

Summary
Viruses are intracellular parasites whose reproduction relies on factors provided by the host. The cellular protein GBF1 is critical for poliovirus replication. Here we show that the contribution of GBF1 to virus replication is different from its known activities in uninfected cells. Normally GBF1 activates the ADP‐ribosylation factor (Arf) GTPases necessary for formation of COPI transport vesicles. GBF1 function is modulated by p115 and Rab1b. However, in polio‐infected cells, p115 is degraded and neither p115 nor Rab1b knock‐down affects virus replication. Poliovirus infection is very sensitive to brefeldin A (BFA), an inhibitor of Arf activation by GBF1. BFA targets the catalytic Sec7 domain of GBF1. Nevertheless the BFA block of polio replication is rescued by expression of only the N‐terminal region of GBF1 lacking the Sec7 domain. Replication of BFA‐resistant poliovirus in the presence of BFA is uncoupled from Arf activation but is dependent on GBF1. Thus the function(s) of this protein essential for viral replication can be separated from those required for cellular metabolism.
Introduction
All known positive strand RNA viruses, regardless of the organism they infect, share a fundamental feature – they remodel pre‐existing cellular membranes into specific structures that support viral RNA replication complexes. The localization of viral RNA replication on membrane surfaces is believed to provide several advantages for the virus: it locally concentrates viral proteins that may initially be present in low concentration, ensures proper orientation of the constituents of multi‐component replication complexes and hides replicating viral RNA templates and products from cellular innate immunity detection and protection mechanisms (Salonen et al., 2005). The subversion of cellular membranes to support virus replication required that these viruses evolve mechanisms to reorganize the normal membrane metabolic pathways.
We recently reported that infection of cells with poliovirus results in a hijacking of the cellular guanine nucleotide exchange factor (GEF) GBF1 to sites of viral RNA replication, where it functions in the viral replication complexes (2007, 2008). GBF1 is one of three mammalian high‐molecular‐weight GEFs responsible for activating the ADP‐ribosylation factor (Arf) GTPases in the Golgi and endosomal membrane systems (Casanova, 2007; Bui et al., 2009). These three GEFs have been shown to activate Arf1 and function in Arf1‐regulated processes, but as they may also use Arf4 and Arf5 as substrates (Claude et al., 1999; Volpicelli‐Daley et al., 2005), we will refer to them as GEFs for Arf rather than a specific Arf isoform. Arf1 cycles between an inactive, cytoplasmic GDP‐bound form and an activated, GTP‐bound membrane‐targeted form. Membrane‐bound Arf1 induces the formation of coated membrane vesicles on cellular organelles; it also regulates cytoskeletal functions and recruits effector proteins to membranes (D'Souza‐Schorey and Chavrier, 2006; Gillingham and Munro, 2007). The Arf activation/nucleotide exchange activity of GBF1 is catalysed by the Sec7 domain, shared by all Arf GEFs. Apart from the catalytic Sec7 domain, this large protein of about 210 kDa contains five other conserved domains (Mouratou et al., 2005) (see Fig. 4A) whose functions are poorly understood. The N‐terminal DCB and HUS domains have been implicated in inter‐ and intra‐molecular interactions and thus are believed to participate in regulation of Arf activation by GBF1 (Ramaen et al., 2007).
Figure 4.

Truncated GBF1 mutants do not rescue cellular secretion from BFA inhibition. A. Schematic map of GBF1 domain organization and of truncated mutants used in this study. Numbers in parenthesis indicate GBF1 amino acids. B. Cells were co‐transfected with pCMV‐Gluc vector expressing secreted Gaussia luciferase and with vectors expressing full‐length YFP‐GBF1A795E, truncated GBF1 fusions or an empty vector (control). Cells were incubated for 5 h in the presence of 1 µg ml−1 BFA or corresponding amount of DMSO. The luciferase activity observed in each sample without BFA was defined as 100%. Western blots show expression of GBF1 species detected with anti‐GFP antibodies. Actin immunoblots are shown as a loading control.
Three cellular proteins have been shown to interact with GBF1: the COPI coat subunit γ‐COP, the membrane tether p115 and the small GTPase Rab1b. γ‐COP interacts directly with GBF1 and is likely involved in specific recruitment of the COPI coat to early Golgi membranes, where GBF1 is localized (Deng et al., 2009). The membrane tethering factor p115, which acts at multiple steps inthe cellular secretory pathway (Sztul and Lupashin, 2006), was reported to interact with a proline‐rich segment at the C‐terminal end of GBF1 (Alvarez et al., 2003).
GBF1 activity is necessary for Arf1‐dependent formation of COPI transport vesicles carrying membranes and escaped ER proteins from the ERGIC and Golgi back to ER (Kawamoto et al., 2002; Zhao et al., 2006). GBF1 also has been implicated in GGA (Golgi‐localized, γ‐ear containing, Arf binding proteins)‐dependent sorting and delivery of cargo proteins to lysosomes and in trafficking of Shiga toxin from the plasma membrane to the Golgi.
GBF1 function is inhibited by a fungal metabolite, brefeldin A (BFA), that traps the Sec7 domains of high‐molecular‐weight GEFs in a non‐productive complex with their substrate Arf‐GDP and thus prevents Arf activation (Mossessova et al., 2003; Renault et al., 2003). BFA also strongly inhibits the replication of poliovirus (Maynell et al., 1992) and some other positive strand RNA viruses (Gazina et al., 2002).
Poliovirus belongs to the family Picornaviridae of positive strand RNA viruses. Picornaviruses are small non‐enveloped viruses whose genome RNA is directly translated after infection into a polyprotein that is processed by viral proteinases into intermediate and final cleavage products, all of which function in the virus life cycle (Fig. 3A). Production of viral non‐structural proteins is sufficient to induce the formation of characteristic clusters of membranous vesicles of ∼200–400 nm diameter, a hallmark morphologic feature of polio‐infected cells (Teterina et al., 1997; Suhy et al., 2000). We have demonstrated that GBF1 inactivation is the cause of poliovirus sensitivity to BFA and that expression of a BFA‐resistant mutant of GBF1 restores virus replication in the presence of the inhibitor. Non‐structural protein 3A of polio and the related Coxsackie B3 virus directly binds the N‐terminal portion of GBF1 (2006a, 2006b), thus providing a mechanism for recruitment of this cellular protein into viral replication complexes. GBF1 was shown to be important for viral RNA replication per se, not for remodelling of cellular membranes into the vesicle‐like membranous scaffold as might have been predicted based on the established role of GBF1 in COPI‐dependent vesicle budding (2007, 2008).
Figure 3.
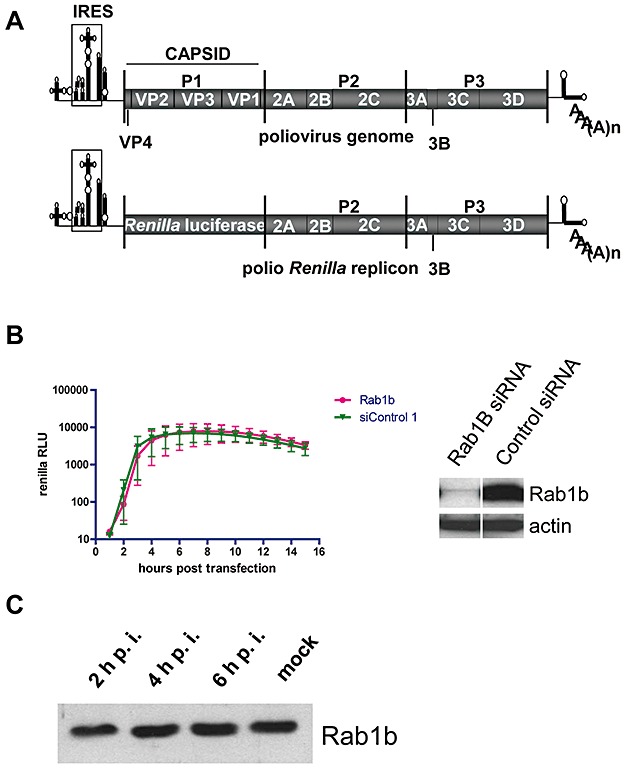
Rab1b does not contribute to polio replication. A. Schematic Map of the poliovirus genome and poliovirus Renilla replicon RNA. B. HeLa cells were treated with anti‐Rab1b siRNA or with a control siRNA for 3 days and then transfected with the polio Renilla luciferase replicon. Luciferase activity was assayed as a measure of polio replication. The Western blot shows the level of Rab1b depletion; actin serves as a loading control. C. HeLa cells infected with poliovirus were collected at the indicated times post infection and the cytoplasmic lysates were resolved by SDS‐PAGE and immunoblotted with anti‐Rab1b antibodies.
In this study we dissected the requirements for GBF1 to contribute to poliovirus replication and found that poliovirus utilizes GBF1 in a manner quite different from the way(s) that GBF1 participates in the cellular secretory pathway. We found that the GBF1 binding partner p115 is specifically degraded in infected cells and that depletion of the N‐terminal GBF1 binding partner Rab1b does not affect virus replication. The Sec7 domain's catalytic activity is not necessary to support virus replication. We provide evidence that polio replication can occur without activation of Arf by GBF1 and that the absence of activated Arf does not disrupt the association of polio proteins with cellular membranes. The entire C‐terminal portion of GBF1 is dispensable for virus replication, and essential functions are provided solely by the N‐terminal region containing the DCB and HUS domains of the protein.
Results
BFA does not induce gross rearrangement of polio protein assembly
Previously we showed that the cellular Arf‐GEF GBF1 is an essential host factor for poliovirus replication. We also demonstrated that poliovirus proteins synthesized in the presence of BFA, a potent inhibitor of GBF1‐dependent activation of Arf, still induced remodelling of cellular membranes indistinguishable from that observed during productive infection. However those membranous structures did not support polio RNA replication even when the BFA block was relieved (Belov et al., 2008). We considered whether the lack of GBF1 activity in the presence of BFA and therefore the absence of activated Arf on membranes affected the overall architecture of the viral replication complex. We examined the association of poliovirus proteins with cellular membranes and their sensitivity to proteinase K digestion. Poliovirus proteins were expressed in HeLa cells by infecting the cells with vaccinia virus producing T7 RNA polymerase and then transfecting them with a plasmid bearing poliovirus cDNA under control of the T7 promotor. BFA (4 ug ml−1) or its solvent, dimethylsulfoxide (DMSO), was added at the time of transfection. This approach allows robust synthesis of polio proteins even in the presence of BFA because this inhibitor does not affect vaccinia virus replication or T7 RNA polymerase activity. At 4.5 h post transfection the cells were treated with digitonin to permeabilize the plasma membrane, and the cells were exposed to proteinase K. Proteins were collected, resolved by SDS‐PAGE and visualized in immunoblots with different anti‐polio protein antibodies. Samples not treated with BFA showed slightly greater levels of viral protein synthesis, because of additional mRNA produced from viral RNA replication (as opposed to T7 RNA polymerase transcription) and/or to reduced transfection efficiency in cells treated with BFA (Fig. 1B, compare lanes 9 and 10). After digitonin treatment, almost all of the viral membrane binding proteins 2C, 2BC and 3A remained in the cells, as expected, because of their association with intracellular membranous structures (Fig. 1B, compare lanes 9, 10 and 5, 6, upper and middle panels respectively). Proteins 3D and 3CD, however, which lack intrinsic membrane binding domains, were only partially recovered from the permeabilized cells (Fig. 1B, compare lanes 9, 10 and 5, 6, lower panel). When the digitonin‐permeabilized cells were treated with proteinase K, poliovirus proteins demonstrated various degrees of resistance to proteolysis with the most resistant 3A (Fig. 1A, compare lanes 1, 2 and 5, 6, middle panel) and the least protected 3CD and 2BC (Fig. 1A, compare lanes 1, 2 and 5, 6, lower and upper panels respectively). Importantly, the amount of polio proteins retained in cells after permeabilization and the level of their accessibility to proteinase K did not dramatically differ in BFA‐treated and non‐treated cells (Fig. 1). Thus the failure to activate Arf did not significantly alter the extent of binding to membranes or the overall protection from proteinase K degradation of these viral proteins.
Figure 1.

Resistance of poliovirus proteins to cell permeabilization and proteinase K digestion. A. HeLa cells infected with vaccinia VT7‐3 virus were transfected with pXpA‐P2P3 plasmid (polio samples) or an empty vector (control samples). BFA (4 µg ml−1) was added where indicated at the time of DNA transfection; other samples contained DMSO (BFA solvent) in equivalent amount. The cells were treated with either digitonin (Dg) and proteinase K (PK) (lanes 1–4), digitonin only (lanes 5–8) or buffer only (lanes 9–12). Material amount corresponding to the same number of cells is loaded in each lane. Small poliovirus proteins 3A (10 kDa) and 3AB (12 kDa) are not resolved on this gel. B. portions of the same immunoblots shown on (A) with digitonin‐treated samples (lanes 5–8) and control samples (lanes 9–12) but with shorter exposure.
p115 is specifically degraded in polio‐infected cells
While strong evidence supports the requirement for GBF1 in picornaviral RNA replication (Belov et al., 2008; Lanke et al., 2009) the specific function(s) provided by GBF1 to support the virus life cycle are not known. The cellular functions of GBF1 are regulated by and depend upon its interactions with other proteins. Therefore, we investigated the fates of known cellular GBF1 binding partners during infection to determine whether these interactions are important for polio replication. The membrane tethering factor p115 was shown to bind to the C‐terminal proline‐rich sequence of GBF1 (Garcia‐Mata and Sztul, 2003). When we analysed endogenous p115 by Western blot of cytoplasmic extracts from poliovirus‐infected HeLa cells at different times after infection, we found that p115 underwent rapid and specific degradation (Fig. 2A, left upper panel). By 6 h post infection, only a small amount of the intact p115 remained. The disappearance of p115 was accompanied by the appearance of a major immunoreactive band(s) of ∼95 kDa. The course of virus infection was monitored by blotting the same membranes with antibodies against the viral protein 2C, a well‐characterized viral replication protein (Fig. 2A, lower panels).
Figure 2.
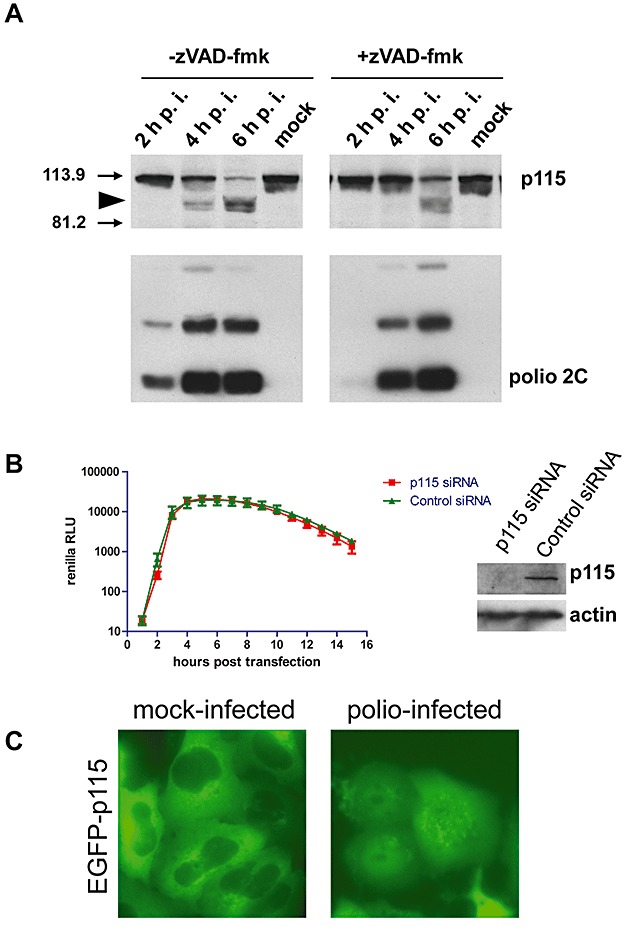
Degradation of p115 in polio‐infected cells. A. HeLa cells infected with poliovirus were incubated in the absence (left panel) or in the presence (right panel) of 100 µM z‐VAD‐fmk. Cells were collected at the indicated times post infection (p.i.) and the cytoplasmic lysates were resolved by SDS‐PAGE prior to immunoblotting with anti‐p115 antibodies. The membrane was stripped and reprobed with anti‐polio 2C antibodies that detect mature 2C protein (indicated) as well as several intermediate products of processing of the poliovirus polyprotein. Arrowhead marks major degradation product(s) of p115. Arrows indicate positions of molecular weight markers. B. HeLa cells were treated with anti‐p115 siRNA or with a control siRNA for 3 days and then transfected with the polio Renilla luciferase replicon. Luciferase activity was assayed as a measure of polio replication. The Western blot shows the level of p115 depletion; actin serves as a loading control. C. HeLa cells expressing EGFP–p115 fusion protein were infected with 50 PFU cell−1 of poliovirus and photographed 4 h post infection. EGFP fluorescence is shown.
To see if p115 or its degradation products are involved in polio replication, we depleted cellular p115 levels by siRNA knock‐down. Subsequent replication of a polio replicon in cells with the severely reduced level of p115 was the same as in cells treated with control non‐specific siRNA (Fig. 2B).
It has been reported that p115 is cleaved by caspases 3 and 8 in cells undergoing apoptosis (Chiu et al., 2002). Poliovirus infection induces the initial stages of the apoptotic response (Belov et al., 2003); thus the observed degradation of p115 might be attributed to activation of the apoptotic programme. To test whether the observed degradation of p115 in poliovirus‐infected cells was caspase‐dependent, we infected HeLa cells in the presence of the broad spectrum, cell‐permeable caspase inhibitor z‐VAD‐fmk. Degradation of p115 in the presence of the drug was delayed about 1.5 h, but not prevented (Fig. 2A, right panels). The slower degradation correlated with the similarly delayed development of infection in the presence of z‐VAD (compare accumulation of virus protein 2C in the presence and the absence of the inhibitor, Fig. 2A, lower panels). When we expressed a fusion of EGFP with p115 of rat origin in mock‐infected cells, fluorescence was localized to ER‐Golgi membranes and was excluded from nuclei, while in infected cells the protein was diffusely spread throughout the entire cell (Fig. 2C), consistent with loss of membrane localization.
The proteolysis of p115 in infected cells could be catalysed by either virus‐specific proteases or by some cellular enzymes activated during infection. The processing cascade of the poliovirus polyprotein generates three proteinases – 2A, 3C and 3CD. All of them contribute to the maturation of viral polypeptides, and they also cleave some specific cellular targets and induce modification of cellular metabolism favourable for the development of infection (Clark et al., 1993; Roehl et al., 1997; Yalamanchili et al., 1997; Ventoso et al., 1998; Kuyumcu‐Martinez et al., 2002; Belov et al., 2004; Kundu et al., 2005). The human p115 sequence contains several potential cleavage sites for poliovirus proteinases 3C or 3CD, which cut between glutamine and glycine residues. Cleavage at the residue Q715 would have been consistent with the appearance of the major degradation product of endogenous p115 observed in infected cells (Fig. 2A). This site is conserved in the rat p115 sequence. To determine if it was targeted by poliovirus 3C or 3CD proteinases, we replaced Q715 with asparagine in the EPFP–p115 fusion. Expression of the wild‐type p115 fusion protein or the Q715N fusion had no noticeable effect on polio RNA replication in the absence or in the presence of BFA (not shown). Infection of cells expressing the EGFP–p115Q715N fusion did not reveal any detectable difference in the cleavage pattern of the protein, compared with cells expressing a wild‐type fusion (not shown).
Thus our results show that p115, an important cellular partner of GBF1, is specifically degraded in polio‐infected cells, most likely by cellular proteases, and that neither p115 itself nor its degradation products are important for virus replication in cell culture.
Rab1b is non‐essential for polio replication
The N‐terminal portion of GBF1 directly interacts with the small cellular GTPase, Rab1b, which modulates GBF1‐dependent COPI recruitment and is important for GBF1 localization to membranes (Monetta et al., 2007). HeLa cells were treated with anti‐Rab1b siRNA and monitored for the subsequent replication of a polio replicon containing the Renilla luciferase reporter gene (Fig. 3A). Severe depletion of Rab1b did not have a significant effect on polio replication (Fig. 3B). We also did not detect any degradation of the Rab1b protein during the time course of poliovirus infection of HeLa cells (Fig. 3C). These results show that Rab1b is likely not important for polio replication in cell culture.
Truncated GBF1 species do not support cell secretion and viability in the presence of BFA but can efficiently rescue polio replication
Experiments described above show that GBF1's contribution to poliovirus replication is not dependent on its interaction with Rab1b and p115 proteins, both of which are important for GBF1 function in uninfected cells. These results prompted us to further compare the functions of GBF1 necessary for supporting normal cellular metabolism from those needed for virus replication. Bioinformatics analysis has identified six conserved domains in the GBF1 sequence (Mouratou et al., 2005), including the Sec7 domain, which is catalytically responsible for guanine nucleotide exchange and Arf activation. Figure 4A shows the domain assembly in the full‐length protein, and a series of truncated GBF1 constructs based on a YFP‐GBF1 expression vector, that were constructed for function analysis. This full‐length GBF1 fusion has been shown to be competent for all known GBF1‐related functions and has been extensively used in studies of both infected and uninfected cells (Niu et al., 2005; 2006a, 2006b; 2007, 2008; Deng et al., 2009). In addition, GBF1 constructs used in this experiment had the A795E BFA‐resistant mutation in the Sec7 domain described previously (Belov et al., 2008). We first tested these constructs for their abilities to support cellular protein secretion and to maintain cell viability in the presence of BFA. Cell secretion was measured by monitoring the activity of secreted Gaussia luciferase, which has a cellular secretion signal sequence (Tannous et al., 2005), in the medium after incubation of cells for 5 h in the presence of 1 µg ml−1 BFA. Expression of the different protein constructs was monitored by SDS‐PAGE. Figure 4B shows that secretion of the reporter protein was efficiently rescued by expression of the full‐length GBF1 fusion protein, but not by any of the truncated mutants even though they were often expressed at higher levels than the full‐length protein, especially the shortest GBF1(1–710) construct (Fig. 4B, right panels). The full‐length fusion protein frequently resolved into a doublet band (see Fig. 4B, upper gel). This may reflect post‐translational modification(s) of the protein, e.g. phosphorylation of GBF1 has been reported previously (Miyamoto et al., 2008).
Expression of the full‐length GBF1 protein efficiently prevented BFA‐induced cell death during prolonged incubation of cells with three different concentrations of BFA (Fig. 5). We monitored several concentrations of BFA to allow detection of even possibly low rescue activity of truncated GBF1 mutants. BFA at concentrations below 25 ng ml−1 was not significantly cytotoxic for our HeLa cells during the 3 days of incubation. Among the truncated constructs, only GBF1(1–1060) showed some level of cell viability rescue, and only at the lowest concentration of BFA tested (25 ng ml−1). None of the other truncated constructs was able to provide BFA resistance to cells. These data show that the deleted portions of the protein are necessary for the normal functioning of GBF1 in cell metabolism.
Figure 5.

Truncated GBF1 mutants do not confer BFA resistance to cell viability. HeLa cells were transfected with plasmids expressing full‐length YFP‐GBF1A795E, truncated GBF1 fusion species or an empty vector (control). Cells were incubated for 3 days with the indicated concentrations of BFA or corresponding amount of DMSO. Cell viability was determined by a luminescent viability assay. The signal obtained from cells incubated in the presence of DMSO was designated as 100% for all panels.
The set of truncated GBF1 proteins, which failed to support viability or protein secretion in HeLa cells, was then evaluated for their abilities to rescue replication of the polio Renilla luciferase replicon in HeLa cells in the presence of BFA. Under these conditions endogenous GBF1 is inhibited and viral RNA replication requires an ectopically expressed GBF1 species. Expression of any of the GBF1 constructs had no effect on viral RNA replication in the absence of BFA (Fig. 6, left panels). Expression of GBF1 species was confirmed by Western blot (not shown). In agreement with our previous data, the small deletion of only 37 amino acids from the N‐terminus of GBF1 completely abrogated its replication rescue capacity (Fig. 6, upper right panel). However, deletion of the C‐terminal domains HDS2 and HDS3 did not interfere with the ability of GBF1 to support polio replication (Fig. 6, middle right panel). Replication rescue dropped sharply with further deletion of the HDS1 domain (Fig. 6, lower right panel). Thus, the N‐terminal portion of GBF1 contains domains essential for function in both cellular and viral metabolic pathways. This region of GBF1 was identified previously to be necessary and sufficient for binding of the 3A protein of Coxsackie B3 virus, a close relative of polio (2006b, 2007). However, in contrast to the essential role of the C‐terminal portion of GBF1 in uninfected cells, the C‐terminal HDS2 and HDS3 domains of GBF1 are completely dispensable for the replication of poliovirus. These results restrict the functional contribution of GBF1 for poliovirus replication to the region between the N‐terminus and somewhere within the HDS1 domain. It is not clear whether the HDS1 domain per se might be required for virus replication, or whether it serves to maintain the conformation or stability of the truncated protein.
Figure 6.

Truncated GBF1 mutants rescues polio replication from BFA inhibition. HeLa cells were transfected with plasmids expressing full‐length YFP‐GBF1A795E, truncated GBF1 fusion species or an empty vector (control). The next day the cells were transfected with the polio Renilla replicon and incubated in the presence of 2 µg ml−1 BFA or corresponding amount of DMSO. Luciferase activity was monitored as a measure of polio replicon replication.
The N‐terminal DCB and HUS domains but not the Sec7 domain catalytic activity of GBF1 are essential for polio replication rescue
Previous data suggested that the Arf‐activating property of GBF1 is important for polio replication as expression of the catalytically inactive form of GBF1 with the dominant‐negative mutation E794K in the Sec7 domain was unable to rescue polio replication in the presence of BFA (Belov et al., 2008). The reduced replication rescue by the GBF1(1–894) construct described above could indicate interference with Sec7 domain activity in the absence of any downstream sequences. To determine whether Sec7 activity contributed to the observed level of rescue by GBF1(1–894), we introduced the dominant‐negative mutation E794K into this construct. Surprisingly, we did not observe any inhibition of the rescue capacity of GBF1(1–894) E794K compared with the construct GBF1(1–894) with the unmodified Sec7 sequence (Fig. 7, upper panels), suggesting that the rescue ability of this construct did not depend on the ability to induce Arf activation. To confirm that Sec7 activity was not required for at least some level of polio replication, we expressed only the N‐terminal sequences of GBF1, including the DCB and HUS domains but excluding the Sec7 domain. Again we saw clearly detectable levels of polio replication rescue from BFA inhibition in HeLa cells expressing this construct (Fig. 7, middle panels). To eliminate any potential interference by the YFP fused to the N‐terminus of GBF1 used in these experiments (or any unlikely contribution of YFP to the rescue of polio replication), we generated a new set of GBF1 expression vectors that produce authentic GBF1 N‐termini. As expected, the GBF1(1–710) fragment without YFP fusion was inactive for rescue of cells from BFA cytotoxicity or for rescue of secretion of Gaussia luciferase (not shown). However this GBF1(1–710) fragment demonstrated a significant level of polio replication rescue (Fig. 7, lower panels). Expression of all GBF1 constructs was verified with Western blots (not shown). Thus, the essential functions for polio replication can be provided by the N‐terminus of GBF1, while the Sec7 domain is not necessary.
Figure 7.

Sec7 domain is not necessary for rescue of polio replication from BFA inhibition. HeLa cells were transfected with plasmids expressing either full‐length YFP‐GBF1A795E (upper panel), truncated GBF1 fusion species lacking the Sec7 domain and all downstream sequences (middle panel) or an empty vector (control). The lower panel shows HeLa cells transfected with pCI‐GBF1 or pCI‐GBF1(1–710) expressing the full‐length GBF1A795E or the N‐terminal fragment of GBF1, both with the authentic GBF1 N‐terminus. Control cells were transfected with an empty vector. The next day the cells were transfected with the polio Renilla replicon and incubated in the presence of 2 µg ml−1 BFA or corresponding amount of DMSO. Luciferase activity was monitored as a measure of polio replicon replication.
Replication of BFA‐resistant poliovirus does not require Arf activation but is dependent on GBF1
We previously showed that Arf activation occurs during replication of polio RNA in infected cells, as well as in vitro upon translation and replication of polio RNA (2005, 2007). The results reported above, however, suggested that functions of GBF1 essential for polio replication could be separated from its Arf‐activating capacity. It was unclear whether the accumulation of activated Arf GTPase that we observed at viral replication sites was a by‐product of GBF1 recruitment, perhaps contributing to virus growth in an unknown way. Crotty et al. have isolated a mutant of poliovirus (DM) that is resistant to inhibition by BFA as a result of two point mutations in the 2C and 3A proteins (Crotty et al., 2004). Robust growth of this virus in the presence of BFA suggested that it could replicate without Arf activation. We examined the state of Arf activation during replication of this BFA‐resistant mutant by using a GGA pull‐down assay (Kawamoto et al., 2002) that detects Arf only in its activated, GTP‐bound form. Figure 8A shows that in the absence of BFA, there was a steady increase in Arf‐GTP accumulation during the course of infection, as seen previously during wild‐type poliovirus infection (Belov et al., 2007). During virus replication in the presence of BFA, however, no Arf activation was detected. Virus replication was slightly delayed compared with infection in the absence of the inhibitor, as shown by measuring the production of proteins containing poliovirus 2C sequences (Fig. 8A, bottom immunoblot). This experiment demonstrates that replication of poliovirus can occur without Arf activation. The ability of the DM mutant to grow in the presence of BFA without Arf activation raised the question of whether this virus still requires GBF1 sequences for replication. We knocked down expression of GBF1 with siRNA treatment. In the absence of BFA, replication of the DM mutant in GBF1 siRNA‐treated cells was inhibited. The level of inhibition showed some variability, from almost complete to about 70%, as observed in this experiment (Fig. 8B, left panel). This variability may reflect different levels of cytotoxicity of GBF1 depletion, reported previously (Belov et al., 2008; Citterio et al., 2008). In spite of the rather modest inhibition of replication of the DM mutant in the absence of BFA in this experiment, GBF1‐deficient cells were almost completely non‐permissive for the DM mutant in the presence of the inhibitor while the control cells supported a high level of replication of this replicon (Fig. 8B, right panel). Thus, GBF1 is mandatory for polio replication, but its GEF activity is dispensable.
Figure 8.

GBF1 but not Arf activation is required for polio replication. A. HeLa cells were infected with the BFA‐resistant DM mutant of poliovirus and incubated in the presence of 2 µg ml−1 BFA or corresponding amount of DMSO. At the indicated times cells were lysed, and activated Arf was collected by GGA pull‐down. Isolated Arf‐GTP was eluted, resolved by SDS‐PAGE and visualized by immunoblot with anti‐Arf antibodies (upper panel). The middle panel shows total Arf in lysates before Arf‐GTP pull‐down. This membrane was stripped and reprobed with anti‐polio 2C antibodies to monitor the development of infection. Mature 2C protein (indicated) as well as several intermediate products of processing of the poliovirus polyprotein are detected (lower panel). B. GBF1 is required for replication of a BFA‐resistant polio mutant. HeLa cells were treated with anti‐GBF1 siRNA or with the control siRNA. On the third day the cells were transfected with the polio Renilla luciferase replicon harbouring BFA‐resistant mutations in the 2C and 3A protein sequences. The cells were incubated with 2 µg ml−1 BFA or corresponding amount of DMSO in the growth medium. Luciferase activity was assayed as a measure of polio replicon replication. The Western blot shows the level of GBF1 depletion; actin immunoblot served as a loading control.
Discussion
Viruses inevitably require cellular machinery to replicate their genomes and produce progeny virions. Previously we identified the cellular protein GBF1 as a host component essential for replication of poliovirus and demonstrated that this protein accumulates on membranes associated with viral replication complexes (2007, 2008). In this study we investigated the GBF1 contribution to viral RNA replication in comparison with its functions required for cellular metabolism. In uninfected cells GBF1 is located at the ER‐Golgi interface; it is responsible for activation of Arf GTPases necessary for formation of COPI‐coated membranous vesicles returning membrane lipids and proteins from the Golgi to ER (Casanova, 2007). The large size and multidomain assembly of the GBF1 protein suggests that this protein might engage in numerous interactions with other cellular proteins. All or some subset of those interactions may participate in virus replication when GBF1 is recruited to the sites of viral replication complexes in infected cells. Of the cellular partners known to interact with GBF1, the membrane tethering protein p115 interacts with the C‐terminus (Garcia‐Mata and Sztul, 2003), while the small cellular GTPase Rab1b was shown to bind to the N‐terminal segment (Monetta et al., 2007). Rab1b and p115 also interact with each other, and this interaction is important for recruitment of a specific set of SNARE proteins that determine proper docking and fusion of COPII‐coated vesicles transferring material from the ER to Golgi (Allan et al., 2000). Thus, Rab1b and p115 seem to modulate GBF1 function in the secretory pathway, and interactions among these three proteins coordinate and regulate COPII‐dependent anterograde and COPI‐dependent retrograde transport (Alvarez et al., 2003; Monetta et al., 2007). It was suggested that COPII‐ and COPI‐dependent vesicular transport mechanisms may be hijacked by picornaviruses and participate in viral replication. Rust et al. demonstrated that poliovirus non‐structural proteins are found in infected cells in COPII‐positive structures on the ER, interpreted to be ER exit sites (Rust et al., 2001). COPI coat proteins were shown to colocalize with viral replication complexes in cells infected with a related picornavirus, echovirus 11 (Gazina et al., 2002). COPI components were also identified in a total genome screen for the factors required for the replication of a picornavirus‐related Drosophila C virus (Cherry et al., 2006). Our data, however, indicate that neither COPII‐ nor COPI‐mediated pathways of normal cellular membrane traffic are subverted in toto for virus replication. We found that replication does not depend on Rab1b or p115 and that the latter protein is degraded in infected cells, while both of these proteins are important for the normal functioning of COPI‐ and COPII‐dependent pathways. Investigation of the ability of truncated GBF1 species to support polio replication and cellular functions revealed that the truncated GBF1(1–1060) construct, lacking the entire C‐terminal HDS2 and HDS3 domains, was indistinguishable from the full‐length construct in supporting virus replication, although it failed to function for cell secretion and viability. The next upstream domain of GBF1, HDS1, seems to significantly contribute to the level of replication, either because of its specific sequence determinants or because it stabilizes structural and conformational properties of the more N‐terminal portion of GBF1. The finding that the C‐terminal domains of GBF1 are dispensable for virus replication is consistent with the observation that p115, which interacts with this region of GBF1, is degraded in polio‐infected cells and thus the intact p115 protein or its degradation fragments are not required for replication. However, it is possible that degradation of p115 may be important for virus growth in the animal host as it can contribute to the shutdown of the secretory pathway believed to facilitate evasion of immune protection mechanisms utilized by poliovirus (Deitz et al., 2000; Dodd et al., 2001; Neznanov et al., 2001). These data provide strong support for the idea that poliovirus does not hijack a complete cellular membrane traffic pathway (COPI‐ or COPII‐dependent) in toto. Rather, it seems that poliovirus combines individual steps representing portions of normally distinct biochemical pathways for building its replication machinery, resulting in severe disturbance and rearrangement of pre‐existing cellular membrane trafficking reactions.
GBF1 is a GEF for the small cellular GTPases of the Arf family. This activity is carried out by the Sec7 domain of GBF1, a centrally located region that makes up only 10% of this large 200 kDa protein. The drug BFA inhibits GBF1 function by binding to a normally transient complex between the Sec7 domain of the GEF and its substrate, Arf‐GDP, thus preventing nucleotide exchange (Mossessova et al., 2003; Renault et al., 2003). When cells are treated with BFA, GBF1 accumulates on membranes up to four times its normal level (Niu et al., 2005). Hence, although a major effect of BFA is blocking Arf activation, BFA also interferes with other GBF1 functions by trapping it in an inactive complex with Arf1‐GDP and inhibiting the dynamic cycle of binding and release of GBF1 from membranes.
One interpretation of the inhibition of polio replication by BFA is that Arf activation is important for virus growth; and indeed it was shown that the levels of activated Arf increased in infected cells during the replication cycle (Belov et al., 2007). In addition, previous experiments showed that GBF1 harbouring an inactivating mutation in Sec7 was unable to rescue polio replication in the presence of BFA and thus supported the notion that Arf activation by GBF1 played an important role in polio replication (2007, 2008). Moreover, two BFA‐resistant variants of GBF1 that have point mutations in their Arf‐activating Sec7 domains, A795E and M832L, efficiently rescue both poliovirus and Coxsackie virus B3 replication from BFA inhibition (Belov et al., 2008; Lanke et al., 2009). However, in this study, we have tested a series of truncated mutants of GBF1 for their abilities to support polio replication in the presence of BFA, and were surprised to see that the Sec7 domain necessary for Arf activation was dispensable for viral replication. Expression of the GBF1 fragment containing just two N‐terminal domains DCB and HUS was sufficient to rescue a significant (albeit not complete) level of replication of the wild‐type polio replicon in the presence of BFA. The separation of the replication‐supporting function of GBF1 from its Arf activation function is consistent with our previously reported failure to rescue polio replication from BFA inhibition by expressing the constitutively activated Arf1Q71L mutant (Belov et al., 2008). Furthermore, we demonstrated that replication of a BFA‐resistant polio mutantin the presence of BFA did not induce detectable Arf activation, but was nevertheless strongly dependent on GBF1. This BFA‐resistant poliovirus carries two mutations located in viral proteins 3A and 2C, shown to have an additive effect on the level of BFA resistance (Crotty et al., 2004). The N‐terminal fragment of GBF1 is known to bind directly to 3A (2006a, 2006b), and functional interaction between 2C and 3A has been implicated by genetic evidence (Teterina et al., 2006).
We propose that inhibition of polio replication by BFA occurs not because BFA prevents Arf activation, but because the BFA‐stabilized complex of GBF1 with Arf‐GDP cannot be bound or used by wild‐type poliovirus as a source of GBF1.
If the Arf exchange activity of GBF1 is dispensable for polio replication, then why does the catalytically inactive E794K mutant within full‐length GBF1 not rescue replication in the presence of BFA? At physiological magnesium concentration, the GBF1‐E794K mutant also forms an abortive complex with Arf1 (Beraud‐Dufour et al., 1998; Renault et al., 2003), and we propose that this inactive complex, like the full‐length GBF1‐BFA‐Arf1‐GDP complex, is equally inaccessible to wild‐type polio for viral replication functions. The DCB and HUS domains within the N‐terminus of GBF1 interact (Ramaen et al., 2007), and there is evidence that this DCB–HUS interaction affects binding of GBF1 to cellular protein partners (Deng et al., 2009). Thus, domain interactions within GBF1 may play important roles in regulating this large protein's activities. One possibility is that BFA may stabilize a GBF1 conformation that is not ideal for interaction with wild‐type polio 3A protein. Mutations in the 2C and 3A proteins in the BFA‐resistant poliovirus mutant, however, may allow poliovirus to utilize GBF1 trapped in these inactive complexes more effectively by modifying the 3A–GBF1 interaction.
No cellular interaction partners are known to bind the N‐terminal region of GBF1 except for Rab1b, which we found to be unimportant for virus replication. Nevertheless the N‐terminal region of two other large Arf1 GEFs, BIG1 and BIG2 were shown to interact with multiple proteins such as AMY‐1, protein kinase A, FKBP13, the Exo70 subunit of the exocyst, the HSC70 chaperone and phosphodiesterase 3A (Li et al., 2003; Padilla et al., 2003; Xu et al., 2005; Ishizaki et al., 2006; Puxeddu et al., 2009). Thus it is very likely that the N‐terminal segment of GBF1 may also be involved in interactions with other cellular factors and in recruiting them to viral replication complexes.
Our results show for the first time that BFA can inhibit not only activation of Arf1, but also a non‐catalytic function of GBF1 carried out by its N‐terminal region. The fact that BFA inhibits additional GBF1 functions beyond activation of Arf1 is a conclusion important not only for virologists, but for all cell biologists using this drug to study membrane trafficking.
It was recently reported that GBF1 is involved in replication of mouse hepatitis coronavirus (Verheije et al., 2008) and hepatitis C virus (Goueslain et al., 2010). The dependence of different groups of positive strand RNA viruses on the same cellular factor suggests that GBF1 may be a part of some conserved step in replication shared by diverse groups of pathogens.
Experimental procedures
Cells and viruses
Monolayer HeLa cells were grown in high‐glucose DMEM supplemented with pyruvate and 10% fetal bovine serum. For infection, the cells were incubated with 50 PFU cell−1 of the Mahoney strain of poliovirus type 1 in DMEM buffered with 50 mM Hepes, pH 7.0. Where indicated, the DM BFA‐resistant mutant (Crotty et al., 2004) was inoculated at 10 PFU cell−1. After 30 min with virus at room temperature, the medium was changed to normal growth medium, and the cells were incubated at 37°C for the indicated times. Control cells (mock‐infected) underwent the same procedures but the medium did not contain virus.
Plasmids
Plasmid pCMV‐Gluc for expression of secreted Gaussia luciferase was from New England Biolabs. Plasmid pXpa‐RenR coding for a poliovirus replicon with the Renilla luciferase gene substituted for the capsid coding sequence was described previously (Belov et al., 2007). Plasmid pXpA‐P2P3 contains the cDNA of poliovirus RNA lacking structural protein coding sequences, under control of a T7 RNA polymerase promotor. Polio‐specific RNA produced from this plasmid is capable of autonomous replication but does not produce infectious virus. Plasmids pYFP‐GBF1, pVenus‐GBF1Δ37, pYFP‐GBF1 E794K (dominant negative mutation), pYFP‐GBF1A795E (BFA‐resistant mutation) and pVenus‐GBF1 for expression of GBF1 derivatives have been previously described (Niu et al., 2005; Belov et al., 2008). Plasmids pVenus‐GBF1(1–1060), pYFP‐GBF1(1–894), pYFP‐GBF1(1–710) for expression of truncated version of GBF1 were constructed by Ting Nui and Marie‐Pierre Golinelli‐Cohen (Deng et al., 2009). To create plasmids encoding truncated GBF1 species with BFA‐resistant mutations, a PciI‐BglII fragment in each plasmid was replaced with the corresponding fragment from pYFP‐GBF1A795E. For plasmid pCI‐GBF1 expressing the full‐length GBF1 with an authentic N‐terminus, the relevant XhoI‐KpnI fragment from pYFP‐GBF1 A795E was cloned into the pCI expression vector (Promega). Plasmid pCI‐GBF1(1–710) contains 710 amino acids of N‐terminal GBF1 fragment from pYFP‐GBF1(1–710) followed by an AU1 tag. All PCR‐derived sequences were verified by sequencing. pEGFP‐C2‐p115 and pYFP‐N1‐p115 were a gift from Elizabeth Sztul (University of Alabama). pEGFP‐C2‐p115Q715N was constructed by introducing the mutation using the QuickExchange II XL mutagenesis kit (Stratagene) into pYFP‐N1‐p115 and then recloning the mutation‐containing fragment into pEGFP‐C2‐p115. The sequence of the transferred fragment sequence was verified. Cloning details are available upon request.
Antibodies
Anti‐p115 rabbit polyclonal antibodies were a gift from Elizabeth Sztul (University of Alabama). Anti‐Rab1b rabbit polyclonal antibodies were from Santa Cruz. Anti‐GBF1 rabbit polyclonal antibodies were a gift from Nihal Altan‐Bonnet (Rutgers University). Anti‐polio 2C and anti‐polio 3A mouse monoclonal antibodies were a gift from Kurt Bienz (Basel University). Anti‐polio 3D polyclonal antibodies were ordered from Chemicon custom antibody production service. Intact recombinant 3D protein, generously provided by Olve Peersen, Colorado State University, was used as immunogen. Mouse monoclonal anti‐GFP antibodies were from Clontech. Mouse monoclonal anti‐Arf antibodies recognizing all human Arf species except Arf4 were from Affinity Bioreagents. Anti‐actin HRP‐conjugated antibodies were from Sigma‐Aldrich. Secondary HRP‐conjugated antibodies were from GE Health Care.
Expression of polio proteins for digitonin and proteinase K treatment in cells infected with recombinant vaccinia virus encoding T7 RNA polymerase
HeLa cells were plated in a 12‐well plate at 400 000 cells well−1 1 day prior to infection with vaccinia virus VT7‐3 expressing T7 RNA polymerase (Fuerst et al., 1986) at a multiplicity of 50 PFU cell−1. At 1 h post infection, cells were transfected with pXpA‐P2P3 plasmid coding for replication proteins of poliovirus with Lipofectamine LTX (Invitrogen) and incubated for 4.5 h, either in the absence or presence of 4 ug ml−1 BFA. For BFA‐treated cultures, BFA was included in all subsequent incubations and washing solutions. Cells were washed once with KHM buffer (110 mM K‐acetate, 2 mM MgCl2, 20 mM Hepes‐KOH, pH 7.4), treated with digitonin (50 µg ml−1) for 5 min at room temperature and washed again twice with KHM buffer. Proteinase K solution (1 µg ml−1) in KHM buffer was added for 5 min at room temperature and the cells were washed once with KHM buffer. Immediately after the cells were lysed with mild lysis buffer (0.1 M Tris‐HCl pH 7.8; 0.5% Triton‐X100) supplemented with protease inhibitors cocktail (Sigma). The material was collected from wells, cleared for 5 min at high speed on a minifuge and analysed by SDS‐PAGE and immunoblot.
Renilla luciferase replicon assay
Polio RNA replication assays were performed as described previously (Belov et al., 2007) with slight modifications. Briefly, HeLa cells grown on opaque 96‐well plates (Costar) were transfected with plasmids with Lipofectamine LTX (Invitrogen) at the time of seeding. The next day the cells were transfected with RenR RNA transcribed from pXpA‐RenR using Mirus Trans‐It mRNA transfection reagent (Mirus Bio LLC). The incubation medium contained 60 µM Endu‐Ren cell‐permeable Renilla luciferase substrate (Promega) and 2 µg ml−1 BFA (or DMSO in controls) where indicated. The plates were maintained in a Molecular Devices M5 plate reader at 37°C. Luciferase readings were recorded every hour for 15 h post transfection from at least 16 replicate wells.
BFA cytotoxicity assay
HeLa cells were transfected with GBF1‐expressing plasmids 1 day prior to addition of BFA at the indicated concentrations. The cells were incubated for 3 days with medium containing BFA replaced twice a day to maintain a constant presence of BFA. Cell viability was assessed with Promega's Cell Titer Glo luciferase‐based viability kit.
Secretion rescue assay
Studies of secretion rescue by GBF1 expression were performed essentially as described (Belov et al., 2008). Briefly, HeLa cells were co‐transfected with pCMV‐Gluc and GBF1‐expressing plasmids. The next day, fresh growth medium with 1 µg ml−1 BFA was added and the cells were incubated for 5 h. Gaussia luciferase activity in the supernatant was assessed with Gaussia Luciferase Assay kit (New England Biolabs) according to the manufacturer's protocol.
siRNA treatment
Anti‐GBF1 CAACACACCUACUAUCUCU, Anti‐Rab1b predesigned siRNA (ID: 120885) and anti‐p115 siRNA (Puthenveedu and Linstedt, 2004) were from Dharmacon. siControl‐1 siRNA was from Ambion. HeLa cells were transfected at the time of seeding in 96‐well plates with Dharmafect 1 according to the manufecturer's guidelines. On the third day after transfection plates were used for replicon replication assays.
Microscopy
HeLa cells grown on coverslips were transfected with pEGFP‐C2‐p115 with Lipofectamine LTX (Invitrogen) and the next day were infected with poliovirus. At 4.5 h post infection the cells were fixed with 4% PFA in PBS for 20 min. Images were taken with a Leica DMIRE microscope. Digital images were processed with Adobe Photoshop software.
GGA Arf‐GTP pull‐down assay
The Arf‐GTP pull‐down assay was performed essentially as described (Belov et al., 2007). Briefly, HeLa cells grown overnight in 10 cm Petri dishes were infected with 10 PFU of poliovirus/cell. BFA (2 mg ml−1) (or DMSO for control cells) was added at the beginning of incubation. At the indicated times the cells were lysed in 1 ml of lysis buffer [50 mM Tris‐Cl (pH 7.5), 100 mM NaCl, 2 mM MgCl2, 0.5% sodium deoxycholate, 1% Triton X‐100, 10% glycerol] containing 100 µM phenylmethylsulfonyl fluoride and a 1/100 of volume of protease inhibitor cocktail (Sigma‐Aldrich) and incubated on ice for 15 min. Lysates were clarified by centrifugation at maximum speed for 20 min in an Eppendorf minifuge at 4°C. The supernatants were collected and stored at −80°C. The Arf‐GTP pull‐down assay was performed as described previously (Kawamoto et al., 2002). Briefly, lysates were precleared by incubation with 50 µl of a 50% slurry of glutathione‐Sepharose 4B beads (Amersham‐Biosciences) for 1 h and centrifuged. Supernatants were incubated with 50 µl of beads that contained 40 µg of prebound GST‐GGA3‐GAT fusion protein (expression construct kindly provided by Julie Donaldson, NHLBI, NIH). Beads were collected by centrifugation and washed three times with 250 µl of (10 times the bed volume of beads) washing buffer. All manipulations with lysates were carried out at 4°C. Proteins were eluted from beads by adding 1× SDS‐PAGE sample buffer and heating the samples at 70°C for 15 min.
Acknowledgements
This work was supported in part by the intramural programme of the NIAID, National Institutes of Health. We thank members of Elizabeth Stzul's laboratory for reagents and helpful discussion of p115 experiments. We thank Marie‐Pierre Golinelli‐Cohen for construction of plasmids and for critical reading of the manuscript.
References
- Allan, B.B. , Moyer, B.D. , and Balch, W.E. (2000) Rab1 recruitment of p115 into a cis‐SNARE complex: programming budding COPII vesicles for fusion. Science 289: 444–448. [DOI] [PubMed] [Google Scholar]
- Alvarez, C. , Garcia‐Mata, R. , Brandon, E. , and Sztul, E. (2003) COPI recruitment is modulated by a Rab1b‐dependent mechanism. Mol Biol Cell 14: 2116–2127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belov, G.A. , Romanova, L.I. , Tolskaya, E.A. , Kolesnikova, M.S. , Lazebnik, Y.A. , and Agol, V.I. (2003) The major apoptotic pathway activated and suppressed by poliovirus. J Virol 77: 45–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belov, G.A. , Lidsky, P.V. , Mikitas, O.V. , Egger, D. , Lukyanov, K.A. , Bienz, K. , and Agol, V.I. (2004) Bidirectional increase in permeability of nuclear envelope upon poliovirus infection and accompanying alterations of nuclear pores. J Virol 78: 10166–10177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belov, G.A. , Fogg, M.H. , and Ehrenfeld, E. (2005) Poliovirus proteins induce membrane association of GTPase ADP‐ribosylation factor. J Virol 79: 7207–7216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belov, G.A. , Altan‐Bonnet, N. , Kovtunovych, G. , Jackson, C.L. , Lippincott‐Schwartz, J. , and Ehrenfeld, E. (2007) Hijacking components of the cellular secretory pathway for replication of poliovirus RNA. J Virol 81: 558–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belov, G.A. , Feng, Q. , Nikovics, K. , Jackson, C.L. , and Ehrenfeld, E. (2008) A critical role of a cellular membrane traffic protein in poliovirus RNA replication. PLoS Pathog 4: e1000216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beraud‐Dufour, S. , Robineau, S. , Chardin, P. , Paris, S. , Chabre, M. , Cherfils, J. , and Antonny, B. (1998) A glutamic finger in the guanine nucleotide exchange factor ARNO displaces Mg2+ and the beta‐phosphate to destabilize GDP on ARF1. EMBO J 17: 3651–3659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bui, Q.T. , Golinelli‐Cohen, M.P. , and Jackson, C.L. (2009) Large Arf1 guanine nucleotide exchange factors: evolution, domain structure, and roles in membrane trafficking and human disease. Mol Genet Genomics 282: 329–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casanova, J.E. (2007) Regulation of Arf activation: the Sec7 family of guanine nucleotide exchange factors. Traffic 8: 1476–1485. [DOI] [PubMed] [Google Scholar]
- Cherry, S. , Kunte, A. , Wang, H. , Coyne, C. , Rawson, R.B. , and Perrimon, N. (2006) COPI activity coupled with fatty acid biosynthesis is required for viral replication. PLoS Pathog 2: e102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu, R. , Novikov, L. , Mukherjee, S. , and Shields, D. (2002) A caspase cleavage fragment of p115 induces fragmentation of the Golgi apparatus and apoptosis. J Cell Biol 159: 637–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Citterio, C. , Vichi, A. , Pacheco‐Rodriguez, G. , Aponte, A.M. , Moss, J. , and Vaughan, M. (2008) Unfolded protein response and cell death after depletion of brefeldin A‐inhibited guanine nucleotide‐exchange protein GBF1. Proc Natl Acad Sci USA 105: 2877–2882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark, M.E. , Lieberman, P.M. , Berk, A.J. , and Dasgupta, A. (1993) Direct cleavage of human TATA‐binding protein by poliovirus protease 3C in vivo and in vitro . Mol Cell Biol 13: 1232–1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claude, A. , Zhao, B.P. , Kuziemsky, C.E. , Dahan, S. , Berger, S.J. , Yan, J.P. , et al (1999) GBF1: a novel Golgi‐associated BFA‐resistant guanine nucleotide exchange factor that displays specificity for ADP‐ribosylation factor 5. J Cell Biol 146: 71–84. [PMC free article] [PubMed] [Google Scholar]
- Crotty, S. , Saleh, M.C. , Gitlin, L. , Beske, O. , and Andino, R. (2004) The poliovirus replication machinery can escape inhibition by an antiviral drug that targets a host cell protein. J Virol 78: 3378–3386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Souza‐Schorey, C. , and Chavrier, P. (2006) ARF proteins: roles in membrane traffic and beyond. Nat Rev Mol Cell Biol 7: 347–358. [DOI] [PubMed] [Google Scholar]
- Deitz, S.B. , Dodd, D.A. , Cooper, S. , Parham, P. , and Kirkegaard, K. (2000) MHC I‐dependent antigen presentation is inhibited by poliovirus protein 3A. Proc Natl Acad Sci USA 97: 13790–13795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng, Y. , Golinelli‐Cohen, M.P. , Smirnova, E. , and Jackson, C.L. (2009) A COPI coat subunit interacts directly with an early‐Golgi localized Arf exchange factor. EMBO Rep 10: 58–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodd, D.A. , Giddings, T.H., Jr , and Kirkegaard, K. (2001) Poliovirus 3A protein limits interleukin‐6 (IL‐6), IL‐8, and beta interferon secretion during viral infection. J Virol 75: 8158–8165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuerst, T.R. , Niles, E.G. , Studier, F.W. , and Moss, B. (1986) Eukaryotic transient‐expression system based on recombinant vaccinia virus that synthesizes bacteriophage T7 RNA polymerase. Proc Natl Acad Sci USA 83: 8122–8126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia‐Mata, R. , and Sztul, E. (2003) The membrane‐tethering protein p115 interacts with GBF1, an ARF guanine‐nucleotide‐exchange factor. EMBO Rep 4: 320–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gazina, E.V. , Mackenzie, J.M. , Gorrell, R.J. , and Anderson, D.A. (2002) Differential requirements for COPI coats in formation of replication complexes among three genera of Picornaviridae . J Virol 76: 11113–11122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillingham, A.K. , and Munro, S. (2007) The small G proteins of the Arf family and their regulators. Annu Rev Cell Dev Biol 23: 579–611. [DOI] [PubMed] [Google Scholar]
- Goueslain, L. , Alsaleh, K. , Horellou, P. , Roingeard, P. , Descamps, V. , Duverlie, G. , et al (2010) Identification of GBF1 as a cellular factor required for hepatitis C virus RNA replication. J Virol 84: 773–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishizaki, R. , Shin, H.W. , Iguchi‐Ariga, S.M. , Ariga, H. , and Nakayama, K. (2006) AMY‐1 (associate of Myc‐1) localization to the trans‐Golgi network through interacting with BIG2, a guanine‐nucleotide exchange factor for ADP‐ribosylation factors. Genes Cells 11: 949–959. [DOI] [PubMed] [Google Scholar]
- Kawamoto, K. , Yoshida, Y. , Tamaki, H. , Torii, S. , Shinotsuka, C. , Yamashina, S. , and Nakayama, K. (2002) GBF1, a guanine nucleotide exchange factor for ADP‐ribosylation factors, is localized to the cis‐Golgi and involved in membrane association of the COPI coat. Traffic 3: 483–495. [DOI] [PubMed] [Google Scholar]
- Kundu, P. , Raychaudhuri, S. , Tsai, W. , and Dasgupta, A. (2005) Shutoff of RNA polymerase II transcription by poliovirus involves 3C protease‐mediated cleavage of the TATA‐binding protein at an alternative site: incomplete shutoff of transcription interferes with efficient viral replication. J Virol 79: 9702–9713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuyumcu‐Martinez, N.M. , Joachims, M. , and Lloyd, R.E. (2002) Efficient cleavage of ribosome‐associated poly(A)‐binding protein by enterovirus 3C protease. J Virol 76: 2062–2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanke, K.H. , Van Der Schaar, H.M. , Belov, G.A. , Feng, Q. , Duijsings, D. , Jackson, C.L. , et al (2009) GBF1, a guanine nucleotide exchange factor for Arf, is crucial for coxsackievirus B3 RNA replication. J Virol 83: 11940–11949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H. , Adamik, R. , Pacheco‐Rodriguez, G. , Moss, J. , and Vaughan, M. (2003) Protein kinase A‐anchoring (AKAP) domains in brefeldin A‐inhibited guanine nucleotide‐exchange protein 2 (BIG2). Proc Natl Acad Sci USA 100: 1627–1632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maynell, L.A. , Kirkegaard, K. , and Klymkowsky, M.W. (1992) Inhibition of poliovirus RNA synthesis by brefeldin A. J Virol 66: 1985–1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyamoto, T. , Oshiro, N. , Yoshino, K. , Nakashima, A. , Eguchi, S. , Takahashi, M. , et al (2008) AMP‐activated protein kinase phosphorylates Golgi‐specific brefeldin A resistance factor 1 at Thr1337 to induce disassembly of Golgi apparatus. J Biol Chem 283: 4430–4438. [DOI] [PubMed] [Google Scholar]
- Monetta, P. , Slavin, I. , Romero, N. , and Alvarez, C. (2007) Rab1b interacts with GBF1 and modulates both ARF1 dynamics and COPI association. Mol Biol Cell 18: 2400–2410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mossessova, E. , Corpina, R.A. , and Goldberg, J. (2003) Crystal structure of ARF1*Sec7 complexed with brefeldin A and its implications for the guanine nucleotide exchange mechanism. Mol Cell 12: 1403–1411. [DOI] [PubMed] [Google Scholar]
- Mouratou, B. , Biou, V. , Joubert, A. , Cohen, J. , Shields, D.J. , Geldner, N. , et al (2005) The domain architecture of large guanine nucleotide exchange factors for the small GTP‐binding protein Arf. BMC Genomics 6: 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neznanov, N. , Kondratova, A. , Chumakov, K.M. , Angres, B. , Zhumabayeva, B. , Agol, V.I. , and Gudkov, A.V. (2001) Poliovirus protein 3A inhibits tumor necrosis factor (TNF)‐induced apoptosis by eliminating the TNF receptor from the cell surface. J Virol 75: 10409–10420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niu, T.K. , Pfeifer, A.C. , Lippincott‐Schwartz, J. , and Jackson, C.L. (2005) Dynamics of GBF1, a Brefeldin A‐sensitive Arf1 exchange factor at the Golgi. Mol Biol Cell 16: 1213–1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padilla, P.I. , Chang, M.J. , Pacheco‐Rodriguez, G. , Adamik, R. , Moss, J. , and Vaughan, M. (2003) Interaction of FK506‐binding protein 13 with brefeldin A‐inhibited guanine nucleotide‐exchange protein 1 (BIG1): effects of FK506. Proc Natl Acad Sci USA 100: 2322–2327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puthenveedu, M.A. , and Linstedt, A.D. (2004) Gene replacement reveals that p115/SNARE interactions are essential for Golgi biogenesis. Proc Natl Acad Sci USA 101: 1253–1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puxeddu, E. , Uhart, M. , Li, C.C. , Ahmad, F. , Pacheco‐Rodriguez, G. , Manganiello, V.C. , et al (2009) Interaction of phosphodiesterase 3A with brefeldin A‐inhibited guanine nucleotide‐exchange proteins BIG1 and BIG2 and effect on ARF1 activity. Proc Natl Acad Sci USA 106: 6158–6163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramaen, O. , Joubert, A. , Simister, P. , Belgareh‐Touze, N. , Olivares‐Sanchez, M.C. , Zeeh, J.C. , et al (2007) Interactions between conserved domains within homodimers in the BIG1, BIG2, and GBF1 Arf guanine nucleotide exchange factors. J Biol Chem 282: 28834–28842. [DOI] [PubMed] [Google Scholar]
- Renault, L. , Guibert, B. , and Cherfils, J. (2003) Structural snapshots of the mechanism and inhibition of a guanine nucleotide exchange factor. Nature 426: 525–530. [DOI] [PubMed] [Google Scholar]
- Roehl, H.H. , Parsley, T.B. , Ho, T.V. , and Semler, B.L. (1997) Processing of a cellular polypeptide by 3CD proteinase is required for poliovirus ribonucleoprotein complex formation. J Virol 71: 578–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rust, R.C. , Landmann, L. , Gosert, R. , Tang, B.L. , Hong, W. , Hauri, H.P. , et al (2001) Cellular COPII proteins are involved in production of the vesicles that form the poliovirus replication complex. J Virol 75: 9808–9818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salonen, A. , Ahola, T. , and Kaariainen, L. (2005) Viral RNA replication in association with cellular membranes. Curr Top Microbiol Immunol 285: 139–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suhy, D.A. , Giddings, T.H., Jr , and Kirkegaard, K. (2000) Remodeling the endoplasmic reticulum by poliovirus infection and by individual viral proteins: an autophagy‐like origin for virus‐induced vesicles. J Virol 74: 8953–8965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sztul, E. , and Lupashin, V. (2006) Role of tethering factors in secretory membrane traffic. Am J Physiol Cell Physiol 290: C11–C26. [DOI] [PubMed] [Google Scholar]
- Tannous, B.A. , Kim, D.E. , Fernandez, J.L. , Weissleder, R. , and Breakefield, X.O. (2005) Codon‐optimized Gaussia luciferase cDNA for mammalian gene expression in culture and in vivo . Mol Ther 11: 435–443. [DOI] [PubMed] [Google Scholar]
- Teterina, N.L. , Gorbalenya, A.E. , Egger, D. , Bienz, K. , and Ehrenfeld, E. (1997) Poliovirus 2C protein determinants of membrane binding and rearrangements in mammalian cells. J Virol 71: 8962–8972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teterina, N.L. , Levenson, E. , Rinaudo, M.S. , Egger, D. , Bienz, K. , Gorbalenya, A.E. , and Ehrenfeld, E. (2006) Evidence for functional protein interactions required for poliovirus RNA replication. J Virol 80: 5327–5337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ventoso, I. , MacMillan, S.E. , Hershey, J.W. , and Carrasco, L. (1998) Poliovirus 2A proteinase cleaves directly the eIF‐4G subunit of eIF‐4F complex. FEBS Lett 435: 79–83. [DOI] [PubMed] [Google Scholar]
- Verheije, M.H. , Raaben, M. , Mari, M. , Te Lintelo, E.G. , Reggiori, F. , Van Kuppeveld, F.J. , et al (2008) Mouse hepatitis coronavirus RNA replication depends on GBF1‐mediated ARF1 activation. PLoS Pathog 4: e1000088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volpicelli‐Daley, L.A. , Li, Y. , Zhang, C.J. , and Kahn, R.A. (2005) Isoform‐selective effects of the depletion of ADP‐ribosylation factors 1–5 on membrane traffic. Mol Biol Cell 16: 4495–4508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wessels, E. , Duijsings, D. , Lanke, K.H. , Van Dooren, S.H. , Jackson, C.L. , Melchers, W.J. , and Van Kuppeveld, F.J. (2006a) Effects of picornavirus 3A proteins on protein transport and GBF1‐dependent COP‐I recruitment. J Virol 80: 11852–11860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wessels, E. , Duijsings, D. , Niu, T.K. , Neumann, S. , Oorschot, V.M. , De Lange, F. , et al (2006b) A viral protein that blocks Arf1‐mediated COP‐I assembly by inhibiting the guanine nucleotide exchange factor GBF1. Dev Cell 11: 191–201. [DOI] [PubMed] [Google Scholar]
- Wessels, E. , Duijsings, D. , Lanke, K.H. , Melchers, W.J. , Jackson, C.L. , and Van Kuppeveld, F.J. (2007) Molecular determinants of the interaction between coxsackievirus protein 3A and guanine nucleotide exchange factor GBF1. J Virol 81: 5238–5245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, K.F. , Shen, X. , Li, H. , Pacheco‐Rodriguez, G. , Moss, J. , and Vaughan, M. (2005) Interaction of BIG2, a brefeldin A‐inhibited guanine nucleotide‐exchange protein, with exocyst protein Exo70. Proc Natl Acad Sci USA 102: 2784–2789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yalamanchili, P. , Banerjee, R. , and Dasgupta, A. (1997) Poliovirus‐encoded protease 2APro cleaves the TATA‐binding protein but does not inhibit host cell RNA polymerase II transcription in vitro . J Virol 71: 6881–6886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao, X. , Claude, A. , Chun, J. , Shields, D.J. , Presley, J.F. , and Melancon, P. (2006) GBF1, a cis‐Golgi and VTCs‐localized ARF‐GEF, is implicated in ER‐to‐Golgi protein traffic. J Cell Sci 119: 3743–3753. [DOI] [PubMed] [Google Scholar]