

Abstract
A new light-sensitive polymer containing multiple light-sensitive triggering groups along the backbone and incorporating a quinone-methide self-immolative moiety was developed and formulated into nanoparticles encapsulating a model dye Nile Red. Triggered burst-release of the payload upon irradiation and subsequent degradation of the nanoparticles was observed. This system is designed to be versatile where the triggering group can be sensitive to a number of wavelengths.
With the rapid progress of nanotechnology over the past decade, there is growing interest in polymeric biomaterials that can be remotely disassembled in a controlled fashion upon an external stimulus, but otherwise are stable under physiological conditions.1 Various internal and external stimuli, such as pH2, specific enzymes3, temperature4–6, ultrasound7,8 and light9 are being explored. Optical stimulus is especially attractive as it can be remotely applied for a short period of time with high spatial and temporal precision. Near-infrared (NIR) light can penetrate deeper into tissue and has many in vivo applications.10 Despite these advantages, there is a dearth of biomaterials that can efficiently respond to light, especially NIR light.
To increase the efficiency of response we designed a polymer using self-immolative monomers.11–14 In such systems the sensitivity to a stimulus is increased due to a domino effect, where a single triggering event leads to multiple output reactions. However, if the triggering is not very efficient, the polymer scission will be incomplete. Another way to increase the sensitivity and to increase the number of fragmented chains is by introducing multiple light-sensitive triggering groups per polymer chain (Figure 1).
Figure 1.

Degradation of light-sensitive polymer 2 upon irradiation.
Herein, we describe a new light-sensitive degradable polymer containing a quinone-methide self-immolative moiety, which can be triggered to degrade through multiple light-sensitive groups along the backbone. We also demonstrate that nanoparticles formulated from this polymer are capable of releasing their small molecule payload upon irradiation. Its design allows for adaptation to a variety of stimuli responsive triggering groups.
The monomer design is based on the self-immolative quinone-methide system.11–14 The cleavage of the triggering group by light induces cyclization of the diamine spacer, which in turn unmasks an unstable quinone-methide moiety. Incorporation of this moiety into a polymer chain causes degradation of the polymer backbone upon irradiation with light.
Monomer 1 was synthesized according to a previously published procedure11 with slight modifications (see Supporting Information). 4,5-Dimethoxy-2-nitrobenzyl alcohol15 was chosen despite its low two-photon uncaging cross-section (0.01 GM)16 compared to 4-bromo-coumarins (1 GM)17 or fluorene-based systems (5 GM)18, because it is well-studied and readily available, making it a good proof-of-concept photolabile group.
Monomer 1 was copolymerized with adipoyl chloride to yield a regular copolymer. The low molecular weight oligomers were removed by repeated precipitation of the crude polymer with cold ethanol, yielding the final product with a molecular weight of 65,000 Da and PDI of 1.54 (characterized by GPC relative to polystyrene standards) with 44% yield.
Cleavage of the triggering groups by irradiation at 350 nm and 750 nm, via one and two-photon processes respectively, was monitored by observing the changes in the absorbance spectrum of polymer 2 in acetonitrile/H2O (9/1). Upon light exposure, the peak at 346 nm, corresponding to 4,5-dimethoxy-2-nitrobenzyl carbamate decreased, while a new peak at 400 nm appeared, corresponding to the cleaved 4,5-dimethoxy-2-nitrosobenzaldehyde16 (Figures S1, S2). The absorption spectrum remained unchanged after 15 minutes of irradiation with 350 nm light, indicating complete deprotection, while it was necessary to irradiate the system for 5 hours at 750 nm to observe changes in the absorption spectrum, consistent with the low two-photon uncaging cross-section of 4,5-dimethoxy-2-nitrobenzyl group. However, more efficient two-photon uncaging coumarin based groups may be used in place of the nitrobenzyl group and are currently under investigation to make the system more sensitive to NIR light and biologically innocuous.
The degradation of polymer 2 was studied by GPC and proton NMR in acetonitrile/water solutions. The polymer solutions were exposed to UV light (350 nm) for various periods of time and incubated at 37°C. Samples were removed and analyzed. The degree of polymer degradation showed strong dependence on the irradiation time (Figure 2). The initial drop in molecular weight in the first few minutes after UV irradiation is likely to be mostly due to the loss of the triggering groups, while further reduction in molecular weight is due to the cleavage of the polymer backbone as a result of cyclization and elimination reactions within the self-immolative monomer unit. The difference in the degradation degree is especially evident in the samples irradiated for 5 and 15 minutes: more triggering groups are cleaved. Consequently, the polymer chains degrade into smaller fragments. Although the estimated molecular weights of the fragments level off at 20,000 Da the molecular weight of monomer 1 (m/z = 544.19) was estimated by GPC to be 3,500 Da, therefore these fragments may be oligomers. Notably, only a small portion of all the triggering groups needs to be cleaved to induce a reduction in the molecular weight of the polymer.
Figure 2.

(A) Degradation of polymer upon exposure to 350 nm light for defined time periods in acetonitrile/H2O (9/1) and (B) GPC traces of polymer 2 before irradiation (red), upon irradiated for 15 min in acetronitrile/H20/Et3N (9/0.5/0.5) (green) and after 24 hr incubation at 37°C after 15 min irradiation (blue).
The proton NMR of polymer 2 before irradiation in CDCl3 showed all the characteristic peaks and splittings (Figure S6). As expected14, in CD3CN/D2O the NMR peaks broadened (Figure S7). Upon irradiation and incubation at 37°C for 18 hours the NMR peaks corresponding to the benzylic protons of 4,5-dimethoxy-2-nitrobenzyl group disappeared and the remaining peaks shifted accordingly (Figure S8). Sharper monomer peaks, indicating the presence of urea and cresol emerged, overlapping with broader polymer and oligomer peaks.
The cyclization of the diamine linker has been shown to be the rate-determining step of the self-immolation within the quinine-methide unit, and it has been shown to accelerate in the presence of triethylamine.11 Thus, we measured polymer degradation in the presence of triethylamine (Figure S15) and observed an increase in the rate of the polymer degradation (Figure S14).
Two-photon irradiation of polymer 2 for 5 hours showed a similar degree of degradation as 5 minute one-photon irradiation (Figure S16).
To evaluate the properties of the new polymer for controlled light-triggered release, nanoparticles were formulated by the single emulsion method (Figure 3), encapsulating the small hydrophobic molecule dye Nile Red. This small molecule was chosen because of its excellent photostability. The Z-average diameter of the nanoparticles was 170 nm and PDI = 0.191, as determined by dynamic light scattering (DLS).
Figure 3.
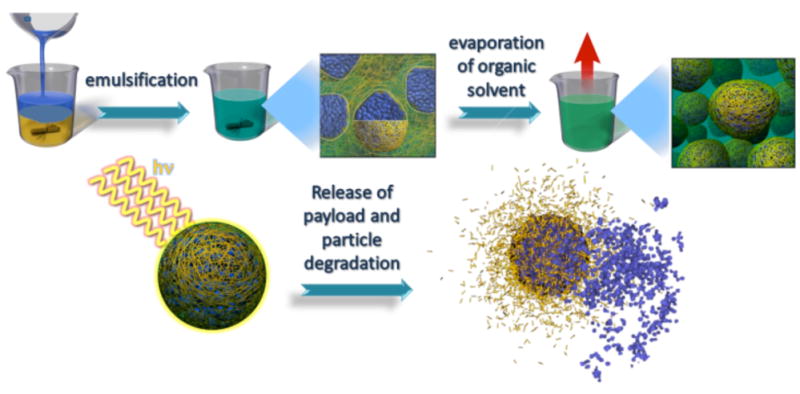
An illustration of the fornulation of nanoparticles, their degradation and light-triggered release of their encapsulated Nile Red payload.
The release of the Nile Red payload upon irradiation was observed by fluorescence spectroscopy. Nanoparticles were redispersed in PBS pH 7.4 and the fluorescence intensity of the suspension was recorded. After irradiation with 350 nm light for 1 min, the fluorescence intensity dropped by 67%, indicating burst release of the dye from the nanoparticles into a more polar medium (Figures 3 and 4).19,20 On the other hand, a suspension of nanoparticles that was not irradiated exhibited unchanged fluorescence intensity over several days. Interestingly, prolonged irradiation of nanoparticles did not result in a further drop of fluorescence signal.
Figure 4.

Fluorescence intensity of Nile Red encapsulated within polymeric nanoparticles and upon irradiation with 350 nm light, monitored at 630 nm with excitation at 540 nm.
Further degradation of nanoparticles of 2 after UV irradiation was observed by DLS at 37°C in PBS buffer at pH 7.4 and pH 10. No particles were detected after 4 days of incubation at pH 10 while in pH 7.4 the particles degraded within 10 days.
We also explored the possibility of triggering the release of Nile Red by NIR light through two-photon absorption. The suspension of nanoparticles in PBS pH 7.4 was irradiated at 750 nm for 20 min intervals followed by 10 min of incubation at 37°C. A gradual decrease in the fluorescence intensity of Nile Red was observed during the 4 hours of irradiation (Figure 5).
Figure 5.

Fluorescence intensity of Nile Red encapsulated within polymeric nanoparticles and after 20 min irradiation increments with 750 nm light, monitored at 630 nm with excitation at 540 nm.
The observation of burst release of Nile Red upon UV irradiation while the polymer degradation is slower suggests the possibility of a secondary mechanism of release. A change in hydrophobicity of the particles upon cleavage of the triggering group may be involved.9 The rapid and efficient unmasking of a large number of the secondary amino groups may make the particles rapidly more permeable to water. This may explain the rapid release of Nile Red upon UV irradiation. However, the two-photon unmasking process is much less efficient which could explain the slower Nile Red release in the NIR two-photon regime. Notably, the final degradation of the nanoparticles is an important property for in vivo biological applications that require materials to degrade into easily excretable fragments.
To rule out the possibility of spontaneous release caused by simple cavitation, poly(lactic-co-glycolic acid) (PLGA) nanoparticles encapsulating Nile Red were formulated by the same method and exposed to UV and NIR light in the same fashion. As expected, no release of Nile Red was observed in this case (Figures S3 and S4).
In conclusion, we have described a novel light-sensitive nanoparticle capable of controlled triggered burst release of small hydrophobic molecules. The versatile design of this system allows the triggering group to be sensitive to internal or remote stimuli. Current efforts are exploring a number of more efficient photo triggers sensitive to a range of wavelengths.
Supplementary Material
Scheme 1.

Synthesis of light-responsive polymer 2.
Acknowledgments
We thank the NIH New Innovator Award (DP20D006499) and the Skaggs School of Pharmacy and Pharmaceutical Sciences for their generous funding of this study.
Footnotes
Supporting Information Available: Experimental details for the synthesis of polymer 2 and formulation of the nanoparticles, absorption spectra of polymer 2 upon photolysis, fluorescence intensity of Nile Red encapsulated in PLGA particles, 1H NMR spectra of polymer 2 before and after irradiation and GPC traces of the degrading polymer 2. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Wang W, Alexander C. Angew Chem Int Ed. 2008;47:7804–7806. doi: 10.1002/anie.200802474. [DOI] [PubMed] [Google Scholar]
- 2.Murthy NXM, Schuck S, Kunisawa J, Shastri N, Frechet JMJ. Proc Natl Acad Sci USA. 2003;100:4995–5000. doi: 10.1073/pnas.0930644100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Veronese FMSO, Pasut G, Mendichi R, Andersson L, Tsirk A, Ford J, Wu G, Kneller S, Davies J, Duncan R. Bioconjugate Chem. 2005;16:775–784. doi: 10.1021/bc040241m. [DOI] [PubMed] [Google Scholar]
- 4.Chung JEYM, Yamato M, Aoyagi T, Sakurai Y, Okano TJ. J Controlled Release. 1999;62:115–127. doi: 10.1016/s0168-3659(99)00029-2. [DOI] [PubMed] [Google Scholar]
- 5.Liu SQ, Tong YW, Yang YY. Biomaterials. 2005;26:5064–5074. doi: 10.1016/j.biomaterials.2005.01.030. [DOI] [PubMed] [Google Scholar]
- 6.Na K, Lee KH, Lee DH, Bae YH. Eur J Pharm Sci. 2006;27:115–122. doi: 10.1016/j.ejps.2005.08.012. [DOI] [PubMed] [Google Scholar]
- 7.Gao ZG, Fain HD, Rapoport N. J Controlled Release. 2005;102:203–222. doi: 10.1016/j.jconrel.2004.09.021. [DOI] [PubMed] [Google Scholar]
- 8.Nelson JL, Roeder BL, Carmen JC, Roloff F, Pitt WG. Cancer Research. 2002;62:7280–7283. [PubMed] [Google Scholar]
- 9.Goodwin AP, Mynar JL, Ma YZ, Fleming GR, Frechet JMJ. J Am Chem Soc. 2005;127:9952–9953. doi: 10.1021/ja0523035. [DOI] [PubMed] [Google Scholar]
- 10.Raghavachari R, editor. Practical Spectroscopy Series. Vol. 25. Marcel Dekker; New York: 2001. Near-Infrared Applications in Biotechnology. [Google Scholar]
- 11.Amir RJ, Pessah N, Shamis M, Shabat D. Angew Chem, Int Ed. 2003;42:4494–4499. doi: 10.1002/anie.200351962. [DOI] [PubMed] [Google Scholar]
- 12.Sagi A, Weinstain R, Karton N, Shabat D. J Am Chem Soc. 2008;130:5434–5435. doi: 10.1021/ja801065d. [DOI] [PubMed] [Google Scholar]
- 13.Weinstain R, Sagi A, Karton N, Shabat D. Chem--Eur J. 2008;14:6857–6961. doi: 10.1002/chem.200800917. [DOI] [PubMed] [Google Scholar]
- 14.DeWit MA, Gillies ER. J Am Chem Soc. 2009;131:18327–18334. doi: 10.1021/ja905343x. [DOI] [PubMed] [Google Scholar]
- 15.Patchornik A, Amit B, Woodward RB. J Am Chem Soc. 1970;92:6333–6335. [Google Scholar]
- 16.Aujard I, Benbrahim C, Gouget M, Ruel O, Baudin JB, Neveu P, Jullien L. Chem Eur J. 2006;12:6865–6879. doi: 10.1002/chem.200501393. [DOI] [PubMed] [Google Scholar]
- 17.Furuta T, Wang SSH, Dantzker JL, Dore TM, Bybee WJ, Callaway EM, Denk W, Tsien RY. Proc Natl Acad Sci USA. 1999;96:1193–1200. doi: 10.1073/pnas.96.4.1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gug S, Bolze F, Specht A, Bourgogne C, Goeldner M, Nicoud JF. Angew Chem, Int Ed. 2008;47:9525–9529. doi: 10.1002/anie.200803964. [DOI] [PubMed] [Google Scholar]
- 19.Sackett DL, Wolff J. Anal Biochem. 1987;167:228–234. doi: 10.1016/0003-2697(87)90157-6. [DOI] [PubMed] [Google Scholar]
- 20.Dutta AK, Kamada K, Ohta K. J Photochem Photobiol, A. 1996;93:57–64. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.