

Abstract
Protein sequences encode both structure and foldability. Whereas the interrelationship of sequence and structure has been extensively investigated, the origins of folding efficiency are enigmatic. We demonstrate that the folding of proinsulin requires a flexible N-terminal hydrophobic residue that is dispensable for the structure, activity, and stability of the mature hormone. This residue (PheB1 in placental mammals) is variably positioned within crystal structures and exhibits 1H NMR motional narrowing in solution. Despite such flexibility, its deletion impaired insulin chain combination and led in cell culture to formation of non-native disulfide isomers with impaired secretion of the variant proinsulin. Cellular folding and secretion were maintained by hydrophobic substitutions at B1 but markedly perturbed by polar or charged side chains. We propose that, during folding, a hydrophobic side chain at B1 anchors transient long-range interactions by a flexible N-terminal arm (residues B1–B8) to mediate kinetic or thermodynamic partitioning among disulfide intermediates. Evidence for the overall contribution of the arm to folding was obtained by alanine scanning mutagenesis. Together, our findings demonstrate that efficient folding of proinsulin requires N-terminal sequences that are dispensable in the native state. Such arm-dependent folding can be abrogated by mutations associated with β-cell dysfunction and neonatal diabetes mellitus.
Keywords: Genetics, Hormones, Metabolism, Protein folding, Protein Structure
Introduction
The efficiency of protein folding poses a fundamental problem at the intersection of biophysics, cell biology, and medicine (1, 2). Because the existence of a unique and accessible ground state is unrepresentative of polypeptides as a class of heteropolymers, foldability is an evolved property of biological sequences (3). Current kinetic models envisage a funnel-shaped free-energy landscape, enabling multiple trajectories to the native state (4–6). What distinguishes foldable from non-foldable sequences (7), and how are bottlenecks avoided (8–10)? The salience of these questions has been reinforced by recognition of proteotoxicity as a general pathological mechanism underlying diverse diseases (11, 12). Here, we describe a cryptic folding element in a protein that is dispensable once the native state has been reached.
A model is provided by insulin, a globular protein central to the regulation of vertebrate metabolism (13). Its impaired biosynthesis causes β-cell dysfunction and permanent neonatal-onset diabetes mellitus (DM)4 (14–17). The insulin gene encodes a single-chain precursor, preproinsulin (Fig. 1A, top) (18). A signal peptide (gray bar) is cleaved on translocation into the endoplasmic reticulum (ER) to yield proinsulin. The precursor contains successive sequence motifs, defining B, C, and A domains (blue, black, and red, respectively, in Fig. 1A) (19). Whereas the translocated polypeptide is reduced and unfolded, oxidative folding in the ER yields a well organized A-B (insulin-like) core and disordered C-domain (dashed black segment in Fig. 1B) (20–26). Folding is coupled to disulfide pairing (A6–A11, A7–B7, and A20–B19; gold in Fig. 1, A and B).5 Proinsulin isomers are formed at low concentrations in β-cells (27), and their accumulation may be linked to β-cell dysfunction (28, 29).
FIGURE 1.

Proinsulin and its biosynthetic pathway. A, pathway of insulin biosynthesis beginning with preproinsulin (top): signal peptide (gray), B-domain (blue), dibasic BC junction (green), C-domain (black), dibasic CA junction (green), and A-domain (red). In the ER the unfolded prohormone undergoes specific disulfide pairing to yield native proinsulin (middle panels). Cleavage of BC and CA junctions (by prohormone convertases PC1 and PC2 and by carboxypeptidase E) leads to mature insulin and the C-peptide (bottom). B, structural model of insulin-like moiety and disordered connecting peptide (dashed line). The A- and B-domains are shown in red and blue, respectively; the disordered connecting domain is shown in dashed black line. Cystines are labeled in yellow boxes. Structural model of proinsulin has recently been verified by heteronuclear NMR studies (26). The flexible C-domain contains a nascent helix (see Fig. 10).
Insulin is obtained from proinsulin by proteolytic processing. After transit through the Golgi apparatus and entry into immature secretory granules (30), specific prohormone convertases excise the C-peptide at conserved dibasic sites (BC and CA junctions; green in Fig. 1, A and B), liberating the bioactive hormone (31–33). Insulin thus contains two chains, designated A (21 residues) and B (30 residues), and is stored as Zn2+-stabilized hexamers within specialized secretory granules (34). The hexamers dissociate on secretion; the circulating hormone functions as a Zn2+-free monomer. Because the structure of insulin is likely to change on binding to the insulin receptor (IR) (35), determinants of foldability may be distinct from (or even at odds with) determinants of activity (36–38).
Three families of insulin hexamers related by concerted conformational changes (designated T6, T3Rf3, and R6) have been defined by x-ray crystallography (39). T and R protomers differ in the secondary structure of the N-terminal segment of the B-chain (residues B1–B8), either extended (T, green in Fig. 2A) or α-helical (R and Rf, blue).6 Alignment of crystallographic protomers highlights multiple possible N-terminal conformations (green and blue in Fig. 2B). The T-state-specific arm, delimited by a β-turn (residues B7–B10), packs variably against a shallow non-polar protein crevice (Fig. 2C). The solution structure of an isolated insulin monomer resembles the T-state (Fig. 2D) (40–42). Flexible packing of PheB1 within this crevice has also been observed in the T-like structure of proinsulin in solution (26). The extended and α-helical states of the B1–B8 arm, linked by the classical TR transition, provide a striking example of a native chameleon sequence in a globular protein (43).
FIGURE 2.

Structure of insulin and conformational variability of the B-chain arm. A, cylinder models of Rf- and T-states (left and right). B, superposition of crystallographic protomers. In each case the N-terminal arm of the B-chain (residues B1–B8) is shown in green (T-state) or blue (Rf-state), and the N-terminal A-chain α-helix (residues A1–A8) in red. A- and B-chains are otherwise shown in light or dark gray. Magenta circles in panel A indicate position of residue B5. Gold balls in panel B indicate positions of the sulfur atoms of cystines A6–A11 (top) and A20–B19 (bottom). Structures in B were obtained from PDB depositions: T-state (entries 4INS, 1APH, 1BPH, 1CPH, 1DPH, 1G7A, 1TRZ, 1TYL, 1TYM, and 1ZNI) and R-state (entries 1EV3, 1EV6, 1MPJ, 1TRZ, 1TYL, 1MPJ, 1ZEG, 1ZEH, and 1ZNI). C and D, residues B1–B6 are shown in green in (C) crystallographic T-state protomers and (D) ensemble of NMR-derived solution structures. The arms are in each case shown as sticks against a space-filling model of one representative structure. The N-terminal A-chain α-helix is shown in red; residues A10 and B14 are shown in light and dark purple. Gold balls represent the sulfur atoms of cystines A6–A11 (center) and B7–A7 (right). A- and B-chains are otherwise shown in light or dark gray. Structures in C were obtained from PDB depositions 4INS and 1BPH; structures in D were obtained from PDB depositions 1TYL and 1G7A.
Structure-function relationships in insulin have been inferred from its pattern of sequence conservation and divergence (39). The classical receptor-binding surface (spanning residues IleA2, ValA3, ValB12, PheB24, and PheB25; Refs. 39, 44, and 45) is invariant (35). Substitutions at this surface markedly impair receptor binding (46–54). Also conserved are side chains integral to the hydrophobic core (LeuA16, TyrA19, LeuB11, and LeuB15 (39)). Altering such core residues hinders disulfide pairing during chemical synthesis (51, 55, 56) and impairs biosynthetic expression (38, 57). Whereas such selected results are readily rationalized, the extent of conservation among vertebrate insulin sequences exceeds the apparent requirements of structure and function. A seeming paradox is posed, for example, by the conservation of residues B1–B5 (sequence FVNQH) despite their dispensability for receptor binding (58) and marked structural variability (59).
Does the N-terminal arm of proinsulin have a hidden biological function? To address this question, the present study has brought together assays of protein folding and trafficking in mammalian cell culture with in vitro studies of protein structure, stability, and activity. Evidence has been obtained that mutations in the arm modulate folding efficiency with possible clinical implications for the genetics of β-cell dysfunction. Strikingly, deletion of PheB1 blocks cellular folding of proinsulin, whereas des-PheB1-insulin retains native-like properties. Three lines of investigation: studies of B1 substitutions, Ala scanning of the arm, and construction of proinsulin/IGF-I chimeras, together demonstrate that foldability requires a flexible N-terminal hydrophobic anchor.
The N-terminal arm of proinsulin provides an example of a cryptic folding element, highlighting foldability as an implicit constraint underlying biological selection of polypeptide sequences. The multistep pathway of insulin biosynthesis from nascent folding to assembly and secretion evidently imposes evolutionary constraints unrelated to the structure and function of the mature hormone. Arm-dependent folding may be abridged by mutations in proinsulin associated with β-cell dysfunction and permanent neonatal-onset diabetes mellitus (60). The arm and its N-terminal hydrophobic anchor thus exemplify the selective advantage of a flexible chameleon sequence in the conformational life cycle of a globular protein.
MATERIALS AND METHODS
Chemical Synthesis
Insulin, KP-insulin (containing substitutions ProB28 → Lys and LysB29 → Pro, which prevent dimerization (41, 61, 62)), proinsulin, and KP-proinsulin were provided by Eli Lilly and Co. (Indianapolis, IN); S-sulfonate B-chain derivatives were obtained by oxidative sulfitolysis (51). A- and B-chain analogs were otherwise prepared by solid-phase synthesis (63). Insulin analogs were prepared by chain combination (63) and purified as described (51). Predicted molecular masses were confirmed by matrix-assisted laser desorption-ionization time-of-flight mass spectrometry (MS). Insulin analogs were monocomponent by reverse phase high-performance liquid chromatography (RP-HPLC). Synthetic yields were calculated based on the mass of product (determined by optical density at 280 nm) relative to control syntheses of wild-type insulin.
Receptor Binding Assays
Dissociation constants for binding of insulin analogs to IR were determined in competitive radioligand binding assays with [125I-TyrA14]human insulin (64). The assay employed the B isoform of IR (IR-B). Assays were performed with isolated IR-B with a C-terminal FLAG tag using a microtiter plate antibody capture technique as described (65). The plates (Nunc Maxisorb) were incubated overnight at 4 °C with FLAG M2 IgG (100 μl/well of 40 μg/ml in phosphate-buffered saline). In all assays the percentage of tracer bound in the absence of competing ligand was <15% to avoid ligand-depletion artifacts. Dissociation constants of analogs were obtained by non-linear regression analysis (66) employing a model describing competitive binding of two different ligands to a receptor. Control studies of cellular extracts in the absence of prior IR-B transfection demonstrated negligible background binding to endogenous cellular proteins.
Mammalian Cell Culture
HEK293T cells were cultured as described (38) at 37 °C in high-glucose Dulbecco's modified Eagle's medium containing 10% fetal bovine serum and 0.1% penicillin/streptomycin with 5% CO2. For transfections, cells were plated into 6-well plates 1 day before transfection. Plasmid DNA (2 μg) was transfected into each well using Lipofectamine (Invitrogen) as described (38, 67).
Metabolic Labeling and PAGE
At 40 h post-transfection cells were preincubated in methionine/cysteine-deficient medium for 30 min, metabolically labeled in the same medium containing 35S-labeled Met and Cys for 1 h, washed once with complete medium, and chased for indicated times. After chase, medium were collected, cells were lysed in 100 mm NaCl, 1% Triton X-100, 0.2% sodium deoxycholate, 0.1% SDS, 10 mm EDTA, and 25 mm Tris-HCl (pH 7.4). Lysates and chase medium were immunoprecipitated with guinea pig anti-insulin antiserum (LINCO Diagnostics) and analyzed by Tris-Tricine-urea-SDS-PAGE under reducing and non-reducing conditions (27, 38, 67).
Spectroscopy
Circular dichroism (CD) spectra, obtained using an Aviv spectropolarimeter equipped with an automated titration unit, were measured at protein concentrations of 25 (far-ultraviolet spectra) or 5 μm (denaturation studies) in 50 mm potassium phosphate (pH 7.4) at 4 °C (56). 1H NMR spectra were obtained at 700 MHz in H2O or D2O solutions containing 10 mm deuterioacetic acid (pH 3.0, direct meter reading) at 25 °C. Spatial relationships within insulin- or proinsulin analogs were probed by nuclear Overhauser effect (NOEs). The following two-dimensional NMR spectra were acquired: double quantum-filtered correlation spectroscopy (DQF-COSY), NOE spectroscopy (NOESY), and total correlation spectroscopy (TOCSY). Resonance assignment of KP-insulin (complete) and KP-proinsulin (partial) was obtained by standard methods (supplemental materials) (68). Presumptive resonance assignment of des-PheB1-insulin (supplemental Table S1) was obtained by analogy to published assignments (69).
Thermodynamic Modeling
CD-detected guanidine denaturation data (as monitored at 222 nm) were fitted by non-linear least squares to a two-state model as described (70). In brief, CD data θ(x) were fitted by a nonlinear least-squares program according to Equation 1,
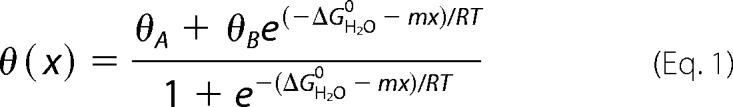 |
where x is the concentration of guanidine hydrochloride and θA and θB are baseline values in the native and unfolded states. These baselines were approximated by pre- and post-transition lines θA(x) = θAH2O + mAx and θB(x) = θBH2O + mBx. Fitting the original CD data and baselines simultaneously circumvents artifacts associated with linear plots of ΔG as a function of denaturant (70, 71).
RESULTS
The Arm of Insulin and Proinsulin Exhibit T-like Conformations
1H NMR studies of engineered insulin monomers have defined a spectroscopic signature of a T-like conformation based on inter-residue NOEs (41, 42). As illustrated in the spectrum of KP-insulin (supplemental Fig. S1), these include long-range contacts between the side chains of PheB1–LeuA13 and HisB5–IleA10 within a shallow inter-chain crevice; additional NOEs between LeuB6 and LeuB11 (not shown) reflect formation of an intervening β-turn at the base of the arm (residues B7–B10). Inconsistent with an R-like α-helical conformation, these and related diagnostic NOEs are retained in the spectrum of proinsulin analogs (supplemental Figs. S1 and S2). In spectra of engineered insulin analogs and proinsulin analogs, NOEs are not observed between the aromatic rings of PheB1 and TyrA14, which among crystallographic T-state protomers of insulin exhibit a broad range of distances (supplemental Fig. S3) (72). Similarly, no NMR evidence has been observed for stable maintenance of a hydrogen bond between the B1 carbonyl oxygen and the side chain NH2 of GlnB4 as observed in a minority of crystal structures (39).
Despite observation of long-range NOEs, the side chains of PheB1 and ValB2 in monomeric analogs of insulin and proinsulin exhibit near random-coil 1H NMR chemical shifts and motional narrowing. These trends hold in spectra acquired under a variety of conditions (in aqueous solution at neutral pH, in 10 mm deuterioacetic acid at pH 3.0, and in 20% deuterioacetic acid, pH 1.9). An example is provided by the ortho and meta 1H NMR secondary chemical shifts of PheB1 in KP-insulin at neutral pH (0.11 and 0.12 ppm, respectively; defined as the difference between observed chemical shifts and tabulated random-coil values): these values are markedly smaller than the corresponding secondary shifts of an analogous Phe ring elsewhere in insulin that stably packs against a nonpolar surface (PheB24, 0.61 and 0.43 ppm).
Additional evidence for the conformational variability of the N-terminal arm has been obtained by heteronuclear NMR spectroscopy. Whereas the main chain 13C NMR chemical shifts of residues B9–B19 in DKP-proinsulin and DKP-insulin (26, 73) exhibit canonical helical values (74), for example, the 13C chemical shifts of residues B1–B3 conform to a pattern associated with segmental flexibility (see “Discussion” and supplemental Table S2) (75). Furthermore, although the amide resonances of PheB1 and ValB2 are not observable due to rapid solvent exchange, those of AsnB3, GlnB4, and HisB5 exhibit a gradient of attenuated {1H}-15N heteronuclear NOEs characteristic of a progressive N-terminal flexibility (26). These findings indicate that N-terminal arm-related NOEs reflect flexible contacts and not fixed spatial relationships within a well organized substructure.
Deletion of PheB1 Impairs Chain Combination but Is Well Tolerated in the Mature Hormone
Classical studies of the total chemical synthesis of insulin have demonstrated that the isolated A- and B-chains contain sufficient information to specify native disulfide pairing (76). Robust to diverse amino acid substitution (56), chain combinations has enabled the preparation of >100 insulin analogs by academic and pharmaceutical laboratories (77). Although yield is limited by side reactions (disulfide-bridged cyclic chains, B-chain dimers and polymers), formation of insulin disulfide isomers is ordinarily negligible (78). Surprisingly, combination of the wild-type A-chain with des-B1 B-chain is perturbed; its yield is reduced 3-fold relative to wild-type chain combination. Although the predominant low molecular weight product is des-B1-insulin (elution time 45.6 min in the HPLC chromatogram shown in Fig. 3A), a major contaminant is a disulfide isomer with elution time delayed by 6 min (arrow in Fig. 3A); the additional elution peaks represent the expected side products. Despite such reduced yield, des-B1-insulin may readily be isolated (Fig. 3B), enabling its characterization.
FIGURE 3.

Chemical synthesis and purification of des-PheB1-DKP-insulin. A, reverse-phase HPLC chromatogram documenting 3-fold reduced yield of des-PheB1 analog following chain combination due to side products, including non-native disulfide isomer (arrow at 51.6 min). B, final purification yielded a single symmetrical peak of appropriate molecular mass and native receptor-binding affinity. Mono-component reverse phase-HPLC profile was obtained under gradient elution conditions employing either acetonitrile or methanol as co-solvents.
Binding of des-B1-insulin to the isolated insulin receptor (isoform B; ▴ and dotted line in Fig. 4A) is essentially indistinguishable from that of wild-type insulin (■ and solid line in Fig. 4A). Curve-fitting yields respective estimates of dissociation constants of 0.068 ± 0.009 (analog) and 0.073 ± 0.010 nm (wild-type). Far UV CD spectra of des-B1- and wild-type insulin are likewise similar (Fig. 4B). The CD-detected guanidine denaturation studies at 4 °C suggest that the two proteins exhibit similar thermodynamic stabilities (Fig. 4C). Application of a two-state model (supplemental Table S3) yields similar estimates of free energies of unfolding (ΔGu) of 4.1 ± 0.1 kcal/mol (des-B1-insulin) versus 4.0 ± 0.1 kcal/mol (wild-type). The analog nonetheless exhibits a small left shift (ΔTmid about 4 °C) in its thermal unfolding transition (i.e. toward lower temperatures in the broad temperature range 30–70 °C) as monitored by mean residue ellipticity at 222 nm (Fig. 4D).
FIGURE 4.

Receptor-binding assay and CD studies. A, competitive displacement assay demonstrated that des-PheB1-insulin (▴; dashed line) binds to the lectin-purified insulin receptor (isoform B) with affinity similar to that of wild-type insulin (■; solid line). B, the far ultraviolet CD spectrum of des-PheB1-insulin (▴; dashed line) at 4 °C is essentially identical to that of wild-type insulin (■; solid line). C, protein denaturation studies in which ellipticity at the helix-sensitive wavelength 222 nm is monitored at 4 °C as a function of the concentration of guanidine hydrochloride. The unfolding transition of des-PheB1-insulin (▴; dashed line) is shifted to the right relative to that of wild-type insulin (■; solid line). D, thermal unfolding of des-PheB1-insulin (▴; dashed line), as monitored at 222 nm, is similar to that of wild-type insulin (■; solid line) from 4 to 30 °C and exhibits a slightly increased slope from 30 to 70 °C.
1H NMR studies of human insulin and des-PheB1-insulin were undertaken as dimers in 10 mm deuterioacetic acid (pH 3.0) at 25 °C (79). Resonance assignments of des-B1-insulin are provided under supplemental Table S1. The aliphatic spectrum of wild-type insulin (Fig. 5B) provides a fingerprint of the T-state-specific packing of PheB1 against the A-chain (Fig. 5A). The spectrum of des-PheB1-insulin (Fig. 5C) exhibits a similar overall envelope of resonances but with selected changes in chemical shift due to the absence of the PheB1 magnetic ring current (ValB2, IleA10, and LeuA13; red lines between panels B and C). These trends in 1H NMR chemical shifts were well resolved in corresponding two-dimensional TOCSY spectra (Fig. 6): whereas the majority of spin systems in insulin are unperturbed by the removal of PheB1 (black labels), selective changes in chemical shift are prominent within the N-terminal arm of the B-chain and within or adjoining its T-state-specific docking site (red labels). Spatial relationships within this docking site (visualized in Fig. 7, A and B) were probed by comparison of two-dimensional NOESY spectra (Fig. 7, C and D). The region shown contains contacts between aliphatic protons (ω1; horizontal axis) and aromatic protons (ω2; vertical axis). Whereas B1-related cross-peaks in the wild-type spectrum (Fig. 7C) are absent as expected in the variant spectrum (Fig. 7D), T-state-specific NOEs from the imidazole ring of HisB5 are retained within an inter-chain crevice (wild-type cross-peaks l–q versus variant cross-peaks r–u). Constraints between the aromatic ring of TyrA19 and neighboring methyl groups (IleA2, LeuB11, and LeuB15; not labeled in the figure), diagnostic of helix-helix packing in the core of insulin (41, 42), are essentially identical in the two proteins.
FIGURE 5.

T-state-specific packing of PheB1 against A-chain and 1H NMR footprint. A, variable position of PheB1 (green) as hydrophobic anchor of the N-terminal arm of the B-chain in collection of 15 crystallographic T-state protomers (PDB entries 4INS, 1APH, 1BPH, 1CPH, 1DPH, 1G7A, 1TRZ, 1TYL, 1TYM, and 1ZNI). B and C, comparison of one-dimensional 1H NMR spectra of human insulin (B) and des-PheB1-insulin (C) as dimers in 10 mm deuterioacetic acid (pD 3.0) at 25 °C. Red lines indicate changes in chemical shift due to the absence of the PheB1 aromatic ring current.
FIGURE 6.

Two-dimensional NMR analysis of des-PheB1-insulin relative to intact insulin. A, TOCSY spectrum of des-B1-insulin; B, corresponding TOCSY spectrum of wild-type insulin. Red labels indicate residues exhibiting significant changes in chemical shift, presumably due to absence of PheB1 aromatic ring current. Conditions were as described in the legend to Fig. 6; TOCSY mixing times were in each case 55 ms.
FIGURE 7.

Binding of B-chain arm within groove and diagnostic long-range NOEs. A, packing of N-terminal portion of arm (residues B1–B5) within groove of T-state structure (PDB accession code 4INS). HisB5 and PheB1 are shown in magenta (left) and green (right); intervening residues are powder blue. The molecular surface is color-coded by electrostatic potential (red, negative and blue, positive). B, corresponding stick model in which identities of neighboring side chains in groove are identified. The B1–B5 coloring is as in panel A; A- and B-chain residues are otherwise shown in gray or black, respectively. C and D, two-dimensional NMR analysis of wild-type insulin (C) and des-PheB1-insulin (D). Comparison of spectra demonstrates that contacts between the imidazole ring of HisB5 and the A-chain are retained in des-PheB1-insulin. Selected resonances positions are as labeled at right. Cross-peak assignments in panel C: a, B1 Hδ-A13 Hβ,γ; b and c, B1 Hδ-B18-Hγ1,2-CH3; d, B1 Hδ-A13 δ-CH3; e and f, B1 Hζ-B18 γ1,2-CH3; g, B1 Hϵ-B14 β-CH3; h, B1 Hϵ-A13 Hβ, Hγ; i and j, B1 Hϵ-B18 γ1,2-CH3; k, B1 Hϵ-A13 δ-CH3; l and m, B5 Hδ-A10 Hβ, Hγ1; n, B5 Hδ-B6 δ-CH3; and o–q, B5 Hδ-A10 γ′-CH3, Hγ2, and δ-CH3. D, cross-peak assignments in panel C: r-u, B5 Hδ-A10 Hβ, Hγ1, γ′-CH3 and δ-CH3. Spectra were obtained at 25 °C in 10 mm deuterioacetic acid (pH 3.0).
Deletion of PheB1 Perturbs Cellular Folding
Transient transfection of human cells with a plasmid expressing proinsulin provides a model for studying its folding within the endoplasmic reticulum, subcellular trafficking, and secretion (Fig. 8A). Following transfection of 293T cells, we thus examined the relative expression of wild-type or variant proinsulins.
FIGURE 8.

Biosynthesis, folding, and secretability of proinsulin variants in cell culture. A, left, nascent proinsulin folds as a monomer in ER wherein zinc-ion concentration is low; in Golgi apparatus zinc-stabilized proinsulin hexamer assembles, which is processed by cleavage of connecting peptide to yield mature insulin. Zinc-insulin crystals are observed in secretory granules. Right, insulin hexamers dissociate in bloodstream to yield active monomers. B–F, analysis of folding and secretability by Tris-Tricine-urea-SDS-PAGE under nonreducing conditions (27). B, HEK293T cells were transfected with an empty vector (control; lanes 1 and 2) or expression plasmids encoding wild-type proinsulin (lanes 3 and 4), variants PheB1 → Asp (lanes 5 and 6), or des-PheB1-proinsulin (lanes 7 and 8). At 48 h cells were pulse-labeled with 35S-amino acids for 1 h and chased for 1 h. Chase medium (lanes marked M) were collected, and cells (lanes marked C) were lysed; each fraction was immunoprecipitated with anti-insulin antiserum. C, control pulse-chase studies of unstable proinsulin analogs [SerA6,SerA11]-proinsulin (lanes 13 and 14) and GlyA2-proinsulin (lanes 15 and 16) as previously described (38). Empty vector and wild-type controls are shown in lanes 9–10 and 11–12, respectively. D and E, corresponding pulse-chase studies of diverse B1 substitutions demonstrated impaired folding by Gly or polar residues (Asn, Gln, Lys, and Ser), whereas foldability is retained on non-polar amino acid substitution (Ala, Leu, Met, Val; asterisks). Imino acid substitution ProB1 also impairs biosynthesis. F, corresponding pulse-chase studies of “arm swap” analogs of proinsulin in which (lanes 41 and 42; hybrid 1) residues B1–B5 were replaced by IGF-I residues 1–4 or (lanes 43 and 44; hybrid 2) residues B1–B4 were replaced by IGF-I residues 1–3. Because IGF-I lacks a residue at canonical position B1, the hybrids are also des-B1 analogs. Empty vector and wild-type controls are shown in lanes 37–38 and 39–40, respectively.
Following pulse labeling of newly synthesized proteins with 35S-amino acids, labeled wild-type or variant proinsulins were immunoprecipitated with polyclonal anti-insulin antiserum and subjected to nonreducing Tris-Tricine-urea-SDS-PAGE, which allows examination of distinct proinsulin disulfide isomers that form within the ER. The absence of endogenous proinsulin in these cells makes detection of the transfected proteins straightforward. As previously demonstrated (27, 38, 67), resolution of discrete proinsulin bands in this gel system provided an assay for extent of native folding, competing disulfide-isomer formation, and efficiency of secretion.7 Fig. 8 (panels B–F) provides a survey of proinsulin variants (radiolabeled for 1 h and chased for 1 h). The portions of the autoradiograms shown contain bands corresponding to monomeric proinsulin or proinsulin disulfide isomers (designated “low molecular weight” region below in contrast to high molecular weight complexes). In Fig. 9 the folding of des-B1-proinsulin was further analyzed (panels A and B), and the analysis was extended to Ala substitutions at positions B2, B3, and B4 (panel C). The polyclonal anti-insulin antiserum employed in these studies was shown by Western blot in control studies to recognize wild-type insulin, des-B1-insulin with similar effectiveness (Fig. 9D).
FIGURE 9.

Pulse-chase studies of proinsulin analogs in HEK293T cells. A, the newly synthesized wild-type or des-PheB1-human proinsulin from transfected HEK293T cells (C) were immunoprecipitated as described in the legend to Fig. 8 and analyzed by Tris-Tricine-urea-SDS-PAGE under non-reducing (lanes 1–3) and reducing (lanes 4–6) conditions. con (lanes 1 and 4) denotes empty vector control. des-PheB1-Human proinsulin presented mostly as misfolded disulfide isomers. Reduction leads to coalescence of native and non-native disulfide isomers as a single band (arrowhead). On reduction, wild-type and variant proinsulin exhibit similar electrophoretic mobilities; extent of expression of variant (lane 6) is reduced relative to wild-type (lane 5). B, aliquots of cells from panel A were chased for 2 h and analyzed under non-reducing and reducing conditions. Whereas the majority of wild-type proinsulin was secreted from cells (C) to medium (M) after a 2-h chase, the des-PheB1-human proinsulin was barely detectable in the cells (lane 10) under non-reducing conditions and was not secreted. Under reducing conditions, however, the variant protein was recoverable (lane 14), suggesting that it formed aberrant disulfide-linked complexes and was retained in the ER. C, corresponding studies of AlaB2, AlaB3, and AlaB4 variant proinsulins analyzed under non-reducing conditions demonstrate a range of perturbations: AlaB4, severe; AlaB2, moderate, and AlaB3, partial impairment of biosynthesis and secretion. D, control Western blot for specificity of antiserum. Aliquots (100 ng) of human insulin (lane 26), des-PheB1-insulin (lane 27), and human proinsulin (lane 28) were resolved under non-reducing conditions, transferred onto a nitrocellular membrane, and blotted with guinea pig anti-porcine insulin antibody. This Western blot indicated that the antibody recognized the three proteins with similar efficiencies.
In accordance with prior studies (27) transfection of the wild-type proinsulin construct gave rise to robust expression, primarily of a fast-migrating species containing native disulfide bonds (Fig. 8B, lanes 3 and 4) relative to an empty vector control (lanes 1 and 2). The most rapidly migrating species (Fig. 8B, arrow) is efficiently secreted from transfected cells (lane 3, “C”) to medium (lane 4, “M”), which typically achieves >95% efficiency by 4 h chase (not shown). In addition, there are less rapidly migrating disulfide isomers that generally represent a minor fraction of proinsulin: these exhibit a lower percentage secretion (highlighted by a bracket in Fig. 8B). Deletion of PheB1 led to multiple intracellular bands with attenuated intensity (Fig. 8B, lane 7), indicating disproportionate disulfide mispairing. Secretion of des-B1-proinsulin was not detectable (Fig. 8B, lane 8).
Perturbed cellular folding of des-B1-proinsulin is in accordance with perturbed chain combination of des-B1-insulin (above). Because band intensities were attenuated after the 1-h chase, the studies were repeated with chase times of 0 and 2 h (Fig. 9, A and B, respectively). Furthermore, because impaired folding could be associated with formation of high molecular weight complexes (due to aberrant formation of mispaired intermolecular disulfide bridges), PAGE analysis was undertaken with and without reduction by dithiothreitol. On reduction a single band was observed, presumably reflecting total expression of the variant proinsulin. Without chase, initial overall expression of des-B1-proinsulin was substantial (Fig. 9A, lane 6) albeit less than that of wild-type proinsulin (lane 5). The distribution of disulfide isomers favored non-native species (Fig. 9A, lane 3). Following an extended chase period of 2 h, a substantial intracellular accumulation of des-B1-proinsulin was observed (reduced band in lane 14 in Fig. 9B) without secretion (lane 15). In the absence of reduction any bands corresponding to low molecular weight species were faint (Fig. 9B, lane 10), implying that the des-B1-proinsulin polypeptides are sequestered in aberrant high molecular weight complexes. Under these conditions wild-type proinsulin predominantly undergoes native folding and secretion (Fig. 9B, lanes 8–9 and 12–13).
Control studies suggest that the impaired foldability of des-B1-proinsulin in 293T cells is unlikely to reflect thermodynamic instability of the protein once folded. Prior biophysical studies have established that two-disulfide insulin- and proinsulin analogs lacking cystine A6–A11 form in vitro partial folds of low stability (ΔΔGu > 2 kcal/mol) (41, 80). In transfected 293T cells removal of the A6–A11 disulfide bridge by pairwise mutation of Cys to Ser did not block expression or secretion (Fig. 8C, lanes 13 and 14). The substitutions likewise caused little detectable change in band mobility (81) despite the presumed loss of structural organization. Folding and secretion are likewise robust to the destabilizing mutation IleA2 → Gly (Fig. 8C, lanes 15 and 16); this substitution in the hydrophobic core was found to impair stability (ΔΔGu) of an insulin analog by at least 1.6 kcal/mol (82). The impaired foldability of des-B1-proinsulin thus stands in marked contrast to the native-like in vitro stability of des-B1-insulin.
Cellular Folding of Proinsulin Requires a Hydrophobic Residue at B1
To further probe the nature of the B1 folding determinant, we next examined multiple substitutions (Fig. 8, B–E). Substitution of PheB1 by Asp, for example, resulted in robust expression with a marked decrease in the fraction of AspB1-proinsulin molecules achieving the native disulfide-bonded form (Fig. 8B, lane 5); secretion of the variant proinsulin was undetectable (lane 6). Whereas nonpolar amino acid side chains at B1 permitted native-like expression and secretion (Ala, Leu, Met, and Val; Fig. 8, D, lanes 23–26, and E, lanes 29–30 and 35–36),8 folding and secretion were impaired by Gly (lanes 21–22) and hydrophilic side chains (Asn, Lys, Gln, Ser; lanes 19–20, 27–28, and 31–34). In each case a nonpolar B1 side chain enabled the variant proinsulin to pass quality control checkpoints within the cells (C) en route to the medium (M). The stringency of the requirement for a nonpolar amino acid side chain at position B1 is surprising given the marked structural variability of PheB1 and its partial solvent exposure as observed among wild-type T-state crystal structures.
Role of the N-terminal Arm
In light of the unexpected contribution of PheB1 to folding efficiency, the overall role of the N-terminal arm of proinsulin was probed by Ala scanning mutagenesis. An extended 2-h chase period was employed as a screen for secretion. An Ala substitution at position B4 imposed a severe block to secretion (Fig. 9C, lane 25, non-reducing conditions), whereas AlaB2 and AlaB3 variants exhibited mild and moderate impairments (lanes 21 and 19, respectively). The corresponding intracellular samples (Fig. 9C, lanes 18 and 20) indicate preferential formation of a disulfide isomer. Previous studies demonstrated that AlaB5 likewise impairs folding and secretion (67).
Proinsulin and IGF-I are homologous proteins with divergent arms and different refolding properties (83–87). IGF-I lacks a B1 residue and exhibits successive arm residues, GPQTLCG; residues B6-B8 (boldface) are conserved in proinsulin. To test whether an IGF-I arm could support the folding of proinsulin, two chimeric analogs were constructed: [des-B1,GlyB2,ProB3,GlnB4]proinsulin (Fig. 8F, lanes 41 and 42, hybrid 1) and [des-B1,GlyB2,ProB3,GlnB4,ThrB5]proinsulin (hybrid 2, lanes 43 and 44). Each of these “arm-swapped” analogs exhibited preferential formation of non-native isomers within the ER with impaired secretion. Native folding of IGF-I in vivo is presumably stabilized by specific IGF-binding proteins that contact its divergent and foreshortened arm (64, 88, 89).
These biological results motivated chemical synthesis [des-B1,GlyB2,ProB3,GlnB4]insulin. As in synthesis of des-B1-insulin, the variant arm impaired the yield of chain combination (also by 3-fold). Although a predominant product was formed, analysis of reverse phase HPLC-resolved side products by MS demonstrated formation of two competing disulfide isomers (presumably containing aberrant disulfide pairings (A6–B7, A7–A11, A20–B19) and (A6–A7, A11–B7, and A20–B19); designated swap and swap2 in Ref. 90. Evidence that the predominant product contained native disulfide bridges was provided by its non-negligible biological activity (IR-B dissociation constant 0.136 ± 0.019 nm; only 3-fold lower than wild-type insulin in the same assay).
DISCUSSION
The recent (and continuing) exponential increase in the size of the data base of protein structures poses the challenge of functional annotation. Whereas residues involved in substrate binding or catalysis may be readily recognizable, determinants of folding efficiency may not be apparent in the native state (3, 7). Such residues may nonetheless impose key evolutionary constraints (38) and emerge as sites of mutation associated with human genetic diseases (60).
The present study has focused on the N-terminal arm of proinsulin (FVNQH; residues B1–B5). In classical studies of insulin (39) a seeming paradox was posed by the conservation of these residues despite their dispensability for receptor binding (58) and marked structural variability (59). Could the arm have a hidden biological function? An interdisciplinary set of studies was thus undertaken to investigate the contribution of the arm to the efficiency of protein folding. Although such a contribution seemed unlikely given that the arm is only partially ordered in an engineered proinsulin monomer (26), we posited that a transient role in folding might be hidden once the native state is reached. Such “hidden” contributions of specific residues to folding efficiency have been extensively explored in a trimeric β-helix (91).
Insulin has been extensively studied since its isolation in 1922 and landmark clinical application (92). It may seem surprising, therefore, that a new function of a sequence motif has only now been recognized. Yet the role of the N-terminal arm of the B-chain has long been enigmatic. Indeed, its structure undergoes a fundamental change in secondary structure, from extended (T) to α-helical (R), as part of a long-range allosteric reorganization of insulin hexamers, designated the TR transition (93). Whereas the conformation of an insulin monomer in solution resembles the T-state (40), adoption of R-like features on receptor binding has been proposed (39, 59, 94). An intriguing hypothesis envisions that induced fit of the arm represents a switch between folding-competent and active conformations (36). This model thus represents the potential biological utility of a chameleon sequence in a globular protein (43).
Proinsulin contains an insulin-like moiety (the A and B domains) and flexible connecting segment (the C domain). A recent heteronuclear NMR study of an engineered proinsulin monomer has provided evidence that its N-terminal arm (residues B1–B8) exhibits partial disorder as indicated by a trend toward N-terminal attenuation of amide-related {1H}-15N heteronuclear NOEs (26). Such studies employed substitutions in the classical dimer interface (ProB28 → Lys and LysB29 → Pro) and trimer interface (HisB10 → Asp) to enable NMR characterization at neutral pH (26, 41). Although such signals at B1 and B2 were not observable, complete 13C NMR resonance assignment was obtained. Trends in main chain 13C secondary chemical shifts (Fig. 10A) provides an estimate of disorder as inferred from a random-coil index (Fig. 10B) and predicted residue-specific order parameters on the picosecond-nanosecond time scale (Fig. 10C) (75). In Fig. 10 chemical-shift index values and predicted dynamic parameters of the B, C, and A domains are shown in blue, black, and red, respectively, in relation to helical segments (spirals at bottom of panel A). Whereas the A domain exhibits consistent low values of the random-coil index (Fig. 10B) and high values of the order parameter (Fig. 10C), the B domain is remarkable for “ramps” from B1–B8 and B25–B30. These trends predict that the B domain should exhibit a progressive increase in disorder on the picosecond-nanosecond time scale toward the N terminus and BC junction, respectively. In the future it would be of interest to obtain quantitative estimates of such disorder by measurement of 15N and 13C NMR relaxation times and their interpretation in relation to spectral density functions.
FIGURE 10.

13C NMR chemical-shift index of proinsulin and predicted protein dynamics. A, 13C NMR chemical shift index (CSI) values of B-domain (blue), C-domain (black), and A-domain (red): upper and lower panels pertain to 13Cα and 13Cβ secondary chemical shifts. Data pertain to an engineered proinsulin monomer at neutral pH as described (26). Helical segments are indicated by spirals at the bottom; red arrow indicates the segmental attenuation of CSI values in the A1–A8 segment, presumed to reflect motions at time scales longer than nanoseconds. Horizontal arrow indicates the B24–B27 β-strand. B and C, CSI values observed in proinsulin may be interpreted in relation to a random-coil index and predicted model-free order parameters (75). B, random-coil index values were calculated based on main chain secondary chemical shifts. C, predicted main chain order parameters as inferred by random-coil index values by the method of Berjanskii and Wishart (75). These parameters pertain to putative subnanosecond fluctuations. Whereas the A domain exhibits near-uniform high order parameters, the B domain N- and C-terminal segments exhibit increasing disorder (“ramps”) at its N terminus (residues B1–B8) and BC junction (residues B25–B30).
Determinants of Foldability
The N-terminal arm of the B-chain consists of residues proximal to the α-helical domain of the hormone (residues B5–B8) and residues distal to this domain (B1–B4). Evidence for the biological importance of the proximal portion of the arm has been provided by clinical observations that mutations at B5, B6, or B8 can cause permanent neonatal-onset diabetes mellitus, presumably due to toxic misfolding of the mutant proinsulin in the ER of pancreatic β-cells (14–17). Studies of insulin chain combination had earlier shown that these residues were critical for disulfide pairing even when receptor binding was not significantly impaired (36, 37, 67, 95).
The pertinence of the T-state to folding and the efficiency of disulfide pairing is supported by stereospecific effects of substitutions of GlyB8. Stabilization (or destabilization) of the T-state-specific β-turn (residues B7–B10) by respective D (or L) amino acid substitutions at B8 is associated with reciprocal effects on foldability and activity. On the one hand, because in the T-state GlyB8 exhibits a negative φ dihedral angle, D-substitutions at B8 are compatible with native-like structure (36, 37). Chiral enforcement of a negative φ angle enhances the efficiency of disulfide pairing in chain combination, but markedly impairs the activity of the insulin analog once formed (36, 37). On the other hand, L-amino acid substitutions, although consistent with the positive B8 φ angle of GlyB8 in the R-state, were observed to impair chain combination as well as yeast expression of single-chain insulin analogs (57, 96). Once formed, such analogs can nonetheless exhibit high activity (37). These reciprocal effects suggest that GlyB8 functions as an “ambidextrous switch” between folding-competent (T-like) and active (perhaps R-like) conformations.
We envisage that, within the partial folds of protein-folding intermediates, nascent T-like local structure in the proximal arm enhances the efficiency and fidelity of disulfide pairing. In particular, the conformation of residues B5–B8 (including inter-domain hydrogen bonding by the imidazole ring of HisB5 (37)) would profoundly alter the position of CysB7, its orientation with respect to CysA6 and CysA7, and in turn the trajectory of the distal arm once a disulfide bond was established. In the T-state residues B1–B4 pack loosely within an inter-chain crevice adjoining CysA6 and CysA7. The present study has shown that deletion of PheB1 blocks cellular folding of proinsulin whereas the two-chain model and mature product, des-PheB1-insulin, retains native-like properties. Ala scanning of the distal arm further demonstrated that each side chain influences the efficiency of folding and secretion (although to varying extents). Because PheB1 (and indeed, residues B1-B4 (58)) may be deleted without loss of activity, the distal arm provides an example of a folding element that is dispensable once the native state has been reached.
We imagine that the pattern of non-polar and polar side chains in the distal arm contributes to the efficiency and fidelity of disulfide pairing.9 General clustering of N-terminal hydrophobic side chains PheB1 and ValB2 with A-chain side chains could, for example, enhance the probability of collisions between the thiol (or thiolate) groups of CysB7 and CysA7. Productive alignment may then be enhanced by the proximal arm as a positive φ angle at B8 enables specific long-range interactions by the side chains of HisB5 and LeuB6. Because the coarseness of our experimental probes nonetheless prevents unambiguous atomic-scale interpretation, multiple molecular models may account for our findings. In the absence of the wild-type arm, the nascent conformational search of the polypeptide leading to native disulfide pairing may be rendered inefficient due to either destabilization of on-pathway intermediates or stabilization of off-pathway intermediates. Furthermore, apparent bottlenecks may reflect thermodynamic or kinetic traps. Arm mutations may even create barriers not pertinent to the folding mechanism of the wild-type protein.
The N-terminal arm of insulin is not conserved among the otherwise homologous growth factors IGF-I and IGF-II (97). The latter proteins lack a B1 residue and contain divergent side chains at positions B2–B5. In particular, IGF-specific residue ThrB5 is incompatible with efficient insulin chain combination (64) and is associated in vitro with formation of a competing IGF-I disulfide isomer on redox-coupled refolding (64, 98). Such divergent folding properties reflect co-evolution of specific IGF-binding proteins (64, 88, 89). The incomplete folding information of IGF-I and IGF-II highlights the breach in the autonomous foldability of proinsulin. We thus speculate that co-evolution of IGF-binding proteins has enabled IGF-I and IGF-II to explore regions of sequence space forbidden to insulin due to constraints of autonomous foldability.
Concluding Remarks
Insulin is one of the most studied globular proteins and yet among the least well understood. Understanding its conformational lifecycle will require extensive future studies, from deciphering the molecular mechanisms by which proinsulin folds to determining structures of hormone-receptor complexes. Recent advances in human genetics have highlighted the direct connection between the biophysical chemistry of proinsulin and the pathogenesis of β-cell dysfunction in monogenic forms of DM (14, 60).
The present study was designed to test the hypothesis that the N-terminal arm of proinsulin functions, despite its conformational variability in the mature hormone, as a cryptic folding element. Experimental design has integrated assays of protein folding and trafficking in mammalian cell culture with in vitro studies of protein structure, stability, and activity. Evidence has been provided that mutations in the arm can cause a broad range of effects on folding efficiency with possible clinical implications. Although no mutations associated with neonatal DM have been found to date in the distal arm (14, 60), it is possible that such mutations would induce less marked ER stress and β-cell dysfunction than those associated with neonatal-onset DM (which cluster in the proximal arm or within the α-helical domain) and so would present later in life (17). Evidence for mutation-specific ages of DM onset associated with the extent of biosynthetic impairment has recently been provided by comparison of mutations at arm position B6 (99). Whereas a neonatal mutation (LeuB6 → Pro) is predicted to distort both packing of the arm and its main chain conformation, a less perturbing substitution (LeuB6 → Met) is associated with onset of DM in adolescence and adulthood (17–36 years of age) (99).10 Distal arm mutations might thus present as a form of maturity-onset diabetes of the young or as polymorphisms conferring susceptibility to adult-onset β-cell dysfunction in the context of obesity (28, 100).
The genetics of neonatal DM and other diseases of misfolding suggest that protein evolution has been constrained not only by structure and function, but also by folding efficiency and, in the breach, the associated risk of toxic misfolding. The present studies of des-B1-insulin and arm analogs of proinsulin have demonstrated that efficient folding may require the transient function of a conserved folding element and that this essential role may be structurally unapparent once the ground state is reached. The contributions of PheB1 and ValB2 to cellular foldability are particularly striking in light of their high thermal B-factors in crystals (39) and 1H NMR motional narrowing in solution (40–42). Whereas these and other nonpolar side chains (including LeuB6, IleA10, and LeuA13) may contribute to hydrophobic collapse near cysteines (at B7, A6, A7, and A11), the efficiency of disulfide pairing is likely to require specific structural features of HisB5 and GlyB8 as visualized in the classical T-state of insulin (39). It is also possible that the aromatic ring of TyrA14 contributes to folding efficiency despite its variable positioning among crystal structures and flexibility in solution (72).
The crystallographic TR transition in zinc insulin hexamers has long provided a model for the transmission of conformational change in proteins (39). Although chameleon sequences analogous to the N-terminal arm of proinsulin are uncommon in the overall crystallographic data base, structures of native states, once reached, may mask the extent of conformational plasticity among protein-folding intermediates. We anticipate that functional annotation of protein structures will in general require multidisciplinary efforts to decipher the folding information hidden in their sequences.
Supplementary Material
Acknowledgments
We thank Eli Lilly and Co. for generously providing materials, S. Yadav and the staff of the peptide core facility at the Cleveland Clinic Foundation, S. Wang for assistance with insulin chain combination, K. Huang and the staff of the Case Center for Proteomics and Structural Biology for assistance with MS assays, L. Whittaker for assistance with receptor-binding studies, M. Lin for plasmid construction, N. B. Phillips and P. G. Katsoyannis for experimental advice, and G. G. Dodson, B. H. Frank, F. Ismail-Beigi, S. B. Kent, and D. F. Steiner for discussion.
This work was supported, in whole or in part, by National Institutes of Health Grants DK48280 (to P. A.) and DK0697674 (to M. A. W.) and a grant from the American Diabetes Association (to M. A. W.). This is a contribution from the Cleveland Center for Membrane and Structural Biology.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Tables S1–S3 and Figs. S1–S3.
Two cystines provide interior struts (A20–B19 and A6–A11) and the third, an external staple (A7–B7). Each is required for stability and activity. Insulin disulfide isomers exhibit molten structures of marginal stability and low activity (90, 101, 102).
The T and R families of insulin protomers also exhibit differences in orientation of the N-terminal A-chain α-helix associated with a change in conformation of the A7–B7 disulfide bridge.
This cellular model enabled assessment of effects of arm substitutions on disulfide pairing in the ER and subsequent trafficking, but does not recapitulate β-cell-specific prohormone processing or formation of microcrystalline insulin storage depots in glucose-regulated secretory vesicles.
The imino-acid substitution ProB1, although non-polar, was observed to impair biosynthesis (Fig. 8D, lanes 17 and 18) in association with perturbed cleavage of the signal peptide (M. Liu and P. Arvan, unpublished results).
Although the present set of substitutions tested at B1 highlights the importance of its non-polar character, ArgB1 occurs as a rare variant in non-mammalian insulin sequences. An example is provided by the divergent insulin of the hagfish (103), a member of a primitive lineage of marine craniates that lack a vertebral column. We speculate that the aliphatic portion of ArgB1 may pack against an analogous arm-related groove, whereas its charged guanidinium moiety projects into solvent. Analogous proximal side chain packing may underlie the incomplete block to folding and secretion imposed by LysB1 relative to AspB1 in the present studies.
A mutation has also been described in the B20–B24 β-turn of proinsulin (ArgB22 → Gln) that is presented in the second decade as maturity-onset diabetes of the young (MODY) (17). Chronic elevation of ER stress in β-cells of this patient presumably led to a slow but progressive loss of β-cell mass during childhood. ER stress may likewise contribute to the pathogenesis of insulin Los Angeles (PheB24 → Ser), a classical insulinopathy of variable penetrance presenting in adulthood (104).
- DM
- diabetes mellitus
- DKP-insulin and DKP-proinsulin
- analogs of insulin and proinsulin containing three substitutions in B chain (AspB10, LysB28, and ProB29)
- ER
- endoplasmic reticulum
- IGF-I and -II
- insulin-like growth factors I and II
- KP-insulin and KP-proinsulin
- analogs of insulin and proinsulin containing two substitutions in B chain (LysB28 and ProB29)
- IR
- insulin receptor
- Tricine
- N-[2-hydroxy-1,1-bis(hydroxymethyl)ethyl]glycine.
REFERENCES
- 1.Onuchic J. N., Luthey-Schulten Z., Wolynes P. G. (1997) Annu. Rev. Phys. Chem. 48, 545–600 [DOI] [PubMed] [Google Scholar]
- 2.Dobson C. M., Karplus M. (1999) Curr. Opin. Struct. Biol. 9, 92–101 [DOI] [PubMed] [Google Scholar]
- 3.Shakhnovich E., Farztdinov G., Gutin A. M., Karplus M. (1991) Phys. Rev. Lett. 67, 1665–1668 [DOI] [PubMed] [Google Scholar]
- 4.Onuchic J. N., Socci N. D., Luthey-Schulten Z., Wolynes P. G. (1996) Fold. Des. 1, 441–450 [DOI] [PubMed] [Google Scholar]
- 5.Dill K. A., Chan H. S. (1997) Nat. Struct. Biol. 4, 10–19 [DOI] [PubMed] [Google Scholar]
- 6.Lazaridis T., Karplus M. (1997) Science 278, 1928–1931 [DOI] [PubMed] [Google Scholar]
- 7.Mirny L. A., Abkevich V. I., Shakhnovich E. I. (1998) Proc. Natl. Acad. Sci. U.S.A. 95, 4976–4981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dinner A. R., Abkevich V., Shakhnovich E., Karplus M. (1999) Proteins 35, 34–40 [DOI] [PubMed] [Google Scholar]
- 9.Tiana G., Broglia R. A., Shakhnovich E. I. (2000) Proteins 39, 244–251 [PubMed] [Google Scholar]
- 10.Oliveberg M., Wolynes P. G. (2005) Q. Rev. Biophys. 38, 245–288 [DOI] [PubMed] [Google Scholar]
- 11.Bucciantini M., Giannoni E., Chiti F., Baroni F., Formigli L., Zurdo J., Taddei N., Ramponi G., Dobson C. M., Stefani M. (2002) Nature 416, 507–511 [DOI] [PubMed] [Google Scholar]
- 12.Dobson C. M. (1999) Trends Biochem. Sci. 24, 329–332 [DOI] [PubMed] [Google Scholar]
- 13.Dodson G., Steiner D. (1998) Curr. Opin. Struct. Biol. 8, 189–194 [DOI] [PubMed] [Google Scholar]
- 14.Støy J., Edghill E. L., Flanagan S. E., Ye H., Paz V. P., Pluzhnikov A., Below J. E., Hayes M. G., Cox N. J., Lipkind G. M., Lipton R. B., Greeley S. A., Patch A. M., Ellard S., Steiner D. F., Hattersley A. T., Philipson L. H., Bell G. I. (2007) Proc. Natl. Acad. Sci. U.S.A. 104, 15040–15044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Colombo C., Porzio O., Liu M., Massa O., Vasta M., Salardi S., Beccaria L., Monciotti C., Toni S., Pedersen O., Hansen T., Federici L., Pesavento R., Cadario F., Federici G., Ghirri P., Arvan P., Iafusco D., Barbetti F.Early Onset Diabetes Study Group of the Italian Society of Pediatric Endocrinology and Diabetes (2008) J. Clin. Invest. 118, 2148–2156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Edghill E. L., Flanagan S. E., Patch A. M., Boustred C., Parrish A., Shields B., Shepherd M. H., Hussain K., Kapoor R. R., Malecki M., MacDonald M. J., Støy J., Steiner D. F., Philipson L. H., Bell G. I., Hattersley A. T., Ellard S. (2008) Diabetes 57, 1034–1042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Molven A., Ringdal M., Nordbø A. M., Raeder H., Støy J., Lipkind G. M., Steiner D. F., Philipson L. H., Bergmann I., Aarskog D., Undlien D. E., Joner G., Søvik O., Bell G. I., Njølstad P. R. (2008) Diabetes 57, 1131–1135 [DOI] [PubMed] [Google Scholar]
- 18.Steiner D. F., Chan S. J. (1988) Horm. Metab. Res. 20, 443–444 [DOI] [PubMed] [Google Scholar]
- 19.Steiner D. F. (1967) Trans. N. Y. Acad. Sci. 30, 60–68 [DOI] [PubMed] [Google Scholar]
- 20.Frank B. H., Veros A. J. (1968) Biochem. Biophys. Res. Commun. 32, 155–160 [DOI] [PubMed] [Google Scholar]
- 21.Pekar A. H., Frank B. H. (1972) Biochemistry 11, 4013–4016 [DOI] [PubMed] [Google Scholar]
- 22.Frank B. H., Pekar A. H., Veros A. J. (1972) Diabetes 21, 486–491 [DOI] [PubMed] [Google Scholar]
- 23.Snell C. R., Smyth D. G. (1975) J. Biol. Chem. 250, 6291–6295 [PubMed] [Google Scholar]
- 24.Brems D. N., Brown P. L., Heckenlaible L. A., Frank B. H. (1990) Biochemistry 29, 9289–9293 [DOI] [PubMed] [Google Scholar]
- 25.Weiss M. A., Frank B. H., Khait I., Pekar A., Heiney R., Shoelson S. E., Neuringer L. J. (1990) Biochemistry 29, 8389–8401 [DOI] [PubMed] [Google Scholar]
- 26.Yang Y., Hua Q. X., Liu J., Shimizu E. H., Choquette M. H., Mackin R. B., Weiss M. A. (2010) J. Biol. Chem. 285, 7847–7851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu M., Li Y., Cavener D., Arvan P. (2005) J. Biol. Chem. 280, 13209–13212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ron D. (2002) J. Clin. Invest. 109, 443–445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu M., Hodish I., Rhodes C. J., Arvan P. (2007) Proc. Natl. Acad. Sci. U.S.A. 104, 15841–15846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Huang X. F., Arvan P. (1994) J. Biol. Chem. 269, 20838–20844 [PubMed] [Google Scholar]
- 31.Steiner D. F., Clark J. L., Nolan C., Rubenstein A. H., Margoliash E., Aten B., Oyer P. E. (1969) Recent Prog. Horm. Res. 25, 207–282 [DOI] [PubMed] [Google Scholar]
- 32.Galloway J. A., Hooper S. A., Spradlin C. T., Howey D. C., Frank B. H., Bowsher R. R., Anderson J. H. (1992) Diabetes Care 15, 666–692 [DOI] [PubMed] [Google Scholar]
- 33.Steiner D. F. (1998) Curr. Opin. Chem. Biol. 2, 31–39 [DOI] [PubMed] [Google Scholar]
- 34.Huang X. F., Arvan P. (1995) J. Biol. Chem. 270, 20417–20423 [DOI] [PubMed] [Google Scholar]
- 35.De Meyts P., Whittaker J. (2002) Nat. Rev. Drug Discov. 1, 769–783 [DOI] [PubMed] [Google Scholar]
- 36.Nakagawa S. H., Zhao M., Hua Q. X., Hu S. Q., Wan Z. L., Jia W., Weiss M. A. (2005) Biochemistry 44, 4984–4999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hua Q. X., Nakagawa S., Hu S. Q., Jia W., Wang S., Weiss M. A. (2006) J. Biol. Chem. 281, 24900–24909 [DOI] [PubMed] [Google Scholar]
- 38.Liu M., Wan Z. L., Chu Y. C., Aladdin H., Klaproth B., Choquette M., Hua Q. X., Mackin R. B., Rao J. S., De Meyts P., Katsoyannis P. G., Arvan P., Weiss M. A. (2009) J. Biol. Chem. 284, 35259–35272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Baker E. N., Blundell T. L., Cutfield J. F., Cutfield S. M., Dodson E. J., Dodson G. G., Hodgkin D. M., Hubbard R. E., Isaacs N. W., Reynolds C. D. (1988) Philos. Trans. R. Soc. Lond. B Biol. Sci. 319, 369–456 [DOI] [PubMed] [Google Scholar]
- 40.Hua Q. X., Shoelson S. E., Kochoyan M., Weiss M. A. (1991) Nature 354, 238–241 [DOI] [PubMed] [Google Scholar]
- 41.Hua Q. X., Hu S. Q., Frank B. H., Jia W., Chu Y. C., Wang S. H., Burke G. T., Katsoyannis P. G., Weiss M. A. (1996) J. Mol. Biol. 264, 390–403 [DOI] [PubMed] [Google Scholar]
- 42.Olsen H. B., Ludvigsen S., Kaarsholm N. C. (1996) Biochemistry 35, 8836–8845 [DOI] [PubMed] [Google Scholar]
- 43.Minor D. L., Jr., Kim P. S. (1994) Nature 367, 660–663 [DOI] [PubMed] [Google Scholar]
- 44.Pullen R. A., Lindsay D. G., Wood S. P., Tickle I. J., Blundell T. L., Wollmer A., Krail G., Brandenburg D., Zahn H., Gliemann J., Gammeltoft S. (1976) Nature 259, 369–373 [DOI] [PubMed] [Google Scholar]
- 45.Liang D. C., Chang W. R., Wan Z. L. (1994) Biophys. Chem. 50, 63–71 [DOI] [PubMed] [Google Scholar]
- 46.Kitagawa K., Ogawa H., Burke G. T., Chanley J. D., Katsoyannis P. G. (1984) Biochemistry 23, 1405–1413 [DOI] [PubMed] [Google Scholar]
- 47.Nakagawa S. H., Tager H. S. (1986) J. Biol. Chem. 261, 7332–7341 [PubMed] [Google Scholar]
- 48.Mirmira R. G., Tager H. S. (1989) J. Biol. Chem. 264, 6349–6354 [PubMed] [Google Scholar]
- 49.Mirmira R. G., Nakagawa S. H., Tager H. S. (1991) J. Biol. Chem. 266, 1428–1436 [PubMed] [Google Scholar]
- 50.Nakagawa S. H., Tager H. S. (1992) Biochemistry 31, 3204–3214 [DOI] [PubMed] [Google Scholar]
- 51.Hu S. Q., Burke G. T., Schwartz G. P., Ferderigos N., Ross J. B., Katsoyannis P. G. (1993) Biochemistry 32, 2631–2635 [DOI] [PubMed] [Google Scholar]
- 52.Nakagawa S. H., Tager H. S., Steiner D. F. (2000) Biochemistry 39, 15826–15835 [DOI] [PubMed] [Google Scholar]
- 53.Xu B., Hua Q. X., Nakagawa S. H., Jia W., Chu Y. C., Katsoyannis P. G., Weiss M. A. (2002) J. Mol. Biol. 316, 435–441 [DOI] [PubMed] [Google Scholar]
- 54.Huang K., Xu B., Hu S. Q., Chu Y. C., Hua Q. X., Qu Y., Li B., Wang S., Wang R. Y., Nakagawa S. H., Theede A. M., Whittaker J., De Meyts P., Katsoyannis P. G., Weiss M. A. (2004) J. Mol. Biol. 341, 529–550 [DOI] [PubMed] [Google Scholar]
- 55.Weiss M. A., Nakagawa S. H., Jia W., Xu B., Hua Q. X., Chu Y. C., Wang R. Y., Katsoyannis P. G. (2002) Biochemistry 41, 809–819 [DOI] [PubMed] [Google Scholar]
- 56.Hua Q. X., Chu Y. C., Jia W., Phillips N. F., Wang R. Y., Katsoyannis P. G., Weiss M. A. (2002) J. Biol. Chem. 277, 43443–43453 [DOI] [PubMed] [Google Scholar]
- 57.Kristensen C., Kjeldsen T., Wiberg F. C., Schäffer L., Hach M., Havelund S., Bass J., Steiner D. F., Andersen A. S. (1997) J. Biol. Chem. 272, 12978–12983 [DOI] [PubMed] [Google Scholar]
- 58.Schwartz G., Katsoyannis P. G. (1978) Biochemistry 17, 4550–4556 [DOI] [PubMed] [Google Scholar]
- 59.Bentley G., Dodson E., Dodson G., Hodgkin D., Mercola D. (1976) Nature 261, 166–168 [DOI] [PubMed] [Google Scholar]
- 60.Weiss M. A. (2009) J. Biol. Chem. 284, 19159–19163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Brems D. N., Alter L. A., Beckage M. J., Chance R. E., DiMarchi R. D., Green L. K., Long H. B., Pekar A. H., Shields J. E., Frank B. H. (1992) Protein Eng. 5, 527–533 [DOI] [PubMed] [Google Scholar]
- 62.Shoelson S. E., Lu Z. X., Parlautan L., Lynch C. S., Weiss M. A. (1992) Biochemistry 31, 1757–1767 [DOI] [PubMed] [Google Scholar]
- 63.Wan Z. L., Huang K., Hu S. Q., Whittaker J., Weiss M. A. (2008) J. Biol. Chem. 283, 21198–21210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sohma Y., Hua Q. X., Liu M., Phillips N. B., Hu S. Q., Whittaker J., Whittaker L. J., Ng A., Roberts C. T., Jr., Arvan P., Kent S. B., Weiss M. A. (2010) J. Biol. Chem. 285, 5040–5055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Whittaker J., Whittaker L. (2005) J. Biol. Chem. 280, 20932–20936 [DOI] [PubMed] [Google Scholar]
- 66.Wang Z. X. (1995) FEBS Lett. 360, 111–114 [DOI] [PubMed] [Google Scholar]
- 67.Hua Q. X., Liu M., Hu S. Q., Jia W., Arvan P., Weiss M. A. (2006) J. Biol. Chem. 281, 24889–24899 [DOI] [PubMed] [Google Scholar]
- 68.Wuthrich K. (1986) NMR of Proteins and Nucleic Acids, John Wiley & Sons, New York [Google Scholar]
- 69.Jorgensen L. N., Dideriksen L. H., Drejer K. (1992) Diabetologia 35, Suppl. 1, A3 [Google Scholar]
- 70.Sosnick T. R., Fang X., Shelton V. M. (2000) Methods Enzymol. 317, 393–409 [DOI] [PubMed] [Google Scholar]
- 71.Pace C. N., Shaw K. L. (2000) Proteins 4, (suppl.) 1–7 [DOI] [PubMed] [Google Scholar]
- 72.Weiss M. A., Nguyen D. T., Khait I., Inouye K., Frank B. H., Beckage M., O'Shea E., Shoelson S. E., Karplus M., Neuringer L. J. (1989) Biochemistry 28, 9855–9873 [DOI] [PubMed] [Google Scholar]
- 73.Yang Y., Petkova A., Huang K., Xu B., Hua Q. X., Ye I. J., Chu Y. C., Hu S. Q., Phillips N. B., Whittaker J., Ismail-Beigi F., Mackin R. B., Katsoyannis P. G., Tycko R., Weiss M. A. (2010) J. Biol. Chem. 285, 10806–10821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wishart D. S., Sykes B. D., Richards F. M. (1992) Biochemistry 31, 1647–1651 [DOI] [PubMed] [Google Scholar]
- 75.Berjanskii M. V., Wishart D. S. (2005) J. Am. Chem. Soc. 127, 14972–14973 [DOI] [PubMed] [Google Scholar]
- 76.Katsoyannis P. G. (1966) Science 154, 1509–1514 [DOI] [PubMed] [Google Scholar]
- 77.Brange J. (1997) Diabetologia 40, S48–S53 [DOI] [PubMed] [Google Scholar]
- 78.Tang J. G., Tsou C. L. (1990) Biochem. J. 268, 429–435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Whittingham J. L., Scott D. J., Chance K., Wilson A., Finch J., Brange J., Guy Dodson G. (2002) J. Mol. Biol. 318, 479–490 [DOI] [PubMed] [Google Scholar]
- 80.Weiss M. A., Hua Q. X., Jia W., Chu Y. C., Wang R. Y., Katsoyannis P. G. (2000) Biochemistry 39, 15429–15440 [DOI] [PubMed] [Google Scholar]
- 81.Liu M., Ramos-Castañeda J., Arvan P. (2003) J. Biol. Chem. 278, 14798–14805 [DOI] [PubMed] [Google Scholar]
- 82.Xu B., Hua Q. X., Nakagawa S. H., Jia W., Chu Y. C., Katsoyannis P. G., Weiss M. A. (2002) Protein Sci. 11, 104–116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Smith M. C., Cook J. A., Furman T. C., Occolowitz J. L. (1989) J. Biol. Chem. 264, 9314–9321 [PubMed] [Google Scholar]
- 84.Hober S., Forsberg G., Palm G., Hartmanis M., Nilsson B. (1992) Biochemistry 31, 1749–1756 [DOI] [PubMed] [Google Scholar]
- 85.Hejnaes K. R., Bayne S., Nørskov L., Sørensen H. H., Thomsen J., Schäffer L., Wollmer A., Skriver L. (1992) Protein Eng. 5, 797–806 [DOI] [PubMed] [Google Scholar]
- 86.Miller J. A., Narhi L. O., Hua Q. X., Rosenfeld R., Arakawa T., Rohde M., Prestrelski S., Lauren S., Stoney K. S., Tsai L., Weiss M. A. (1993) Biochemistry 32, 5203–5213 [DOI] [PubMed] [Google Scholar]
- 87.Hober S., Lundström Ljung J., Uhlén M., Nilsson B. (1999) FEBS Lett. 443, 271–276 [DOI] [PubMed] [Google Scholar]
- 88.Hober S., Hansson A., Uhlén M., Nilsson B. (1994) Biochemistry 33, 6758–6761 [DOI] [PubMed] [Google Scholar]
- 89.Hober S., Uhlén M., Nilsson B. (1997) Biochemistry 36, 4616–4622 [DOI] [PubMed] [Google Scholar]
- 90.Hua Q. X., Jia W., Frank B. H., Phillips N. F., Weiss M. A. (2002) Biochemistry 41, 14700–14715 [DOI] [PubMed] [Google Scholar]
- 91.Betts S., King J. (1999) Structure 7, R131–R139 [DOI] [PubMed] [Google Scholar]
- 92.Bliss M. (1982) The Discovery of Insulin, University of Chicago Press, Chicago, IL [Google Scholar]
- 93.Brader M. L., Dunn M. F. (1991) Trends Biochem. Sci. 16, 341–345 [DOI] [PubMed] [Google Scholar]
- 94.Derewenda U., Derewenda Z., Dodson E. J., Dodson G. G., Reynolds C. D., Smith G. D., Sparks C., Swenson D. (1989) Nature 338, 594–596 [DOI] [PubMed] [Google Scholar]
- 95.Nakagawa S. H., Tager H. S. (1991) J. Biol. Chem. 266, 11502–11509 [PubMed] [Google Scholar]
- 96.Guo Z. Y., Zhang Z., Jia X. Y., Tang Y. H., Feng Y. M. (2005) Acta Biochim. Biophys. Sin. 37, 673–679 [DOI] [PubMed] [Google Scholar]
- 97.Guo Z. Y., Shen L., Feng Y. M. (2002) Biochemistry 41, 1556–1567 [DOI] [PubMed] [Google Scholar]
- 98.Chen Y., You Y., Jin R., Guo Z. Y., Feng Y. M. (2004) Biochemistry 43, 9225–9233 [DOI] [PubMed] [Google Scholar]
- 99.Meur G., Simon A., Harun N., Virally M., Dechaume A., Bonnefond A., Fetita S., Tarasov A. I., Guillausseau P. J., Boesgaard T. W., Pedersen O., Hansen T., Polak M., Gautier J. F., Froguel P., Rutter G. A., Vaxillaire M. (2010) Diabetes 59, 653–661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Florez J. C. (2008) Diabetologia 51, 1100–1110 [DOI] [PubMed] [Google Scholar]
- 101.Sieber P. S., Eisler K., Kamber B., Riniker B., Rittel W., Märki F., de Gasparo M. (1978) Hoppe Seyler's Z. Physiol. Chem. 359, 113–123 [DOI] [PubMed] [Google Scholar]
- 102.Hua Q. X., Gozani S. N., Chance R. E., Hoffmann J. A., Frank B. H., Weiss M. A. (1995) Nat. Struct. Biol. 2, 129–138 [DOI] [PubMed] [Google Scholar]
- 103.Cutfield J. F., Cutfield S. M., Dodson E. J., Dodson G. G., Emdin S. F., Reynolds C. D. (1979) J. Mol. Biol. 132, 85–100 [DOI] [PubMed] [Google Scholar]
- 104.Shoelson S. E., Polonsky K. S., Zeidler A., Rubenstein A. H., Tager H. S. (1984) J. Clin. Invest. 73, 1351–1358 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.