

Abstract
The extensive genetic diversity of HIV-1 presents a significant barrier to the development of an effective and durable HIV vaccine. This variability not only makes it difficult to identify the targets against which immune responses should be directed, but it also confers on the virus the capacity for rapid escape from effective immune responses. Here, we describe recent investigations of the genetic diversity of HIV-1 at transmission and of the evolution of the virus as it adapts to the host immune environment during the acute phase of HIV-1 infection. These studies increase our understanding of the virology of the earliest stages of HIV-1 infection and provide critical insights into the mechanisms underlying viral replication and immune control of diverse HIV-1 strains. Such knowledge will inform the design of smarter, more effective, vaccines capable of inducing immune control of HIV-1.
Introduction
The acute phase of HIV-1 infection encompasses the initial interaction between virus and host and is of critical interest because the outcome strongly influences the pathogenesis and clinical course of the infection. Acute HIV-1 infection resulting from sexual transmission begins with the breaching of the mucosal barrier by infectious virions and the establishment of a localized focus of infection in tissue-associated immune cells in the mucosa. As the infection spreads to the regional lymph nodes and is disseminated to other lymphocyte-rich compartments throughout the body, particularly the spleen and the gut-associated lymphoid tissue (GALT), the virus encounters high densities of primary target CD4+ T cells causing a massive burst of viral replication. An ensuing peak of viremia is closely followed by a decrease in viral load, in association with the concomitant induction of HIV-1-specific CD8+ T cells and depletion of susceptible CD4+ T cells. The viral load ultimately levels to a steady-state “set-point” viremia that then remains broadly stable throughout much of the chronic phase of infection. The immune control of HIV-1 is determined by the intrinsic qualities of both the immune response and the HIV-1 population. The effectiveness of early CD8+ T cell responses in controlling HIV-1 replication is a critical determinant of the set-point viral load [1], and their importance is further supported by the association between particular HLA class I alleles and improved course of HIV-1 infection [2]. However, the exact mechanisms by which some individuals achieve control of viral replication remain unclear, yet their elucidation would provide valuable insight into the immune environment that should be induced by an HIV-1 vaccine. Recent investigations of the virology of acute HIV-1 infection have started to yield critical insights into i) the virus lineage(s) that establishes infection, ii) the earliest viral adaptive changes that enable escape from host immune responses, and iii) the impact of immune escape mutations in HIV-1 on viral replication and pathogenesis. Here, we summarize some of these recent advances and discuss the application of these findings to the development of more effective HIV-1 vaccine approaches.
Characterization of Transmitted/Founder Viruses
The extensive genetic diversity of HIV-1 poses a significant challenge to the development of broadly effective prophylactic interventions because the majority of circulating strains are likely to differ significantly from the antigenic insert in the prophylactic agent. As such, the identification of unique viral genetic characteristics that are associated with the transmission of HIV-1 would provide important information about the mechanisms of transmission/establishment of infection and may narrow the choices among potential HIV-1 targets. Although a small number of studies have successfully identified viral characteristics associated with successful transmission, e.g., shorter V1–V2 length and fewer N-linked glycosylation sites in Env of non-B subtypes [3, 4], the practical difficulties associated with obtaining samples from the earliest days of HIV-1 infection have hampered their characterization. Recent efforts have focused on sampling virus populations during acute infection, especially prior to seroconversion, in order to infer the genetic sequence of the founder virus using a combination of sequence analysis, evolutionary modeling, and phylogenetic analyses [5–10]. These studies have demonstrated that approximately 75% of sexual transmissions of HIV-1 are attributable to infection with a single viral genetic lineage, whether for HIV-1B transmission in men who have sex with men (MSM) [7, 10] or for HIV-1A and HIV-1C transmission in heterosexual couples [5, 6, 8]. In the cases in which multiple founder variants were deduced there were usually two and typically no more than five founder lineages, and such cases were often associated with factors that can disrupt the mucosal barrier, e.g., sexually transmitted disease (STD)-associated genital inflammation [6]. More recently, a study by Bar et al. illustrated that injection drug use (IDU) was associated with the transmission of a high frequency of multiple sequence variants, suggesting that bypassing the normal mucosal barrier may facilitate transmission of multiple variants [11].
Recently this approach has been extended to known transmission pairs which allows for the comparison of the deduced founder virus in the recipient to the entire pool of potential founder lineages that were present in the donor quasispecies. In their study of 10 HIV-1A and HIV-1C heterosexual transmission pairs, Haaland et al.[6] analyzed approximately 250 amino acids from the V1 loop to the V4 loop of env and identified 2/10 pairs in which the founder virus was an exact genetic match of a virus present in the donor, 6/10 pairs in which the founder virus differed from the most closely related virus in the donor by 5 or fewer amino acids (~98% identity), and 2/10 pairs in which the founder virus and the most closely related donor virus differed by 10–11 amino acids (~96% identity). Interestingly, in 9 of the10 transmission pairs the donor variant most closely related (>98% identity) to the founder virus in the recipient comprised only a small fraction of the total donor virus population (<5%). A similar pattern was observed when whole virus genomes were analyzed in a recent study of 3 transmission pairs in which the transmitter was in chronic infection, and thus had a highly genetically diverse viral population [12].
In addition to these studies of HIV-1 transmission, recent investigations of low dose, intra-rectal infection of rhesus macaques with SIVmac251, a non-clonal SIV inoculum, demonstrated that the majority of infections were founded by a single genetic lineage. Moreover, the genetic relatedness between the inferred founder virus and the most closely related variant in the inoculum was similar to that described above for HIV-1 transmission [13]. In addition to supporting the HIV-1 results, these data suggest the potential value of mucosal challenge approaches for the accurate modeling of HIV-1 transmission for vaccine testing.
The observation that HIV-1 infection is typically established by a single genetic lineage during sexual transmission offers hope that HIV-1 vaccines may have to block a limited number of viral strains in most cases. Unfortunately, to date no unique characteristics of transmitted variants have been identified that would permit more specific targeting.
Next-Generation Sequencing of Acute/Early Viruses
The population of genetically diverse viruses that develops in each infected individual confers on HIV-1 the capacity to rapidly and relentlessly adapt to host immune responses. Such immune adaptation contributes to ineffective immune control of viral replication [14, 15], and experiments in the SIV/macaque model underline that CTL escape will pose a significant threat to the durability and efficacy of vaccine-induced CD8+ T cell responses [16]. To properly understand immune adaptation of HIV-1, it is critical to characterize the dynamics of viral evolution during acute infection when numerous adaptive changes take place. Recent studies examining near full-length genomes [12, 17, 18] showed that at peak viremia the virus population remains virtually homogeneous with significant evolution occurring by week 6 of infection, including a dramatic increase in CTL immune-adaptive mutations, particularly in highly variable proteins such as Env and Nef [12, 17]. Interestingly, multiple mutations in targeted CTL epitopes were observed in both studies. Herbeck et al. [12] noted that such mutations were often mutually exclusive, which suggests that the virus population can explore multiple adaptive pathways but that there are limits to the plasticity of individual viruses, even in highly variable proteins. Although such intensive longitudinal analyses of viral evolution during acute/early infection have been limited to a small number of individuals, next-generation sequencing technologies promise to scale up these efforts and to increase our understanding of early HIV-1 adaptations to host selection pressures. Chip-based sequencing technologies such as pyrosequencing (“454”) [19] and four-color cyclic reversible termination sequencing (CRT) (“Illumina”) [20] offer rapid and cost-effective production of up to 10,000-fold sequencing read coverage of the HIV-1 genome. This represents a substantial improvement over the costly and time-consuming amplification and sequencing of individual amplicons for the analysis of viral genetic variation. Indeed, the depth of coverage provided by these approaches allows the quantification of genetic variants at frequencies that are undetectable by standard sequencing approaches, and the increased sensitivity provides an unprecedented view of the earliest events in the adaptation of HIV-1 to frontline immune responses mounted during acute infection. The relatively short read lengths may, however, limit the utility of next-generation sequencing technologies for establishing linkage of mutations except over short regions of the genomes.
To date, next-generation sequencing of HIV-1 has predominantly been used to identify and quantify antiretroviral drug resistance mutations at frequencies below the limit of detection of commercial drug resistance genotyping assays (to approximately a 1% level), in order to understand the impact of minor variant resistance mutations on treatment outcomes [21–24]. Recent efforts have been applied to HIV-1 evolution during the acute/early phase of infection. For example, by comparing chip-based pyrosequencing to conventional sequencing for the analysis of the evolution of specific CTL epitopes during acute/early infection in longitudinal samples from three patients, Fischer et al. [25] showed that next-generation sequencing captured a greater degree of genetic diversity and detected CTL escape variants earlier than conventional approaches. The detection by “deep” sequencing of viral CTL escape variants earlier in infection than previously appreciated, e.g., as early as 17 days post-infection, was also recently reported in the SIV/macaque system [26]. Next-generation sequencing also allows the characterization of CD8+ T cell epitopes that “shatter” during viral escape, i.e., exhibit multiple, highly variable, low frequency escape mutations, before coalescing on a single escape pathway, versus epitopes that are limited to escape at a single residue [27]. As such, deep sequencing approaches are now capable of distinguishing between the early pathways of viral escape within different CTL epitopes, revealing critical information about the ease with which HIV-1 can evade these responses.
The recent development of novel assembly, alignment, and analysis tools has enabled high-throughput, next-generation sequencing of near full-length viral genomes (Henn and Allen, unpublished; Deng and Mullins, unpublished). This approach revealed that the earliest escaping epitopes were in more variable proteins of the virus (Vif, Env and Nef) [27], and corresponded to highly immunodominant responses (Henn and Allen, unpublished). These data suggest that viral escape may be tied more closely to the kinetics and specificity of the immune response, than the functionality of the response. These advances will allow future studies to extend beyond analyses of the evolution of specific epitopes in a small number of samples to full-scale, unbiased screening of HIV-1 evolution in large, cross-sectional and longitudinal cohorts. Although individual-based studies are critical for providing fine detail of the interactions between specific viruses and specific host immune responses, population-based studies are necessary to identify common patterns of HIV-1 escape from the earliest immune responses, and more specifically, to define attributes associated with effective control of HIV-1.
Immune Escape-Associated Viral Fitness Costs
The dynamics of immune escape are predominantly governed by the impact of the escape mutation on relative viral fitness, a composite parameter representing the net contribution of all mutation-associated benefits and costs. Although the evasion of immune responses conferred by escape mutations represents a clear fitness benefit to the virus, the HIV-1 proteome is not endlessly plastic and the same mutations can carry associated fitness costs. The existence of such costs is evidenced by the reversion of transmitted escape mutations during acute and early HIV-1 infection [12, 15, 17, 18, 28], and a number of CTL escape mutations have been identified that disrupt normal virus protein structure and/or function [29–32]. The majority of deleterious escape mutations have been identified in the relatively conserved Gag protein, whereas Troyer et al. [33] recently showed that CTL escape mutations in Env did not typically carry an associated fitness cost and in several cases enhanced competitive viral fitness. This result is consistent with the lack of a fitness cost associated with virus escape from neutralizing antibodies [34] and suggests that differential functional constraints play a role in determining escape mutation-associated fitness costs. In addition, a recent analysis of the impact on viral replication capacity of twenty CTL escape mutations in Gag epitopes identified only three escape mutations that caused substantial reductions in viral replication capacity, suggesting that high-cost escape mutations are relatively rare [35]. Interestingly, the three high-cost CTL escape mutations occurred in epitopes dominantly targeted by protective HLA class I alleles during acute infection [1]. This suggests that the protection afforded by particular HLA class I alleles may result from either maintenance of a dominant and effective CD8+ T cell response because the barrier to viral escape in the targeted epitope is high, and/or from selection of high-cost escape mutations that attenuate virus replication.
Additional evidence of a role for CTL escape-associated fitness costs in control of HIV-1 replication comes from the examination of viruses derived from HIV-1 controllers (individuals who maintain long-term control of HIV-1 viremia) in which rare or novel CTL escape mutations are commonly observed and which exhibit generally reduced replication capacities [36–39]. However, the source of the CTL escape variants in these patients remains unclear; they could result either from early immune selection in the contemporary host, or from infection with a founder virus that already contains the escape mutations. Indeed, two recent reports describe improved early clinical correlates associated with the transmission of viruses expressing multiple CTL escape mutations known to impair in vitro replication capacity [40, 41]. Importantly, the improved early outcomes associated with such transmissions have been observed to fade as the transmitted escape mutations revert to wild type [32], and the long-term clinical outcome of these transient effects remains uncertain.
As the virology of acute HIV-1 infection is better characterized, it becomes possible to model how differences in founder virus replication and in viral escape from dominant CD8+ T cell responses may contribute to observed variability in the immune control of HIV-1 replication. One hypothesis is that differences in intrinsic founder virus replication, caused by carry-over mutations (whether immune escape-derived or otherwise), and differences in the rate of escape from contemporary dominant CD8+ T cell responses due to escape-associated fitness costs, combine to influence set-point viral load and early clinical disease course (Figure 1). For example, the transmission of a virus attenuated by accumulated carry-over mutations to a host that mounts dominant CD8+ T cell responses against epitopes with high escape-associated fitness costs is proposed to contribute to elite control of viral replication. In contrast, transmission of an attenuated virus to a host with dominant CD8+ T cell responses against epitopes with low, or no, barrier to escape is proposed to result in a more typical set-point viral load and clinical course. Such models may provide a useful framework for considering the relative roles of multiple, highly variable, host and virus characteristics.
Figure 1. Differential founder virus replication and immune escape from dominant CD8+ T cell responses combine to influence the HIV-1 set-point viral load and clinical course.
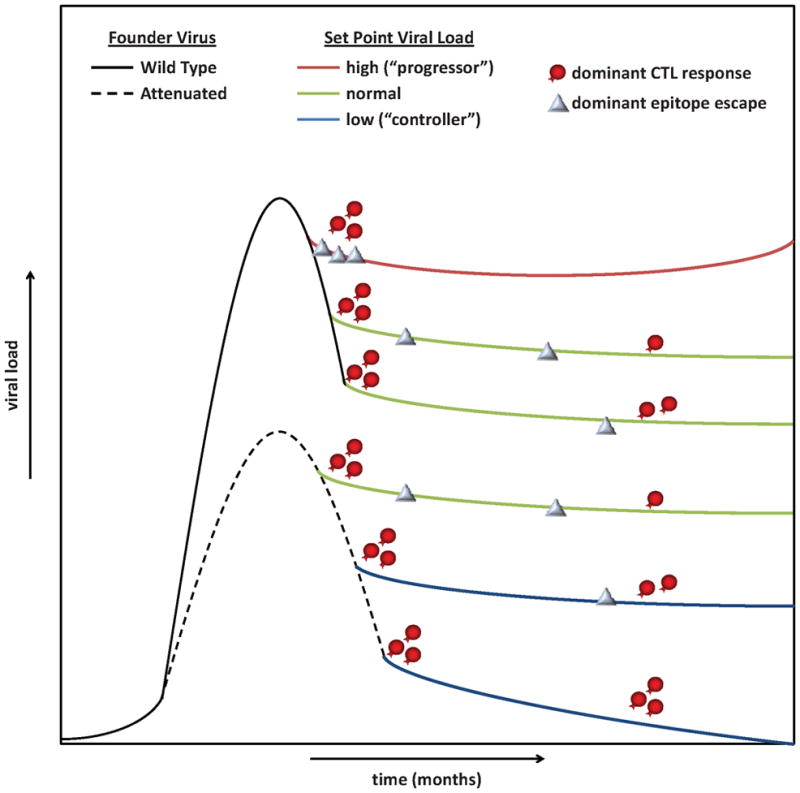
The differential replication of wild type (solid line) and attenuated (dashed line) founder viruses results in different levels of peak viremia and long-term control of HIV-1. The impact of dominant CD8+ T cell responses (red cells) on the viral population is determined by the relative rate of escape in the targeted epitopes (grey diamonds) which ranges from immediate escape (top set-point line) as the result of the escape mutations being transmitted in the founder virus to never (bottom set-point line) as the result of high escape-associated fitness costs and low viral replication. The differential founder virus replication and dominant CD8+ T cell response effectiveness result in poor control of acute HIV-1 replication and high set-point viral load (red set-point line), normal control and set-point (green lines), and protective control and low to undetectable set-point (blue set-point lines).
Escape and Fitness Guided Vaccine Approaches
The development of an effective vaccine against HIV-1, whether capable of inducing sterilizing or therapeutic immunity, is essential. The consideration of CTL escape in the design of vaccine immunogens will be of particular importance for vaccines that induce effective antiviral immune responses but fail to provide either sterilizing immunity or complete suppression of viral replication. Indeed, there is clear evidence from both human and macaque studies that HIV-1 can escape from narrow vaccine-induced immune responses and that such escape can erode vaccine efficacy [16, 42]. Thus, an increased understanding of barriers to transmission, the nature of transmitted viruses, viral immune escape pathways, and immune escape-associated impairment of replication is required for the development of rational strategies for vaccine design. Variability-inclusive approaches such as the “mosaic” [43] and the “center-of-tree” approach with high frequency variants included (COT+) [44–46], seek to induce immune responses against a broad range of naturally occurring strains and to block common CTL escape states by creating composite immunogens that maximize the coverage of 9-mers found in circulating HIV-1 isolates. Other designs seek to compile the most highly conserved and most immunodominant regions of the HIV-1 proteome into a single antigen to focus immune responses against regions of the virus in which CTL escape carries a severe cost to the virus [47–49]. For example, the immunogen in the ‘“conserved elements” (CE) design is generated by an iterative process that includes the most conserved regions of the HIV-1 proteome and known protective CTL epitopes, organizes the elements of the construct to maximize protein expression and epitope processing, and integrates CTL escape-specific modifications on the basis of escape-associated fitness cost data [50]. Finally, other approaches attempt to overcome natural immunodominance hierarchies to focus responses against more conserved sub-dominant CD8+ T cell epitopes in which there are high escape-associated viral fitness costs rather than against current immunodominant epitopes in which escape occurs with little or no cost [47] (Boutwell et al., unpublished). The common characteristic of all of these approaches is that they seek to leverage our knowledge of the global and intra-host evolution of HIV-1 to overcome the challenges posed by the diversity and adaptive capacity of the virus in order to elicit responses less susceptible to viral escape.
Conclusion
In summary, careful characterization of the early events of acute HIV-1 infection has provided key insights into viral-host interactions that influence viral pathogenesis and immune control of viral replication. Continued study of HIV-1 sequence evolution, in particular during acute and early infection and in situations of natural control of HIV-1 replication, is required to refine vaccine design approaches and to identify novel mechanisms of immune control. The recent application of next-generation sequencing approaches to the study of HIV-1 should speed this process substantially.
Acknowledgments
This article was submitted as part of a supplement detailing the Acute HIV-1 Infection Meeting held on September 22–23, 2009 in Boston, MA. This meeting was funded by NIH-NIAID, Harvard University’s Center for AIDS Research and the Ragon Institute of MGH, MIT and Harvard.
Footnotes
No author has a commercial or other association that might pose a conflict of interest.
References
- 1.Streeck H, Jolin JS, Qi Y, et al. Human immunodeficiency virus type 1-specific CD8+ T-cell responses during primary infection are major determinants of the viral set point and loss of CD4+ T cells. J Virol. 2009;83:7641–8. doi: 10.1128/JVI.00182-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Carrington M, O’Brien SJ. The influence of HLA genotype on AIDS. Annu Rev Med. 2003;54:535–51. doi: 10.1146/annurev.med.54.101601.152346. [DOI] [PubMed] [Google Scholar]
- 3.Chohan B, Lang D, Sagar M, et al. Selection for human immunodeficiency virus type 1 envelope glycosylation variants with shorter V1–V2 loop sequences occurs during transmission of certain genetic subtypes and may impact viral RNA levels. J Virol. 2005;79:6528–31. doi: 10.1128/JVI.79.10.6528-6531.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rong R, Bibollet-Ruche F, Mulenga J, Allen S, Blackwell JL, Derdeyn CA. Role of V1V2 and other human immunodeficiency virus type 1 envelope domains in resistance to autologous neutralization during clade C infection. J Virol. 2007;81:1350–9. doi: 10.1128/JVI.01839-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Abrahams MR, Anderson JA, Giorgi EE, et al. Quantitating the multiplicity of infection with human immunodeficiency virus type 1 subtype C reveals a non-poisson distribution of transmitted variants. J Virol. 2009;83:3556–67. doi: 10.1128/JVI.02132-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Haaland RE, Hawkins PA, Salazar-Gonzalez J, et al. Inflammatory genital infections mitigate a severe genetic bottleneck in heterosexual transmission of subtype A and C HIV-1. PLoS Pathog. 2009;5:e1000274. doi: 10.1371/journal.ppat.1000274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Keele BF, Giorgi EE, Salazar-Gonzalez JF, et al. Identification and characterization of transmitted and early founder virus envelopes in primary HIV-1 infection. Proc Natl Acad Sci U S A. 2008;105:7552–7. doi: 10.1073/pnas.0802203105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Salazar-Gonzalez JF, Bailes E, Pham KT, et al. Deciphering human immunodeficiency virus type 1 transmission and early envelope diversification by single-genome amplification and sequencing. J Virol. 2008;82:3952–70. doi: 10.1128/JVI.02660-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lee HY, Giorgi EE, Keele BF, et al. Modeling sequence evolution in acute HIV-1 infection. J Theor Biol. 2009;261:341–60. doi: 10.1016/j.jtbi.2009.07.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gottlieb GS, Heath L, Nickle DC, et al. HIV-1 variation before seroconversion in men who have sex with men: analysis of acute/early HIV infection in the multicenter AIDS cohort study. J Infect Dis. 2008;197:1011–5. doi: 10.1086/529206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bar KJ, Li H, Chamberland A, et al. Wide Variation in the Multiplicity of HIV-1 Infection Among Injection Drug Users. J Virol. doi: 10.1128/JVI.00077-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Herbeck JT, Rolland MT, Deng WC, Collier AC, Mullins JI. AIDS Vaccine, P07–06. Paris, France: 2009. HIV-1 transmission and early evolution: whole genome analysis. [Google Scholar]
- 13.Keele BF, Li H, Learn GH, et al. Low-dose rectal inoculation of rhesus macaques by SIVsmE660 or SIVmac251 recapitulates human mucosal infection by HIV-1. J Exp Med. 2009;206:1117–34. doi: 10.1084/jem.20082831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Feeney ME, Tang Y, Roosevelt KA, et al. Immune escape precedes breakthrough human immunodeficiency virus type 1 viremia and broadening of the cytotoxic T-lymphocyte response in an HLA-B27-positive long-term-nonprogressing child. J Virol. 2004;78:8927–30. doi: 10.1128/JVI.78.16.8927-8930.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Leslie AJ, Pfafferott KJ, Chetty P, et al. HIV evolution: CTL escape mutation and reversion after transmission. Nat Med. 2004;10:282–9. doi: 10.1038/nm992. [DOI] [PubMed] [Google Scholar]
- 16.Barouch DH, Kunstman J, Kuroda MJ, et al. Eventual AIDS vaccine failure in a rhesus monkey by viral escape from cytotoxic T lymphocytes. Nature. 2002;415:335–9. doi: 10.1038/415335a. [DOI] [PubMed] [Google Scholar]
- 17.Goonetilleke N, Liu MK, Salazar-Gonzalez JF, et al. The first T cell response to transmitted/founder virus contributes to the control of acute viremia in HIV-1 infection. J Exp Med. 2009;206:1253–72. doi: 10.1084/jem.20090365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li B, Gladden AD, Altfeld M, et al. Rapid reversion of sequence polymorphisms dominates early human immunodeficiency virus type 1 evolution. J Virol. 2007;81:193–201. doi: 10.1128/JVI.01231-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Margulies M, Egholm M, Altman WE, et al. Genome sequencing in microfabricated high-density picolitre reactors. Nature. 2005;437:376–80. doi: 10.1038/nature03959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bentley DR, Balasubramanian S, Swerdlow HP, et al. Accurate whole human genome sequencing using reversible terminator chemistry. Nature. 2008;456:53–9. doi: 10.1038/nature07517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Eriksson N, Pachter L, Mitsuya Y, et al. Viral population estimation using pyrosequencing. PLoS Comput Biol. 2008;4:e1000074. doi: 10.1371/journal.pcbi.1000074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mitsuya Y, Varghese V, Wang C, et al. Minority human immunodeficiency virus type 1 variants in antiretroviral-naive persons with reverse transcriptase codon 215 revertant mutations. J Virol. 2008;82:10747–55. doi: 10.1128/JVI.01827-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Varghese V, Shahriar R, Rhee SY, et al. Minority variants associated with transmitted and acquired HIV-1 nonnucleoside reverse transcriptase inhibitor resistance: implications for the use of second-generation nonnucleoside reverse transcriptase inhibitors. J Acquir Immune Defic Syndr. 2009;52:309–15. doi: 10.1097/QAI.0b013e3181bca669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Le T, Chiarella J, Simen BB, et al. Low-abundance HIV drug-resistant viral variants in treatment-experienced persons correlate with historical antiretroviral use. PLoS One. 2009;4:e6079. doi: 10.1371/journal.pone.0006079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fischer W, Keele B, Bhattacharya T, et al. AIDS Vaccine, P09–21 LB. Paris, France: 2009. Deepsequencing of HIV-1 from acute infection: low initial diversity, and rapid but variable CTL escape. [Google Scholar]
- 26.Bimber BN, Burwitz BJ, O’Connor S, et al. Ultradeep pyrosequencing detects complex patterns of CD8+ T-lymphocyte escape in simian immunodeficiency virus-infected macaques. J Virol. 2009;83:8247–53. doi: 10.1128/JVI.00897-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Henn MR, Boutwell CL, Lennon N, et al. AIDS Vaccine, P09–20 LB. Paris, France: 2009. Ultra-deep sequencing of full-length HIV-1 genomes identifies rapid viral evolution during acute infection. [Google Scholar]
- 28.Kearney M, Maldarelli F, Shao W, et al. Human immunodeficiency virus type 1 population genetics and adaptation in newly infected individuals. J Virol. 2009;83:2715–27. doi: 10.1128/JVI.01960-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Boutwell CL, Rowley CF, Essex M. Reduced viral replication capacity of human immunodeficiency virus type 1 subtype C caused by cytotoxic-T-lymphocyte escape mutations in HLA-B57 epitopes of capsid protein. J Virol. 2009;83:2460–8. doi: 10.1128/JVI.01970-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brockman MA, Schneidewind A, Lahaie M, et al. Escape and compensation from early HLA-B57-mediated cytotoxic T-lymphocyte pressure on human immunodeficiency virus type 1 Gag alter capsid interactions with cyclophilin A. J Virol. 2007;81:12608–18. doi: 10.1128/JVI.01369-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schneidewind A, Brockman MA, Sidney J, et al. Structural and functional constraints limit options for cytotoxic T-lymphocyte escape in the immunodominant HLA-B27-restricted epitope in human immunodeficiency virus type 1 capsid. J Virol. 2008;82:5594–605. doi: 10.1128/JVI.02356-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Crawford H, Lumm W, Leslie A, et al. Evolution of HLA-B*5703 HIV-1 escape mutations in HLA-B*5703-positive individuals and their transmission recipients. J Exp Med. 2009;206:909–21. doi: 10.1084/jem.20081984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Troyer RM, McNevin J, Liu Y, et al. Variable fitness impact of HIV-1 escape mutations to cytotoxic T lymphocyte (CTL) response. PLoS Pathog. 2009;5:e1000365. doi: 10.1371/journal.ppat.1000365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bunnik EM, Lobbrecht MS, van Nuenen AC, Schuitemaker H. Escape from autologous humoral immunity of HIV-1 is not associated with a decrease in replicative capacity. Virology. 2009 doi: 10.1016/j.virol.2009.11.009. [DOI] [PubMed] [Google Scholar]
- 35.Boutwell CL, Schneidewind A, Brumme Z, et al. AIDS Vaccine, P09–19 LB. Paris, France: 2009. CTL escape mutations in Gag epitopes restricted by protective HLA class I alleles cause substantial reductions in viral replication capacity. [Google Scholar]
- 36.Miura T, Brockman MA, Brumme CJ, et al. Genetic characterization of humanimmunodeficiency virus type 1 in elite controllers: lack of gross genetic defects or common amino acid changes. J Virol. 2008;82:8422–30. doi: 10.1128/JVI.00535-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Miura T, Brumme CJ, Brockman MA, et al. HLA-associated viral mutations are common in human immunodeficiency virus type 1 elite controllers. J Virol. 2009;83:3407–12. doi: 10.1128/JVI.02459-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Miura T, Brockman MA, Schneidewind A, et al. HLA-B57/B*5801 human immunodeficiency virus type 1 elite controllers select for rare gag variants associated with reduced viral replication capacity and strong cytotoxic T-lymphocyte [corrected] recognition. J Virol. 2009;83:2743–55. doi: 10.1128/JVI.02265-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Miura T, Brockman MA, Brumme ZL, et al. HLA-associated alterations in replication capacity of chimeric NL4–3 viruses carrying gag-protease from elite controllers of human immunodeficiency virus type 1. J Virol. 2009;83:140–9. doi: 10.1128/JVI.01471-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chopera DR, Woodman Z, Mlisana K, et al. Transmission of HIV-1 CTL escape variants provides HLA-mismatched recipients with a survival advantage. PLoS Pathog. 2008;4:e1000033. doi: 10.1371/journal.ppat.1000033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Goepfert PA, Lumm W, Farmer P, et al. Transmission of HIV-1 Gag immune escape mutations is associated with reduced viral load in linked recipients. J Exp Med. 2008;205:1009–17. doi: 10.1084/jem.20072457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Betts MR, Exley B, Price DA, et al. Characterization of functional and phenotypic changes in anti-Gag vaccine-induced T cell responses and their role in protection after HIV-1 infection. Proc Natl Acad Sci U S A. 2005;102:4512–7. doi: 10.1073/pnas.0408773102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fischer W, Perkins S, Theiler J, et al. Polyvalent vaccines for optimal coverage of potential T-cell epitopes in global HIV-1 variants. Nat Med. 2007;13:100–6. doi: 10.1038/nm1461. [DOI] [PubMed] [Google Scholar]
- 44.Nickle DC, Rolland M, Jensen MA, et al. Coping with viral diversity in HIV vaccine design. PLoS Comput Biol. 2007;3:e75. doi: 10.1371/journal.pcbi.0030075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fischer W, Liao HX, Haynes BF, Letvin NL, Korber B. Coping with viral diversity in HIV vaccine design: a response to Nickle et al. PLoS Comput Biol. 2008;4:e15. doi: 10.1371/journal.pcbi.0040015. author reply e25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nickle DC, Jojic N, Heckerman D, et al. Comparison of immunogen designs that optimize peptide coverage: reply to Fischer et al. PLoS Comput Biol. 2008;4:e25. doi: 10.1371/journal.pcbi.0040025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Altfeld M, Allen TM. Hitting HIV where it hurts: an alternative approach to HIV vaccine design. Trends Immunol. 2006;27:504–10. doi: 10.1016/j.it.2006.09.007. [DOI] [PubMed] [Google Scholar]
- 48.Rolland M, Nickle DC, Mullins JI. HIV-1 group M conserved elements vaccine. PLoS Pathog. 2007;3:e157. doi: 10.1371/journal.ppat.0030157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Letourneau S, Im EJ, Mashishi T, et al. Design and pre-clinical evaluation of a universal HIV-1 vaccine. PLoS One. 2007;2:e984. doi: 10.1371/journal.pone.0000984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rolland MM, Swain VJ, Lanxon-Cookson E, et al. AIDS Vaccine, P17–14. Paris, France: 2009. Fitness-informed HIV-1 Gag-p24 vaccine design. [Google Scholar]