

Abstract
This study explores the general utility of a new class of biosensor that allows one to selectively visualize molecules of a chosen membrane protein that are at the cell surface. These biosensors make use of recently described bipartite fluoromodules comprised of a fluorogen-activating protein (FAP) and a small molecule (fluorogen) whose fluorescence increases dramatically when noncovalently bound by the FAP(1).
Cells that express single-pass recombinant membrane proteins, each presenting a FAP on the exterior face of the plasma membrane and a standard fluorescent protein (EGFP or mRFP) on the interior face, were generated and examined by fluorescence microscopy. In each case, fluorescent signal was observed exclusively at the cell surface when the FAP domain was imaged using membrane-impermeant fluorogen, but was observed in additional intracellular locations when the fluorescent protein domain was imaged. Cells that expressed external N-terminal FAP-fusions to three well-studied human membrane proteins - the β2 adrenergic receptor (β2AR), the insulin-regulated glucose transporter (GLUT4), and the cystic fibrosis transmembrane conductance regulator (CFTR) - were also generated and examined; these too showed fluorescent signal exclusively at the cell surface after exposure to membrane-impermeant fluorogen. Further, when endocytosis of tagged β2AR was stimulated by agonist treatment in the presence of fluorogen, fluorescent signal was seen to transit from the surface to the cell interior. FAP-tagging thus provides a means for selectively visualizing plasma membrane proteins and for monitoring the trafficking of these proteins to and from the cell surface.
Keywords: Reporter protein, Membrane protein, Live cell assay, Cell surface labeling, Green fluorescent protein, GFP, Fluorogen activating protein, FAP, β2AR, GLUT4, CFTR
INTRODUCTION
Plasma membrane proteins play roles in thousands of cellular processes and are the targets of more than half of all therapeutic drugs. Many of these proteins exhibit regulated translocation between the cell surface and the cell interior. For example, most G-protein coupled receptors (GPCRs) and receptor tyrosine kinases (RTKs) are internalized by endocytosis after exposure to agonists (2-3), and numerous ion and metabolite transporters traffic to the membrane in response to particular physiological stimuli (4).
Many methods are available to selectively label particular plasma membrane proteins in living cells with fluorescent molecules so as to study membrane protein trafficking. These include immunofluorescence with specific antibodies or antibodies to epitope tags, labeling of tetracysteine (TC) tagged fusion proteins with FlAsH or ReAsH reagents, covalent attachment of fluorophores using SNAP-tag or Halo Tag fusions, biotinylation of acceptor tagged proteins using purified biotin ligase, and labeling acyl carrier proteins with phosphopantetheine transferases (ACP-PPTase) (5-13). While these approaches are valuable for studies of many kinds, all are limited by the fact that they require multiple wash steps and reagents. The aim of the studies reported here was to explore the experimental utility of a new approach that enables fluorescent labeling of proteins by the addition of a single soluble reagent, with no subsequent wash or separation steps.
Our approach employs the genetically encoded biosensor system recently described by Szent Gyorgyi et al.(1). In this system, fluorescent signal depends on the interaction between a reporter polypeptide and a small molecule (the fluorogen) that is only fluorescent when bound to the reporter. These polypeptides, which were selected from a library of single chain antibodies (scFv's) and improved by directed evolution, were given the name FAPs, for fluorogen activating proteins. The FAP system consists of a genetically encoded FAP and chemical label, the fluorogen. Each FAP has specificity to a particular fluorogen. The HL4-MG FAP is capable of activating malachite green fluorogens, cell impermeable MG-11P and cell permeable MG-ester. HL1.1-TO1 activates the cell impermeable thiazole orange fluorogen, TO1-2P (Table 1)
Table 1.
Properties of FAPs and fluorogens. Fluorogens are derivatives of malachite green (MG) or thiazole orange (TO). FAPs are comprised of hypervariable heavy (H) and light chains (L) joined by a flexible linker. All information taken from (1).
| Molecular Weight (kDa) |
FAP | Fluorogen | Structure |
|---|---|---|---|
| 26.1 | HL4-MG | MG-11P |
|
| 26.1 | HL4-MG | MG-ester |
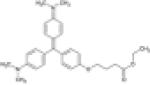
|
| 26.9 | HL1.1-TO1 | TO1-2P |
|
When fused to an extracellular terminus or loop of a plasma membrane protein and visualized using membrane-impermeant fluorogen, FAP reporters provide an opportunity to selectively observe only those protein molecules that are present in the plasma membrane, with molecules elsewhere in the cell remaining dark because they cannot contact the fluorogen.
The recombinant constructs described by Szent Gyorgyi et al. were not fusions to full-length natural proteins; rather they consisted of a FAP moiety anchored at the cell surface by a minimal transmembrane domain. In the experiments reported here, a number of complete proteins with FAP's at their extracellular N-termini were expressed in mammalian cells. Some of these proteins were EGFP or mRFP reporters linked to the FAP by a single transmembrane domain; others were well-characterized multipass plasma membrane proteins. To demonstrate the utility of FAP tags, we studied FAP fusion proteins with relevance to human diseases, GLUT4, CFTR and β2AR. FAP-tagged membrane proteins were visualized selectively at the cell surface using cell impermeable fluorogens, or throughout the endomembrane system using a cell permeable fluorogen. Further, the utility of FAP-tagging for monitoring dynamic processes such as membrane protein trafficking was demonstrated by the observation of agonist stimulated internalization of tagged β2AR.
MATERIALS AND METHODS
Reagents
(−)-Isoproterenol(+)bitartrate and DL-Propranolol HCL were from Sigma-Aldrich (St. Louis, MO). Cellstripper was purchased from Mediatech (Manassas, VA).
Plasmids and retroviral vectors
Expression vector pDisplaySacLac2 was constructed as follows. The SacB gene was PCR-amplified from the vector pDNR-1r (Clontech, Mountain View CA) using primers TATATAGGCCCAGCCGGCCCCACATATACCTGCCGTTCAC and TATATAGGCCCCTGCGGCCACGTCAATGCCAATAGGATATCG. The amplicon was cut with SfiI and cloned into SfiI-digested pDisplayBlue (Szent-Gyorgyi et al, 2008) to produce pDisplaySac. Primers AGAGGATCTGAATGCTGTGG and CTCGAGCTAACGCCACCTGCTGGCATCGTCCAGGCTGTGGACGTGGCTTCTTCTGCCAA were used to generate an amplicon from a pDisplayBlue template that contained the PDGFR transmembrane-domain; the amplicon was cut with BsmI and XhoI and ligated into BsmI/XhoI-digested pDisplaySac. The resulting construct was digested with PflMI. Primers CCACAGCCTGGGTTAGCTCACTCATTAGGCA and CCACCTGCTGGCTAACGCCAGTTTGAGGGGACGACGA were used to generate an amplicon from a pDisplayBlue template that contained the lacZα-complementing fragment, and PflMI-digested amplicon was ligated into the construct to create pDisplaySacLac2.
Retroviral expression vector pBabeSacLac2 was constructed as follows. pBabe-puro-H-Ras-V12 (Addgene, Cambridge MA), which carries an SfiI site upstream of the puromycin resistance gene, was linearized with SfiI, treated with T4 polymerase to create blunt ends, and self-ligated to create a derivative without an SfiI site. This plasmid was digested with BamHI and SalI to produce a recipient fragment. pDisplaySacLac2 was digested with BamHI and XhoI, and a 650bp BamHII/XhoI fragment was recovered and ligated into the recipient fragment to produce pBabeSacLac2.
Generation of vector inserts
SfiI sites were added to the ends of open reading frames by PCR. 5′ primers added the 13-nt sequence GGCCCAGCCGGCC to the extreme 5′ end of the open reading frame (ORF), and 3′ primers added the 13-nt sequence GGCCGCAGGGGCC to the extreme 3′ end of the ORF. Amplicons were cloned into the vectors pCR2.1-TOPO or pCR-Blunt II-TOPO (Invitrogen, Carlsbad CA), sequenced to confirm that no mutations were introduced during amplification, and removed from the vector by SfiI digestion. Molecules were generated in this way for six ORFs: EGFP (template: pStealth,(12)), mRFP (template: pACT-mRFP-Mem, gift of Dr. Y. Saeki, Ohio State University), HL1.1-TO1 (template: pNL6-HL1.1-TO19 (1)), HL4-MG (template pNL6-HL4-MG,(1)), human GLUT4 (template: cDNA clone 726246, Open Biosystems, Huntsville AL), and human CFTR (template: pcDNA3.1 CFTR, gift of Dr. R.Frizzell, University of Pittsburgh. These two amplicons also included the template stop codon; in the other cases a stop codon was not included. A seventh fragment containing a complete ORF and stop codon from the ADRB2 gene, encoding the β2AR receptor (template: fosmid WI2-2202O9/G248P86156H5, BACPAC Resources Center, Oakland CA) was prepared in an equivalent manner, but with the sequence flanked by BsmI sites.
Cell lines
NIH 3T3 cells were obtained from the American Type Culture Collection (Manassas, VA). C2C12 cells were the gift of Dr. P. Campbell, Carnegie Mellon University. HEK 293 cells were the gift of Dr. R. Frizzell, University of Pittsburgh School of Medicine.
Transfection
Transfections were performed using Mirus TransIT®-LT1 Transfection Reagent (Mirus, Madison WI) and pDisplaySacLac2 vectors. For 35mm dishes: DNA (2.5μL, concentration of 1μg/mL) was added to 7.5 μL TransIT®-LT1 in 250 μL of DMEM. Transfection complexes were allowed to form for 30 minutes and subsequently added to cells grown in antibiotic free DMEM with 10% calf serum. Transfection complexes were removed and medium changed after 24 hours. After two or more weeks of growth, stable transfectants were obtained by FACS as described previously (1).
Transduction
Transducing particles for pBabeSacLac2 constructs were generated using the Phoenix Ecotropic Packaging System (Nolan laboratory, Stanford University). Phoenix-Ecotropic cells were plated at 1.3×106 cells/ 75cm2 flask in DMEM with calf serum without antibiotics. pBabeSacLac2 DNA was transfected as described above scaled to surface area. After 24 hours, transfection complexes were removed and replaced with 8mL of DMEM with calf serum and incubated for 48 hours at 32°C/5% CO2. Medium was removed and filtered through Millex-HV 0.45μm syringe filter and flash frozen in liquid nitrogen. Recipient cells were plated at 2×105 cells/ 35mm dish 24 hours prior to transduction. Cells were infected by adding viral supernatant and 6μg/mL of hexadimethrine bromide and incubated for 24 hours at 32°C/5% CO2. Cells were replated in 75cm2 flasks and screened for expression 48 hours later.
Fluorescence microscopy
Cells were grown in DMEM plus 10% calf serum in 23 mm glass-bottom dishes (Mattek, part no p35G-1.5-14-C) and imaged with Carl Zeiss LSM 510 Meta/ UV DuoScan Inverted Spectral Confocal Microscope at 63x objective magnification. For cells expressing FAP HL1.1-TO1, 40nM TO1-2P, a membrane-impermeant fluorogen, was added approximately 5 minutes prior to imaging. For cells expressing HL4-MG, 40nM MG-11P, a membrane-impermeant fluorogen or 40nM MG-ester, a membrane-permeant fluorogen, were added approximately 10 minutes prior to imaging. Times (5 minutes and 10 min) were shown in pilot experiments to be sufficient to reach saturation with regard to signal intensity and location. Excitation and emission wavelengths were 488nm and 505-550nm for EGFP, 561nm and 575-615nm for mRFP, 488nm and 505-550nm for HL1.1-TO1, and 633nm and LP 650nm for HL4-MG. Illumination intensity and detector gain settings on the microscope were held constant for all observations.
β2AR internalization
Imaging was performed using an Olympus IX50 microscope equipped with a spinning disk confocal imaging system (Solamere Technology Group, Salt Lake City, UT). For TO1-2P, excitation was a 488 Argon laser and a 500 nm long pass filter for emission (HQ500LP; Chroma Technologies). NIH 3T3 cells expressing HL1.1-TO1- β2AR were incubated in the presence of 100nM TO1-2P and stimulated with 10μM isoproterenol or untreated (control) for 40 minutes, followed by confocal image acquisition at 37°C with 10% CO2.
RESULTS
Expression of membrane-spanning “dumbbell” proteins with FAP and FP domains
To facilitate the expression of membrane-associated proteins carrying FAP or fluorescent protein domains, we constructed two new expression vectors, the plasmid vector pDisplaySacLac2 and the retroviral vector pBabeSacLac2. In pDisplaySacLac2 transgene expression is driven by the CMV immediate early promoter. In pBabeSacLac2 expression is driven by the MMLV LTR promoter.
Both vectors express a translation unit that includes (from 5′ to 3′) a leader sequence derived from a murine Igκ gene, an HA epitope, a SacB stuffer flanked by SfiI restriction sites, a c-Myc epitope, a BsmI restriction site, a transmembrane domain derived from the human platelet derived growth factor receptor B gene (PDGFRB), and a LacZα stuffer flanked by PflMI restriction sites. Cleavage of the vector with SfiI or PflMI releases the respective stuffer sequences and leaves the same noncomplementary 3′ three-base overhangs. Thus, if one prepares an ORF of interest with appropriate 3-base overhangs, it can be readily cloned between either the SfiI or the PflMI sites in a directed fashion (14). A schematic view of the relevant translation unit in the vectors is shown in Figure 1.
Figure 1.
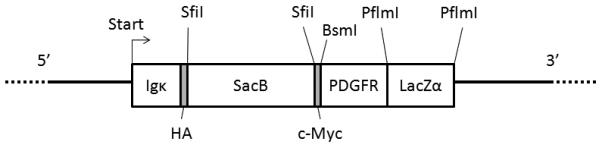
Schematic representation of the expression unit in the pDisplaySacLac2 and pBabe SacLac2 vectors. From 5′ to 3′, the unit begins at ATG start codon followed by the murine Igκ leader sequence and an HA epitope tag. Next, is the bacterial selection marker SacB is flanked by SfiI restriction sites, followed by a c-Myc epitope. This is followed by a membrane spanning segment derived from the human PDGFR gene, and then a LacZα gene flanked by PflmI restriction sites.
ORFs encoding the FAP's HL1.1-TO1 and HL4-MG (Table 1) or the fluorescent proteins EGFP or mRFP were cloned between the SfiI or PflMI sites in pDisplaySacLac2 or pBabeSacLac2 to give “dumbbell” constructs that express recombinant single-pass transmembrane proteins with their N-terminal domains outside the cell and their C-terminal domains inside. NIH 3T3 cells that stably express the constructs were isolated and examined by fluorescence microscopy after fluorogen addition, if appropriate.
Images of cells expressing five different dumbbell proteins are shown in Figure 2. When visualized with membrane-impermeant fluorogens (Table 1), both the FAP-mRFP and FAP-EGFP dumbbells showed signal exclusively at the cell surface (Figure 2A,F). RFP and GFP signal was also present at the cell surface, but in marked contrast to the FAP patterns, much of the signal came from inside the cell. Extensive internal signal was also observed for cells expressing the HL4-MG-EGFP dumbbell that were incubated with membrane-permeant fluorogen (Figure 2J).
Figure 2.

Fluorescence images of dumbbell constructs in NIH 3T3 cells. The left column shows representations of the topology of each fusion protein with respect to the plasma membrane, where top is extracellular and bottom is intracellular. The next three columns show green, red and merge fluorescence channels. The last column shows histograms generated from the fluorescence images by plotting the pixel intensity across a specified region (indicated by the white line). Image analysis was performed using the Image J software by specifying a linear region of interest and using the plot profile operation. Panels (A-H) are FAP-FP expressing cells in the presence of impermeable fluorogen. Cell permeable fluorogen labeling of FAP-FP cells are shown in panels I-L. Scale bars represent 10μm.
Expression of human membrane proteins with N-terminal FAP-tags
Beta2 adrenergic receptor
The human β-2 adrenergic receptor (β2AR, encoded by the gene ADRB2) is a G-protein coupled receptor (GPCR) with an extracellular N-terminus, seven transmembrane domains, and a cytoplasmic C-terminus. β2AR responds to the hormones epinephrine and norepinephrine as well as a large number of known agonists and antagonists, including several important drugs used to treat asthma and other respiratory conditions(15). To create vectors that express β2AR with FAP or FP domains at their N-termini, an ORF encoding human β2AR was ligated into the BsmI site of pBabeSacLac2, and ORFs encoding HL1.1-TO1, HL4-MG, or EGFP were ligated between the SfiI sites in place of the SacB stuffer.
Fluorescence micrographs of cells expressing β2AR fusion proteins are shown in Figure 3. When imaged after exposure to nonpermeant fluorogen, HL4-MG-β2AR cells showed signal exclusively at the cell surface (Figure 3A). In contrast, cells expressing HL4-MG-β2AR that were exposed to membrane-permeant fluorogen (Figure 3B), and cells expressing EGFP- β2AR protein, (Figure 3C) showed extensive internal signal as well as surface labeling.
Figure 3.

Fluorescence images of HL4-MG and EGFP fusions to transmembrane proteins. Left column shows representations of fusion protein topology in the plasma membrane, where top is extracellular and bottom is intracellular. (A-C) NIH 3T3 cells expressing human β2AR with FAP or EGFP fused to the N-terminus. (D-F) C2C12 cells expressing human GLUT4 with extracellular FAP or EGFP, plus new transmembrane domain, fused to the N-terminus. (G-I) HEK 293 cells expressing CFTR with FAP or EGFP, plus new transmembrane domain, fused to the N-terminus. . (A, D, G) Cells are imaged in the presence of impermeable fluorogen, MG-11P. (B, E, H) Membrane permeant fluorogen, MG-ester, labels intracellular fusion proteins. Scale bars represent 10μm.
Glucose transporter 4
Human glucose transporter 4 (GLUT4, encoded by the SLC2A4 gene) is a type II 12-pass transmembrane protein. GLUT4 is the primary insulin-responsive glucose transporter in muscle and fat cells and plays a central role in the pathology of Type 1 and Type 2 diabetes (16).
To create vectors that express recombinant GLUT4 with extracellular N-terminal FAP or FP domains, an ORF encoding mature human GLUT4 was ligated between the PflMI sites of pBabeSacLac2, and ORFs encoding HL1.1-TO1, HL4-MG, or EGFP were ligated between the SfiI sites. The proteins expressed from these recombinant genes were expected to have 13 transmembrane domains with the FAP or FP exposed extracellularly.
Fluorescence micrographs of C2C12 cells expressing GLUT4 constructs are shown in Figure 3. When imaged after exposure to nonpermeant fluorogen, FAP-GLUT4 cells showed signal exclusively at the cell surface (Figure 3D). And, as with the β2AR expressing cells, FAP-GLUT4 expressing cells exposed to membrane-permeant fluorogen (Figure 3E), and EGFP-GLUT4 cells (Figure 3F), showed extensive internal signal as well as surface labeling.
Cystic fibrosis transmembrane conductance regulator
The cystic fibrosis transmembrane conductance regulator (encoded by the CFTR gene) is a type II 12-pass membrane protein of the ABC transporter class. Mutations in CFTR are responsible for the human genetic disease, cystic fibrosis (17).
To create vectors that express CFTR with extracellular FAP or FP domains at their N-termini, an ORF encoding mature human CFTR was ligated between the PflMI sites of pBabeSacLac2, and ORFs encoding HL1.1-TO1, HL4-MG, or EGFP were ligated between the SfiI sites. These vector constructs were expected to express proteins with topologies identical to those of the GLUT4 constructs.
Fluorescence micrographs of HEK 293 cells expressing FAP or EGFP CFTR constructs are shown in Figure 3. When imaged after exposure to nonpermeant fluorogen, the FAP construct showed signal exclusively at the cell surface (Figure 3G). And, as with the β2AR and GLUT4 expressing cells, FAP-CFTR expressing cells exposed to membrane-permeant fluorogen (Figure 3H) showed internal signal as well as surface labeling, just as did cells expressing EGFP-CFTR (Figure 3I).
β2AR internalization in response to agonist
For many GPCR's including β2AR, stimulation with agonist leads to G-protein activation in seconds followed by internalization of the receptor over the following minutes through a process known as receptor down-regulation (18). As shown in Figure 4, treatment of FAP-tagged β2AR with a standard dose of the agonist isoproterenol in the presence of membrane-impermeant fluorogen led to signal internalization, indicating that FAP-tagged proteins can be used in concert with membrane-impermeant fluorogens to monitor membrane protein trafficking in living cells.
Figure 4.

Internalization of FAP-tagged β2AR in response to agonist treatment. A: Cells expressing HL1.1- β2AR were incubated in membrane impermeant fluorogen (100 nM TO1-2P) for 40 minutes and imaged by confocal microscopy. B: Cells expressing HL1.1- β2AR were incubated in membrane impermeant fluorogen (100 nM TO1-2P) plus 10 μM isoproterenol for 40 minutes and imaged by confocal microscopy. Scale bars represent 10μm.
DISCUSSION
FAP reporters enable selective visualization of surface-exposed protein
The need to precisely identify the subcellular location of proteins is increasingly important in fluorescence imaging. Subtle changes in protein localization can have a profound effect on its function, especially those located at the plasma membrane. Small changes in protein trafficking like insertion into the plasma membrane, may induce widespread changes on the cell such as transporting ions or glucose across the cell or propagating signals in response to extracellular stimuli. Traditional fluorescence imaging methods are unable to resolve proteins that are at the cell surface. GFP-tagged proteins provide a well-established approach for tracking the movements of specific proteins, including membrane proteins in living cells by light microscopy (12,19-20). However, because GFP-tagged proteins exhibit fluorescence regardless of cellular location, this approach is limited in its ability to distinguish surface-localized protein from internal protein. Indeed, even the most sensitive generally available light microscopic method, total internal reflection (TIRF) microscopy, cannot distinguish protein molecules that are in the plasma membrane from those that are nearby - e.g., in vesicles within ~100nm of the surface.(21) This limitation has inspired the creation of alternative methods to facilitate protein localization. Chemical labeling of genetically encoded tags with small-molecules allows for spatial and temporal control over protein visualization with the ability to utilize chemically diverse and environmentally sensitive probes. This approach has been demonstrated by intracellular labeling methods such as the biarsenical systems and TC motif (FlAsH and ReAsH), fluorescent O6-benzylguanine labeling of hAGT fusions (Halo tag) and fluorescently labeled ligands for FKBP12 fusions(7-8,22). Techniques for selective chemical labeling of cell surface proteins, ACP-PPTase and biotin-ligase based methods have proven useful, however these labels are irreversible and require multiple wash steps with prolonged incubation times(5-6). By using FAP-based imaging, we have combined the ability to visualize dynamic biological processes with the specificity of chemical labeling methods.
Each of the FAP-tagged proteins expressed in this study was visualized exclusively at the cell surface when cultures were exposed to membrane-impermeant fluorogen. Signal appeared inside the cell only when tagged protein bound to fluorogen was internalized by endocytosis (Figure 4). In contrast, when cells expressing FAP-tagged membrane proteins were exposed to membrane-permeant fluorogen (MG-ester, Table 1), fluorescence was observed not just at the cell surface but also in the nuclear membrane, the Golgi, and in vesicles of various sizes. Given that the nuclear membrane is contiguous with the membrane of the endoplasmic reticulum, it is likely that the signal observed at the nuclear membrane represents fusion-protein molecules that have arrived there by lateral diffusion from the ER membrane. Signal in the Golgi likely represents proteins passing through that organelle on their way to the cell surface. And the punctate signal seen throughout the cytoplasm likely represents exocytic and/or endocytic vesicles carrying fusion proteins to or from the plasma membrane.
The orientation of the fusion proteins used in this study is such that the FAP domain projects into the lumen of any intracellular membrane-bounded compartment that it may occupy. Thus MG-ester molecules would need to transit both the plasma membrane and the organelle membrane to reach the FAP and yield fluorescent signal. The observation of such signal implies to us that after entering the cytoplasm, MG-ester molecules are not immediately converted to charged membrane-impermeable molecules by the action of cellular esterases. Our results with the EGFP-HL4 dumbbell protein provide particularly strong support for this interpretation, in that the recombinant protein shows essentially the same location pattern whether visualized with MG-ester or by EGFP fluorescence (Figure 2K).
We have also shown that the SacLac modular cloning system facilitates generation of FAP tagged fusion proteins. β2AR, GLUT4 and CFTR have been successfully tagged and demonstrated that they can be detected at the cell surface. Furthermore, the dynamic trafficking of proteins in living cells was shown by agonist induced endocytosis of HL1.1-TO1- β2AR. These FAP fusion proteins will provide a platform for further studies to examine trafficking properties specific to each protein.
Our results provide a basis for developing new and superior assays that detect translocation of proteins to or from the cell surface in response to specific stimuli. Thus for example, to assay for protein internalization, one might incubate cells in membrane-impermeant fluorogen before and after stimulation and look for a reduction in fluorescent signal consequent to stimulation. Or to assay for protein delivery to the plasma membrane, one might incubate cells in membrane-impermeable fluorogen and look for an increase in signal consequent to stimulation. Data could be acquired by fluorescence microscopy or by higher throughput means such as flow cytometry or fluorimetry in a microwell format.
ACKNOWLEDGEMENTS
We thank Drs. Ray Frizzell, James Fitzpatrick and Marcel Bruchez for materials and useful discussions. This work was supported by grant U54 RR022241 from the National Center for Research Resources, National Institutes of Health.
Supported in part by grant U54 RR022241 from the National Center for Research Resources, National Institutes of Health.
REFERENCES
- 1.Szent-Gyorgyi C, Schmidt BF, Creeger Y, Fisher GW, Zakel KL, Adler S, Fitzpatrick JA, Woolford CA, Yan Q, Vasilev KV. Fluorogen-activating single-chain antibodies for imaging cell surface proteins. Nat Biotechnol. 2008;26(2):235–40. doi: 10.1038/nbt1368. and others. [DOI] [PubMed] [Google Scholar]
- 2.Kohout TA, Lefkowitz RJ. Regulation of G protein-coupled receptor kinases and arrestins during receptor desensitization. Mol Pharmacol. 2003;63(1):9–18. doi: 10.1124/mol.63.1.9. [DOI] [PubMed] [Google Scholar]
- 3.Bache KG, Slagsvold T, Stenmark H. Defective downregulation of receptor tyrosine kinases in cancer. EMBO J. 2004;23(14):2707–12. doi: 10.1038/sj.emboj.7600292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ma D, Jan LY. ER transport signals and trafficking of potassium channels and receptors. Curr Opin Neurobiol. 2002;12(3):287–92. doi: 10.1016/s0959-4388(02)00319-7. [DOI] [PubMed] [Google Scholar]
- 5.Chen I, Howarth M, Lin W, Ting AY. Site-specific labeling of cell surface proteins with biophysical probes using biotin ligase. Nat Methods. 2005;2(2):99–104. doi: 10.1038/nmeth735. [DOI] [PubMed] [Google Scholar]
- 6.George N, Pick H, Vogel H, Johnsson N, Johnsson K. Specific labeling of cell surface proteins with chemically diverse compounds. J Am Chem Soc. 2004;126(29):8896–7. doi: 10.1021/ja048396s. [DOI] [PubMed] [Google Scholar]
- 7.Griffin BA, Adams SR, Tsien RY. Specific covalent labeling of recombinant protein molecules inside live cells. Science. 1998;281(5374):269–72. doi: 10.1126/science.281.5374.269. [DOI] [PubMed] [Google Scholar]
- 8.Keppler A, Gendreizig S, Gronemeyer T, Pick H, Vogel H, Johnsson K. A general method for the covalent labeling of fusion proteins with small molecules in vivo. Nat Biotechnol. 2003;21(1):86–9. doi: 10.1038/nbt765. [DOI] [PubMed] [Google Scholar]
- 9.Keppler A, Pick H, Arrivoli C, Vogel H, Johnsson K. Labeling of fusion proteins with synthetic fluorophores in live cells. Proc Natl Acad Sci U S A. 2004;101(27):9955–9. doi: 10.1073/pnas.0401923101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Los GV, Encell LP, McDougall MG, Hartzell DD, Karassina N, Zimprich C, Wood MG, Learish R, Ohana RF, Urh M. HaloTag: a novel protein labeling technology for cell imaging and protein analysis. ACS Chem Biol. 2008;3(6):373–82. doi: 10.1021/cb800025k. others. [DOI] [PubMed] [Google Scholar]
- 11.Roche KW, Standley S, McCallum J, Dune Ly C, Ehlers MD, Wenthold RJ. Molecular determinants of NMDA receptor internalization. Nat Neurosci. 2001;4(8):794–802. doi: 10.1038/90498. [DOI] [PubMed] [Google Scholar]
- 12.Telmer CA, Berget PB, Ballou B, Murphy RF, Jarvik JW. Epitope tagging genomic DNA using a CD-tagging Tn10 minitransposon. Biotechniques. 2002;32(2):422–4, 426. doi: 10.2144/02322rr04. 428-30. [DOI] [PubMed] [Google Scholar]
- 13.Wang Q, Khayat Z, Kishi K, Ebina Y, Klip A. GLUT4 translocation by insulin in intact muscle cells: detection by a fast and quantitative assay. FEBS Lett. 1998;427(2):193–7. doi: 10.1016/s0014-5793(98)00423-2. [DOI] [PubMed] [Google Scholar]
- 14.Telmer CA, Retchless AC, Kinsey AD, Conley Y, Rigatti B, Gorin MB, Jarvik JW. Detection and assignment of mutations and minihaplotypes in human DNA using peptide mass signature genotyping (PMSG): application to the human RDS/peripherin gene. Genome Res. 2003;13(8):1944–51. doi: 10.1101/gr.995103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Moore RH, Khan A, Dickey BF. Long-acting inhaled beta2-agonists in asthma therapy. Chest. 1998;113(4):1095–108. doi: 10.1378/chest.113.4.1095. [DOI] [PubMed] [Google Scholar]
- 16.Hou JC, Pessin JE. Ins (endocytosis) and outs (exocytosis) of GLUT4 trafficking. Curr Opin Cell Biol. 2007;19(4):466–73. doi: 10.1016/j.ceb.2007.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ameen N, Silvis M, Bradbury NA. Endocytic trafficking of CFTR in health and disease. J Cyst Fibros. 2007;6(1):1–14. doi: 10.1016/j.jcf.2006.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McLean AJ, Milligan G. Ligand regulation of green fluorescent protein-tagged forms of the human beta(1)- and beta(2)-adrenoceptors; comparisons with the unmodified receptors. Br J Pharmacol. 2000;130(8):1825–32. doi: 10.1038/sj.bjp.0703506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Groves JT, Parthasarathy R, Forstner MB. Fluorescence imaging of membrane dynamics. Annu Rev Biomed Eng. 2008;10:311–38. doi: 10.1146/annurev.bioeng.10.061807.160431. [DOI] [PubMed] [Google Scholar]
- 20.Verveer PJ, Wouters FS, Reynolds AR, Bastiaens PI. Quantitative imaging of lateral ErbB1 receptor signal propagation in the plasma membrane. Science. 2000;290(5496):1567–70. doi: 10.1126/science.290.5496.1567. [DOI] [PubMed] [Google Scholar]
- 21.Axelrod D. Total internal reflection fluorescence microscopy in cell biology. Traffic. 2001;2(11):764–74. doi: 10.1034/j.1600-0854.2001.21104.x. [DOI] [PubMed] [Google Scholar]
- 22.Robers M, Pinson P, Leong L, Batchelor RH, Gee KR, Machleidt T. Fluorescent labeling of proteins in living cells using the FKBP12 (F36V) tag. Cytometry A. 2009;75A(3):207–24. doi: 10.1002/cyto.a.20649. [DOI] [PubMed] [Google Scholar]