

Abstract
Although it is well known that multiple genes may influence prostate cancer risk, most current efforts at identifying prostate cancer risk variants rely on single-gene approaches. In previous work using mostly single-gene approaches, we observed significant associations (P < 0.05) for 6 of 46 polymorphisms in five genes in a Swedish prostate cancer case-control study population. We now report on the higher-order gene-gene interactions among those 46 genetic variants and the combined effect of the six polymorphisms with significant main effects for association with prostate cancer risk in 795 controls and 1,461 cases. Classification and regression tree analysis was used to evaluate higher-order gene-gene interactions. No interactions were confirmed by the result from logistic regressions. For the combined analysis, we tested the hypothesis that individuals carrying multiple copies of risk variants are at increased risk for prostate cancer. Individuals carrying more than eight copies of any risk variant were almost twofold more likely to get prostate cancer (OR = 1.99, P = 0.0014). A significant trend relationship was observed (P < 0.0001). In the present study, additive effects but not multiplicative effects among these six polymorphisms with significant main effects were observed.
Keywords: interaction, prostate cancer, association, SNPs
INTRODUCTION
Results from a large number of case-control studies, twin studies, and segregation analyses have consistently suggested that there is a genetic susceptibility to prostate cancer [1]. Sequence variants in multiple genes have been hypothesized to account for this genetic susceptibility. Most current efforts in evaluating these prostate cancer risk variants rely on single-gene approaches; however, it is more important to understand the combined effect of multiple risk variants on prostate cancer susceptibility, because many genes in multiple biological pathways interact to regulate tumor initiation and progression. Simultaneous evaluation of multiple risk variants likely improves the ability and accuracy in assessing individual risk to prostate cancer.
We previously performed a literature review of prostate cancer association studies and selected 46 polymorphisms reported to influence prostate cancer risk for evaluation in a large Swedish population-based case-control prostate cancer population [2]. Our initial effort aimed to systematically study the individual effect of these variants on prostate cancer risk in this homogeneous study population; from that work we found significant associations (P < 0.05) for 6 of the 46 polymorphisms, in five genes. For the present study, we were interested in exploring the gene–gene interactions, as well as the combined effects of multiple risk variants.
First, we used classification and regression tree (CART) analysis to evaluate higher-order gene–gene interactions and used logistic regression analysis to further examine the interactions among the markers selected by CART analysis. Second, we examined the combined effects of the six significant polymorphisms. Our hypothesis is that individuals carrying multiple copies of risk variants have increased risk for prostate cancer.
MATERIALS AND METHODS
Study Population
The study design and description were described in detail elsewhere [3]. Briefly, this is a large-scale population-based case-control study in Sweden, named CAPS (CAncer Prostate in Sweden). Prostate cancer patients were identified and recruited from regional cancer registries in Sweden. The inclusion criterion for cases was pathological or cytological verified adenocarcinoma of the prostate, diagnosed between July 1, 2001 and September 30, 2002. Control subjects were randomly selected from the continuously updated Swedish Population Registry and frequency matched according to age (within 5 years) and geographic origin of the cases. In total, 1,470 cases and 866 controls were recruited. Among them, DNA samples and questionnaires were available for 1,461 cases and 795 controls. Characteristics of the study population are summarized in Table I of Lindström et al [2]. All participants gave full informed consent.
Variants associated with prostate cancer risk
The method for selecting reported prostate cancer risk variants was described in detail elsewhere [2]. Briefly, we systematically searched PubMed for all prostate cancer association studies published in English before March 2004 using the following combinations of key words: (prostate, cancer, polymorphism), (prostate, cancer, association, genetic), (prostate, cancer, SNP), (prostate, cancer, sequence, variants), (prostate, cancer, association), (prostate, cancer, microsatellite). Only studies with a minimum of 100 cases and 100 controls were included. We identified a total of 79 polymorphisms that had been reported at least once to be significantly associated with prostate cancer risk, among which 46 were genotyped in CAPS. The remaining variants were excluded for various reasons, including (i) findings were based on a relatively small sample size (n = 11) or (ii) the single-nucleotide polymorphisms (SNPs) had already been evaluated in CAPS (n = 5), they were monomorphic (n = 2), had an unknown genomic location (n = 1), or had genotyping difficulty (n = 14). The distribution of allele frequencies among these 46 polymorphisms (35 SNPs and 11 microsatellite markers) in cases and controls was presented in Table 3 in Lindström et al. [2].
Genotyping Methods
The genotyping methods for these 46 variants were described in detail elsewhere [2]. Briefly, a MassARRAY system (Sequenom, San Diego, CA) was used for SNP genotyping, and an ABI 3730 DNA analyzer (Applied Biosystems, Foster City, CA) was used to identify nucleotide repeat variants.
Statistical Analysis
The quality control and single marker association analyses have been described in detail [2]. Briefly, all markers were checked for consistency with Hardy–Weinberg equilibrium using the GENETICS package implemented in the R programming language. All of the microsatellite markers have been categorized into two distinct alleles, as detailed previously [2]. To confirm the individual effect of each marker on prostate cancer risk, logistic regression with adjustment of age and geographical region was applied.
Higher-order gene-gene interactions
We further explored the higher-order of interactions of markers with prostate cancer risk using a data mining approach, classification and regression tree analysis (CART) [4]. To minimize the effect of missing data, individuals with 5% missing data were deleted, which left 1,918 individuals in this analysis. CART is a data-mining tool for tree-structured nonparametric data analysis based on binary recursive partitioning methodology; it partitions data into terminal nodes, which are relatively homogeneous with regard to the disease status.
Briefly, the methodology consists of three parts. First, a maximal tree is grown that overfits the data. Instead of trying to decide whether a node is a terminal or not, CART will continue splitting until some prespecified rules are satisfied; in our case, we limited the minimum number of individuals in the terminal node to 10. This tree is called a maximal tree. Second, the overfitting tree is pruned back to a sequence of subtrees. The pruning process begins after the maximal tree is achieved and continues all the way back to the root node. Weakest nodes, those which add the least to the overall accuracy of the tree, will be pruned away first. Third, an optimal tree is determined through pruning, one that minimizes the relative cost (average misclassification error/0.50 estimated by 10 fold of cross-validation) while maintaining the model parsimony. Under parsimony, the simplest possible model is desirable. Thus, a 1SE rule is used to select the final tree; that is, the smallest tree is chosen as the final tree among trees with relative cost within one standard error of the minimum relative cost. To minimize the effect of variability due to the random division data into 10 parts, we repeated the 10-fold cross-validation 10 times and averaged the results. A commercially available CART program was used in this analysis [5].
To gain insight into the potential mechanism for the increased prostate cancer risk with markers selected by CART analysis, we also used the logistic regression analysis to test interactions among the markers in the final model selected by CART. Each marker with three possible genotypes or categories was regrouped into two categories, based on the CART classification results.
Combined analysis of multiple markers
The present hypothesis is that individuals carrying multiple copies of risk variants have increased risk for prostate cancer. We focused on just the six polymorphisms that had previously shown significant single-gene association [2]. These included the CAG repeat in exon 1 of the androgen receptor gene (AR), one SNP in the CYP17A1 gene, two SNPs in the SRD5A2 gene, deletion of the GSTT1 gene, and one SNP (IVS5-59C>A) in MSR1. The risk allele was identified from single SNP association analysis. The allele with main effect showing a positive association with prostate cancer risk (i.e., odds ratio OR > 1) was selected as the risk allele. We also confirmed these selected risk alleles with the literature. To test the hypothesis, we used logistic regression analysis to detect the association. Two modeling approaches were used: (i) modeling the number of individuals carrying risk alleles and (ii) modeling the percentage of individuals carrying risk alleles.
With the six polymorphisms, individuals may carry up to 12 risk alleles. From our data, the numbers ranged from 0 to 11. We collapsed 0, 1, 2, and 3 risk alleles as one lowest group and 9, 10, and 11 risk alleles as one highest group, to avoid the sparse cell problem. Missing data (3–13%) were treated as noncarried risk alleles. The total number of individuals carrying risk alleles was treated as an ordinal variable in the regression model.
Each individual may have a different number of polymorphisms genotyped due to missing data. Instead of estimating the absolute number of carried risk variants, we also estimated the percentage of carried risk alleles of these six markers for subjects. The percentage is defined as the total number of carried risk alleles across the six markers divided by twice the number of nonmissing markers. We then used logistic regression to test for association between the percentage and prostate cancer risk. SAS software (version 9.1; SAS Institute, Cary, NC) was used for all of these analyses.
RESULTS
The chromosomal location and risk allele frequency in cases and controls for the full set of 46 polymorphisms is given in Table 3 of Lindström et al. [2]. The six significant polymorphisms selected for the present study are given in Table 1. All markers were in Hardy–Weinberg equilibrium among cases and controls.
Table 1.
Six markers included in the analysis of prostate cancer risk
| Gene | Polymorphism | Chromosomal location | Variation | Risk allele in the “first half” data |
||
|---|---|---|---|---|---|---|
| Allele | Frequency in cases | Frequency in controls | ||||
| ARa | rs4045402 | X, Xq11.2-q12 | ≤22/>22 | >22 | 0.44 | 0.37 |
| CYP17 | rs743572 | 10, 10q24.3 | A/G | A | 0.62 | 0.59 |
| GSTT1 | del. of gene | 22, 22q11.23 | A/- | A | 0.66 | 0.61 |
| MSR1 | IVS5-59C>A | 8, 8p22 | C/A | C | 0.98 | 0.96 |
| SRD5A2 | rs676033 | 2, 2p23 | C/T | T | 0.36 | 0.32 |
| SRD5A2 | rs523349 | 2, 2p23 | C/G | G | 0.34 | 0.32 |
Higher-order gene-gene interactions
Figure 1 shows the final model selected by CART after pruning. For each node, the splitting criteria, the number and percent of cases (class = 1) and controls (class = 0) are presented. The final model includes a combination of three markers in three genes: GSTT1 deletion, SRD5A_rs523349, and NAT1_rs15561. The first split is according to GSTT1 deletion genotype. Those who carry one copy or two copies of deletions were further split according to SRD5A2_rs523349 genotypes. Those who carry the SRD5A2_rs523349 genotypes CG or GG were further split based on NAT1 rs15561 genotype (terminal nodes 2 and 3). The training accuracy using this model was 57.94% for cases and 53.68% for controls; the testing accuracy, however, was only 53.87% for cases and 49.62% for controls.
Fig. 1.
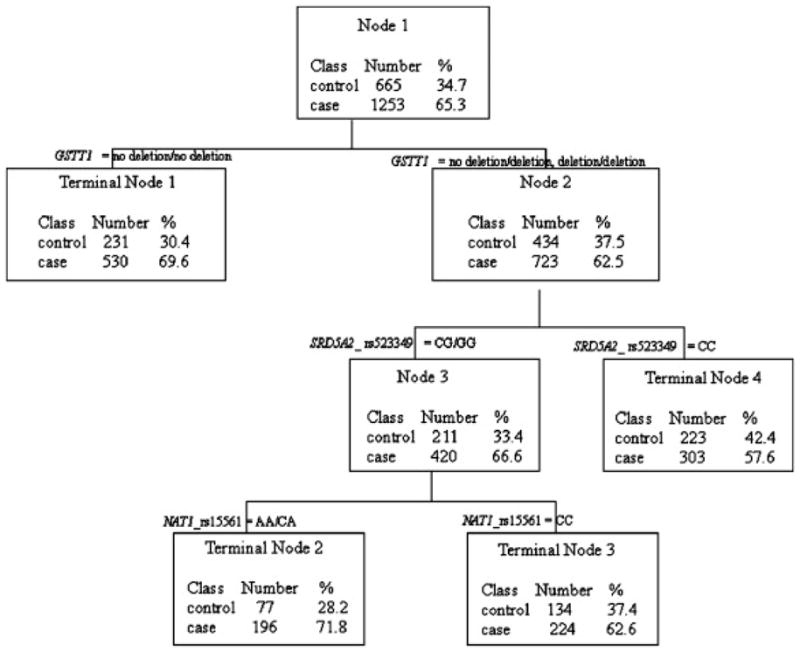
Classification and regression tree (CART) analysis: results of higher-order interactions among 46 polymorphisms.
We tested the multiplicative interaction among the three markers identified from CART (GSTT1 deletion, SRD5A_rs523349, and NAT1_rs15561) using logistic regressions. After adjusting for age and geographic region, none of the three-way or two-way interactions showed significant results. The final model included the main effects for the three markers. Marginal significance for GSTT1 deletion and significance for SRD5A_rs523349 and NAT1_rs15561 were observed (P= 0.0623, 0.0251, and 0.0402, respectively, after adjusting for age, geographic region, and the other two polymorphisms) (data not shown).
Combined analysis of multiple markers
Table 2 presents the combined effect of risk variants. Individuals with fewer than four high-risk alleles were combined as the reference group. Individuals carrying more than four risk alleles had a higher risk of getting prostate cancer, compared with the reference group. Carrying additional high-risk alleles was associated with an 11% increase in risk. A significant linear trend relationship was observed (P < 0.0001). As expected, modeling percentage also showed the trend effect. Individuals in the fourth quartile of the percentage were 1.61 times more likely to have prostate cancer than those in the first quartile (OR = 1.61, P < 0.0001).
Table 2.
Combined effects of risk variants
| Model | Case/control | OR (95% CI)a | p-valuea |
|---|---|---|---|
| Number of risk allele | |||
| 0–3 | 151/115 | Reference | - |
| 4 | 183/127 | 1.16 (0.83 – 1.63) | 0.3762 |
| 5 | 290/156 | 1.47 (1.08 – 2.02) | 0.0160 |
| 6 | 305/155 | 1.58 (1.15– 2.17) | 0.0044 |
| 7 | 244/121 | 1.60 (1.15 – 2.22) | 0.0057 |
| 8 | 168/74 | 1.82 (1.26 – 2.64) | 0.0015 |
| 9–11 | 118/46 | 1.99 (1.30 – 3.04) | 0.0014 |
| Per allele | 1.11 (1.05 – 1.16) | < 0.0001 | |
| P-value for trend | < 0.0001 | ||
| Percentage of carrying risk allele | |||
| Q1 (≤ 0.417) | 464/319 | Reference | - |
| Q2 (> 0.417 and ≤ 0.5) | 346/176 | 1.35 (1.07 – 1.71) | 0.0114 |
| Q3 (> 0.5 and ≤ 0.6) | 259/133 | 1.36 (1.05 – 1.76) | 0.0190 |
| Q4 (> 0.6 ) | 390/166 | 1.61 (1.28 – 2.04) | < 0.0001 |
| P-value for trend | < 0.0001 | ||
Adjusted for age and geographical region using the logistic regression
DISCUSSION
CART analysis did not reveal higher-order interactions among the 46 markers. The prediction accuracy of the best model using three markers was only 53.87% and 49.62% for cases and controls, respectively. The follow-up logistic regression analysis of these three markers also did not show any multiplicative interaction effects among the three markers in the best model identified by CART. This suggests a potential lack of multiplicative interaction effects among the polymorphisms studied. We further tested the multiplicative interaction effects among the six significant polymorphisms identified by Lindström et al. [2], using logistic regression. No significant multiplicative interactions were observed. However, while testing whether individuals carrying multiple copies of risk variants have increased risk for prostate cancer (additive effect), we found a clear trend effect in the test, and carrying an additional high-risk allele was associated with an 11% increase in risk.
The odds ratios for the single polymorphism associations in Lindström et al. [2] were generally small (~1.20). In this study, the odds ratios for subjects carrying five, six, seven, eight, and more than eight risk alleles all had OR > 1.47, compared with those who carried fewer than four risk alleles. There was no significant association between those who carried four risk alleles and those who carried fewer than four risk alleles. The results from percentage modeling analysis also had higher significant associations (OR > 1.35). These were qualitatively higher than the odds ratios in the single marker association. Since the etiology of a complex disease like prostate cancer usually involves multiple factors, such as multiple genes in multiple biological pathways and environmental factors, the effect of each individual polymorphism is likely to be moderate [6] and [7]. Thus, a single polymorphism may have limited predictive value in assessing prostate cancer risk. A combined effect of multiple risk variants may give more precise delineation of risk groups and may suggest future directions for association studies [8].
This population-based case-control study has several strengths, including the large sample size and well-characterized phenotype. It is a relatively homogeneous population. All the polymorphisms have been studied elsewhere. It is an ideal sample for replication studies. We are also aware of limitations of this study. First, the high-risk allele we selected may not be accurate in the absence of knowledge of the biological background. If the selection of high-risk alleles is incorrect, the results will be greatly affected and the interpretations will be incorrect as well. Second, the way we calculated the percentage assumes that each variant contributes the same effect on prostate cancer risk. This may not reflect the true biological effect of each gene. It may be more appropriate to use a reasonable weight in calculating the percentage by incorporating the known functional impacts of each variant.
In summary, the result of gene–gene interactions using CART was not confirmed by logistic regression. The combined analyses show that individuals carrying multiple copies of risk variants have increased risk for prostate cancer. Thus, additive effects but not multiplicative effects among these genes were observed in the study.
Acknowledgments
The authors thank all study participants in the Cancer Prostate in Sweden study (CAPS). We thank Ulrika Lund for coordinating the study at Karolinska Institute, and also all urologists who recruited their patients to this study and provided clinical data to the national registry of prostate cancer. We thank Karin Andersson, Susan Okhravi-Lindh, Gabriella Thorén-Berglund, and Margareta Åswärd at the Regional Cancer Registries in Umeå, Uppsala, Stockholm-Gotland, and Lindköping. In addition, we thank Sören Holmgren and the personnel at the Medical Biobank in Umeå for skillfully handling the blood samples. This study has been supported by grants from the Swedish Cancer Foundation and a Spear grant from the Umeå University Hospital, Umeå, Sweden. This study was also funded in part by U.S. National Cancer Institute grants to J.X. (CA95052, CA105055, and CA106523).
References
- 1.Schaid DJ. The complex genetic epidemiology of prostate cancer. Hum Mol Genet. 2004;13(Spec1):R103–R121. doi: 10.1093/hmg/ddh072. [DOI] [PubMed] [Google Scholar]
- 2.Lindström S, Zheng SL, Wiklund F, Jonsson BA, Adami HO, Bälter KA, Brookes AJ, Sun J, Chang BL, Liu W, Li G, Isaacs WB, Adolfsson J, Grönberg H, Xu J. Systematic replication study of reported genetic associations in prostate cancer: strong support for genetic variation in the androgen pathway. Prostate. 2006;66:1729–1743. doi: 10.1002/pros.20489. [DOI] [PubMed] [Google Scholar]
- 3.Zheng SL, Augustsson-Bälter K, Chang B, Hedelin M, Li L, Adami HO, Bensen J, Li G, Johnasson JE, Turner AR, Adams TS, Meyers DA, Isaacs WB, Xu J, Grönberg H. Sequence variants of toll-like receptor 4 are associated with prostate cancer risk: results from the Cancer Prostate in Sweden Study. Cancer Res. 2004;24:2918–2922. doi: 10.1158/0008-5472.can-03-3280. [DOI] [PubMed] [Google Scholar]
- 4.Breiman L, Friedman JH, Stone CJ, Olshen RA. Wadsworth statistics/probability series. Chapman & Hall/CRC; Boca Raton: 1984. Classification and regression trees. [Google Scholar]
- 5.Steinberg D, Colla P. CART—classification and regression trees. Salford Systems; San Diego: 1997. [Google Scholar]
- 6.Moore JH. The ubiquitous nature of epistasis in determining susceptibility to common human diseases. Hum Hered. 2003;56:73–82. doi: 10.1159/000073735. [DOI] [PubMed] [Google Scholar]
- 7.Thornton-Wells TA, Moore JH, Haines JL. Genetics, statistics and human disease: analytical retooling for complexity. Trends Genet. 2004;20:640–647. doi: 10.1016/j.tig.2004.09.007. [DOI] [PubMed] [Google Scholar]
- 8.Wu X, Gu J, Grossman HB, Amos CI, Etzel C, Huang M, Zhang Q, Millikan RE, Lerner S, Dinney CP, Spitz MR. Bladder cancer predisposition: a multigenic approach to DNA-repair and cell-cycle-control genes. Am J Hum Genet. 2006;78:464–479. doi: 10.1086/500848. [DOI] [PMC free article] [PubMed] [Google Scholar]