tompa P., Csermely P. (2004) FASEB J. 18, 1169–1175 [DOI] [PubMed] [Google Scholar]
- 39.Weber F., Hayer-Hartl M. (2000) Methods Mol. Biol. 140, 111–115 [DOI] [PubMed] [Google Scholar]
- 40.Kurganov B. I. (2002) Biochemistry 67, 409–422 [DOI] [PubMed] [Google Scholar]
- 41.Minami Y., Höhfeld J., Ohtsuka K., Hartl F. U. (1996) J. Biol. Chem. 271, 19617–19624 [DOI] [PubMed] [Google Scholar]
- 42.Minami Y., Minami M. (1999) Genes Cells 4, 721–729 [DOI] [PubMed] [Google Scholar]
- 43.Goswami A. V., Chittoor B., D'Silva P. (2010) J. Biol. Chem. 285, 19472–19482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lu Z., Cyr D. M. (1998) J. Biol. Chem. 273, 27824–27830 [DOI] [PubMed] [Google Scholar]
- 45.Nishikori S., Yamanaka K., Sakurai T., Esaki M., Ogura T. (2008) Genes Cells 13, 827–838 [DOI] [PubMed] [Google Scholar]
- 46.Hendrick J. P., Langer T., Davis T. A., Hartl F. U., Wiedmann M. (1993) Proc. Natl. Acad. Sci. U.S.A. 90, 10216–10220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kudlicki W., Odom O. W., Kramer G., Hardesty B. (1996) J. Biol. Chem. 271, 31160–31165 [DOI] [PubMed] [Google Scholar]
- 48.Thoms S. (2002) FEBS Lett. 520, 107–110 [DOI] [PubMed] [Google Scholar]
- 49.Gliubich F., Gazerro M., Zanotti G., Delbono S., Bombieri G., Berni R. (1996) J. Biol. Chem. 271, 21054–21061 [DOI] [PubMed] [Google Scholar]
- 50.Okada S., Okada T., Aimi T., Morinaga T., Itoh T. (2000) FEBS Lett. 485, 153–156 [DOI] [PubMed] [Google Scholar]
- 51.Artigues A., Iriarte A., Martinez-Carrion M. (2002) J. Biol. Chem. 277, 25047–25055 [DOI] [PubMed] [Google Scholar]
- 52.Bhangoo M. K., Tzankov S., Fan A. C., Dejgaard K., Thomas D. Y., Young J. C. (2007) Mol. Biol. Cell 18, 3414–3428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Egebjerg J., Christiansen J., Brown R. S., Larsen N., Garrett R. A. (1989) J. Mol. Biol. 206, 651–668 [DOI] [PubMed] [Google Scholar]
- 54.Kuliñski T., Bratek-Wiewiórowska M. D., Wiewiórowski M., Zielenkiewicz A., Zółkiewski M., Zielenkiewicz W. (1991) Nucleic Acids Res. 19, 2449–2455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Toots I., Misselwitz R., Böhm S., Welfle H., Villems R., Saarma M. (1982) Nucleic Acids Res. 10, 3381–3389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lescoute A., Westhof E. (2006) RNA 12, 83–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ban N., Nissen P., Hansen J., Moore P. B., Steitz T. A. (2000) Science 289, 905–920 [DOI] [PubMed] [Google Scholar]
- 58.Tarassov I., Kamenski P., Kolesnikova O., Karicheva O., Martin R. P., Krasheninnikov I. A., Entelis N. (2007) Cell Cycle 6, 2473–2477 [DOI] [PubMed] [Google Scholar]
- 59.Bouzaidi-Tiali N., Aeby E., Charrière F., Pusnik M., Schneider A. (2007) EMBO J. 26, 4302–4312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tarassov I., Entelis N., Martin R. P. (1995) EMBO J. 14, 3461–3471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tarassov I., Entelis N., Martin R. P. (1995) J. Mol. Biol. 245, 315–323 [DOI] [PubMed] [Google Scholar]
- 62.Brandina I., Graham J., Lemaitre-Guillier C., Entelis N., Krasheninnikov I., Sweetlove L., Tarassov I., Martin R. P. (2006) Biochim. Biophys. Acta 1757, 1217–1228 [DOI] [PubMed] [Google Scholar]
- 63.Entelis N., Brandina I., Kamenski P., Krasheninnikov I. A., Martin R. P., Tarassov I. (2006) Genes Dev. 20, 1609–1620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Salinas T., Duchêne A. M., Delage L., Nilsson S., Glaser E., Zaepfel M., Maréchal-Drouard L. (2006) Proc. Natl. Acad. Sci. U.S.A. 103, 18362–18367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Batey R. T., Williamson J. R. (1998) RNA 4, 984–997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kolesnikova O., Kazakova H., Comte C., Steinberg S., Kamenski P., Martin R. P., Tarassov I., Entelis N. (2010) RNA 16, 926–941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Skibinska L., Banachowicz E., Gapiñski J., Patkowski A., Barciszewski J. (2004) Biopolymers 73, 316–325 [DOI] [PubMed] [Google Scholar]
- 68.Fox J. W., Wong K. P. (1979) J. Biol. Chem. 254, 10139–10144 [PubMed] [Google Scholar]
- 69.Osterberg R., Sjöberg B., Garrett R. A. (1976) Eur. J. Biochem. 68, 481–487 [DOI] [PubMed] [Google Scholar]
- 70.Funari S. S., Rapp G., Perbandt M., Dierks K., Vallazza M., Betzel C., Erdmann V. A., Svergun D. I. (2000) J. Biol. Chem. 275, 31283–31288 [DOI] [PubMed] [Google Scholar]
- 71.Westhof E., Romby P., Romaniuk P. J., Ebel J. P., Ehresmann C., Ehresmann B. (1989) J. Mol. Biol. 207, 417–431 [DOI] [PubMed] [Google Scholar]
- 72.Mueller F., Sommer I., Baranov P., Matadeen R., Stoldt M., Wöhnert J., Görlach M., van Heel M., Brimacombe R. (2000) J. Mol. Biol. 298, 35–59 [DOI] [PubMed] [Google Scholar]
- 73.Yusupov M. M., Yusupova G. Z., Baucom A., Lieberman K., Earnest T. N., Cate J. H., Noller H. F. (2001) Science 292, 883–896 [DOI] [PubMed] [Google Scholar]
- 74.Harms J., Schluenzen F., Zarivach R., Bashan A., Gat S., Agmon I., Bartels H., Franceschi F., Yonath A. (2001) Cell 107, 679–688 [DOI] [PubMed] [Google Scholar]