

Abstract
A combination of recombinant FKP and α-(1→3)-fucosyltransferase allows the facile synthesis of the sialyl Lewis X tetrasaccharide glycan and its derivatives in excellent yield. In this system, the universal fucosyl donor, guanidine 5′-diphosphate-β-L-fucose (GDP-fucose), or its analogues can be generated in situ by cofactor recycling using pyruvate kinase.
Keywords: Chemoenzymatic synthesis, transferases, fucose, sialic acid
1. Introduction
The kaleidoscopic functions of cell-surface glycans are governed by their diverse structures and their dynamic interactions with glycan-binding proteins (i.e., lectins).1 Multivalent glycan–lectin interactions either convey changes in the extracellular environment or modulate cell–cell/cell–pathogen communications. For example, sialyl Lewis X (sLex), a tetrasaccharide glycan constitutively expressed on leukocytes, mediates the recruitment of these cells from the bloodstream into surrounding tissues during the early stage of inflammation.2-4 SLex binds to E- and P-selectins, upregulated on the surface of endothelial cells, leading to leukocyte tethering and extravasation.5 In addition, the sLex–E-selectin interaction is also crucial for directing the migration of hematopoietic stem cells through the blood, across the endothelial vasculature to different organs and to their bone marrow niches, the first and essential step in clinical stem cell transplantation.6
As revealed by NMR spectroscopyand X-ray crystallography, the Lex domain of the sLex tetrasaccharide assumes a very rigid structure, displaying two surfaces along the NeuAc-Gal-Fuc axis of opposite hydrophility.7 The stability of this highly compact, bipolar structure stems from the stacking of its fucose ring on top of the galactose residue with its exocyclic C-5 methyl group forming key van der Waals contacts with the hydrophobic surface of the galactose. Studies have shown the formation of this stack to be important in the binding of sLex to target lectins. Removal of the methyl group leads to a five-fold decrease in binding affinity of sLex to its target protein, E-selectin.8
In inflammatory diseases (i.e., rheumatoid arthritis, asthma and transplant rejection), the body’s immune system inappropriately triggers an inflammatory signal and causes damage to its own tissues. One approach to treat these diseases is to disrupt the recruitment of leukocytes, a process mediated by sLex–selectin interactions. The possibility that inhibitors of selectin-mediated cell adhesion could serve as broad-spectrum anti-inflammatory agents has sparked significant efforts in both the pharmaceutical industry and academic laboratories to design sLex-based small-molecule inhibitors as selectin antagonists.5 In order to evaluate the therapeutic value of sLex-based selectin antagonists, it is necessary to develop efficient methods for the synthesis of sLex and its derivatives bearing unnatural functionalities in each of the monosaccharide building blocks for structure–activity relationship studies.
Chemical syntheses of sLex have been pursued intensively,5, 9 and have permitted the elucidation of the key functional groups and structural features that contribute to the sLex–selectin interaction. Despite recent advances in glycosylation methodologies, the chemical synthesis of complex fucosides is still complicated by tedious protecting group manipulations, and the use of harsh reagents and stringent anhydrous conditions.10 This problem has been elegantly addressed by several groups using alternative approaches based on enzymatic glycosylation. Pioneering studies on the chemoenzymatic synthesis of the sLex tetrasaccharide were performed by Wong and co-workers who employed a recombinant human α-(1→3)-fucosyltransferase produced in eukaryotic cells.7, 11 In this process, the universal fucosyl donor, guanidine 5′-diphosphate-β-L-fucose (GDP-fucose), was generated from mannose-1-phosphate (Man-1-P) via the combination of three enzymes: GDP-mannose pyrophosphorylase, GDP-mannose 4,6-dehydrase (GMD) and GDP-keto-6-deoxymannose 3,5-epimerase/4-reductase (GMER). The latter two enzymes are found in the de novo biosynthetic pathway of GDP-fucose.12 The human α-(1→3)-fucosyltransferase, being a type II transmembrane glycoprotein, is difficult to produce in large quantities. Additionally, Man-1-P, although commercially available, is prohibitively expensive for large-scale synthesis.
We recently reported a facile and cost-effective method for the chemoenzymatic synthesis of GDP-fucose and Lex derivatives.13 This method exploits FKP (L-fucokinase/GDP-fucose pyrophosphorylase), a bifunctional enzyme isolated from Bacteroides fragilis 9343, which converts L-fucose to fucose-1-phosphate (Fuc-1-P) and thence to GDP-fucose.14, 15 This transformation is found in the salvage pathway of B. fragilis 9343 GDP-fucose production and is conserved in all Bacteroides species. We found that a His6-tagged recombinant FKP, expressed in E. coli, has relaxed specificity toward fucose analogues bearing unnatural substituents at the C-5 position and is capable of generating GDP-fucose derivatives in vitro with high efficiency. Furthermore, we demonstrated that the activities of FKP can be combined with a Helicobacter pylori α-(1→3)-fucosyltransferase for preparative-scale syntheses of Lex trisaccharide glycans and its structurally related derivatives.13 Herein, we report a novel chemoenzymatic method for the synthesis of the sLex tetrasaccharide glycan and its derivatives on a preparative scale using the recombinant FKP and the α-(1→3)-fucosyltransferase (Scheme 1). Importantly, this approach regiospecifically incorporates fucose or its synthetic analogues to the acceptor glycan sialyl N-acetyllactosamine (sLacNAc). Moreover, we demonstrate that the atom economy of this synthetic process can be improved by simply introducing a biologically inspired cofactor recycling system, in which both ATP and GTP are formed in situ using a commercially available pyruvate kinase.
Scheme 1.

A chemoenzymatic approach for the synthesis of the sLex tetrasaccharide derivatives.
2. Results and discussion
Our synthetic route is based on previous reports that bacterial fucosyltransferases act efficiently on sialylated glycans with good activity.16 To confirm the activity of the recombinant H. pylori α-(1→3)-fucosyltransferase toward sLacNAc, we prepared this acceptor trisaccharide glycan using the chemoenzymatic approach developed by Chen and co-workers for synthesizing α-(2→3)-linked sialosides.17 In this method, sialic acid or its analogues are converted to the corresponding sialylated trisaccharides in a one-pot reaction using a combination of two enzymes: CMP-sialic acid synthetase and α-(2→3)-sialyltransferase. We cloned a Neisseria meningitides CMP-sialic acid synthetase18 and a Pasteurella multocida α-(2→3)-sialyltransferase19 and expressed them in E. coli. With these recombinant enzymes in hand, we successfully synthesized sLacNAc using the system shown in Scheme 1, in which CMP-sialic acid was generated in situ by the N. meningitides CMP-sialic acid synthetase.
To compare the activity of the α-(1→3)-fucosyltransferase toward LacNAc and sLacNAc, we measured the kcat and KM of the fucosylation reaction for both acceptor substrates using a coupled enzyme assay (Supplementary Data). Both of these substrates bear a short 2-azidoethyl spacer that allows fast copper-free click modification via azide–alkyne cycloaddition20 to fabricate glycan microarrays for high-throughput screening of sLex–lectin interactions.21 To our satisfaction, sialylation of the LacNac afforded a 50% increase in turnover number of the fucosyltransferase (Table 1). However, the incorporation of the bulky and charged sialic acid residue hampered the formation of the enzyme–substrate complex. The KM value determined for 2-azidoethyl sLacNAc was 11-fold higher than that determined for 2-azidoethyl LacNAc.
Table 1.
Activity and specificity of the α-(1→3)-fucosyltransferase
| Substrate | KM (mM) | kcat (min-1) | kcat/KM (mM-1 s-1) |
|---|---|---|---|
| 0.34 ± 0.05 | 320 ± 20 | 15.7 | |
| 1.3 ± 0.1 | 442 ± 10 | 5.7 | |
| 3.9 ± 0.2 | 483 ± 10 | 2.1 | |
| 0.18 ± 0.02 | 265 ± 10 | 24.5 |
The fucosylation activity of α-(1→3)-fucosyltransferase was measured using a coupled enzyme assay. All kinetic measurements were performed in 100 mM Tris-HCl, pH 7.5 at 37 °C. Each data point was collected in triplicate. Error margins were obtained from non-linear regression analyses of pooled triplicate measurements. The kinetic paramenters for GDP-L-fucose were measured using LacNAcCH2CH2N3 as the acceptor substrate.
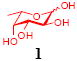
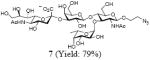
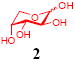
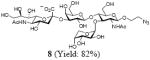
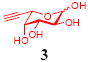
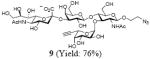
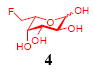
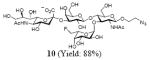
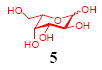
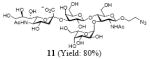
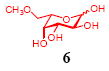
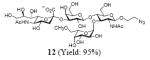
After we confirmed the activity of the recombinant α-(1→3)-fucosyltransferase toward sLacNAc, the stage was set for the one-pot synthesis of the sLex tetrasaccharides. This task was accomplished by combining a fucose analogue and the acceptor trisaccharide, 2-azidoethyl sLacNAc with the recombinant FKP and α-(1→3)-fucosyltransferase. An inorganic pyrophosphatase (Sigma) was included to hydrolyze the pyrophosphate byproduct generated in the reaction and to drive the reaction to completion. Using this method, we synthesized a series of sLex tretrasaccharide derivatives (7–12, 17–22 mg each) bearing a variety of substituents at the fucose C-5 position (Table 2). SLex derivative 3 was functionalized with an alkynyl group that could enhance the van der Waals contacts between the fucose residue and the galactose. Fucose analogues 4, 5 and 6 were modified with fluoride, hydroxyl and methoxyl groups, respectively. These groups might form additional hydrogen bonds with the neighboring galactose, thereby enforcing different conformational constraints on the final sLex tetrasaccharides. The resulting sLex derivatives bearing these functionalities will be valuable tools for probing the influence of the conformational constraints in sLex–selectin binding. The unnatural tetrasaccharide glycans in Table 2 were typically isolated in yields greater than 75%. In a few cases nearly quantitative formation of the desired tetrasaccharide was achieved.
Table 2.
Synthesis of sLex tetrasaccharide derivatives
| l-Fucose analogues | sLex derivatives |
|---|---|
 |
 |
 |
 |
 |
 |
 |
 |
 |
 |
 |
 |
Though this chemoenzymatic process is straightforward and highly efficient, the generation of one molecule of sLex product is accompanied by the formation of an equal amount of ADP and GDP byproducts. To overcome this drawback, we developed a regeneration system in which the ADP and GDP byproducts can be recycled for in situ generation of the universal fucosyl donor—GDP-fucose. During glycolysis, ADP is reconverted into ATP through the transfer of phosphate from phosphoenolpyruvate in a process catalyzed by pyruvate kinase. Wong and Thiem applied this principle to produce sugar nucleotides in situ using a coupled-enzymatic approach.22, 23 Inspired by these precedents, we introduced readily available pyruvate kinase into the fucosylation system for cofactor recycling (Scheme 2). We supplied the reaction system with a catalytic amount of ADP and GDP. In the presence of pyruvate kinase and phosphoenolpyruvate (2 equiv to fucose), ADP and GDP were converted in situ into ATP and GTP, respectively, as the substrates for FKP to produce GDP-fucose. As reported previously, pyruvate kinase has similar maximum velocities for ADP and GDP. However, its KM for ADP is four-fold smaller than its KM for GDP.24 Since fucose phosphorylation is the rate-limiting step of the FKP-catalyzed GDP-fucose formation, the in situ regeneration of GDP-Fuc is sensitive to the ADP to GDP ratio.13 In order to identify an optimal condition for the recycling system, we screened a combination of ADP and GDP at different molar ratios while maintaining the concentration of pyruvate kinase and fucosylation enzymes constant. We discovered that 1:2 ADP–GDP afforded the fastest reaction rate when the ADP loading was 10 mol % relative to fucose. Using this recycling system, the fucosylation reaction finished in four hours and afforded sLex in 83% yield, which is comparable to the yield reported above.
Scheme 2.

Synthesis of the sLex tetrasaccharide with in situ cofactor regeneration.
3. Conclusions
In conclusion, the chemoenzymatic method described here offers a practical and versatile approach for the synthesis of the sLex tetrasaccharide and its derivatives. As we routinely express the FKP and fucosyltransferase with high activity on a 100-miligram scale in a single day, this procedure can be easily extended for multigram synthesis (The specific activities of FKP and α-(1→3)-fucosyltransferase were determined to be 4.5 U mg-1 protein and 6–10 U mg-1 protein, respectively. One unit is defined as the amount of enzyme that is required to produce 1 μmol of product per minute at 37 °C). Not only does this method provide a facile means to produce sLex bearing neo-substituents at the fucose C-5 position, it is also directly applicable to the generation of sLex derivatives with unnatural functional groups incorporated at the sialic acid C-5 or C-9 position.17, 25 Unnatural sLex binds to all three selectins (E-, L-, P-selectins) with similar affinity. By incorporating unnatural functionalities of various stereoelectronic properties, we may be able to generate sLex derivatives that are selective for a particular selectin. Currently, we are using this method to produce a sLex library for fabricating glycan microarrays to profile sLex–selectin interactions.
4. Experimental
4.1 Kinetic measurements
Initial velocity experiments were performed at various concentrations of one substrate in the presence of a fixed, saturating concentration of the second substrate. Initial velocities of the α-(1→3)-fucosyltransferase-catalyzed fucosylation reactions were assayed spectrophotometrically by coupling the formation of GDP to the reaction of pyruvate kinase and lactate dehydrogenase. The decrease in absorbance of NADH at 340 nm (ε = 6220 M-1 cm-1) was measured at 37 °C using a BioTek Synergy 4 microplate reader. The standard reaction contained 100 mM Tris (pH 7.5), 1.0 mM MnCl2, 1.0 mM phosphoenolpyruvate, 225 μM NADH, 3.73 units pyruvate kinase, and 4.53 units lactate dehydrogenase in addition to the substrates in a final volume of 200 μL. After incubation for 15 min at 37 °C, the reactions were initiated by the addition of α-(1→3)-fucosyltransferase (18.5 nM). The α-(1→3)-fucosyltransferase activities were corrected for background activity. The rate of product formation is proportional to the rate of NADH oxidation, where one molecule of NADH is oxidized for each molecule of Lex or sLex formed. Individual substrate saturation kinetic data were fitted to Eq. 1 Graphpad Prism 5.00:
| (1) |
where Vmax is the maximal velocity, [A] is the substrate concentration, and KM is the Michaelis–Menten constant (KM).
4.2 General procedure for preparative-scale (0.059 mmol) synthesis of the sLacNAc derivatives
Reactions were typically carried out in a 15-mL centrifuge tube with 5.0 mL Tris-HCl buffer (200 mM, pH 8.8) containing sialic acid sodium salt (27.7 mg, 0.088 mmol, 1.5 equiv), CTP disodium salt (46.4 mg, 0.088 mmol, 1.5 equiv), N-acetyllactosamine or 2-azidoethyl N-acetyllactosamine (22.6 or 26.5 mg, 0.059 mmol, 1 equiv), MgCl2 (20 mM), Neisseria meningitides CMP-sialic acid synthetase (3.0 units), and Pasteurella multocida α2,3 sialyltransferase (1.5 units). The reaction mixture was incubated at 37 °C for 3 h with shaking (225 rpm). The reaction was monitored by TLC analysis using 4:2:1:0.1 EtOAc–MeOH–H2O–HOAc as the developing solvent and the plates were stained with 5% H2SO4 in EtOH. After adding the same volume of ice-cold EtOH to quench the reaction, the alcoholic mixture was incubated on ice for 15 min. Insoluble material was removed by centrifugation (8,000 × g, 30 min), and the supernatant was concentrated in vacuo. The aqueous residues were lyophilized to dryness. The crude products were purified by Bio-Gel P2 gel-filtration chromatography (1.5 × 120 cm) eluted with aq NH4HCO3 (50 mM). Only the fractions containing the product were collected, lyophilized, and further purified using Bio-Gel P2 gel-filtration chromatography (1.5 × 120 cm) eluted with NH4HCO3 (50 mM). Lyophilized sialyl lactosamines were characterized by NMR spectroscopy and HRMS. Yield: sLacNAc, 35.9 mg (90%); 2-azidoethyl sLacNAc, 39.6 mg (90 %).
4.3 General procedure for preparative-scale (0.025 mmol) synthesis of the sLex tetrasaccharide derivatives
One-pot reactions were performed in 15-mL centrifuge tubes with 5.0 mL Tris-HCl buffer (100 mM, pH 7.5) containing L-fucose or its C-5 substituted analogs (2.0 equiv, 0.05 mmol), 2-azidoethyl sLacNAc (18.6 mg, 0.025 mmol, 1.0 equiv,), ATP (25.4mg, 0.05 mmol, 2.0 equiv), GTP (26.2, 0.05 mmol, 2.0 equiv), MnSO4 (20 mM). The enzymes inorganic pyrophosphatase (100 units, lyophilized form containing MgCl2), FKP (9 units), and α-(1→3)-fucosyltransferase (2.4 units) were added to the solution. The reaction mixture was incubated at 37 °C for 3 h with vigorous shaking (225 rpm). The reaction was monitored by TLC analysis using 4:2:2:0.1 EtOAc–EtOH–H2O–HOAc as the developing solvent, and the plates were stained with 5% H2SO4 in EtOH. After adding the same volume of ice-cold EtOH to quench the reaction, the alcoholic mixture was incubated on ice for 15 min. Insoluble material was removed by centrifugation (8,000 × g, 30 min), and the supernatant was concentrated in vacuo. The aqueous residues were lyophilized to dryness. Crude products were purified by Bio-Gel P2 gel-filtration chromatography (1.5 × 120 cm) eluted with aq NH4HCO3 (50 mM). Only the fractions containing the product were collected, lyophilized, and further purified using Bio-Gel P2 gel-filtration chromatography (1.5 × 120 cm) eluted with NH4HCO3 (50 mM). Lyophilized sLex derivatives were characterized by NMR spectroscopy and HRMS. Yield: 17.1–21.9 mg (76–95 %).
4.4 General procedure for preparative-scale (0.025 mmol) synthesis of the 2-azidoethyl sLex tetrasaccharide with in situ regeneration of GDP-fucose
The synthesis was performed in 15-mL centrifuge tubes with 5.0 mL of Tris-HCl buffer (100 mM, pH 7.5) containing L-fucose (8.20 mg, 0.05 mmol, 2.0 equiv), 2-azidoethyl sLacNAc (18.6 mg, 0.025 mmol, 1.0 equiv), ADP (2.13 mg, 0.005 mmol, 0.2 equiv), GDP (4.4 mg, 0.01 mmol, 0.4 equiv), MnSO4 (20 mM), phosphoenolpyruvate potassium salt (20.6 mg, 0.10 mmol, 4.0 equiv), KCl (80 mM). The enzymes pyruvate kinase (400 units), inorganic pyrophosphatase (100 units, lyophilized form containing MgCl2), FKP (9 units), and α-(1→3)-fucosyltransferase (2.4 units) were added to the solution. The reaction mixture was incubated at 37 °C for 4.5 h with vigorous shaking (225 rpm). The reaction was monitored by TLC analysis using 4:2:2:0.1 EtOAc–EtOH–H2O–HOAc as the developing solvent, and the plates were stained with 5% H2SO4 in EtOH. After adding the same volume of ice-cold EtOH to quench the reaction, the alcoholic mixture was incubated on ice for 15 min. Insoluble material was removed by centrifugation (8,000 × g, 30 min) and the supernatant was concentrated in vacuo. The aqueous residues were lyophilized to dryness. Crude product was purified by Bio-Gel P2 gel filtration chromatography (1.5 × 120 cm) eluted with aq NH4HCO3 (50 mM). Only the fractions containing the product were collected, lyophilized, and further purified using Bio-Gel P2 gel filtration chromatography (1.5 × 120 cm) eluted with NH4HCO3 (50 mM). Lyophilized 2-azidoethyl sLex was characterized by NMR spectroscopy and HRMS. Yield: 18.6 mg (83 %).
4.4.1 2-Azidoethyl (5-acetamido-3,5-dideoxy-d-glycero-α-d-galacto-2-nonulo pyranosylonic acid)-(2→3)-β-D-galactopyranosyl-(1→4)[(1→3)-α-l-fucopyranosyl]-2-acetamido-2-deoxy-β-D-glucopyranoside (7)
This compound was synthesized according to general procedure for synthesis of the sLex tetrasaccharide derivatives (17.5 mg, yield: 79%). 1H NMR (600 MHz, D2O): δ 1.18 (d, J = 5.8 Hz, 3H), 1.80 (t, J = 12.0 Hz, 1H), 2.04 (s, br, 6H), 2.77 (d, J = 11.6 Hz, 1H), 3.41-3.55 (m, 3H), 3.57-3.63 (m, 3H), 3.64-3.74 (m, 3H), 3.75-3.82 (m, 3H), 3.84-4.00 (m, 10H), 4.02-4.10 (m, 4H), 4.53 (d, J = 7.5 Hz, 1H), 4.61 (d, J = 8.0 Hz, 1H), 5.11 (s, 1H). 13C NMR (150 MHz, D2O): δ 15.3, 22.0, 22.3, 39.8, 50.4, 51.7, 55.7, 59.7, 60.9, 61.5 62.6, 66.7, 67.3, 67.7, 68.1, 68.3, 68.7, 69.2, 69.3, 71.9, 72.9, 73.3, 74.85, 74.91, 75.3, 75.6, 98.6, 99.7, 100.8, 101.6, 173.9, 174.4, 175.0. HRMS: Calcd for [C33H55N5O23+H]+: 890.3363. Found: 890.3367.
4.4.2 2-Azidoethyl (5-acetamido-3,5-dideoxy-d-glycero-α-d-galacto-2-nonulopyranosylonic acid)-(2→3)-β-D-galactopyranosyl-(1→4)[(1→3)-α-D-arabinopyranosyl]-2-acetamido-2-deoxy-β-D-glucopyranoside (8)
This compound was synthesized according to general procedure for synthesis of the sLex tetrasaccharide derivatives (18.0 mg, yield: 82%). 1H NMR (600 MHz, D2O):δ 1.80 (d, J = 12.2 Hz, 1H), 2.04 (s, 3H), 2.05 (s, 3H), 2.77 (d, J = 12.5, 4.6 Hz, 1H), 3.41-3.45 (m, 1H), 3.47-3.50 (m, 1H), 3.52-3.58 (m, 2H), 3.59-3.63 (m, 3H), 3.65-3.72 (m, 4H), 3.74-3.79 (m, 2H), 3.84-3.91 (m, 6H), 3.93-3.95 (m, 2H), 3.96-4.07 (m, 4H), 4.10 (dd, J = 9.8, 3.1 Hz, 1 H), 4.55 (d, J = 7.8 Hz, 1H), 4.61 (d, J = 8.3 Hz, 1H), 4.65 (d, J = 12.5 Hz, 1H), 5.18 (d, J = 3.7 Hz, 1H). 13C NMR (150 MHz, D2O):δ 22.0, 22.3, 39.7, 50.4, 51.7, 55.6, 59.7, 60.0, 60.6, 61.4, 62.6, 63.6, 67.3, 68.0, 68.3, 68.4, 68.7, 69.1, 69.4, 71.9, 72.9, 73.2, 74.9, 75.1, 75.3, 75.6, 98.9, 99.7, 100.9, 101.5, 173.9, 174.4, 175.0. HRMS: Calcd for [C32H53N5O23+H]+: 876.3210. Found: 876.3213.
4.4.3 2-Azidoethyl (5-acetamido-3,5-dideoxy-d-glycero-α-d-galacto-2-nonulopyranosylonic acid)-(2→3)-β-D-galactopyranosyl-(1→4)[(1→3)-6,7-deoxy-α-L-galacto-hept-6-ynopyranosyl]-2-acetamido-2-deoxy-β-D-glucopyranoside (9)
This compound was synthesized according to general procedure for synthesis of the sLex tetrasaccharide derivatives (17.1 mg, yield: 76%). 1H NMR (600 MHz, D2O): δ 1.82 (t, J = 12.2 Hz, 1H), 2.04 (s, 6H), 2.77 (dd, J = 12.3, 4.3 Hz, 1H), 2.01 (d, J = 2.0 Hz, 1H), 3.41-3.45 (m, 1H), 3.47-3.51 (m, 1H), 3.59-3.62 (m, 3H), 3.64-3.74 (m, 6H), 3.75-3.79 (m, 1H), 3.82-3.92 (m, 7H), 3.94-4.00 (m, 3H), 4.01-4.07 (m, 3H), 4.10 (dd, J = 9.8, 2.6 Hz, 1H), 4.54 (d, J = 7.9 Hz, 1 H), 4.62 (d, J = 8.4 Hz, 1H), 5.15 (d, J = 3.9 Hz, 1H), 5.58 (s, 1H). 13C NMR (150 MHz, D2O): δ 22.0, 22.3, 39.8, 50.4, 51.7, 55.5, 59.6, 61.5, 62.6, 63.2, 67.2, 67.3, 68.1, 68.28, 68.34, 68.7, 69.1, 71.0, 71.8, 72.9, 73.3, 75.0, 75.3, 75.7, 76.1, 79.4, 99.0, 99.6, 100.9, 101.8, 173.8, 174.4, 175.0. HRMS: Calcd for [C34H53N5O23+H]+: 900.3210. Found: 900.3189.
4.4.4 2-Azidoethyl (5-acetamido-3,5-dideoxy-d-glycero-α-d-galacto-2-nonulopyranosylonic acid)-(2→3)-β-D-galactopyranosyl-(1→4)[(1→3)- 6-deoxy-6-fluoro-α-l-galactopyranosyl]-2-acetamido-2-deoxy-β-D-glucopyranoside (10)
This compound was synthesized according to general procedure for synthesis of the sLex tetrasaccharide derivatives (20.0 mg, yield: 88%). 1H NMR (600 MHz, D2O): δ 1.80 (t, J = 12.1 Hz, 1H), 2.03 (s, 3H), 2.04 (s, 3H), 2.77 (dd, J = 12.1, 3.7 Hz, 1H), 3.41-3.51 (m, 2H), 3.59-3.63 (m, 4H), 3.64-3.67 (m, 2H), 3.69-3.71 (m, 3H), 3.72-3.78 (m, 2H), 3.83-3.87 (m, 2H), 3.88-3.91 (m, 3H), 3.92-3.97 (m, 4H), 4.00-4.09 (m, 4H), 4.52 (d, J = 7.8 Hz, 1 H), 4.54-4.72 (m, 2H), 4.69 (d, J = 10.3 Hz, 1H), 5.07 (dd, J = 15.4, 5.4 Hz, 1H), 5.22 (d, J = 3.6 Hz, 1H). 13C NMR (150 MHz, D2O): δ 22.0, 22.3, 39.8, 50.4, 51.7, 55.7, 59.6, 61.4, 62.6, 67.3, 67.8, 68.1, 68.3, 68.7, 69.0, 69.1, 69.20, 69.25, 69.4, 71.8, 72.9, 73.6, 74.8, 75.3, 75.7, 83.5, 84.6, 98.6, 99.7, 100.8, 101.9, 173.8, 174.3, 174.9. HRMS: Calcd for [C33H54FN5O23+H]+: 908.3272. Found: 908.3252.
4.4.5 2-Azidoethyl (5-acetamido-3,5-dideoxy-d-glycero-α-d-galacto-2-nonulopyranosylonic acid)-(2→3)-β-D-galactopyranosyl-(1→4)[(1→3)-α-l-galactopyranosyl]-2-acetamido-2-deoxy-β-D-glucopyranoside (11)
This compound was synthesized according to general procedure for synthesis of the sLex tetrasaccharide derivatives (18.1 mg, yield: 80%). 1H NMR (600 MHz, D2O): δ 1.80 (t, J = 12.2 Hz, 1H), 2.04 (s, 3H), 2.05 (s, 3H), 2.78 (dd, J = 12.5, 4.6 Hz, 1H), 3.42-3.51 (m, 2H), 3.56-3.58 (m, 1H), 3.59-3.67 (m, 5H), 3.68-3.73 (m, 6H), 3.74-3.79 (m, 2H), 3.84-3.88 (m, 2H), 3.89-3.92 (m, 2H), 3.93-3.98 (m, 5H), 4.00-4.01 (m, 1H), 4.03-4.07 (m, 2H), 4.11 (dd, J = 9.9, 3.1 Hz, 1 H), 4.54 (d, J = 7.8 Hz, 1 H), 4.61 (d, J = 7.8 Hz, 1H), 4.75-4.77 (m, 1H), 5.21 (d, J = 3.9 Hz, 1 H). 13C NMR (150 MHz, D2O): δ 22.0, 22.4, 39.8, 50.4, 51.7, 55.7, 59.7, 60.8, 61.3, 62.6, 67.4, 68.05, 68.12, 68.3, 68.7, 69.0, 69.2, 69.3, 70.0, 71.9, 72.9, 73.9, 74.6, 74.9, 75.4, 75.6, 89.4, 99.7, 100.9, 102.0, 173.8, 174.6, 175.1. HRMS: Calcd for [C33H55N5O24+H]+: 906.3315. Found: 906.3320.
4.4.6 2-Azidoethyl (5-acetamido-3,5-dideoxy-d-glycero-α-d-galacto-2-nonulopyranosylonic acid)-(2→3)-β-D-galactopyranosyl-(1→4)[(1→3)-6-O-methyl-α-l-galactopyranosyl]-2-acetamido-2-deoxy-β-D-glucopyranoside (12)
This compound was synthesized according to general procedure for synthesis of the sLex tetrasaccharide derivatives (21.9 mg, yield: 95%). 1H NMR (600 MHz, D2O): δ 1.82 (t, J = 12.2 Hz, 1H), 2.04 (s, 3H), 2.05 (s, 3H), 2.60 (s, 2H), 2.77 (dd, J = 12.5, 4.6 Hz, 1H), 3.40 (s, 3H), 3.42-3.45 (m, 1H), 3.46-3.52 (m, 1H), 3.57-3.65 (m, 8H), 3.66-3.68 (m, 1H), 3.69-3.74 (m, 4H), 3.75-3.79 (m, 1H), 3.84-3.92 (m, 4H), 3.93-3.98 (m, 5H), 4.01-4.06 (m, 2H), 4.09 (dd, J = 9.9, 3.2 Hz, 1H), 4.51 (d, J = 7.8 Hz, 1 H), 4.61 (d, J = 7.1 Hz, 1H), 4.86 (dd, J = 7.6, 4.2 Hz, 1H), 5.21 (d, J = 3.8 Hz, 1H). 13C NMR (150 MHz, D2O): δ 22.04, 22.34, 24.53, 39.77, 50.39, 51.70, 55.73, 58.57, 59.66, 61.38, 62.61, 67.45, 67.92, 68.12, 68.33, 68.65, 68.73, 69.00, 69.22, 71.88, 71.91, 72.91, 73.49, 74.27, 74.88, 75.30, 75.62, 98.22, 99.73, 100.88, 101.89, 173.88, 174.35, 175.02. HRMS: Calcd for [C34H57N5O24+H]+: 920.3472. Found: 920.3470.
Supplementary Material
Acknowledgments
This work was supported by the National Institutes of Health (4R00 GM080585-03) and startup funds from Albert Einstein College of Medicine. Yizheng He was supported by the Albert Einstein Summer Undergraduate Research Program. BL21 cells harbouring fkp in pET-16b were provided by Professor Laurie E. Comstock. We thank Professor John S. Blanchard and Ms. Alison Sikora for assistance in kinetics measurements.
Footnotes
Supplementary data Supplementary data for this paper (general experimental procedures and spectral data for all sLex derivatives) are available online at doi: xxxx.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Varki A, Cummings RD, Esko JD, Freeze HH, Stanley P, Bertozzi CR, Hart GW, Etzler ME. Eseentials of Glycobiology. 2. Cold Spring Harbor; New York: 2008. [PubMed] [Google Scholar]
- 2.Phillips ML, Nudelman E, Gaeta FC, Perez M, Singhal AK, Hakomori S, Paulson JC. Science. 1990;250:1130–1132. doi: 10.1126/science.1701274. [DOI] [PubMed] [Google Scholar]
- 3.Walz G, Aruffo A, Kolanus W, Bevilacqua M, Seed B. Science. 1990;250:1132–1135. doi: 10.1126/science.1701275. [DOI] [PubMed] [Google Scholar]
- 4.Lowe JB, Stoolman LM, Nair RP, Larsen RD, Berhend TL, Marks RM. Cell. 1990;63:475–485. doi: 10.1016/0092-8674(90)90444-j. [DOI] [PubMed] [Google Scholar]
- Simanek EE, McGarvey GJ, Jablonowski JA, Wong CH. Chem Rev. 1998;98:833–862. doi: 10.1021/cr940226i. [DOI] [PubMed] [Google Scholar]
- 6.Lapidot T, Dar A, Kollet O. Blood. 2005;106:1901–1910. doi: 10.1182/blood-2005-04-1417. [DOI] [PubMed] [Google Scholar]
- 7.Ichikawa Y, Lin YC, Dumas DP, Shen GJ, Garciajunceda E, Williams MA, Bayer R, Ketcham C, Walker LE, Paulson JC, Wong CH. J Am Chem Soc. 1992;114:9283–9298. [Google Scholar]
- 8.Ramphal JY, Zheng ZL, Perez C, Walker LE, DeFrees SA, Gaeta FC. J Med Chem. 1994;37:3459–3463. doi: 10.1021/jm00047a003. [DOI] [PubMed] [Google Scholar]
- 9.Examples of chemical synthesis of sLex, see Kameyama A, Ishida H, Kiso M, Hasegawa A. Carbohydr Res. 1991;209:c1–c4. doi: 10.1016/0008-6215(91)80171-i.Nicolaou KC, Hummel CW, Bockovich NJ, Wong CH. J Chem Soc Chem Comm. 1991:870–872.Danishefsky SJ, Gervay J, Peterson JM, Mcdonald FE, Koseki K, Griffith DA, Oriyama T, Marsden SP. J Am Chem Soc. 1995;117:1940–1953.Yan L, Kahne D. J Am Chem Soc. 1996;118:9239–9248.
- 10.a Boltje TJ, Buskas T, Boons G. Nat Chem. 2009;1:611–622. doi: 10.1038/nchem.399. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Seeberger PH. Nat Chem Biol. 2009;5:368–372. doi: 10.1038/nchembio0609-368. [DOI] [PubMed] [Google Scholar]
- 11.Hanson S, Best M, Bryan MC, Wong CH. Trends Biochem Sci. 2004;29:656–663. doi: 10.1016/j.tibs.2004.10.004. [DOI] [PubMed] [Google Scholar]
- 12.Ma B, Simala-Grant JL, Taylor DE. Glycobiology. 2006;16:158R–184R. doi: 10.1093/glycob/cwl040. [DOI] [PubMed] [Google Scholar]
- 13.Wang W, Hu T, Frantom PA, Zheng T, Gerwe B, Del Amo DS, Garret S, Seidel RD., 3 Proc Natl Acad Sci U S A. 2009;106:16096–16101. doi: 10.1073/pnas.0908248106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Coyne MJ, Reinap B, Lee MM, Comstock LE. Science. 2005;307:1778–1781. doi: 10.1126/science.1106469. [DOI] [PubMed] [Google Scholar]
- 15.Yi W, Liu X, Li Y, Li J, Xia C, Zhou G, Zhang W, Zhao W, Chen X, Wang PG. Proc Natl Acad Sci U S A. 2009;106:4207–4212. doi: 10.1073/pnas.0812432106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rabbani S, Miksa V, Wipf B, Ernst B. Glycobiology. 2005;15:1076–1083. doi: 10.1093/glycob/cwj004. [DOI] [PubMed] [Google Scholar]
- 17.Yu H, Chokhawala HA, Huang S, Chen X. Nat Protoc. 2006;1:2485–2492. doi: 10.1038/nprot.2006.401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yu H, Yu H, Karpel R, Chen X. Bioorg Med Chem. 2004;12:6427–6435. doi: 10.1016/j.bmc.2004.09.030. [DOI] [PubMed] [Google Scholar]
- 19.Yu H, Chokhawala H, Karpel R, Yu H, Wu B, Zhang J, Zhang Y, Jia Q, Chen X. J Am Chem Soc. 2005;127:17618–17619. doi: 10.1021/ja0561690. [DOI] [PubMed] [Google Scholar]
- 20.Baskin JM, Prescher JA, Laughlin ST, Agard NJ, Chang PV, Miller IA, Lo A, Codelli JA, Bertozzi CR. Proc Natl Acad Sci U S A. 2007;104:16793–16797. doi: 10.1073/pnas.0707090104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Feizi T, Fazio F, Chai W, Wong CH. Curr Opin Struct Biol. 2003;13:637–645. doi: 10.1016/j.sbi.2003.09.002. [DOI] [PubMed] [Google Scholar]
- 22.Ichikawa Y, Wang R, Wong CH. Methods Enzymol. 1994;247:107–127. doi: 10.1016/s0076-6879(94)47009-x. [DOI] [PubMed] [Google Scholar]
- 23.Stiller R, Thiem J. Liebigs Ann Chem. 1992:467–471. [Google Scholar]
- 24.Plowman KM, Krall AR. Biochemistry. 1965;4:2809–2814. doi: 10.1021/bi00888a035. [DOI] [PubMed] [Google Scholar]
- 25.Blixt O, Han S, Liao L, Zeng Y, Hoffmann J, Futakawa S, Paulson JC. J Am Chem Soc. 2008;130:6680–6681. doi: 10.1021/ja801052g. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.